






Inflammation-associated genomic instability in cancer
Review Article
Khian Hong Pua, Chen Li Chew, David Philip Lane & Vinay Tergaonkar
Genome Instability & Disease 1, 1–9(2020)
Abstract
Inflammation is essential in protecting us against foreign pathogens. However when left unchecked, it becomes chronic inflammation, a potential precursor to carcinogenesis. While inflammatory signalling provides pro-survival stimuli, it also causes genomic instability and allows mutant cells to escape cell cycle arrest and apoptosis. This occurs through the release of reactive oxygen/nitrogen species (ROS/RNS), the increased expression of activation-induced cytidine deaminase (AID), inhibition of p53 function and the reactivation of TERT expression. Because chronic inflammation can ultimately lead to genomic instability, there is a need to target chronic inflammation, through the suppression of effector pathways with biologics or small molecules. More recently, therapies have also focused on stimulating physiological resolution, which offers a promising, tissue-specific alternative. As we better understand the triggers of chronic inflammation, therapies can be more specific, without compromising the overall functions of our immune system.
Introduction
Inflammation is the body’s natural response to bacterial or viral infection (Medzhitov and Horng 2009). While acute inflammation provides a critical defence against foreign organisms for survival and healing, it becomes chronic when unchecked. Chronic inflammation is known to drive the development of diseases including cancers like prostate cancer, hepatocellular carcinoma and colon cancer. This largely stems from cytokines, chemokines, growth factors and other immune regulating proteins produced during chronic inflammation (Medzhitov and Horng 2009). Not only do these factors provide pro-survival stimuli, but they also can lead to DNA damage and instability, which promote carcinogenesis (Irelan et al. 2007; Li Chew et al. 2015; Siveen et al. 2014; Wang et al. 2014).
In this review, we will provide a brief introduction to the various inflammatory pathways, and focus on how chronic inflammation leads to genomic instability. Finally, we will also discuss strategies to target genomic instability caused by chronic inflammation.
Pathways in chronic inflammation and how they lead to genomic instability
Introduction to inflammatory pathways
Inflammation is the body’s intrinsic response to invading pathogens. The three main inflammatory pathways include the NF-κB, JAK-STAT and MAPK signalling pathways.
NF-κB signalling
The two component arms of NF-κB signalling—the canonical and non-canonical pathways (Fig. 1a, b), are both crucial in the inflammatory response. Activation of the canonical pathway involves the engagement of cytokine receptors such as the TNF receptor (TNFR) and innate immune receptors such as Toll-like receptors (TLR) (Chew et al. 2018; Correa et al. 2005) (Fig. 1b). This leads to the consequent activation of IκB kinase (IKK) complex, which phosphorylates IκBα, triggering its ubiquitin-dependent degradation and the consequent release of p50-RelA heterodimers into the nucleus, where they activate the transcription of NF-κB target genes (Fig. 1b) (Chew et al. 2018).
Fig. 1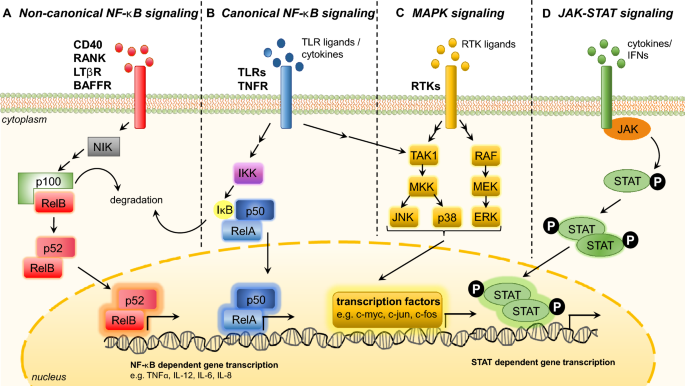
Major signalling pathways regulating inflammation. a Non-canonical nuclear factor-κB (NF-κB) signalling is activated through the ligands of CD40, RANK, BAFFR and LTβR. Receptor activation stabilizes NF-κB-inducing kinase (NIK), which phosphorylates p100, leading to its subsequent processing to p52. The p52-RelB heterodimer then translocate into the nucleus to activate transcription of target genes. b Canonical nuclear factor-κB (NFκB) pathway is activated by tumor necrosis factor receptors (TNFR) or toll-like receptors (TLR), which leads to an activation of IKK. IKK phosphorylates IκB which consequently dissociates from the p50/RelA complex and is degraded. Nuclear translocation of the p50/RelA heterodimer leads to expression of inflammatory target genes. c Mitogen-activated protein kinase (MAPK) starts with the activation of receptor tyrosine kinases by RTK ligands. The subsequent activation of MAPK kinase kinases like TAK1 and RAF lead to activation of MAPK kinases (e.g. MKK/MEK), which result in the phosphorylation and activation of the MAPK extracellular signal-regulated kinase-1 (ERK1), c-Jun N-terminal kinase (JNK) and p38. The subsequent phosphorylation of downstream proteins (e.g. c-myc) leads to increased expression of inflammation target genes. d JAK-STAT signalling is activated by various cytokines and interferons, which results in receptor dimerization, JAK tyrosine kinase auto phosphorylation, phosphorylation of receptors and of recruited STAT proteins. Dimerization and subsequent nuclear translocation of phosphorylated STAT molecules take place, activating the transcription of inflammatory target genes
Full size imageOn the other hand, the non-canonical NF-κB pathway is activated through the ligands of CD40, RANK, BAFFR, Fn-14 and LTβR (Sun 2011). Receptor activation leads to the degradation of TRAF3 ubiquitin ligase, leading to the stabilization of NF-κB-inducing kinase (NIK). NIK is critical in p100 phosphorylation and subsequent processing to p52. The p52-RelB heterodimer then translocates into the nucleus to activate transcription of target genes (Fig. 1a) (Cildir et al. 2016).
MAPK signalling
MAPKs include p38, JNK and ERK which are serine–threonine kinases activated by a variety of extracellular and intracellular stimuli (Kim and Choi 2010). The binding of TNF-α or lipopolysaccharide (LPS) activates MAP kinase kinase kinases (MAP3Ks) (e.g., TAK1), which go on to activate MAP kinase kinases (MAP2Ks) (e.g., MKK4), leading to the subsequent activation of JNK and p38 MAP kinase (Kim and Choi 2010) (Fig. 1). The ERK signalling starts with receptor tyrosine kinase (RTK) dimerization and subsequent phosphorylation of its cytoplasmic domains. This recruits Grb2 and SOS, and activates Ras GTPase, which triggers the sequential phosphorylation of Raf, MEK and ERK. Phosphorylated ERK translocates to the nucleus where it phosphorylates transcription factors such as c-myc (Fig. 1c) (Kim and Choi 2010).
JAK-STAT signalling
The JAK-STAT pathway is activated by receptor dimerization, upon binding of cytokines and Type I and Type II IFNs (Aaronson and Horvath 2002). This activates receptor-associated JAK tyrosine kinases, resulting in self-phosphorylation and phosphorylation of the receptors. STATs are consequently recruited and phosphorylated by JAKs. The phosphorylated STAT molecules then dimerize and translocate into the nucleus to activate the transcription of target genes (Shuai and Liu 2003) (Fig. 1d).
The aberrant activation of these pathways, in particular the NFκB pathway, has been implicated in the development of cancers including breast cancer and glioblastoma (Li et al. 2015; Shin et al. 2014), providing evidence that inflammation plays a causal role in the progression of cancer. While pro-inflammatory pathways can provide pro-survival stimuli, they also cause genomic instability and mutations, which contribute to carcinogenesis.
Inflammation and genomic instability
During chronic inflammation, there is sustained release of reactive oxygen/nitrogen species, increased AID expression, inhibition of p53 and reactivation of TERT expression. These factors, acting alone or in combination, contribute to genomic instability.
Inflammation and the release of reactive oxygen/nitrogen species (ROS/RNS)
Inflammation results in the release of reactive oxygen/nitrogen species (ROS/RNS) like nitric oxide (NO) from epithelial and immune cells such as neutrophils or macrophages, which can cause damage to genomic material (Fig. 2a). ROS/RNS results in the formation of mutagenic lesions like 8-oxo-7, 8-dihydro-2′-deoxyguanosine (8-oxoDG) and 8-nitroguanine (Kawanishi et al., 2017). For example, 8-nitroguanine is thermodynamically unstable, and can lead to single base substitutions, in particular G:C to T:A transversions (Kawanishi et al. 2017).
Fig. 2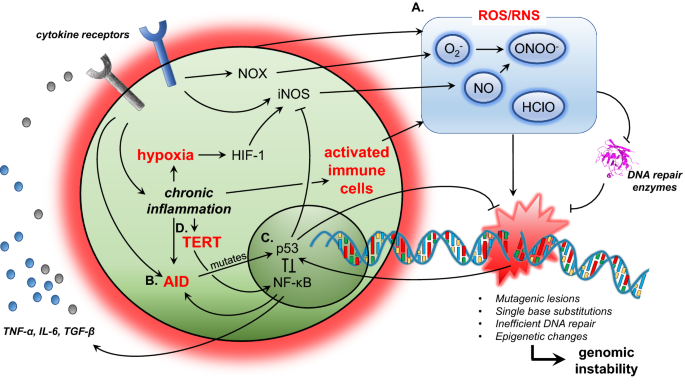
Mechanisms of inflammation-associated genomic instability. a Release of reactive oxygen/nitrogen species (ROS/RNS). Activated immune cells, NOX or iNOS expression and hypoxia can lead to the release of ROS/RNS. Not only do ROS/RNS lead to mutagenic lesions in the DNA, they also inhibit DNA repair enzymes, leading to genomic instability. b Aberrant AID expression during chronic inflammation could potentially lead to mutations in p53, which maintains genomic stability. c Chronic inflammation induces the expression of both NF-κB and p53. NF-κB signalling generally opposes p53’s function to maintain genomic stability. d The reactivation of TERT expression by NF-κB signalling prevents replicative senescence, which allows for tumorigenic cells to survive and proliferate. Red text depicts major mechanisms that lead to genomic instability
Full size imageThe infectious agents Helicobacter pylori, hepatitis B virus, human papillomavirus, Epstein–Barr virus, Schistosoma haematobium and Opisthorchis viverrini have been classified by the International Agency for Research on Cancer (IARC) as group 1 carcinogens. These infections have been correlated with the expression of inducible nitric oxide synthase (iNOS) and subsequent 8-nitroguanine expression (Kawanishi et al. 2016).
Inflammation, particularly through the NFκB pathway, results in the release of cytokines like TNFα and IL6 (Fig. 2a). TNFα is shown in mouse knockout models to induce NAD(P)H oxidase (NOX) in tumour cells (Fig. 2a) (Oshima et al. 2014). NOX results in the generation of oxides in cancer cells, which reacts with NO to form peroxynitrites, leading to DNA damage and instability (Kawanishi et al. 2017). Further, TNFα and IL6 have been shown to induce iNOS expression (Kawanishi et al. 2017).
Inflammation can also lead to hypoxia, which increases the expression of hypoxia-inducible factor-1 (HIF-1) (Fig. 2a) (Kawanishi et al. 2017). HIF-1 promotes the expression of iNOS, which has been associated with increased 8-nitroguanine accumulation in cells of patients with malignant fibrous histiocytoma (Hoki et al. 2007).
The release of RNS can inhibit certain DNA repair enzymes. S-nitration at the active cysteine of O-6-methylguanine-DNA methyltransferase (MGMT) has been shown to inhibit MGMT in mouse hepatocytes (Fig. 2a) (Wei et al. 2011).
Hypochlorous acid, generated by activated neutrophils during inflammation, is a ROS (Jiang et al. 2003). In rats where inflammation was induced, elevated hypochlorous acid levels resulted in the formation of 5-chlorocytosines (5ClC), which undergo chloramine conversion and/or deamination to 5-chlorouracils, which can serve as a marker of DNA damage (Jiang et al. 2003). Not only does 5ClC lead to epigenetic changes, it also has been associated with C–T transitions in in vitro mutagenesis assays conducted in E. coli (Fedeles et al., 2015).
Inflammation and AID
Activation-induced cytidine deaminase (AID) deaminates cytidine. This results in base-excision or mismatch repair, which leads to mutations and consequent genomic instability (Kumar et al. 2014). While this process is important in B-cell somatic hypermutation and class switch recombination, deregulation of AID expression, especially under chronic inflammation, can lead to cancer (Kumar et al. 2014).
Under normal physiological conditions, expression of AID is restricted to B cells undergoing class-switch recombination. The constitutive expression of AID in mice results in T-cell lymphoma, micro-adenoma in lung alveoli (Okazaki et al., 2007) and hepatocellular carcinoma (Endo et al., 2008). Further, chronic inflammation in hepatocellular carcinoma and colorectal cancer has been shown to lead to the secretion of cytokines, which induce AID expression that leads to p53 mutations (Fig. 2b) (Endo et al. 2008; Kou et al. 2007). TGF-β expression is associated with liver inflammatory conditions like chronic hepatitis or liver cirrhosis, while TNF-α expression is increased in chronic inflammatory bowel diseases (Endo et al. 2008; Kou et al. 2007). TGF-β stimulation in human liver hepatocytes and TNF-α stimulation in human colonic epithelial cells have been shown to upregulate AID expression, which is associated with consequent mutations in TP53 in human HepG2 (liver) and LoVo (colon) cells (Endo et al. 2008; Kou et al. 2007).
p53 in inflammation
The tumor suppressor protein p53 is critical in the sensing and maintenance of genetic stability (Lane and Levine 2010). DNA damage activates p53, which results in apoptosis or cell cycle arrest (Huang et al. 1996). The simultaneous induction of both p53 and NFκB has been shown to occur under certain conditions like DNA damage or TNFα stimulation (Wang et al. 1999; Wu and Lozano 1994). p53 and NFκB have largely opposing functions in the promotion of cell survival, and this opposition becomes particularly pertinent in chemotherapy, where genetic damage induces the apoptosis of cancer cells via p53 (Fig. 2) (Tergaonkar et al. 2002). Inhibition of NFκB through overexpressing IκBα in a xenograft model or the genetic deletion of Ikk1/2 in mouse embryonic fibroblasts re-sensitizes cells to chemotherapeutic agents (Tergaonkar et al. 2002; Wang et al. 1999).
Further, competition between p53 and RelA for the limited pool of transcriptional coactivators p300/CBP allow for them to modulate each other’s activities (Webster and Perkins 1999). On a different level, while IKK2 is involved in NFκB activation, it has been shown to phosphorylate p53, which then targets it for ubiquitination by β-TrCP1 (Xia et al. 2009).
In chronic inflammation, the general opposition to p53’s function exacerbates the effects of mutagenic ROS/RNS and AID, preventing the apoptosis of mutant cells which are potentially tumorigenic.
Inflammation and telomerase
Telomerase is an enzyme which maintains telomere length, allowing cells to bypass replicative senescence (Greider and Blackburn 1989). The reactivation of telomerase in cancer cells can also result in non-canonical effects, which promote cancer cell survival (Akincilar et al. 2015; Khattar et al. 2016). While TERT is a direct transcriptional target of MYC in tumors, TERT also stabilizes DNA binding of MYC and regulates its degradation in human Burkitt lymphoma cells, contributing to cancer cell survival (Koh et al. 2015).
Telomerase has also been shown to bind to NFκB to regulate the expression of cytokines like IL6 and TNFα, which drives inflammation (Fig. 2) (Ghosh et al. 2012). In a feed-forward positive loop, inflammatory pathways have in turn been shown to regulate the expression of telomerase. Non-canonical NFκB signaling has also been shown to drive the expression of TERT in glioblastoma (Li et al. 2015), while RAS-ERK signaling has been shown to activate TERT promoter in human melanomas where BRAF is mutated (Li et al. 2016).
With the reactivation of TERT, cells which would normally undergo replicative senescence can now bypass cell cycle arrest. This, coupled with the non-canonical effects of TERT on cancer cells, allow for tumorigenic cells to survive and proliferate.
Targeting chronic inflammation which leads to genomic instability
There are two broad principles governing therapeutic approaches to chronic inflammation: (1) Suppression of effector pathways; (2) Stimulation of physiological resolution (Schett and Neurath 2018).
Suppressing effector pathways
Proinflammatory effector cytokines
One way of targeting cytokines like TNF-α and IL-6 would be through monoclonal antibodies or using soluble receptors against these cytokines (Fig. 3a). Approved monoclonal antibody (mAb) therapies against TNF-α include Infliximab, Adalimumab, Certolizumab pegol and Golimumab (Croft and Siegel 2017). Soluble receptors approved include Etanercept, which is a TNFR2-Fc fusion protein that antagonizes TNF-α function (Croft and Siegel 2017). mAbs antagonizing IL-6 function include Siltuximab (against IL-6) (van Rhee et al. 2010) and Tocilizumab (against IL6 receptor) (Fig. 3a) (Choy et al. 2002).
Fig. 3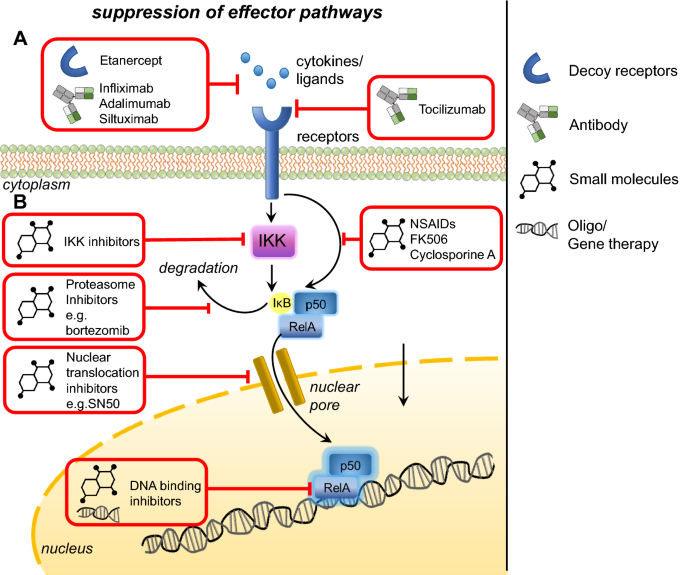
Therapies targeting effector pathways in inflammatory signalling. a Targeting proinflammatory effector cytokines through antibodies or soluble receptors against these cytokines and their receptors. b Targeting proinflammatory signalling pathways with small molecules against kinases, proteasomes, nuclear translocation or DNA binding
Full size imageProinflammatory signalling pathways
Small molecules inhibiting proinflammatory signalling and cytokine effector pathways represent an alternative therapeutic option (Fig. 3b). Inhibition of protein kinases can be achieved through adenosine triphosphate (ATP) analogs (e.g., β-carboline against IKK), allosteric inhibition (e.g. BMS-345541 against IKK) or gene therapy (e.g. dominant negative kinases) (Gupta et al. 2010). Small molecules like cytosine arabinoside have been used to activate protein phosphatase 2A and consequently lead to the dephosphorylation of IKKβ (Sreenivasan et al. 2003). Proteasome inhibitors like Bortezomib prevent IκBα degradation and hence suppress NFκB activation (Fig. 3b) (Sunwoo et al. 2001). Small molecules can also inhibit nuclear translocation of signalling molecules (e.g. SN50 against NFκB p50) (Fig. 3b) (Lin et al. 1995), or inhibit post-translational modifications of signalling molecules (e.g. Anacardic acid which inhibits p65 acetylation) (Sung et al. 2008). Small molecules can also inhibit NFκB binding to DNA promoter regions, for example sesquiterpene lactones (Garcia-Pineres et al. 2001) or decoy oligonucleotides which sequester NFκB (Morishita et al. 1997) (Fig. 3b). For a comprehensive review on small molecule inhibition of the NFκB pathway, please refer to Gupta et al. (2010).
More generally, anti-inflammatory drugs like non-steroid anti-inflammatory drugs (NSAIDs) and immunosuppressive agents have also been developed to inhibit NFκB activation. NSAIDs like aspirin inhibit IκBα phosphorylation and hence subsequent NFκB activation (McDade et al. 1998) (Fig. 3). Immunosuppressants, including immunophilins like cyclosporine A and FK506, inhibit calcineurin, which consequently suppress NFκB activation (Liu et al. 1991) (Fig. 3). Current research on immunophilins focus on target identification of alternative immunosuppressive macrocyclic compounds like Sanglifehrin A (Pua et al. 2017) to identify proteins which could be potential therapeutic targets. Further, labs are also working to synthesize novel macrocyclic libraries in vitro, which could be screened for novel compounds that can target the immune system in a specific manner with minimal side effects.
Inflammatory effector molecules mediating genomic instability
Therapies could potentially target inflammatory effector molecules which mediate genomic instability (e.g., iNOS and AID), although such an approach might potentially affect the overall immune and physiological systems.
While there are no drugs in the clinic which specifically inhibit AID activity, AID expression can be suppressed by DNMT inhibitors like 5-aza-deoxycytidine in human B-cell lines (Tsai et al. 2013). Iron has also been shown to inhibit AID activity and consequent B-cell class-switch recombination in mouse splenic B cells (Li et al. 2012). There are current efforts to identify potential small molecules which can inhibit AID catalytic activity (Alvarez-Gonzalez et al. 2018), which could prove to be a valuable therapeutic agent if we can target its action to cells or tissues where there is undesired AID activity.
Small molecule inhibitors of iNOS are comprised of mimics of arginine and of interactors of cofactors tetrahydrobiopterin, heme, calmodulin or Flavin (Vitecek et al. 2012). These include N-[[3-(aminomethyl)phenyl]methyl]-ethanimidamide (1400 W) (Garvey et al. 1997) and tilarginine (L-NMMA) (Aisaka et al. 1989). However, these inhibitors are not specific towards the NOS isoforms, and could lead to cardiovascular and other systemic effects (Vitecek et al. 2012).
Over the past decade, p53 targeting is primarily focused on pharmacological targeting of the MDM2/X-p53 axis. Clinical trials are ongoing for molecules developed to restore p53’s tumour suppressor activity via modulation of its negative regulator, the E3 ubiquitin ligase, MDM2/X, particularly for oncological indications (Tisato et al. 2017). Reactivators of wild-type p53 include the cis-imidazolines, belonging to the “nutlins” family of compounds, piperidinone-derived AMG232, as well as the stapled peptide, ALRN-6924, which concomitantly targets both MDM2 and MDMX (Tisato et al. 2017). Given the complexity of the relationship between p53 and inflammation, it remains to be seen if direct targeting of p53 would lead to sustained anti-inflammatory therapeutic benefits.
While anti-inflammatory approaches are effective, there is an indiscriminate suppression of the overall immune system, which would increase susceptibility to infections. For example, AID deficiency in mice has been associated with apoptosis of germinal center B cells and autoimmune disease (Hsu et al. 2011; Li et al. 2012; Tan et al. 2018). Further, there could be off-target effects, for example, liver toxicity as a result of cyclosporine A (Hu et al. 2014). An immune response could also develop against biologics (antibodies or fusion proteins) used in therapy. In addition, there is a lifetime dependence on these therapies, as they do not promote physiological resolution (Schett and Neurath 2018). Thus, in recent years, people are looking increasingly to inflammation resolution as a viable alternative therapeutic option.
Stimulating physiological resolution
To encourage resolution of inflammation, we could promote cessation of neutrophil influx through lipid mediators like prostaglandin D2 and lipoxin A4 (LXA4), which downregulate chemokine receptors like CXCR2, rendering neutrophils unresponsive to neutrophil activating substances such as leukotriene B4 (Schett and Neurath 2018).
Another approach could be to promote death and removal of dead neutrophils with death ligands including TRAIL or FasL, or TGFβ that can induce apoptosis of neutrophils, which are subsequently cleared by macrophages (Schett and Neurath 2018).
Lastly, we know that M2-like or pro-resolution macrophages facilitate the inflammation resolution process through phagocytosis of pathogens and apoptotic cells, release of lipids which facilitate resolution, and expression of anti-inflammatory receptors like TGFR2 (Schett and Neurath 2018). Due to the varying metabolic requirements of M1 (proinflammatory) versus M2-like macrophages, manipulation of metabolite availability could potentially alter the function of macrophages. For example, itaconate treatment of human macrophages has been shown to inhibit the production of proinflammatory cytokines like IL1β and TNF (Mills et al. 2018). Cytokines like IL4, IL13 and TGFβ have also been shown to trigger a pro-resolution switch in macrophages (Doherty et al. 1993; Gong et al. 2012; Stein et al. 1992).
While the promotion of inflammation resolution is promising, the ability to promote inflammation resolution in a tissue-specific manner by precisely targeting specific pathways and molecules would represent the next frontier in inflammation resolution.
Conclusion
While inflammation is important in defending our system from foreign pathogens, chronic inflammation can be an undesirable outcome of unchecked pro-inflammatory signalling. Research has established a link between inflammation and the pathophysiology of cancer, and this stems largely from the genomic instability that results from inflammatory cytokines and signalling. Inflammation also leads to a suppression of cell cycle arrest and apoptosis, further allowing for the proliferation of mutant cells. There is, thus, a need to therapeutically target chronic inflammation, either through the suppression of effector pathways or through the promotion of inflammation resolution. Nonetheless, there remains a desperate need to better understand the drivers of chronic inflammation, and how these differ from acute inflammation. This would help us to design better therapies which can specifically target chronic inflammation.
References
Aaronson, D. S., & Horvath, C. M. (2002). A road map for those who don’t know JAK-STAT. Science,296, 1653–1655.
Aisaka, K., Gross, S. S., Griffith, O. W., & Levi, R. (1989). NG-Methylarginine, an inhibitor of endothelium-derived nitric oxide synthesis, is a potent pressor agent in the guinea pig: Does nitric oxide regulate blood pressure in vivo? Biochemical and Biophysical Research Communications,160, 6.
Akincilar, S. C., Low, K. C., Liu, C. Y., Yan, T. D., Oji, A., Ikawa, M., et al. (2015). Quantitative assessment of telomerase components in cancer cell lines. FEBS Letters,589, 974–984.
Alvarez-Gonzalez, J. A., Maul, R., Kohli, R. M., & Gearhart, P. J. (2018). Small molecule inhibitors of Activation-induced deaminase. Journal of Immunology,200(48), 18.
Chew, C. L., Conos, S. A., Unal, B., & Tergaonkar, V. (2018). Noncoding RNAs: master regulators of inflammatory signaling. Trends in Molecular Medicine,24, 66–84.
Choy, E. H., Isenberg, D. A., Garrood, T., Farrow, S., Ioannou, Y., Bird, H., et al. (2002). Therapeutic benefit of blocking interleukin-6 activity with an anti-interleukin-6 receptor monoclonal antibody in rheumatoid arthritis: a randomized, double-blind, placebo-controlled, dose-escalation trial. Arthritis and Rheumatism,46, 3143–3150.
Cildir, G., Low, K. C., & Tergaonkar, V. (2016). Noncanonical NF-kappaB signaling in health and disease. Trends in Molecular Medicine,22, 414–429.
Correa, R. G., Matsui, T., Tergaonkar, V., Rodriguez-Esteban, C., Izpisua-Belmonte, J. C., & Verma, I. M. (2005). Zebrafish IkappaB kinase 1 negatively regulates NF-kappaB activity. Current Biology,15, 1291–1295.
Croft, M., & Siegel, R. M. (2017). Beyond TNF: TNF superfamily cytokines as targets for the treatment of rheumatic diseases. Nature Reviews Rheumatology,13, 217–233.
Doherty, T. M., Kastelein, R., Menon, S., Andrade, S., & Coffman, R. L. (1993). Modulation of murine macrophage function by IL-13. The Journal of Immunology,151, 10.
Endo, Y., Marusawa, H., Kou, T., Nakase, H., Fujii, S., Fujimori, T., et al. (2008). Activation-induced cytidine deaminase links between inflammation and the development of colitis-associated colorectal cancers. Gastroenterology,135, 889–898. (898 e881-883).
Fedeles, B. I., Freudenthal, B. D., Yau, E., Singh, V., Chang, S. C., Li, D., et al. (2015). Intrinsic mutagenic properties of 5-chlorocytosine: A mechanistic connection between chronic inflammation and cancer. Proceedings of the National Academy of Sciences USA,112, E4571–E4580.
Garcia-Pineres, A. J., Castro, V., Mora, G., Schmidt, T. J., Strunck, E., Pahl, H. L., et al. (2001). Cysteine 38 in p65/NF-kappaB plays a crucial role in DNA binding inhibition by sesquiterpene lactones. Journal of Biological Chemistry,276, 39713–39720.
Garvey, E. P. O. J. A., Furfine, E. S., Kiff, R. J., Laszlo, F. W. B. J. R., & Knowles, R. G. (1997). 1400 W is a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide synthase in Vitro and in Vivo. Journal of Biological Chemistry,272, 5.
Ghosh, A., Saginc, G., Leow, S. C., Khattar, E., Shin, E. M., Yan, T. D., et al. (2012). Telomerase directly regulates NF-kappaB-dependent transcription. Nature Cell Biology,14, 1270–1281.
Gong, D., Shi, W., Yi, S. J., Chen, H., Groffen, J., & Heisterkamp, N. (2012). TGFbeta signaling plays a critical role in promoting alternative macrophage activation. BMC Immunology,13, 31.
Greider, C. W., & Blackburn, E. H. (1989). A telomeric sequence in the RNA of tetrahymena telomerase required for telomere repeat. Nature,337, 7.
Gupta, S. C., Sundaram, C., Reuter, S., & Aggarwal, B. B. (2010). Inhibiting NF-kappaB activation by small molecules as a therapeutic strategy. Biochimica et Biophysica Acta,1799, 775–787.
Hoki, Y., Murata, M., Hiraku, Y., Ma, N., Matsumine, A., Uchida, A., et al. (2007). 8-Nitroguanine as a potential biomarker for progression of malignant fibrous histiocytoma, a model of inflammation-related cancer. Oncology Reports,18, 5.
Hsu, H. C., Yang, P., Wu, Q., Wang, J. H., Job, G., Guentert, T., et al. (2011). Inhibition of the catalytic function of activation-induced cytidine deaminase promotes apoptosis of germinal center B cells in BXD2 mice. Arthritis and Rheumatism,63, 2038–2048.
Hu, G., Wang, K., Groenendyk, J., Barakat, K., Mizianty, M. J., Ruan, J., et al. (2014). Human structural proteome-wide characterization of Cyclosporine A targets. Bioinformatics,30, 3561–3566.
Huang, L., Clarkin, K. C., & Wahl, G. M. (1996). Sensitivity and selectivity of the DNA damage sensor responsible for activating p53-dependent G1 arrest. Proceedings of the National Academy of Sciences USA,93, 6.
Irelan, J. T., Murphy, T. J., DeJesus, P. D., Teo, H., Xu, D., Gomez-Ferreria, M. A., et al. (2007). A role for IkappaB kinase 2 in bipolar spindle assembly. Proceedings of the National Academy of Sciences USA,104, 16940–16945.
Jiang, Q., Blount, B. C., & Ames, B. N. (2003). 5-Chlorouracil, a marker of DNA damage from hypochlorous acid during inflammation. A gas chromatography-mass spectrometry assay. Journal of Biological Chemistry,278, 32834–32840.
Kawanishi, S., Ohnishi, S., Ma, N., Hiraku, Y., & Murata, M. (2017). Crosstalk between DNA damage and inflammation in the multiple steps of carcinogenesis. International Journal of Molecular Sciences,18, 1808.
Kawanishi, S., Ohnishi, S., Ma, N., Hiraku, Y., Oikawa, S., & Murata, M. (2016). Nitrative and oxidative DNA damage in infection-related carcinogenesis in relation to cancer stem cells. Genes Environment,38, 26.
Khattar, E., Kumar, P., Liu, C. Y., Akincilar, S. C., Raju, A., Lakshmanan, M., et al. (2016). Telomerase reverse transcriptase promotes cancer cell proliferation by augmenting tRNA expression. The Journal of Clinical Investigation,126, 4045–4060.
Kim, E. K., & Choi, E. J. (2010). Pathological roles of MAPK signaling pathways in human diseases. Biochimica et Biophysica Acta,1802, 396–405.
Koh, C. M., Khattar, E., Leow, S. C., Liu, C. Y., Muller, J., Ang, W. X., et al. (2015). Telomerase regulates MYC-driven oncogenesis independent of its reverse transcriptase activity. The Journal of Clinical Investigation,125, 2109–2122.
Kou, T., Marusawa, H., Kinoshita, K., Endo, Y., Okazaki, I. M., Ueda, Y., et al. (2007). Expression of activation-induced cytidine deaminase in human hepatocytes during hepatocarcinogenesis. International Journal of Cancer,120, 469–476.
Kumar, R., DiMenna, L. J., Chaudhuri, J., & Evans, T. (2014). Biological function of activation-induced cytidine deaminase (AID). Biomedical Journal,37, 269–283.
Lane, D., & Levine, A. (2010). p53 Research: the past thirty years and the next thirty years. Cold Spring Harb Perspectives in Biology,2, a000893.
Li, Y., Cheng, H. S., Chng, W. J., & Tergaonkar, V. (2016). Activation of mutatnt TERT promoter by RAS-ERK signaling is a key step in malignant progression of BRAF-mutant human melanomas. Proceedings of the National Academy of Sciences USA,113, 6.
Li Chew, C., Lunardi, A., Gulluni, F., Ruan, D. T., Chen, M., Salmena, L., et al. (2015). In vivo role of INPP4B in tumor and metastasis suppression through regulation of PI3 K-AKT signaling at endosomes. Cancer Discovery,5, 740–751.
Li, G., Pone, E. J., Tran, D. C., Patel, P. J., Dao, L., Xu, Z., et al. (2012). Iron inhibits activation-induced cytidine deaminase enzymatic activity and modulates immunoglobulin class switch DNA recombination. Journal of Biological Chemistry,287, 21520–21529.
Li, Y., Zhou, Q. L., Sun, W., Chandrasekharan, P., Cheng, H. S., Ying, Z., et al. (2015). Non-canonical NF-kappaB signalling and ETS1/2 cooperatively drive C250T mutant TERT promoter activation. Nature Cell Biology,17, 1327–1338.
Lin, Y. S. Y., Veach, Y. R. A., Torgerson, T. R., & Hawiger, J. (1995). Inhibition of Nuclear Translocation of Transcription Factor NF-kB by a synthetic peptide containing a cell membrane-permeable motif and nuclear localization sequence. Journal of Biological Chemistry,270, 4.
Liu, J., Farmer, J. D., Lane, W. S., Friedman, J., Weissman, I., & Schreiber, S. L. (1991). Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell,66, 9.
McDade, T. P., Perugini, R. A., Vittimberga, F. J., Carrigan, R. C., & Callery, M. P. (1998). Salicylates inhibit NF-kB activation and enhance TNF-a-induced apoptosis in human pancreatic cancer cells. Journal of Surgical Research, 83, 56–61.
Medzhitov, R., & Horng, T. (2009). Transcriptional control of the inflammatory response. Nature Reviews Immunology,9, 692–703.
Mills, E. L., Ryan, D. G., Prag, H. A., Dikovskaya, D., Menon, D., Zaslona, Z., et al. (2018). Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature,556, 113–117.
Morishita, R., Sugimoto, T., Aoki, M., Kida, I., Tomita, N., Moriguchi, A., et al. (1997). In vivo transfection of cis element “decoy” against nuclear factor-kB binding site prevents mycardial infarction. Nature,3, 6.
Okazaki, I. M., Kotani, A., & Honjo, T. (2007). Role of AID in tumorigenesis. Advances in Immunology,94, 245–273.
Oshima, H., Ishikawa, T., Yoshida, G. J., Naoi, K., Maeda, Y., Naka, K., et al. (2014). TNF-alpha/TNFR1 signaling promotes gastric tumorigenesis through induction of Noxo1 and Gna14 in tumor cells. Oncogene,33, 3820–3829.
Pua, K. H., Stiles, D. T., Sowa, M. E., & Verdine, G. L. (2017). IMPDH2 is an intracellular target of the cyclophilin A and sanglifehrin A complex. Cell Reports,18, 432–442.
Schett, G., & Neurath, M. F. (2018). Resolution of chronic inflammatory disease: universal and tissue-specific concepts. Nature Communications,9, 3261.
Shin, E. M., Hay, H. S., Lee, M. H., Goh, J. N., Tan, T. Z., Sen, Y. P., et al. (2014). DEAD-box helicase DP103 defines metastatic potential of human breast cancers. The Journal of Clinical Investigation,124, 18.
Shuai, K., & Liu, B. (2003). Regulation of JAK-STAT signalling in the immune system. Nature Reviews Immunology,3, 900–911.
Siveen, K. S., Nguyen, A. H., Lee, J. H., Li, F., Singh, S. S., Kumar, A. P., et al. (2014). Negative regulation of signal transducer and activator of transcription-3 signalling cascade by lupeol inhibits growth and induces apoptosis in hepatocellular carcinoma cells. British Journal of Cancer,111, 1327–1337.
Sreenivasan, Y., Sarkar, A., & Manna, S. K. (2003). Mechanism of cytosine arabinoside-mediated apoptosis: role of Rel A (p65) dephosphorylation. Oncogene,22, 4356–4369.
Stein, M., Keshav, S., Harris, N., & Gordon, S. (1992). Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. Journal of Experimental Medicine,176, 287–292.
Sun, S. C. (2011). Non-canonical NF-kappaB signaling pathway. Cell Research,21, 71–85.
Sung, B., Pandey, M. K., Ahn, K. S., Yi, T., Chaturvedi, M. M., Liu, M., et al. (2008). Anacardic acid (6-nonadecyl salicylic acid), an inhibitor of histone acetyltransferase, suppresses expression of nuclear factor-kappaB-regulated gene products involved in cell survival, proliferation, invasion, and inflammation through inhibition of the inhibitory subunit of nuclear factor-kappaBalpha kinase, leading to potentiation of apoptosis. Blood,111, 4880–4891.
Sunwoo, J. B., Chen, Z., Dong, G., Yeh, N., Bancroft, C. C., Sausville, E., et al. (2001). Novel Proteasome Inhibitor PS-341 inhibits activation of nuclear factor-kB, cell survival, tumor growth, and angiogenesis in squamous cell carcinoma. Clinical Cancer Research,7, 10.
Tan, Q., Tai, N., Li, Y., Pearson, J., Pennetti, S., Zhou, Z., Wong, F.S., & Wen, L. (2018). Activation-induced cytidine deaminase deficiency accelerates autoimmune diabetes in NOD mice. JCI Insight 3.
Tergaonkar, V., Pando, M., Vafa, O., Wahl, G., & Verma, I. (2002). p53 stabilization is decreased upon NFkB activation: A role for NFkB in acquisition of resistance to chemotherapy. Cancer Cell,1, 11.
Tisato, V., Voltan, R., Gonelli, A., Secchiero, P., & Zauli, G. (2017). MDM2/X inhibitors under clinical evaluation: perspectives for the management of hematological malignancies and pediatric cancer. Journal of Hematology & Oncology,10, 133.
Tsai, C. Y. P., Chern, T., Chuang, S., Lin, J., Klemm, L., Muschen, M., et al. (2013). AID downregulation is a novel function of the DNMT inhibitor 5-aza-deoxycytidine. Oncotarget,5, 13.
van Rhee, F., Fayad, L., Voorhees, P., Furman, R., Lonial, S., Borghaei, H., et al. (2010). Siltuximab, a novel anti-interleukin-6 monoclonal antibody, for Castleman’s disease. Journal of Clinical Oncology,28, 3701–3708.
Vitecek, J., Lojek, A., Valacchi, G., & Kubala, L. (2012). Arginine-based inhibitors of nitric oxide synthase: therapeutic potential and challenges. Mediators of Inflammation,2012, 318087.
Wang, C., Cusack, J. C. J., Liu, R., & Baldwin, A. S. J. (1999). Control of inducible chemoresistance: Enhanced anti-tumor therapy through increased apoptosis by inhibition of NF-kB. Nature Medicine,5, 6.
Wang, C. Q., Krishnan, V., Tay, L. S., Chin, D. W., Koh, C. P., Chooi, J. Y., et al. (2014). Disruption of Runx1 and Runx3 leads to bone marrow failure and leukemia predisposition due to transcriptional and DNA repair defects. Cell Reports,8, 767–782.
Webster, G. A., & Perkins, N. D. (1999). TRanscriptional cross talk between NF-kB and p53. Molecular and Cellular Biology,19, 11.
Wei, W., Yang, Z., Tang, C. H., & Liu, L. (2011). Targeted deletion of GSNOR in hepatocytes of mice causes nitrosative inactivation of O6-alkylguanine-DNA alkyltransferase and increased sensitivity to genotoxic diethylnitrosamine. Carcinogenesis,32, 973–977.
Wu, H., & Lozano, G. (1994). NF-kB activation of p53. Journal of Biological Chemistry,269, 8.
Xia, Y., Padre, R. C., De Mendoza, T. H., Bottero, V., Tergaonkar, V. B., & Verma, I. M. (2009). Phosphorylation of p53 by IkappaB kinase 2 promotes its degradation by beta-TrCP. Proceedings of the National Academy of Sciences USA,106, 2629–2634.
Author information
Khian Hong Pua and Chen Li Chew have contributed equally to the manuscript.
Affiliations
p53 Laboratory, Agency for Science, Technology and Research (A-STAR), 8A Biomedical Grove, Singapore, 138648, Singapore
Khian Hong Pua & David Philip Lane
Institute of Molecular and Cell Biology (IMCB), Agency for Science, Technology and Research (A-STAR), #03-17, 61 Biopolis Drive, Singapore, 138673, Singapore
Chen Li Chew & Vinay Tergaonkar
Department of Biochemistry, Yong Loo Lin School of Medicine, National University of Singapore, Singapore, Singapore
Vinay Tergaonkar
Corresponding author
Correspondence to Vinay Tergaonkar.
Rights and permissions
About this article
Cite this article
Pua, K.H., Chew, C.L., Lane, D.P. et al. Inflammation-associated genomic instability in cancer. GENOME INSTAB. DIS. 1, 1–9 (2020). https://doi.org/10.1007/s42764-019-00006-6
Received07 June 2019
Revised01 August 2019
Accepted05 August 2019
Published17 September 2019
Issue DateJanuary 2020
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
Inflammation
Mutation
NF-κB
Telomerase
用户登录
还没有账号?
立即注册