






Post-translational modifications of nuclear sirtuins
Review Article
Genome Instability & Disease 1, 34–45(2020)
Abstract
Silent information regulator proteins (SIRT), or sirtuins, are evolutionarily conserved NAD+-dependent deacetylases and ADP-mono-ribosyltransferases. In mammalian, seven sirtuins have been identified, namely SIRT1–7, with different subcellular localization. Nuclear sirtuins, including SIRT1, SIRT6 and SIRT7, localize predominantly in the nucleus and are implicated in many vital biological processes, including stress response, transcription, genome maintenance, tumorigenesis and aging. Dysregulation of nuclear sirtuins is associated with the development of many diseases, including cancer and metabolic disorders. Therefore, the activities of nuclear sirtuins must be properly regulated. In this review, we summarize the current knowledge on the post-translational modifications of nuclear sirtuins and discuss how these modifications modulate their functions.
Introduction
Acetylation of lysine is a dynamic process of highly regulated post-translational modification, which was initially only found on histones (Vidali et al. 1968). The first non-histone protein identified to carry lysine acetylation is p53 (Gu and Roeder 1997). After that, with the help of high-resolution mass spectrometry, thousands of proteins have been identified to be acetylated at the lysine residues (Choudhary et al. 2009; Kim et al. 2006). Like other post-translational modifications, acetylation on specific lysine residues can regulate a spectrum of protein properties ranging from enzyme activity, protein–protein interaction, protein stability, and subcellular localization, etc. Given its important roles, the protein acetylation must be tightly regulated.
The enzymes responsible for fine-tuning the acetylation-deacetylation dynamics on proteins are lysine acetyltransferases (KATs) and lysine deacetylases (KDACs). For KDACs, it can be generally divided into two major groups: the metal ion-dependent classical histone deacetylases (HDACs) and the NAD+-dependent sirtuins. The main difference between the two groups of KDACs is that they use different co-factors to catalyze deacetylation reaction. Catalysis by classical HDACs requires a metal ion, for example Zn2+ or Fe2+. The catalytic mechanism proposed for classical HDACs is based on the structure study of histone deacetylase-like protein (HDLP) from Aquifex aeolicus and HADC8, which belongs to class I HDAC (Finnin et al. 1999; Somoza et al. 2004). Most of the key residues within the active site and substrate binding pocket of HDAC8 are conserved across class I HDAC. In brief, the metal ion is firstly coordinated by residue D178, H180 and D267 (HDAC8 residues). The coordinated metal ion then polarizes the carbonyl group at the acetyl lysine residue of the substrate together with the hydroxyl of Y360. Subsequently, a nucleophilic attack is carried out by the metal ion-activated water molecule on the carbonyl carbon to form a tetrahedral intermediate. Residues H142 and H143 of HDAC8 also help to activate the water molecule for nucleophilic attack. Finally, the collapse of the tetrahedral intermediate will generate acetate and deacetylated lysine products (Dowling et al. 2008; Finnin et al. 1999; Gantt et al. 2010; Somoza et al. 2004; Vannini et al. 2004). Sirtuins (silent information regulator proteins, SIRTs) are NAD+-dependent deacetylases or ADP-mono-ribosyltransferases (Ahuja et al. 2007; Imai et al. 2000; Liszt et al. 2005). More recently, it was found that some sirtuin family members also show deacylation activity (Du et al. 2011; Peng et al. 2011; Tan et al. 2014). The feature to employ NAD+ as a co-factor distinguishes sirtuins from other KDACs. For deacetylation reaction catalyzed by sirtuins, the acetyl oxygen of the substrate first carries out a nucleophilic attack towards the 1′-carbon of NAD+ ribose to generate nicotinamide and C1′-O-alkylamidate intermediate. Subsequently, a conserved histidine residue in sirtuins activates the 2′-hydroxyl group on the NAD+ ribose. Activated 2′-hydroxyl group then attacks the carbon of O-alkylamidate to generate 1′,2′-cyclic intermediate. Finally, the carbonyl carbon of 1′,2′-cyclic intermediate is attacked by base-activated water molecule to form deacetylated lysine products and 2′-O-acetyl-ADP-ribose (OAADPr) (Jackson and Denu 2002; Min et al. 2001; Sauve et al. 2001; Smith and Denu 2006).
Sirtuin proteins are evolutionarily conserved among species. Seven sirtuins (SIRT1–7) have been identified in mammals so far. While SIRT1, SIRT6 and SIRT7 are localized predominantly in the nucleus, SIRT3, SIRT4 and SIRT5 are mainly present in the mitochondria. SIRT2 is reside prominently in the cytoplasm (Michishita et al. 2005). Structurally, the seven mammalian sirtuins share a conserved catalytic core domain flanking by additional N- and C-terminal extensions in different length. The nuclear sirtuins (SIRT1, SIRT6 and SIRT7) have been implicated in numerous key biological processes, including stress response, energy metabolism, gene transcription, maintenance of genome stability, tumorigenesis and aging (Chalkiadaki and Guarente 2015; Ghosh and Zhou 2015; Yu and Auwerx 2009). Therefore, the activity of nuclear sirtuins needs to be well controlled. Despite intensive studies on the targets and the functional networks of nuclear sirtuins, little is known about how themselves are regulated, with even less information on their post-translational modifications. In this review, we summarize the current available knowledge on the post-translational modifications of nuclear sirtuins and how these modifications contribute to the regulation of their functions.
SIRT1
SIRT1 is the most extensively studied nuclear sirtuins. Its crucial roles in a variety of fundamental biological processes, such as stress resistance, metabolic regulation, DNA repair, tumor suppression and aging, have garnered immense interest. SIRT1 achieves its far-reaching regulatory effects through targeting and deacetylating many of its well-defined substrates, including tumor suppressor protein p53, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), fork-head box transcription factors (FOXOs) and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) (Rahman and Islam 2011). In this context, it is not surprising that SIRT1 activity must be carefully controlled in response to various cellular stimuli under different physiologic conditions.
The regulation of SIRT1 in cells can be achieved at multiple levels. At transcriptional and post-transcriptional level, SIRT1 expression is modulated by the activity of related transcription factors and microRNAs (miRNAs), which have been well summarized in previous reviews (Buler et al. 2016; Revollo and Li 2013; Yamakuchi 2012). At post-translational level, the activity of SIRT1 can be regulated through interacting with other proteins. For instance, active regulator of SIRT1 (AROS) interacts with SIRT1 and enhances SIRT1 deacetylase activity towards p53. Residues 114–217, which localize at the N-terminal of SIRT1, were responsible for their interaction. However, it is unclear how this interaction contributes to enhance SIRT1 activity (Kim et al. 2007). On the other hand, deleted in breast cancer-1 (DBC1) was suggested to act as an endogenous inhibitor of SIRT1 (Kim et al. 2008; Zhao et al. 2008). DBC1 can directly interact with SIRT1. The leucine zipper motif contained region (residues 243–264) of DBC1 and catalytic core domain of SIRT1 are responsible for their interaction (Kim et al. 2008). Overexpression of DBC1 in cells impairs SIRT1 deacetylase activity and results in hyperacetylation of several well-defined SIRT1 substrates, including p53 and FOXO3. In line with this, depletion of DBC1 in cells cause p53 hypoacetylation and potentiates SIRT1-dependent anti-apoptosis upon genotoxic stress (Kim et al. 2008; Zhao et al. 2008). The mechanism underlying DBC1-mediated regulation of SIRT1 activity was not fully understood until the discovery of ESA motif within SIRT1. The ESA (essential for SIRT1 activity) motif is a 25-amino acid sequence located at the C-terminal extension of SIRT1 (residue 641–665 for human SIRT1, Fig. 1). ESA motif can bind to the core catalytic domain to activate enzymatic activity of SIRT1 and its affinity for substrate binding. DBC1 also binds to the deacetylase core and competes with ESA domain for the binding site on SIRT1, therefore “switching off” the SIRT1 self-activation and impairing SIRT1 activity (Kang et al. 2011). Moreover, under cellular stress condition, DBC1 can be phosphorylated at T454 by ataxia telangiectasia mutated (ATM) protein, which creates a novel binding site for SIRT1 and reinforces the interaction between SIRT1 and DBC1. The enhanced DBC1-SIRT1 interaction was suggested to be an important event for cell fate determination upon stress (Yuan et al. 2012).
Fig. 1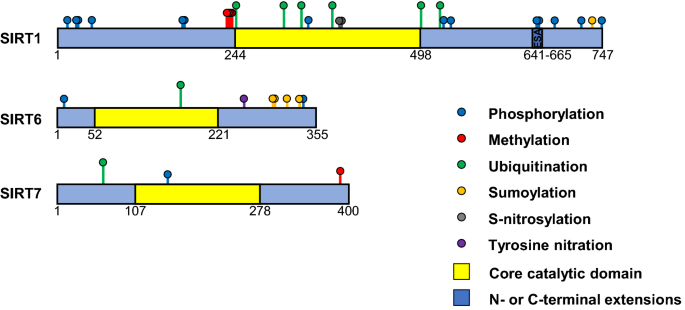
Overview of human nuclear sirtuins and their PTMs. Human nuclear sirtuins, SIRT1, SIRT6, and SIRT7 are schematically presented. Yellow boxes indicates the core catalytic domain. Different length of N- and C-terminal extensions flanking the core catalytic domain indicates as blue boxes. The essential for SIRT1 activity (ESA) sequence is shown as dark blue box. Different types of post-transcriptional modifications are indicated. The precise amino acids modified by the different post-transcriptional modifications are listed in Table 1
Full size imageApart from protein–protein interaction, SIRT1 activity can also be regulated through post-translational modification. Identification of the post-translational modifications and their regulatory effect towards SIRT1 have been developed rapidly. Several recent reviews have already focused on this topic (Flick and Luscher 2012; Revollo and Li 2013). However, with recent discoveries of novel modifiers and modification sites on SIRT1, our understanding of post-translational modifications on SIRT1 and their function needs to be updated. In this review, we summarized the most recent findings on the post-translational modifications of SIRT1 and how the SIRT1 activity and functions are regulated (Table 1).
| Sirtuin | Target site | Modification type | Modifier | PTM function | References |
|---|---|---|---|---|---|
| SIRT1 | S14 | Phosphorylation | Potentially enhance deacetylase activity | Sasaki et al. (2008) | |
| S26 | Phosphorylation | Potentially enhance deacetylase activity | Sasaki et al. (2008) | ||
| S27 | Phosphorylation | JNK1 | Enhance deacetylase activity and nuclear localization | Nasrin et al. (2009), Sasaki et al. (2008) | |
| JNK2 | Enhance protein stability | Ford et al. (2008) | |||
| CaMKKβ | Decreased protein stability and deacetylase activity | Wen et al. (2013) | |||
| S47 | Phosphorylation | JNK1 | Enhance deacetylase activity and nuclear localization | Nasrin et al. (2009), Sasaki et al. (2008) | |
| mTOR | Suppress deacetylase activity | Back et al. (2011) | |||
| CDK5 | Enhance nuclear localization | Bai et al. (2012) | |||
| CaMKKβ | Decreased protein stability and deacetylase activity | Wen et al. (2013) | |||
| S172 | Phosphorylation | Potentially enhance deacetylase activity | Sasaki et al. (2008) | ||
| S173 | Phosphorylation | Potentially enhance deacetylase activity | Sasaki et al. (2008) | ||
| K233 | Methylation | SET7/9 | Potentially disrupt interaction with p53 | Liu et al. (2011) | |
| K235 | Methylation | SET7/9 | Potentially disrupt interaction with p53 | Liu et al. (2011) | |
| K236 | Methylation | SET7/9 | Potentially disrupt interaction with p53 | Liu et al. (2011) | |
| K238 | Methylation | SET7/9 | Potentially disrupt interaction with p53 | Liu et al. (2011) | |
| K254 | Ubiquitination | USP22 | Decreased protein stability | Lin et al. (2012) | |
| K311 | Ubiquitination | MDM2 | Decreased protein stability and enhance nuclear localization | Peng et al. (2015) | |
| K335 | Ubiquitination | USP22 | Decreased protein stability | Lin et al. (2012) | |
| T344 | Phosphorylation | AMPK | Enhance or suppress deacetylase activity | Lau et al. (2014), Lee et al. (2012) | |
| K377 | Ubiquitination | USP22 | Decreased protein stability | Lin et al. 2012) | |
| S387 | S-nitrosylation | SNO–GAPDH | Suppress deacetylase activity | Kornberg et al. (2010) | |
| S390 | S-nitrosylation | SNO–GAPDH | Suppress deacetylase activity | Kornberg et al. (2010) | |
| K499 | Ubiquitination | USP22 | Decreased protein stability | Lin et al. (2012) | |
| K523 | Ubiquitination | USP22 | Decreased protein stability | Lin et al. (2012) | |
| T530 | Phosphorylation | Cyclin B/CDK1 | Promote cell cycle progression | Sasaki et al. (2008) | |
| JNK1 | Enhance deacetylase activity and nuclear localization | Nasrin et al. (2009) | |||
| S540 | Phosphorylation | Cyclin B/CDK1 | Promote cell cycle progression | Sasaki et al. (2008) | |
| S659 | Phosphorylation | CK2 | Enhance substrate binding and deacetylase activity | Zschoernig and Mahlknecht (2009) | |
| S661 | Phosphorylation | CK2 | Enhance substrate binding and deacetylase activity | Zschoernig and Mahlknecht (2009) | |
| S682 | Phosphorylation | HIPK2 | Suppress deacetylase activity | Conrad et al. (2016) | |
| S719 | Phosphorylation | Potentially enhance deacetylase activity | Sasaki et al. (2008) | ||
| K734 | Sumoylation | SENP1 | Enhance deacetylase activity | Yang et al. (2007) | |
| S747 | Phosphorylation | Potentially enhance deacetylase activity | Sasaki et al. (2008) | ||
| SIRT6 | S10 | Phosphorylation | JNK | Enhanced ADP-mono-ribosyltransferase activity on PARP1 | Van Meter et al. (2016) |
| K170 | Ubiquitination | CHIP | Enhanced protein stability | Ronnebaum et al. (2013) | |
| Y257 | Tyrosine nitration | Decrease deacetylase activity | Hu et al. (2015) | ||
| K296 | Sumoylation | SUMO1 | Decrease deacetylase activity towards H3K56ac | Cai et al. (2016) | |
| K300 | Sumoylation | SUMO1 | Decrease deacetylase activity towards H3K56ac | Cai et al. (2016) | |
| K316 | Sumoylation | SUMO1 | Decrease deacetylase activity towards H3K56ac | Cai et al. (2016) | |
| K332 | Sumoylation | SUMO1 | Decrease deacetylase activity towards H3K56ac | Cai et al. (2016) | |
| S338 | Phosphorylation | AKT1 | Decreased protein stability | Thirumurthi et al. (2014) | |
| SIRT7 | K63 | Ubiquitination | USP7 | Enhance deacetylase activity | Jiang et al. (2017) |
| T153 | Phosphorylation | AMPK | Promote subcellular redistribution and degradation | Sun et al. (2016) | |
| R388 | Methylation | PRMT6 | Suppress deacetylase activity | Yan et al. (2018) |
Table 1 Summary of post-translational modification sites and their corresponding modifiers on human nuclear sirtuins
Phosphorylation
Phosphorylation is one of the most extensively studied post-translational modifications in a variety of proteins. In an early report, at least 13 phosphorylated serine/threonine residues in human SIRT1 were identified by mass spectrometry. All these phosphorylated residues locate at either N- or C-terminal extension of SIRT1. The phosphorylation seems to have a positive effect on SIRT1 as removal of SIRT1 phosphorylation by phosphatase treatment decreases its deacetylase activity (Sasaki et al. 2008). Further analysis showed that among the detected phosphorylation sites, T530 and S540 are targeted by cyclin B/cyclin-dependent kinase 1 (CDK1) protein complex, and phosphorylation on these two residues is crucial for cell cycle progression. Specifically, re-introduction of wild-type SIRT1 back into SIRT1 knockout cells can restore the proliferation defect, while SIRT1 carrying T530A/S540A double mutations fails to rescue such defect (Sasaki et al. 2008). In addition, C-Jun N-terminal kinase 1 (JNK1) can interact with and phosphorylate SIRT1 at S27, S47, and T530 upon oxidative stress. Phosphorylation by JNK1 promotes the nuclear localization of SIRT1 and enhances its deacetylase activity towards histone substrates but not p53 (Nasrin et al. 2009). This indicates that phosphorylation at specific sites may selectively affect SIRT1 enzymatic activity towards certain substrates. Interestingly, JNK2, a member from the same kinase family, was reported to phosphorylate SIRT1 at S27 and promote SIRT1 stability, whereas the protein stability of SIRT1 is not affected by JNK1 (Ford et al. 2008). On the other hand, mTOR can phosphorylate SIRT1 at S47 and inhibit its deacetylase activity in human epidermoid squamous carcinoma cells (Back et al. 2011). Enhanced S47 phosphorylation on SIRT1 was also found in senescent porcine aortic endothelial cells. SIRT1 tends to accumulate in the nucleus and its anti-inflammatory effect is abolished. The protein responsible for SIRT1-S47 hyperphosphorylation is cyclin-dependent kinase 5 (CDK5). Inhibition of CDK5 promotes SIRT1 nuclear exportation and ameliorates senescence phenotypes in porcine aortic endothelial cells, leading to the attenuation of cellular senescence and atherosclerosis development in hyper-cholesterolemic ApoE−/− mice (Bai et al. 2012). Moreover, in an independent study, Ca2+/calmodulin-dependent protein kinase kinase β (CaMKKβ) was identified to phosphorylate SIRT1 at S27 and S47 in response to atheroprotective flow in vascular endothelial cells (Wen et al. 2013). Phosphorylation at these two sites can increase SIRT1 protein stability and deacetylase activity. The CaMKKβ-SIRT1 axis appears to have an anti-atherogenic role since depletion of either CaMKKβ or endothelial SIRT1 increases atherosclerosis lesions in ApoE−/− mice (Wen et al. 2013). Taken together, it seems that in different cell types, phosphorylation of SIRT1 by different protein kinases may have distinct effects on SIRT1 protein functions.
In addition to the kinases mentioned above, other protein kinases can also phosphorylate SIRT1 and regulate its function. Casein kinase 2 (CK2) can phosphorylate mouse SIRT1 on S154, S649, S651 and S683 upon ionizing radiation (Kang et al. 2009). Two of these sites were also identified to be phosphorylated by CK2 in human SIRT1 corresponding to sites S659 and S661 (Zschoernig and Mahlknecht 2009). Phosphorylation on these sites increases the substrate binding affinity (at least towards p53) and enzymatic activity of SIRT1, therefore attenuating the p53-mediated apoptosis upon DNA damage. In this regard, CK2 was proposed to exert anti-apoptotic function upon stress through phosphorylating and activating SIRT1 (Kang et al. 2009). It is worth noting that S659 and S661 localize in the functional ESA motif of SIRT1 (Fig. 1). Considering the two phosphorylation sites are just flanking one of the two key residues that required for ESA motif function, it is reasonable to speculate that phosphorylation at these sites regulates the interaction between ESA motif and core domain of SIRT1, thus modulating SIRT1 activity (Flick and Luscher 2012). More recently, another CK2 targeting site on SIRT1, S164, was identified in mouse liver. SIRT1-S164 phosphorylation seems to be associated with obesity because of its very high level in obese mice but hardly detectable in lean mice. Mechanistically, S164 phosphorylation prevents SIRT1 from nuclear localization and modestly decreased its deacetylase activity. Adenovirus-mediated expression of wild-type or phosphor-defective mutant SIRT1 (S164A) in diet-induced obese mice improve the glucose intolerance and liver steatosis, while expression of S164D SIRT1, which is a phosphor-mimic mutant, failed to do so. Moreover, both SIRT1 S164 phosphorylation and CK2 levels were found to increase in liver samples of non-alcoholic fatty liver disease (NAFLD) patients and correlated with progression of the disease. It was, therefore, proposed that inhibition of CK2-mediated SIRT1 S164 phosphorylation may provide a potential therapeutic strategy to treat NAFLD and other obesity-related diseases (Choi et al. 2017).
Besides CK2, two of the dual specificity tyrosine-phosphorylated and regulated kinase (DYRK) family proteins, DYRK1A and DYRK3, can phosphorylate mouse SIRT1 on T522 (corresponding to human residue T530). Under stress condition, phosphorylation at T522 increases SIRT1 deacetylase activity towards its substrate p53, thus inhibiting apoptosis. The phosphorylation at T522 increases the release of deacetylated protein products, therefore promoting SIRT1 association with its substrates (Guo et al. 2010). In addition, cyclic AMP/protein kinase A (cAMP/PKA) signaling cascade-dependent phosphorylation of SIRT1 at a highly conserved residue S434 (corresponding to human residue S442) has recently been reported. This modification significantly increases SIRT1 activity towards PGC-1α independent of NAD+ change, resulting in PGC-1α activation and enhanced fatty acid oxidation in response to several metabolic stresses (Gerhart-Hines et al. 2011). On the other hand, two independent studies showed that AMP-activated protein kinase (AMPK) can phosphorylate SIRT1 on T334. However, the regulatory effect of this modification seems opposite in two studies (Lee et al. 2012; Lau et al. 2014). Lee et al. found that in liver cancer cells, AMPK phosphorylates SIRT1 at T344 and inhibits its catalytic activity towards p53. As a result, p53 transcriptional activity is enhanced and apoptosis in hepatocellular carcinoma cells is promoted (Lee et al. 2012). On the contrary, Lau et al. claimed that SIRT1 T344 phosphorylation by AMPK helps SIRT1 to be released from its endogenous inhibitor DBC1 (deleted in breast cancer 1), thus promoting SIRT1 deacetylase activity and inhibiting p53 function in bone tumor cells (U2OS cells) (Lau et al. 2014). The reason why they observed opposite regulatory effect by phosphorylation on the same site is unclear, probably due to the different cell lines used in their studies. Finally, a most recent study found that in response to DNA damage, SIRT1 can be phosphorylated by HIPK2 (homeodomain interacting protein kinase 2) at S682. S682 phosphorylation decreases SIRT1 activity and potentiates p53 downstream gene expression which in turn promotes apoptosis. The proposed underlying mechanism is that DNA damage-induced phosphorylation of SIRT1 at S682 disrupted the interaction between SIRT1 and its endogenous activator AROS, resulting in SIRT1 inactivation (Conrad et al. 2016).
Methylation
Methylation is a post-translational modification featured by adding methyl groups to lysine or arginine residues on a protein. Currently, the understanding of SIRT1 methylation is still limited, only one study reported that SIRT1 can be methylated by lysine methyltransferase SETD7 (or SET7/9) at K233, K235, K236 and K238 in its N-terminal extension. However, the functional relevance of these modifications is still missing since they did not affect SIRT1 deacetylase activity, at least towards p53. Nevertheless, the interaction between SIRT1 and SET7/9 is increased upon DNA damage and this interaction is sufficient to abrogate the SIRT1 association with p53, enhanced p53 acetylation level and its transcriptional activity (Liu et al. 2011). Therefore, it is possible that methylation modulates SIRT1 function through affecting its accessibility towards certain substrates.
SUMOylation
SUMOylation is a post-translational modification featuring by the attachment of small ubiquitin-like modifier proteins to lysine residues of targeted protein. Human SIRT1 can be SUMOylated at K734 and this modification increases SIRT1 enzymatic activity towards p53. When exposure to UV radiation or H2O2, sentrin-specific protease 1(SENP1) interacts with and deSUMOylates SIRT1, inhibiting SIRT1 activity and consequently activating p53 to promote apoptosis and cell death. Supporting these observations, H1299 cells depleted with Senp1 are more resistant to UV- and H2O2-induced apoptosis (Yang et al. 2007). Despite all these findings, the exact mechanism by which SUMOylation promotes SIRT1 activity remains unclear.
Ubiquitination
Like SUMOylation, protein ubiquitination involves the addition of single ubiquitin or ploy-ubiquitin chains to targeted protein. SIRT1 was reported to be ubiquitinated potentially at K254, K335, K377, K499 and K523. Ubiquitin-specific peptidase 22 (USP22) can positive regulate SIRT1 through deubiquitination. Mechanistically, by removing polyubiquitin chains from SIRT1, USP22 promotes SIRT1 protein stability and thereby inhibits p53 acetylation and its transcriptional activity in colon cancer cell line HCT116. Moreover, deletion of Usp22 in mouse embryonic fibroblasts (MEF) results in decreased SIRT1 stability and p53-dependent apoptosis. In addition, Usp22 knockout mice showed early embryonic lethality which indicates that USP22 may play a crucial role during early embryogenesis, probably through the USP22-SIRT1-p53 pathway (Lin et al. 2012). More recently, SIRT1 was shown to be ubiquitinated at K311 upon DNA damage by E3 ligase MDM2. Although K311 ubiquitination does not affect SIRT1 enzymatic activity under normal conditions, it decreases SIRT1 stability and promotes SIRT1 nuclear localization in response to DNA damage. As a result, ubiquitination of SIRT1 at K311 affects its anti-apoptotic function during DNA damage repair process (Peng et al. 2015).
Acetylation
A recent study revealed an unexpected mechanism that SIRT1 can augment its own activity through self-deacetylation at K230 (mouse SIRT1 residue). Deacetylation of SIRT1 at K230 promotes its activity towards several of its established substrates, including p53, H3K9ac and H4K16ac. SIRT7, which is also a nuclear sirtuin, interacts with SIRT1 and restrains SIRT1 activity by inhibiting its autodeacetylation (Fang et al. 2017). Functionally, SIRT7-mediated restriction of SIRT1 activity is essential during adipogenesis. In support of this, SIRT1 showed a much stronger interaction with SIRT7 and decreased activity in differentiating adipocytes. Moreover, adipocyte differentiation is impaired in SIRT7 knockout MEF cells, coupled with increasing SIRT1 activity, and knockdown of SIRT1 in same cell line can rescue such phenotype. In addition, SIRT7 knockout mice showed a defective white adipose tissue deposit phenotype, and removal of one allele of SIRT1 within adipocyte tissue in SIRT7 knockout mice can significantly restore the adipocyte differentiation (Fang et al. 2017). Therefore, post-translational acetylation and self-deacetylation of SIRT1 act as a novel way to regulate SIRT1 activity, and the crosstalk between SIRT1 and SIRT7 in modulating SIRT1 autodeacetylation may serve as an important mechanism in controlling differentiation and maintenance of adipose tissue.
Nitrosylation
S–Nitrosylation is a regulatory modification involving covalent addition of a nitric oxide (NO) group to a cysteine residue of targeted protein (Nakamura and Lipton 2013). Nuclear SIRT1 was reported to be nitrosylated by nitrosylated glycer-aldehyde-3-phosphate dehydrogenase (SNO–GAPDH) at S387 and S390. The two amino acids reside within the core catalytic domain of SIRT1 and their nitrosylation suppresses SIRT1 deacetylase activity. This leads to enhanced acetylation of PGC-1α, a well-established substrate of SIRT1, and decreased its transcriptional activity. Considering the critical role of SIRT1–PGC1α pathway in metabolic regulation, SNO–GAPDH-mediated SIRT1 nitrosylation may present clinical relevance in metabolic dysfunction-associated diseases (Kornberg et al. 2010).
Carbonylation
Protein carbonylation is an irreversible post-translational modification featured by adding a reactive carbonyl group, such as aldehyde and ketone, to targeted protein and is regarded as a major hallmark of oxidative stress (Dalle-Donne et al. 2006). Treatment of human lung epithelial cells with H2O2 and cigarette smoke extract induces SIRT1 carbonylation. The modification probably happens at cysteine residues between amino acid 467–492 on SIRT1. As a consequence, SIRT1 protein stability and activity are reduced, and it is expulsed from the nucleus. More importantly, mice exposed to cigarette smoke also show increased carbonylation and decreased protein level of SIRT1, which indicates that carbonylation may serve as a general mechanism both in vitro and in vivo to regulate SIRT1 function in response to oxidative stress (Caito et al. 2010).
SIRT6
SIRT6 is a chromatin-associated protein, its multifaceted roles in maintaining genome stability, inflammation, tumor suppression and aging have already been extensively characterized. SIRT6 can act as deacetylase or ADP-mono-ribosyltransferase. SIRT6 can also catalyze deacylation to remove long-chain fatty-acyl groups from lysine residues on histone and none-histone proteins (Feldman et al. 2013; Jiang et al. 2013). The first identified SIRT6 substrates are acetylated lysine 9 and 56 of histone H3 (H3K9ac and H3K56ac), deacetylation of these histone marks plays a vital role in chromatin compaction and DNA damage repair signaling (Michishita et al. 2008; Yang et al. 2009). Most recently, acetylated lysine 18 of histone H3 (H3K18ac) was identified as a novel target of SIRT6. SIRT6 deacetylates H3K18ac at pericentric heterochromatin to help local transcriptional repression and prevents cellular senescence (Tasselli et al. 2016). SIRT6 is also regarded as a “longevity protein”, largely due to its anti-aging properties. SIRT6-knockout mice showed a relatively shorter lifespan and copied many phenotypes associated with premature aging syndrome (Mostoslavsky et al. 2006). On the other hand, overexpression of SIRT6 can significantly increase lifespan of male mice only not in female mice, though the underlined mechanism remains unclear (Kanfi et al. 2012). Moreover, SIRT6 facilitates DNA repair signaling and thereby promotes genome stability through modulating several targets. For example, upon DNA damage, SIRT6 helps to stabilize DNA-dependent protein kinase (DNA-PK) at the break sites to promote DNA damage repair (McCord et al. 2009). SIRT6 also deacetylates C-terminal binding protein interacting protein (CtIP) in response to DNA damage and facilitates CtIP-mediated DNA double-strand break (DSB) repair via homologous recombination (HR) (Kaidi et al. 2010). Additionally, SIRT6 helps to recruit chromatin remodeler SNF2H to break sites and increase the chromatin accessibility to promote DSB repair (Toiber et al. 2013). Under oxidative stress, SIRT6 can mono-ADP-ribosylate and activate PARP1 to facilitate efficient DSB repair (Mao et al. 2011). Despite the progresses in the understanding of SIRT6 functions, little is known about how SIRT6 itself is regulated. Recent advances in the regulation of SIRT6 via post-translational modifications are summarized below:
Phosphorylation
SIRT6 was found to be phosphorylated at S338 in several breast cancer cell lines by kinase AKT1. SIRT6-S338 phosphorylation increases the interaction between SIRT6 and E3 ligase MDM2, therefore promoting the degradation of SIRT6. Ectopic expression of phosphorylation mutant SIRT6 (SIRT6-S338A) inhibited breast cancer cells proliferation in culture and injecting these cells into nude mice decreased the breast tumor formation. In addition, SIRT6 S338 phosphorylation level is inversely correlated with the disease progression in breast cancer patient (Thirumurthi et al. 2014). Thus, SIRT6-S338 phosphorylation may serve as a bio-marker to evaluate breast cancer progression. In a more recently published work, Michael et al. revealed a mechanism through which SIRT6 is activated in response to oxidative stress and promotes DSB repair. Upon oxidative stress, c-Jun N-terminal kinase (JNK) phosphorylates SIRT6 on S10, and this modification facilitates SIRT6 recruitment to DNA damage sites. Furthermore, SIRT6 S10 phosphorylation activates SIRT6 ADP-mon-ribosyltransferase activity towards PARP1 and is crucial for efficient PARP1 recruitment to DSB sites. Interestingly, in the same study, the authors observed that although PARP1 recruitment is nearly abolished in SIRT6-deficient cells, the recruitment of SIRT6 to damage sites was hardly affected in PARP1 knockout cells, indicating that SIRT6 recruitment to DSB sites is a very early event during DNA repair in response to oxidative stress (Van Meter et al. 2016). Together, these results suggested that SIRT6 activity can be post-translationally regulated in response to certain biological stimuli.
It is worth noting that in addition to the two sites mentioned above, other residues on SIRT6, like T294, S303 and S330, can also be potentially phosphorylated as identified by several proteomic studies (Bian et al. 2014; Dephoure et al. 2008; Miteva and Cristea 2014; Olsen et al. 2010). However, the functional relevance of these phosphorylation needs further investigation.
SUMOylation
The regulatory role of SUMOylation on SIRT6 was found only recently. Specifically, SIRT6 is SUMOylated by SUMO1 at four lysine residues K296, K300, K316 and K332 without affecting its nuclear localization. SIRT6 carrying SUMOylation-deficiency mutations selectively decreases its deacetylase activity towards H3K56ac, but not another histone substrate, H3K9ac. SIRT6 has previously been shown to act as a tumor suppressor at least partially through repressing the expression of c-Myc downstream genes (Sebastian et al. 2012). However, SIRT6 with SUMOylation-deficiency mutations decreases its recruitment to c-Myc target gene locus, therefore attenuating SIRT6 tumor suppressive function (Cai et al. 2016). In summary, SUMOylation at specific sites can suppress SIRT6 activity towards certain substrate and affect its function in tumorigenesis.
Ubiquitination
Two independent studies reported that SIRT6 can be regulated through ubiquitination. Ronnebaum et al. found that carboxyl terminus of Hsp70-interacting protein (CHIP) can non-canonically ubiquitinate SIRT6 at K170, which inhibits SIRT6 ubiquitination by other proteins, thereby preventing SIRT6 from proteasomal degradation. The authors further pointed out that CHIP improves SIRT6 stability to potentiate SIRT6 function in DNA repair, inflammation and metabolic regulation (Ronnebaum et al. 2013). In another report, ubiquitin-specific peptidase 10 (USP10) was identified to deubiquitinate SIRT6, thus stabilizing it in colon cancer cells. By increasing SIRT6 stability, USP10 decreases oncogene c-Myc transcriptional activity, reduces cancer cell proliferation and growth and, therefore, inhibits tumor formation (Lin et al. 2013). Together, current evidence suggests that ubiquitination can modulate SIRT6 function, mainly through affecting its protein stability.
Nitration
Protein tyrosine nitration involves addition of a nitro group to tyrosine residue on targeted protein. Recently, Hu et al. reported that when treating HEK293 cells or human retinal microvascular endothelial cells with 3-morpholinosydnonimine (SIN-1, a peroxynitrite donor which can produce nitric oxide and superoxide), SIRT6 showed an increased tyrosine nitration level. This modification occurs at SIRT6 Y257 and increased tyrosine nitration results in decreased catalytic activity of SIRT6. In addition, significantly increased SIRT6 nitration and decreased deacetylase activity of SIRT6 were also found in an endotoxin-induced retinal inflammation mice model, indicating that SIRT6 nitration is correlated to retinal inflammation. In summary, tyrosine nitration may act as a novel regulatory mechanism to negatively control SIRT6 activity (Hu et al. 2015).
SIRT7
Out of three nuclear sirtuins, SIRT7 is the least understood one. Its roles in maintaining genomic integrity, rDNA transcription, tumorigenesis and aging process just emerged recently. SIRT7 exhibits deacetylase activity specifically toward H3K18ac. H3K18ac deacetylation by SIRT7 at specific promotors represses the transcription of targeting genes (Barber et al. 2012). As a nuclear sirtuin, SIRT7 is predominantly enriched in nucleoli, where it is associated with RNA polymerase I (Pol-I) and upstream binding factor UBF to promote rDNA transcription (Ford et al. 2006; Grob et al. 2009). A study recently also reported that SIRT7 facilitates Pol-I-mediated rDNA transcription by deacetylating polymerase-associated factor 53 (PAF53), one of the RNA polymerase I subunits (Chen et al. 2013). Mechanistically, deacetylation of PAF53 leads to increased occupancy of Pol-I on rDNA and promotes rDNA transcription. Under energy stress conditions, SIRT7 is relocated into nucleoplasm resulting in hyperacetylation of PAF53 and inhibits pre-rRNA synthesis (Chen et al. 2013). In addition, SIRT7 also play an essential role in pre-rRNA processing. SIRT7 was reported to associate with small nuclear ribonucleoproteins (snoRNPs), an RNA–protein complex which is required for pre-rRNA processing and rRNA maturation. Specifically, SIRT7 can deacetylate U3–55k, a core component of snoRNPs, and deacetylation of U3–55k promotes its interaction with U3 snoRNA, which is an essential step for pre-rRNA processing. Re-localization of SIRT7 into nucleoplasm upon stress upregulates U3–55k acetylation and disrupts its association with U3 snoRNA, therefore inhibiting the pre-rRNA processing (Chen et al. 2016). These studies revealed multiple roles for SIRT7 in ribosome biogenesis, regulating both pre-rRNA synthesis and processing. On the other hand, SIRT7 also involves in the genome maintenance. SIRT7 was found to be recruited to DNA damage sites through a PARP1-dependent manner and it deacetylates local H3K18ac to help p53 binding protein 1 (53BP1) recruitment, therefore promoting efficient DNA damage repair through NHEJ (Vazquez et al. 2016). In addition, SIRT7 can also mediate desuccinylation of H3K122 at DNA damage sites to promote condensation of local chromatin and DNA repair (Li et al. 2016). Our understanding on the post-translational modifications of SIRT7 and their functional relevance currently is very limited, as summarized below.
Phosphorylation
The C-terminal extension of SIRT7 contains a CDK1 (cyclin-dependent kinase 1) phosphorylation site at residue 344–348 (ATPLR). It has been shown that SIRT7 can be phosphorylated and inactivated during mitosis through CDK1/cyclin B pathway. As cell cycle processes, cyclin B is gradually degraded resulting in CDK1 inactivation. SIRT7 is then dephosphorylated by an unknown phosphatase, regains its function upon exit from mitosis and the rDNA transcription resumes. These results suggest that the CDK1/cyclin B pathway can modulate ribosomal gene transcription by post-translationally regulating SIRT7 (Grob et al. 2009). Recently, another study reported that AMPK can phosphorylate SIRT7 at T153 under energy starvation condition and promote cell survival by limiting SIRT7 function (Sun et al. 2016). Upon glucose deprivation, SIRT7-T153 phosphorylation level is increased and promotes the association between SIRT7 and a member of the 11S proteasome activators, REGγ. Binding to REGγ promotes SIRT7 subcellular redistribution from nucleolus to nucleoplasm and protein degradation, therefore limiting rDNA transcription and preventing over-consumption of energy. In summary, AMPK-dependent phosphorylation of SIRT7 acts as an important mechanism for REGγ-mediated SIRT7 subcellular redistribution and degradation, contributing to proper regulation of metabolic homeostasis under energy stress conditions (Sun et al. 2016). SIRT7 phosphorylation is also increased upon treatment with angiotensin-II (Ang-II), which stimulates cardiac fibroblasts conversion to myofibroblasts, thereby leading to cardiac fibrosis and impaired cardiac functions. The authors provided evidence that the enhanced SIRT7 phosphorylation level may promote cardiac fibrosis by activating SMAD2 and ERK pathways (Wang et al. 2017). However, the exact mechanism by which SIRT7 phosphorylation contributes to the activation of these two pathways to promote cardiac fibrosis is undefined in the study, which needs further investigation.
Methylation
SIRT7 was found to be methylated by protein arginine methyltransferase 6 (PRMT6) in a recent study (Yan et al. 2018). Specifically, PRMT6 can directly interact with and methylate SIRT7 at R388 both in vivo and in vitro. SIRT7 methylation at R388 leads to the decrease in its deacetylase activity towards H3K18ac without affecting its subcellular localization. Functionally, PRMT6-dependent methylation of SIRT7 at R388 impairs SIRT7 deacetylase activity and maintains H3K18ac level in the promoter region of genes related to mitochondria biogenesis, therefore regulating mitochondria function and respiration. Moreover, the methylation level of SIRT7 seems to be controlled by glucose availability or activity of AMPK. High glucose leads to increased SIRT7 R388 methylation in MEF cells and liver tissue. On the contrary, glucose deprivation suppresses SIRT7 R388 methylation in MEF cells. Further experiments showed that glucose starvation activates AMPK, and AMPK activation disrupts the PRMT6-SIRT7 interaction resulting in SIRT7 hypomethylation and decreased mitochondria function to balance energy consumption. Taken together, PRMT6-mediated methylation of SIRT7 may serve as an important mechanism in energy homeostasis by coupling glucose sensing and mitochondria biogenesis (Yan et al. 2018).
Ubiquitination
In a recent study, Jiang et al. found that ubiquitin-specific peptidase 7 (USP7) can remove polyubiquitin chains from residue K63 of SIRT7. Deubiquination by USP7 does not affect SIRT7 protein stability but represses its deacetylase activity. Further functional analysis shows that USP7 and SIRT7 coordinate to regulate the expression of genes involved in gluconeogenesis, including gene-encoded glucose-6-phosphatase catalytic subunit (G6PC) protein. Specifically, SIRT7 targets G6PC promoter loci through ETS domain-containing protein Elk-4 (ELK4), deacetylates local H3K18ac and promotes expression of G6PC. Deubiquination by USP7 can antagonize SIRT7 function during this process. In summary, USP7 acts as a negative modulator of SIRT7 through deubiquitination and inhibit SIRT7-mediated gluconeogenesis-related gene expression (Jiang et al. 2017).
Concluding remarks
In this review, we summarize the current available knowledge on how nuclear sirtuins are regulated through post-translational modifications. It should be noticed that most of the regulatory effects mentioned above are only examined in certain cell types or under specific biological conditions. Whether these mechanisms are also applicable to other cell types or cellular conditions remains largely unknown and need to be further validated. In addition, except SIRT1, our understanding to the post-translational modifications and their potential impacts on SIRT6 and SIRT7 is quite limited. Considering the emerging roles of these two sirtuins in many vital cellular events, it is reasonable to speculate that there are still unknown post-translational modifications existing that may regulate their functions, which warrant investigation in the future. As dysregulation of nuclear sirtuins is often associated with development of several major diseases, such as metabolic syndrome, cancer and aging, further understanding of how they are regulated, both before and after translation, will not only provide new insights into their fundamental physiological functions, but also have important clinical implication.
References
Ahuja, N., Schwer, B., Carobbio, S., Waltregny, D., North, B. J., Castronovo, V., et al. (2007). Regulation of insulin secretion by SIRT4, a mitochondrial ADP-ribosyltransferase. The Journal of Biological Chemistry,282, 33583–33592.
Back, J. H., Rezvani, H. R., Zhu, Y., Guyonnet-Duperat, V., Athar, M., Ratner, D., et al. (2011). Cancer cell survival following DNA damage-mediated premature senescence is regulated by mammalian target of rapamycin (mTOR)-dependent Inhibition of sirtuin 1. The Journal of Biological Chemistry,286, 19100–19108.
Bai, B., Liang, Y., Xu, C., Lee, M. Y., Xu, A., Wu, D., et al. (2012). Cyclin-dependent kinase 5-mediated hyperphosphorylation of sirtuin-1 contributes to the development of endothelial senescence and atherosclerosis. Circulation,126, 729–740.
Barber, M. F., Michishita-Kioi, E., Xi, Y., Tasselli, L., Kioi, M., Moqtaderi, Z., et al. (2012). SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature,487, 114–118.
Bian, Y., Song, C., Cheng, K., Dong, M., Wang, F., Huang, J., et al. (2014). An enzyme assisted RP-RPLC approach for in-depth analysis of human liver phosphoproteome. Journal of Proteomics,96, 253–262.
Buler, M., Andersson, U., & Hakkola, J. (2016). Who watches the watchmen? Regulation of the expression and activity of sirtuins. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology,30, 3942–3960.
Cai, J., Zuo, Y., Wang, T., Cao, Y., Cai, R., Chen, F. L., et al. (2016). A crucial role of SUMOylation in modulating Sirt6 deacetylation of H3 at lysine 56 and its tumor suppressive activity. Oncogene,35, 4949–4956.
Caito, S., Rajendrasozhan, S., Cook, S., Chung, S., Yao, H., Friedman, A. E., et al. (2010). SIRT1 is a redox-sensitive deacetylase that is post-translationally modified by oxidants and carbonyl stress. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology,24, 3145–3159.
Chalkiadaki, A., & Guarente, L. (2015). The multifaceted functions of sirtuins in cancer. Nature Reviews Cancer,15, 608–624.
Chen, S., Blank, M. F., Iyer, A., Huang, B., Wang, L., Grummt, I., et al. (2016). SIRT7-dependent deacetylation of the U3–55k protein controls pre-rRNA processing. Nature Communications,7, 10734.
Chen, S., Seiler, J., Santiago-Reichelt, M., Felbel, K., Grummt, I., & Voit, R. (2013). Repression of RNA polymerase I upon stress is caused by inhibition of RNA-dependent deacetylation of PAF53 by SIRT7. Molecular Cell,52, 303–313.
Choi, S. E., Kwon, S., Seok, S., Xiao, Z., Lee, K.-W., Kang, Y., et al. (2017). Obesity-linked phosphorylation of SIRT1 by casein kinase 2 inhibits its nuclear localization and promotes fatty liver. Molecular and Cellular Biology,37, e00006–00017.
Choudhary, C., Kumar, C., Gnad, F., Nielsen, M. L., Rehman, M., Walther, T. C., et al. (2009). Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science,325, 834–840.
Conrad, E., Polonio-Vallon, T., Meister, M., Matt, S., Bitomsky, N., Herbel, C., et al. (2016). HIPK2 restricts SIRT1 activity upon severe DNA damage by a phosphorylation-controlled mechanism. Cell Death and Differentiation,23, 110–122.
Dalle-Donne, I., Aldini, G., Carini, M., Colombo, R., Rossi, R., & Milzani, A. (2006). Protein carbonylation, cellular dysfunction, and disease progression. Journal of Cellular and Molecular Medicine,10, 389–406.
Dephoure, N., Zhou, C., Villen, J., Beausoleil, S. A., Bakalarski, C. E., Elledge, S. J., et al. (2008). A quantitative atlas of mitotic phosphorylation. Proceedings of the National Academy of Sciences of the United States of America,105, 10762–10767.
Dowling, D. P., Gantt, S. L., Gattis, S. G., Fierke, C. A., & Christianson, D. W. (2008). Structural studies of human histone deacetylase 8 and its site-specific variants complexed with substrate and inhibitors. Biochemistry,47, 13554–13563.
Du, J., Zhou, Y., Su, X., Yu, J. J., Khan, S., Jiang, H., et al. (2011). Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science,334, 806–809.
Fang, J., Ianni, A., Smolka, C., Vakhrusheva, O., Nolte, H., Krüger, M., et al. (2017). Sirt7 promotes adipogenesis in the mouse by inhibiting autocatalytic activation of Sirt1. Proceedings of the National Academy of Sciences,114, E8352–E8361.
Feldman, J. L., Baeza, J., & Denu, J. M. (2013). Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. The Journal of Biological Chemistry,288, 31350–31356.
Finnin, M. S., Donigian, J. R., Cohen, A., Richon, V. M., Rifkind, R. A., Marks, P. A., et al. (1999). Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature,401, 188–193.
Flick, F., & Luscher, B. (2012). Regulation of sirtuin function by posttranslational modifications. Frontiers in Pharmacology,3, 29.
Ford, E., Voit, R., Liszt, G., Magin, C., Grummt, I., & Guarente, L. (2006). Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes & Development,20, 1075–1080.
Ford, J., Ahmed, S., Allison, S., Jiang, M., & Milner, J. (2008). JNK2-dependent regulation of SIRT1 protein stability. Cell Cycle,7, 3091–3097.
Gantt, S. L., Joseph, C. G., & Fierke, C. A. (2010). Activation and inhibition of histone deacetylase 8 by monovalent cations. The Journal of Biological Chemistry,285, 6036–6043.
Gerhart-Hines, Z., Dominy, J. E., Jr., Blattler, S. M., Jedrychowski, M. P., Banks, A. S., Lim, J. H., et al. (2011). The cAMP/PKA pathway rapidly activates SIRT1 to promote fatty acid oxidation independently of changes in NAD(+). Molecular Cell,44, 851–863.
Ghosh, S., & Zhou, Z. (2015). SIRTain regulators of premature senescence and accelerated aging. Protein & Cell,6, 322–333.
Grob, A., Roussel, P., Wright, J. E., McStay, B., Hernandez-Verdun, D., & Sirri, V. (2009). Involvement of SIRT7 in resumption of rDNA transcription at the exit from mitosis. Journal of Cell Science,122, 489–498.
Gu, W., & Roeder, R. G. (1997). Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell,90, 595–606.
Guo, X., Williams, J. G., Schug, T. T., & Li, X. (2010). DYRK1A and DYRK3 promote cell survival through phosphorylation and activation of SIRT1. The Journal of Biological Chemistry,285, 13223–13232.
Hu, S., Liu, H., Ha, Y., Luo, X., Motamedi, M., Gupta, M. P., et al. (2015). Posttranslational modification of Sirt6 activity by peroxynitrite. Free Radical Biology & Medicine,79, 176–185.
Imai, S., Armstrong, C. M., Kaeberlein, M., & Guarente, L. (2000). Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature,403, 795–800.
Jackson, M. D., & Denu, J. M. (2002). Structural identification of 2'- and 3'-O-acetyl-ADP-ribose as novel metabolites derived from the Sir2 family of beta -NAD+-dependent histone/protein deacetylases. The Journal of Biological Chemistry,277, 18535–18544.
Jiang, H., Khan, S., Wang, Y., Charron, G., He, B., Sebastian, C., et al. (2013). SIRT6 regulates TNF-alpha secretion through hydrolysis of long-chain fatty acyl lysine. Nature,496, 110–113.
Jiang, L., Xiong, J., Zhan, J., Yuan, F., Tang, M., Zhang, C., et al. (2017). Ubiquitin-specific peptidase 7 (USP7)-mediated deubiquitination of the histone deacetylase SIRT7 regulates gluconeogenesis. The Journal of Biological Chemistry,292, 13296–13311.
Kaidi, A., Weinert, B. T., Choudhary, C., & Jackson, S. P. (2010). Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science,329, 1348–1353.
Kanfi, Y., Naiman, S., Amir, G., Peshti, V., Zinman, G., Nahum, L., et al. (2012). The sirtuin SIRT6 regulates lifespan in male mice. Nature,483, 218–221.
Kang, H., Jung, J. W., Kim, M. K., & Chung, J. H. (2009). CK2 is the regulator of SIRT1 substrate-binding affinity, deacetylase activity and cellular response to DNA-damage. PLoS One,4, e6611.
Kang, H., Suh, J. Y., Jung, Y. S., Jung, J. W., Kim, M. K., & Chung, J. H. (2011). Peptide switch is essential for Sirt1 deacetylase activity. Molecular Cell,44, 203–213.
Kim, E. J., Kho, J. H., Kang, M. R., & Um, S. J. (2007). Active regulator of SIRT1 cooperates with SIRT1 and facilitates suppression of p53 activity. Molecular Cell,28, 277–290.
Kim, J. E., Chen, J., & Lou, Z. (2008). DBC1 is a negative regulator of SIRT1. Nature,451, 583–586.
Kim, S. C., Sprung, R., Chen, Y., Xu, Y., Ball, H., Pei, J., et al. (2006). Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Molecular Cell,23, 607–618.
Kornberg, M. D., Sen, N., Hara, M. R., Juluri, K. R., Nguyen, J. V., Snowman, A. M., et al. (2010). GAPDH mediates nitrosylation of nuclear proteins. Nature Cell Biology,12, 1094–1100.
Lau, A. W., Liu, P., Inuzuka, H., & Gao, D. (2014). SIRT1 phosphorylation by AMP-activated protein kinase regulates p53 acetylation. American Journal of Cancer Research,4, 245–255.
Lee, C. W., Wong, L. L., Tse, E. Y., Liu, H. F., Leong, V. Y., Lee, J. M., et al. (2012). AMPK promotes p53 acetylation via phosphorylation and inactivation of SIRT1 in liver cancer cells. Cancer Research,72, 4394–4404.
Li, L., Shi, L., Yang, S., Yan, R., Zhang, D., Yang, J., et al. (2016). SIRT7 is a histone desuccinylase that functionally links to chromatin compaction and genome stability. Nature Communications,7, 12235.
Lin, Z., Yang, H., Kong, Q., Li, J., Lee, S. M., Gao, B., et al. (2012). USP22 antagonizes p53 transcriptional activation by deubiquitinating Sirt1 to suppress cell apoptosis and is required for mouse embryonic development. Molecular Cell,46, 484–494.
Lin, Z., Yang, H., Tan, C., Li, J., Liu, Z., Quan, Q., et al. (2013). USP10 antagonizes c-Myc transcriptional activation through SIRT6 stabilization to suppress tumor formation. Cell Reports,5, 1639–1649.
Liszt, G., Ford, E., Kurtev, M., & Guarente, L. (2005). Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. The Journal of Biological Chemistry,280, 21313–21320.
Liu, X., Wang, D., Zhao, Y., Tu, B., Zheng, Z., Wang, L., et al. (2011). Methyltransferase Set7/9 regulates p53 activity by interacting with Sirtuin 1 (SIRT1). Proceedings of the National Academy of Sciences of the United States of America,108, 1925–1930.
Mao, Z., Hine, C., Tian, X., Van Meter, M., Au, M., Vaidya, A., et al. (2011). SIRT6 promotes DNA repair under stress by activating PARP1. Science,332, 1443–1446.
McCord, R. A., Michishita, E., Hong, T., Berber, E., Boxer, L. D., Kusumoto, R., et al. (2009). SIRT6 stabilizes DNA-dependent Protein Kinase at chromatin for DNA double-strand break repair. Aging,1, 109–121.
Michishita, E., McCord, R. A., Berber, E., Kioi, M., Padilla-Nash, H., Damian, M., et al. (2008). SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature,452, 492–496.
Michishita, E., Park, J. Y., Burneskis, J. M., Barrett, J. C., & Horikawa, I. (2005). Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Molecular Biology of the Cell,16, 4623–4635.
Min, J., Landry, J., Sternglanz, R., & Xu, R. M. (2001). Crystal structure of a SIR2 homolog-NAD complex. Cell,105, 269–279.
Miteva, Y. V., & Cristea, I. M. (2014). A proteomic perspective of Sirtuin 6 (SIRT6) phosphorylation and interactions and their dependence on its catalytic activity. Molecular & Cellular Proteomics,13, 168–183.
Mostoslavsky, R., Chua, K. F., Lombard, D. B., Pang, W. W., Fischer, M. R., Gellon, L., et al. (2006). Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell,124, 315–329.
Nakamura, T., & Lipton, S. A. (2013). Emerging role of protein-protein transnitrosylation in cell signaling pathways. Antioxidants & Redox Signaling,18, 239–249.
Nasrin, N., Kaushik, V. K., Fortier, E., Wall, D., Pearson, K. J., de Cabo, R., et al. (2009). JNK1 phosphorylates SIRT1 and promotes its enzymatic activity. PLoS One,4, e8414.
Olsen, J.V., Vermeulen, M., Santamaria, A., Kumar, C., Miller, M.L., Jensen, L.J., Gnad, F., Cox, J., Jensen, T.S., Nigg, E.A., et al. (2010). Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Science signaling3, ra3.
Peng, C., Lu, Z., Xie, Z., Cheng, Z., Chen, Y., Tan, M., et al. (2011). The first identification of lysine malonylation substrates and its regulatory enzyme. Molecular & Cellular Proteomics,10(M111), 012658.
Peng, L., Yuan, Z., Li, Y., Ling, H., Izumi, V., Fang, B., et al. (2015). Ubiquitinated sirtuin 1 (SIRT1) function is modulated during DNA damage-induced cell death and survival. The Journal of Biological Chemistry,290, 8904–8912.
Rahman, S., & Islam, R. (2011). Mammalian Sirt1: insights on its biological functions. Cell Communication and Signaling,9, 11.
Revollo, J. R., & Li, X. (2013). The ways and means that fine tune Sirt1 activity. Trends in Biochemical Sciences,38, 160–167.
Ronnebaum, S. M., Wu, Y., McDonough, H., & Patterson, C. (2013). The ubiquitin ligase CHIP prevents SirT6 degradation through noncanonical ubiquitination. Molecular and Cellular Biology,33, 4461–4472.
Sasaki, T., Maier, B., Koclega, K. D., Chruszcz, M., Gluba, W., Stukenberg, P. T., et al. (2008). Phosphorylation regulates SIRT1 function. PLoS One,3, e4020.
Sauve, A. A., Celic, I., Avalos, J., Deng, H., Boeke, J. D., & Schramm, V. L. (2001). Chemistry of gene silencing: the mechanism of NAD+-dependent deacetylation reactions. Biochemistry,40, 15456–15463.
Sebastian, C., Zwaans, B. M., Silberman, D. M., Gymrek, M., Goren, A., Zhong, L., et al. (2012). The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell,151, 1185–1199.
Smith, B. C., & Denu, J. M. (2006). Sir2 protein deacetylases: evidence for chemical intermediates and functions of a conserved histidine. Biochemistry,45, 272–282.
Somoza, J.R., Skene, R.J., Katz, B.A., Mol, C., Ho, J.D., Jennings, A.J., Luong, C., Arvai, A., Buggy, J.J., Chi, E., et al. (2004). Structural snapshots of human HDAC8 provide insights into the Class I histone deacetylases. Structure 12, 1325–1334.
Sun, L., Fan, G., Shan, P., Qiu, X., Dong, S., Liao, L., et al. (2016). Regulation of energy homeostasis by the ubiquitin-independent REGgamma proteasome. Nature Communications,7, 12497.
Tan, M., Peng, C., Anderson, K. A., Chhoy, P., Xie, Z., Dai, L., et al. (2014). Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metabolism,19, 605–617.
Tasselli, L., Xi, Y., Zheng, W., Tennen, R. I., Odrowaz, Z., Simeoni, F., et al. (2016). SIRT6 deacetylates H3K18ac at pericentric chromatin to prevent mitotic errors and cellular senescence. Nature Structural & Molecular Biology,23, 434.
Thirumurthi, U., Shen, J., Xia, W., LaBaff, A.M., Wei, Y., Li, C.W., Chang, W.C., Chen, C.H., Lin, H.K., Yu, D., et al. (2014). MDM2-mediated degradation of SIRT6 phosphorylated by AKT1 promotes tumorigenesis and trastuzumab resistance in breast cancer. Science signaling7, ra71.
Toiber, D., Erdel, F., Bouazoune, K., Silberman, D. M., Zhong, L., Mulligan, P., et al. (2013). SIRT6 recruits SNF2H to DNA break sites, preventing genomic instability through chromatin remodeling. Molecular Cell,51, 454–468.
Van Meter, M., Simon, M., Tombline, G., May, A., Morello, T. D., Hubbard, B. P., et al. (2016). JNK Phosphorylates SIRT6 to Stimulate DNA Double-Strand Break Repair in Response to Oxidative Stress by Recruiting PARP1 to DNA Breaks. Cell Reports,16, 2641–2650.
Vannini, A., Volpari, C., Filocamo, G., Casavola, E. C., Brunetti, M., Renzoni, D., et al. (2004). Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proceedings of the National Academy of Sciences of the United States of America,101, 15064–15069.
Vazquez, B. N., Thackray, J. K., Simonet, N. G., Kane-Goldsmith, N., Martinez-Redondo, P., Nguyen, T., et al. (2016). SIRT7 promotes genome integrity and modulates non-homologous end joining DNA repair. The EMBO Journal,35, 1488–1503.
Vidali, G., Gershey, E. L., & Allfrey, V. G. (1968). Chemical studies of histone acetylation. The distribution of epsilon-N-acetyllysine in calf thymus histones. The Journal of Biological Chemistry,243, 6361–6366.
Wang, H., Liu, S., Liu, S., Wei, W., Zhou, X., Lin, F., et al. (2017). Enhanced expression and phosphorylation of Sirt7 activates smad2 and ERK signaling and promotes the cardiac fibrosis differentiation upon angiotensin-II stimulation. PLoS One,12, e0178530.
Wen, L., Chen, Z., Zhang, F., Cui, X., Sun, W., Geary, G. G., et al. (2013). Ca2+/calmodulin-dependent protein kinase kinase beta phosphorylation of Sirtuin 1 in endothelium is atheroprotective. Proceedings of the National Academy of Sciences of the United States of America,110, E2420–2427.
Yamakuchi, M. (2012). MicroRNA Regulation of SIRT1. Frontiers in Physiology,3, 68.
Yan, W.W., Liang, Y.L., Zhang, Q.X., Wang, D., Lei, M.Z., Qu, J., He, X.H., Lei, Q.Y., Wang, Y.P. (2018). Arginine methylation of SIRT7 couples glucose sensing with mitochondria biogenesis. EMBO Reports, e46377.
Yang, B., Zwaans, B. M., Eckersdorff, M., & Lombard, D. B. (2009). The sirtuin SIRT6 deacetylates H3 K56Ac in vivo to promote genomic stability. Cell Cycle (Georgetown, Tex),8, 2662–2663.
Yang, Y., Fu, W., Chen, J., Olashaw, N., Zhang, X., Nicosia, S. V., et al. (2007). SIRT1 sumoylation regulates its deacetylase activity and cellular response to genotoxic stress. Nature Cell Biology,9, 1253–1262.
Yu, J., & Auwerx, J. (2009). The role of sirtuins in the control of metabolic homeostasis. Annals of the New York Academy of Sciences,1173(Suppl 1), E10–19.
Yuan, J., Luo, K., Liu, T., & Lou, Z. (2012). Regulation of SIRT1 activity by genotoxic stress. Genes & development,26, 791–796.
Zhao, W., Kruse, J. P., Tang, Y., Jung, S. Y., Qin, J., & Gu, W. (2008). Negative regulation of the deacetylase SIRT1 by DBC1. Nature,451, 587–590.
Zschoernig, B., & Mahlknecht, U. (2009). Carboxy-terminal phosphorylation of SIRT1 by protein kinase CK2. Biochemical and biophysical research communications,381, 372–377.
Author information
Affiliations
School of Biomedical Sciences, LKS Faculty of Medicine, The University of Hong Kong, Pok Fu Lam, Hong Kong
Kaiqiang Zhao & Zhongjun Zhou
Shenzhen Institute of Research and Innovation, The University of Hong Kong, Pok Fu Lam, Hong Kong
Kaiqiang Zhao & Zhongjun Zhou
Corresponding author
Correspondence to Zhongjun Zhou.
Rights and permissions
About this article
Cite this article
Zhao, K., Zhou, Z. Post-translational modifications of nuclear sirtuins. GENOME INSTAB. DIS. 1, 34–45 (2020). https://doi.org/10.1007/s42764-019-00001-x
Received01 November 2018
Revised17 January 2019
Accepted31 January 2019
Published08 March 2019
Issue DateJanuary 2020
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
Nuclear sirtuins
Post-translational modification
Deacetylation
regulation
用户登录
还没有账号?
立即注册