






ATM, DNA-PKcs and ATR: shaping development through the regulation of the DNA damage responses
Review Article
Demis Menolfi & Shan Zha Genome Instability & Disease 1,47–68(2020)
Abstract
Genomic integrity is critical for normal development, healthy aging and suppressing oncogenic transformation. The DNA damage response (DDR) is a complex network that is activated by DNA structural changes to preserve genome integrity. Situated at the apex of the mammalian DDR are three PI3-kinase-related protein kinases—ATM, DNA-PKcs and ATR. They are activated by different DNA lesions via direct binding to their unique sensor protein complexes (MRE11-RAD50-NBS1 for ATM, Ku70-Ku80/86 for DNA-PKcs and ATRIP-RPA for ATR) and phosphorylate a large number of partially overlapping substrates, including themselves and each other to promote DNA repair and regulate cell cycle checkpoints and tissue homeostasis. This review focuses on mouse models with deletion and point mutations of ATM, DNA-PKcs and ATR, and discusses how their activation mechanism and their kinase activity contribute to their unique, yet interactive roles in DNA repair in general and during tissue-specific development processes and how their deficiency leads to specific physiological and pathophysiological consequences.
Introduction
Genomic stability is essential for proper development, healthy aging and for the prevention of cancer. DNA double-strand breaks (DSBs) are the most severe type of DNA damages. However, DSBs are also unavoidable as obligatory intermediates during lymphocyte and germ cell development and as byproducts of normal DNA replication and responses to environmental insults. Unrepaired DSBs lead to cell cycle arrest, apoptosis and prevent functional maturation of lymphocytes and germ cells. Mis-repair of DSBs leads to genomic instability, including deletions, amplifications or chromosomal translocations, which serve as the basis for oncogenic selections. In addition to DSBs, replicating cells face additional challenges while trying to copy through repetitive sequences, secondary structures, protein road blocks and pre-existing DNA lesions. In mammalian cells, DNA damage response (DDR) network senses DNA lesions and coordinates DNA repair with other cellular events (e.g., transcription, replication, mitosis) to ensure genomic stability and eliminate cells with severe damages. At the pinnacle of the mammalian DDR are three phosphoinositide 3-kinase (PI3K)-related protein kinases (PIKKs)—ataxia–telangiectasia mutated (ATM), DNA-dependent protein kinase catalytic subunit (DNA-PKcs) and ATM and RAD3 related (ATR). They share similar domain organizations, a HEAT-repeat rich N-terminal segment followed by the conserved FRAP-ATM-TRRAP (FAT) domain, the kinase domain, the PIKK regulatory domain (PRD) and the FAT C-terminal motif (FATC) (Perry and Kleckner 2003; Blackford and Jackson 2017) (Fig. 1a). ATM and ATR are conserved in all eukaryotes and DNA-PKcs has recently evolved in vertebrates. As serine/threonine protein kinases, ATM, DNA-PKcs and ATR preferentially phosphorylate a serine or threonine residue followed by a glutamine (S/T-Q motif) (Kim et al. 1999). In addition to phosphorylate other substrates, activated ATM, DNA-PKcs and ATR also phosphorylate themselves (termed auto-phosphorylation) and each other. Activation of ATM, DNA-PKcs and ATR requires direct recruitment to DNA through their own sensor protein complex. In general, DNA-PKcs is activated by Ku70/Ku80 (KU86 in humans) at the end of DSBs (Singleton et al. 1999), ATM via MRE11-RAD50-NBS1 (MRN) complex in response to DSB and other DNA lesions (Ansel et al. 2004; Block and Lees-Miller 2005) and ATR through ATRIP-RPA at extended single strand DNA (ssDNA) (Zou and Elledge 2003) (Fig. 1b). The complete spectrum of DNA structures that can activate ATM is yet to be determined. In vitro, long DNA stretches (> 1 kb) and various other DNA structures activate ATM through MRE11-RAD50-NBS1 (MRN) complex (Ansel et al. 2004; Block and Lees-Miller 2005). Their unique activators and partially overlapping substrate pools contribute to the specific yet complementary functions in DNA repair, development and disease prevention. Here, we first discuss animal models with deletion or mutations in ATM, DNA-PKcs and ATR, including the newly generated kinase-dead models for all three PI3KKs, in an attempt to link their mechanism of action to physiological consequences and highlight both their signaling roles as well as their structural functions in maintaining genomic integrity in a tissue and biological processes specific manner (Tables 1, 2, 3). We will conclude by comparing their redundant vs unique functions uncovered by double deficiency or specific kinase inhibitors.
Fig. 1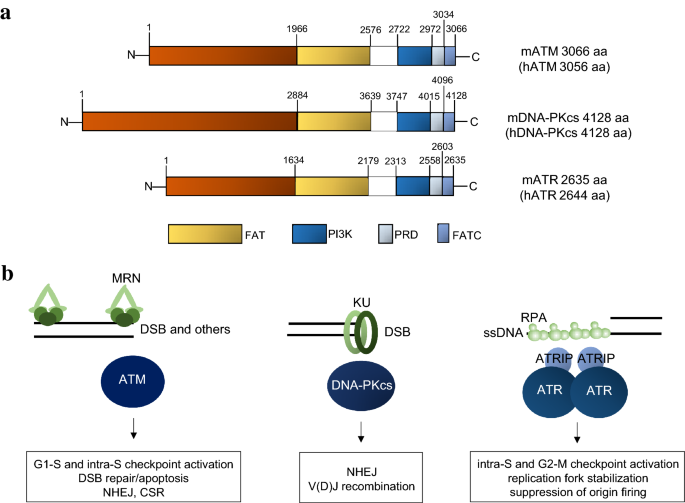
a Structural common domains of mouse and human ATM, DNA-PKcs and ATR with aminoacid position indicated. b DNA structures and factors required for ATM, DNA-PKcs and ATR recruitment in response to DNA damage
Full size image
Table 1 ATM mouse models

Table 2 DNA-PKcs mouse models

Table 3 ATR mouse models
ATM
ATM functions in the DNA double-strand break repair pathway
ATM is the master regulator of the cellular response to DSBs and is also activated by other abnormal DNA structures. MRN complex recognizes DSBs and other structural changes on DNA, and in turn recruits and activates ATM through direct interaction between the C-terminal of Nbs1 and ATM (Ansel et al. 2004; Block and Lees-Miller 2005; Uziel et al. 2003). Upon activation, ATM dimers undergo auto-phosphorylation (S367, S1893, S1981, S2996 and potentially other sites of human ATM) and become active monomers (Bakkenist and Kastan 2003; Lee and Paull 2005). Auto-phosphorylation at S1981 is widely used as a sensitive marker of ATM activation. However, the function of ATM auto-phosphorylation at specific sites seems to differ in different experimental systems (see below). Activated ATM phosphorylates hundreds of substrates, including, but not limited to, the C-terminal tail of histone H2AX, the cohesion subunit SMC1, the tumor suppressor protein p53, the checkpoint kinase Chk2. Together ATM and its substrates promote efficient and accurate DNA repair and coordinate other cellular functions (e.g., cell cycle, transcription) during repair to minimize genomic instability (Shiloh and Ziv 2013) (Fig. 2a).
Fig. 2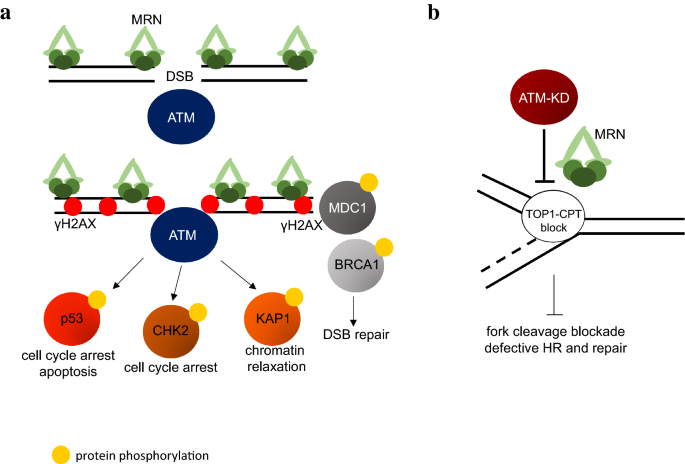
a Canonical functions of ATM during DSB repair. After DSB formation, MRN complex is recruited to the site of damage and recruits ATM protein, which undergoes activation (dimer to monomer transition) and phosphorylates several targets (e.g., p53, CHK2, KAP1, MDC1) necessary for cell cycle arrest, DNA repair, apoptosis. b ATM-KD protein has been shown to physically block strand cleavage at CPT-induced DSBs in an MRN-dependent manner, severely affecting downstream repair and homologous recombination (HR)
Full size imageAT patients: phenotypical and molecular characterization
ATM kinase was initially identified as the gene inactivated in patients with the ataxia–telangiectasia (A–T) syndrome. A–T is an autosomal recessive disorder that has an estimated prevalence of 1:40,000 to 1:100,000 in the world population (Swift et al. 1986). A–T patients are characterized by cerebellar ataxia, oculocutaneous telangiectasia (i.e., dilated blood vessels primarily in the ocular sclerae), and extreme sensitivity to ionizing radiation (IR) (Amirifar et al. 2019). Primary immunodeficiency affecting T cell development and antibody isotype switch are also prevalent (Amirifar et al. 2019). About 25% of A–T patients develop lymphoid malignancies, with a skew toward T cell leukemias in comparison to sporadic lymphomas (Boder 1985). Diseases of respiratory system, including recurrent sinopulmonary infections, also cause significant morbidity in the A–T population and were thought to be the result of declined neurological function and primary immunodeficiency (Schroeder et al. 2005; Schroeder and Zielen 2014).
Cells derived from A–T patients are hypersensitive to DNA DSB generating agents (e.g., etoposide and radiation), but can tolerate crosslinking agents (e.g., cisplatin), which have been successfully used to treat leukemia in A–T patients. Upon IR, A–T cells fail to activate the G2/M checkpoint and also show a much reduced G1/S arrest, in part due to the inability to phosphorylate p53 (Canman et al. 1998). Furthermore, A–T lymphocytes have an elevated amount of chromosomal translocations and recurrent inversions involving immunoglobulin (Ig) and T cell receptor (TCR) loci (Kojis et al. 1991; Meyn 1993).
ATM knockout mouse: similarities and differences with human patients
Since the cloning of Atm gene (Savitsky et al. 1995), four independent groups generated mouse models with complete loss of Atm (Barlow et al. 1996; Borghesani et al. 2000; Elson et al. 1996; Xu et al. 1996). They all have very similar phenotypes and, therefore, are referred to as the Atm−/− mice together. Atm−/− mice are born at the expected ratio and viable. Atm−/− mice recapitulate many clinical phenotypes of the A–T patients, including growth retardation (~ 25% smaller), male and female infertility associated with complete lack of mature gametes, and immunodeficiency characterized by a partial block at double positive (DP) to single positive transition in T cells (Borghesani et al. 2000) and defects in B cell class switch recombination (CSR) (Lumsden et al. 2004; Reina-San-Martin et al. 2004). Mechanistically, the infertility of male Atm−/− mice has been linked to accumulation of spo11 oligonucleotides, suggesting excessive endonuclease cleavage during the initiation of meiotic recombination (Di Giacomo et al. 2005). The lymphocyte development defects have been attributed to the critical role of ATM in DSB repair, which is necessary for the somatic assembly and subsequent modifications of the antigen receptor gene products. Atm−/− mice are also extremely sensitive to ionizing radiation and die shortly after (within 5 days) due to acute gastrointestinal toxicity (Barlow et al. 1996). Like A–T patient-derived cells, primary cells from Atm-deficient mice are also hypersensitive to IR, defective in cell cycle checkpoint controls and display premature senescence in culture (Barlow et al. 1996). But, despite a clear role of ATM in radiation-induced neuronal cell death in mice (Herzog et al. 1998), Atm-deficient mouse models do not develop spontaneous ataxia unless in other susceptibility backgrounds (Katyal et al. 2014; Lee et al. 2014). Mild alteration in Purkinje cells (Borghesani et al. 2000) and behavioral defects (Campbell et al. 2015) were noted in a subset of ATM null mice models.
It is worth noting that mice deficient in NBS1, necessary for ATM recruitment to and activation by DSBs, or mice expressing a humanized mutant NBS1 allele (recurrent in Nijmegen breakage syndrome patients) or a mutated MRE11 allele (in ATLD patients) suffer from severe developmental defects, but do not develop spontaneous cancer, likely due to essential and ATM-independent roles of NBS1, MRE11, and by extent MRN complex, in DNA repair (Difilippantonio et al. 2005; Theunissen et al. 2003; Williams et al. 2002). In this context, complete loss of MRE11, NBS1 or RAD50 leads to early embryonic lethality (Buis et al. 2008; Luo et al. 1999; Zhu et al. 2001).
Auto-phosphorylation of ATM is dispensable for mouse development and DNA repair
ATM auto-phosphorylation is a widely used marker for ATM activation. In human cells, alanine substitution at one or several ATM auto-phosphorylation sites compromise ATM activation (Bakkenist and Kastan 2003; Kozlov et al. 2003). Purified ATM activated in vitro undergoes the dimer to monomer transition independent of S1981 phosphorylation (Guo et al. 2010; Dupré 2006 #635). In an attempt to determine the physiological functions of auto-phosphorylation, Nussenzweig and colleagues generated transgenic mouse models that express mutated ATM with alanine substitution at the single S1987 (S1981 in humans) or three proposed auto-phosphorylation sites S367/S1899/S1987 (S367/S1893/S1981 in humans) from a bacterial artificial chromosome (BAC) (Aebersold et al. 2009; Daniel et al. 2008; Pellegrini et al. 2006). Mice lacking the endogens ATM and only expressing ATM from the BAC transgene (AtmTgS1987AAtm−/− or AtmTg3SAAtm−/−, single or triple S/A substitution) are fertile and display no lymphocytes development defects characteristic of Atm-deficient mice. Furthermore, the S/A mutant ATM is able to accumulate at sites of DNA damage, phosphorylates its substrate (e.g., Kap1) and activates the G2/M cell cycle checkpoints, suggesting that auto-phosphorylation at S367/S1893/S1981 is not required for ATM activation and activity in mice. It remains to be determined whether this dependency on auto-phosphorylation for full ATM activation in human cells, but not in mouse models, is due to difference between human vs mouse ATM or to adaptations during development that were not available in cell lines or other yet-to-be determined differences.
Mouse model expressing kinase-dead ATM is embryonic lethal
Given the large size of ATM and the existence of other well-characterized auto-phosphorylation sites, including Ser2996, within the kinase domain (Kozlov et al. 2011), mouse model expressing kinase-dead ATM was generated using BACs engineering (Daniel et al. 2012) (D2889A or Q2730P) or by knockin catalytically inactivating mutation (aspartate to alanine) in the catalytic site (D2880 in mouse corresponding to D2870 in human) (Yamamoto et al. 2012). Notably, knockin D2880A-mutated ATM is expressed at a slightly lower level than endogenous WT ATM, while the transgene alleles (D2889A and Q2730P) were selected to have near normal ATM protein levels. In contrast to normal development of Atm−/− mice, expression of kinase-dead ATM protein solely leads to early embryonic lethality (E9.5–10.5) in both knockin and transgenic mouse models. This finding provides an explanation for why the vast majority (~ 90%) of A–T patients carry truncating mutations with little or no detectable ATM protein expression. Missense ATM mutation with stable protein expression is quite rare in A–T patients and most leads to low protein expression with a few exceptions discussed below. Yet, Atm+/KD mice are fertile, of normal sized and do not show any defect in lymphocyte development, consistent with the lack of classical dominant negative effect in the presence of WT ATM. Somatic inactivation of the ATM conditional allele in AtmC/KD mice generated AtmKD/− lymphocytes, which display defects in both V(D)J recombination and CSR similar to that of Atm−/− cells, suggesting that ATM kinase activity, regardless of the presence or absence of ATM protein, is important for lymphocyte-specific gene rearrangements. Analyses of the AtmKD/− and AtmTgD2899AAtm−/− cells reveal severe genomic instability, especially increases of replication- or post-replication-associated chromosomal instabilities (Yamamoto et al. 2016), suggesting that the physical presence of the catalytically inactive ATM protein might block the access of other proteins to the DNA damage lesions, thereby impeding other repair events (Fig. 2b). The intermolecular auto-phosphorylation of the ATM-KD by the WT-ATM might explain why WT-ATM can relieve the inhibitory effects of ATM-KD in Atm+/KD mice and cells.
Malignancy risk in ATM deficient mouse models
Like A–T patients, Atm-null mice have greatly increased risk for T cell malignancies. By 4 months of age, almost all Atm-null mice succumb aggressive thymic lymphomas with recurrent t(12;14) translocations that are syntenic to the chromosome 14 inversions often developed in the peripheral T cells of A–T patients (Liyanage et al. 2000; Zha et al. 2010b). The discovery of aberrant TCR δ arrangement at the junction of the t(12;14) translocations and chromosomal 14 amplification (Jiang et al. 2015b) suggest an important role of ATM in the faithful repair of the V(D)J recombination generated DNA breaks. The critical role of ATM in V(D)J recombination is further revealed in mouse model lacking XLF, a non-essential member of the non-homologous end-joining (NHEJ) DNA pathway (Zha et al. 2011a). As such, while XLF-deficient mice have normal lymphocyte development, Atm and XLF double-deficient lymphocytes cannot complete V(D)J recombination and are blocked in early development. Correspondingly, somatic deletion of ATM in DP T cells, via Lck-Cre, completely prevents thymic lymphomas (Zha et al. 2010a) and inactivation of RAG recombinase that initiates V(D)J recombination significantly delays thymic lymphomas in Atm−/− mice (Petiniot et al. 2000, 2002). In addition to DNA repair, the checkpoint function of ATM also contributes to its tumor suppression ability, including, but not limited to, p53-independent checkpoint functions of ATM at the G2/M boundary and phosphorylation of p53 tumor suppressor at Serine 15 and possibly other sites to enforce the G1/S checkpoint (Canman et al. 1998). TP53-deficient mice also succumb to thymic lymphomas, but often with aneuploidy, not recurrent translocations (Bassing et al. 2003), consistent with the additional role of ATM in DNA repair during V(D)J recombination. In human cancers, TP53 inactivation is often mutually exclusive with ATM inactivation, supporting their partially overlapping function in DNA damage responses. Heterozygous Atm+/− mice also have increased sensitivity to IR, manifested by premature aging and decreased survival (Barlow et al. 1999b), consistent with the increased cancer risk in A–T carriers (Renwick et al. 2006).
While Atm−/− mice die exclusively from thymic lymphomas, mouse model carrying homozygous in frame deletion (7636del9) found in A–T patient expresses ATM protein and has a much delayed tumor kinetics and a prevalence of sarcoma in addition to thymic lymphomas (Spring et al. 2002). A recently described ATM null mouse model (caused by early frameshift by exon 4 deletion) seems to have a relatively decreased incidence of cancers and extended survival (Campbell et al. 2015). The penetration and the onset of lymphomas in Atm−/− mice can also be affected by the uptake of anti-oxidants (Reliene and Schiestl 2006). This complex heterogeneity at both genetic and environmental levels likely contributes to the broad and often variable spectrum of clinical phenotypes in A–T patients.
Although expression of ATM-KD solely is not compatible with embryonic development, analyses of TCGA database suggest that kinase domain point mutations are highly enriched in human cancer. Indeed, nearly 75% of cancer-associated ATM mutations are missense mutations and the majorities of them are in the kinase domain (Liu et al. 2012). In mouse model, inactivation of ATM conditional allele in AtmC/KD mice is more oncogenic than complete loss of ATM, resulting in a higher lymphoma frequency and earlier onset (Liu et al. 2012). Furthermore, AtmKD/− cells, including cancers, are hypersensitive to Topoisomerase I inhibitors (e.g., camptothecin, CPT), in part because ATM-KD protein physically blocks CPT-induced DSBs formation in an MRN-dependent manner, revealing one way by which ATM-KD can compromise replication-related DNA repair beyond the loss of ATM. In this context, while loss of ATM does not affect homologous recombination measured by the DR-GFP reporter, expression of kinase-dead ATM or ATM inhibition significantly suppresses HR (Chen et al. 2017; Rass et al. 2013), highlighting the structural function of ATM and the difference between ATM inhibition vs ATM deletion, which is also evident for DNA-PKcs and ATR.
It is worth noting that the cancer-associated ATM mutations, although enriched in the kinase domain, might have diverse impact on ATM protein levels and kinase activity. The c-terminal FATC domain, as well as the PI3K regulatory domain following the kinase domain, have all been implicated in regulating ATM kinase activity, protein stability and activation. Additional structural and functional characterizations would be necessary to understand the full spectrum and the physiological impact of ATM mutations in human cancer. Given the prevalence of ATM germline heterozygous mutations and their implication in cancer risk (Choi et al. 2016), understanding the biological impact of ATM mutations would provide the basis for guided therapy.
Beyond DSB repair: physiological functions of ATM during oxidative stress and signaling
Besides its important function in DSBs repair, emerging evidences suggest that ATM also regulates the responses to oxidative stresses that are independent of DNA damages. Using purified ATM protein, Paull and colleagues found that oxidative stress (e.g., hydrogen peroxide, H2O2) activates ATM in a dimeric conformation, independent of MRN and DNA (Guo et al. 2010). This is in contrast with the MRN-dependent activation of ATM in the presence of linear DNA, where the activation of ATM is often accompanied by the dimer to monomer transition (Lee and Paull 2004, 2005). Furthermore, they identified Cys2991, localized at the PI3Kinase regulatory domain immediately after the kinase domain, which forms intermolecular disulfide bond upon oxidative stresses. A point mutation of ATM (C2991L) effectively prevents oxidative stress-dependent activation of purified ATM without affecting MRN- and DNA-dependent activation (Guo et al. 2010). These studies provide the mechanistic basis for a DNA damage-independent activation of ATM by oxidative stresses.
Early on, it was noticed that tissues and cells from A–T patients have evidence of excessive protein and lipid oxidation (Barlow et al. 1999a; Reichenbach et al. 2002) and fibroblasts derived from A–T patients are hypersensitive to oxidizing agents (Shackelford et al. 2001; Yi et al. 1990). Atm−/− mice also have high reactive oxygen species (ROS) levels measured by fluorescent sensor in brain and hematological system (Kamsler et al. 2001; Quick and Dugan 2001). Hematopoietic stem cell dysfunction in Atm−/− mice was partially rescued by oral antioxidant N-acetyl-L-cysteine (NAC) (Ito et al. 2004). NAC also delayed the onset of lymphoma in ATM-deficient mice, suggesting a connection between increased/abnormal ROS and tumor development (Schubert et al. 2004). Based on these observations, chemo-preventive use of antioxidant has been proposed for A–T patients. Notably, the leading cause of lethality in A–T patients is pulmonary fibrosis and chronic obstructive pulmonary disease (COPD) (Schroeder et al. 2005; Schroeder and Zielen 2014). It remains unclear whether the role of ATM in regulating oxidative stress response contributes to the vulnerability of lung, an organ with high oxidative tension. Atm−/− mice succumb to lymphomas before the development of any spontaneous lung disease. But Atm-deficient alveolar epithelial cells have higher oxidative stress levels and oropharyngeal administration of bleomycin, a chelating agent that generates free radicals, induces severe inflammation, fibrosis and eventually irreversible damage in the lung of Atm−/− mice (Duecker et al. 2018). Since the ability for oxidative damage to trigger DNA-strand breaks, it remains challenging to determine how much of the lung or even neurological phenotype of A–T patients can be attributed to defects in oxidative stresses responses alone.
To understand where and how oxidative stress activated ATM functions in cells, phospho-proteomic studies have been performed upon H2O2 treatment (Kozlov et al. 2016). While the nuclear targets of ATM could potentially be caused by indirect DNA damage, this and other studies have identified a number of cytoplasmic targets of ATM that are phosphorylated upon oxidative stress (e.g., oxidative stress responsive 1, ORF1, and hepatoma-derived growth factor, HDGF). A study using xenopous extracts suggests that ATM can directly regulate the cytoplasmic pentose phosphate pathway (Cosentino et al. 2011). In mammalian cells, ATM is found to regulate tuberous sclerosis complex 2 (TSC2) in the cytoplasm to modulate mTORC1 function (Tripathi et al. 2013) and to phosphorylate PEX5 at Ser141 to promote autophagy of the peroxisome (Zhang et al. 2015). To complicate the picture, nuclear activation of ATM by DNA damage is also known to regulate cytoplasmic functions. For example, ATM contributes to IR-induced activation of NF-κB pathway through Nemo, MEK and several other proposed modalities (Fang et al. 2014; Li et al. 2001; Wu et al. 2010; Yang et al. 2011). ATM has also been reported to regulate Akt pathway in response to insulin stimulation and IR treatment (Viniegra et al. 2005) and to phosphorylate 4EBP1 protein, though promoting cell growth, in response to insulin signaling (Yang and Kastan 2000). Thus, it remains unclear how much of the cytoplasmic activation of ATM by H2O2 is caused by direct activation of ATM by oxidative stresses. What is also unknown is how the oxidative stress role of ATM contributes to physiological and pathophysiological function of ATM in preventing neuronal degeneration and immunodeficiency and in suppressing malignant transformation. Ideally, separation of function mutations that can distinguish the DNA damage vs oxidative stress induced activation of ATM would be helpful.
DNA-PKcs
The role of DNA-PKcs during classical non-homologous end-joining (cNHEJ)
In addition to its role in DNA damage responses, DNA-PKcs is best known as the vertebrate specific member of the cNHEJ. Classical NHEJ is one of the two major DNA DSB repair pathways in mammalian cells, with the other being homologous recombination (HR). In contrast to HR, which requires a homology template, often the sister chromatid, for repair, cNHEJ directly ligates DNA ends together with minimal processing and thus operates throughout the cell cycle. In mammalian cells, cNHEJ can be loosely divided into three steps: end-synapsis, end-processing and end-ligation. DNA-PKcs has been implicated in all three steps of cNHEJ.
Ku70 and Ku80 (Ku86 in humans) initiate cNHEJ by binding to the double-stranded DNA ends and, therefore, recruit the evolutionary conserved DNA ligase complex—XRCC4, DNA Ligase 4, XRCC4-like factor (XLF), for end-ligation. In vertebrates, DNA bound KU also recruits and activates DNA-PKcs. Among other roles, activated DNA-PKcs recruits and activates Artemis endonuclease to process DNA ends, including hairpins (Lieber 2010) (Fig. 3a). Consistent with this mode of activation, complete loss of DNA-PKcs abrogates end-processing, but not direct end-ligation in mouse models (Gao et al. 1998a; Kurimasa et al. 1999; Priestley et al. 1998). Mouse model expressing kinase-dead DNA-PKcs died in uterus with severe end-ligation defects (Jiang et al. 2015a), suggesting that once assembled to the DNA ends, the kinase activity of DNA-PK is necessary to license end-ligation (Fig. 3b). In addition, DNA-PKcs has also been implicated in the synaptic phase of NHEJ, since DNA-PKcs null cells have reduced ligation fidelity (Gao et al. 1998a; Jiang et al. 2015a) and in single molecule experiments, purified DNA-PKcs brings two DNA ends together independent of end-ligation (Graham et al. 2016; Reid et al. 2015). Two newer cNHEJ factors, PAXX and MRI were discovered recently. They are not absolutely essential for end-ligation or end-processing in otherwise wildtype cells, but XLF-deficient cells and animal models depend on PAXX and MRI for end-ligation during cNHEJ (Grundy et al. 2016; Hung et al. 2018; Kumar et al. 2016; Lescale et al. 2016; Liu et al. 2017; Ochi et al. 2015; Roy et al. 2015; Xing et al. 2015).
Fig. 3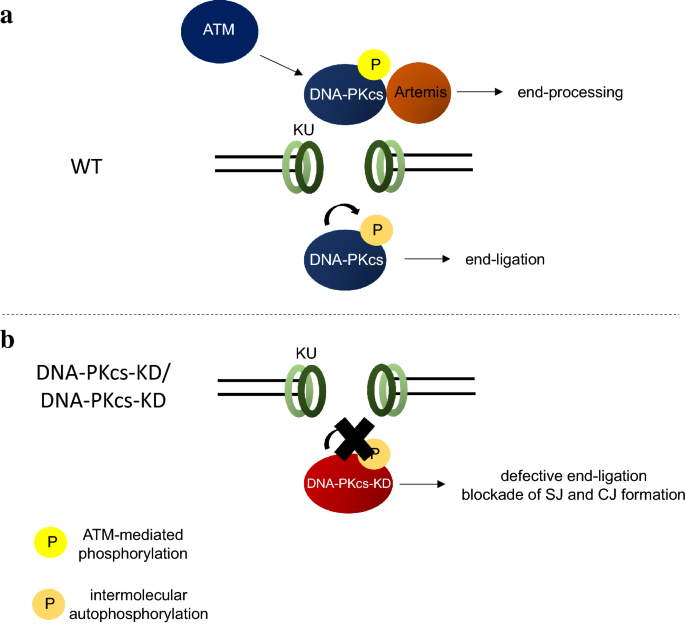
a KU recruits DNA-PKcs protein to DSBs. Different phosphorylation events have been shown to regulate DNA-PKcs functions. ATM (and DNA-PKcs) phosphorylation regulates DNA-PKcs recruitment of Artemis allowing proper end-processing during NHEJ, while DNA-PKcs auto-phosphorylation has been shown to control end-ligation (see text for details). b DNA-PKcs-KD protein is defective in auto-phosphorylation and this consequently leads to impaired end-ligation and both signal join (SJ) and coding join (CJ) formation
Full size imageThe critical role of DNA-PKcs and cNHEJ in lymphocyte development and maturation
In addition to general DSB repair, cNHEJ is exclusively required for the somatic assembly and subsequent modifications of the antigen receptor gene products in developing lymphocytes. Specifically, V(D)J recombination is a site-specific recombination process, restricted to the G1 phase of the cell cycle (Jiang et al. 2005), in which the variable regions of immunoglobulin (Ig) and T cell receptor (TCR), in B and T cells, respectively, are assembled by the juxtaposition of germline variable (V), diversity (D) and joining (J) gene segments. Lymphocyte-specific recombination activating genes (RAG1 and RAG2) initiate V(D)J recombination by introducing DSBs between the participating V/D/J gene segments and their flanking recombination signals sequences (RSS). RAG cleavage generates a pair of blunt recombination signal ends (SEs) and a pair of covalently sealed hairpin coding ends (CEs). While the two blunt SEs are directly and precisely ligated to each other via cNHEJ to form signal join (SJ), the hairpin coding ends have first to be opened by Artemis with the help of DNA-PKcs and then joined in a process that results in gain or loss of nucleotides to form the coding join (CJ), which encode the variable region exon of the Ig and TCR. While the joining of both SEs and CEs is exclusively mediated by the cNHEJ pathway, only CEs need hairpin opening, therefore, providing a physiological system that can distinguish end-ligation or end-processing defects during cNHEJ.
In peripheral lymphoid organs (e.g., spleen, lymph node and Peyer’s patch), naïve B cells undergo class switch recombination (CSR)—a rearrangement involving the constant region of the Ig heavy chain (IgH) gene to generate antibody with different effector functions (isotypes). Activation-induced deaminase (AID) initiates CSR by introducing base mismatch at the repetitive switching (S) region preceding each set of constant region exons. These mismatches are subsequently converted to DSBs. A DSB in the upstream Sµ region is joined to the one in the downstream switching region (e.g., IgG1) via cNHEJ to complete CSR. In contrast to V(D)J recombination, CSR can also be mediated by the alternative end-joining (A-EJ) pathway that preferentially uses microhomology (MH) at the junction, in cells lacking essential components of cNHEJ pathway (e.g., XRCC4, Ligase 4 or KU) (Boboila et al. 2010; Boulton and Jackson 1996; Kabotyanski et al. 1998; Wang et al. 2003). DNA-PKcs is not known to have an active role in A-EJ. DNA-PKcs-deficient B cells preferentially use MH for CSR (Crowe et al. 2018), suggesting cNHEJ defects, potentially at the level of synapsis.
Human patients with DNA-PKcs mutations
Given the functions of cNHEJ described above, patients deficient in NHEJ factors usually display severe combined immunodeficiency (SCID), radio sensitivity, and development defects in the post-mitotic neurons. Rare patients with hypomorphic inactivation of Ligase 4, XLF and Artemis suffer from microcephaly and severe developmental defects, along with SCID (Enders et al. 2006; Murray et al. 2014; Plowman et al. 1990; Woodbine et al. 2014). Despite the large size of DNA-PKcs (Prkdc gene), only two DNA-PKcs mutations were reported in human patients (van der Burg et al. 2009; Woodbine et al. 2013). The L3062R missense mutation in the FAT domain was discovered in a patient with isolated SCID with no signs of microcephaly or mental retardation (van der Burg et al. 2009). L3062R-DNA-PKcs has normal kinase activity and is able to interact with Artemis, but it has been proposed that L3062R-DNA-PKcs cannot activate Artemis. The second patient (Woodbine et al. 2013) carries a compound heterozygous mutation in Prkdc, causing low DNA-PKcs protein levels and reduced kinase activity. This patient has severe growth failure and microcephaly in addition to SCID, suggesting that DNA-PKcs might have a role in neuronal development.
DNA-PKcs null mouse models
As the predominant DSB repair pathway in non-dividing somatic cells (e.g., post-mitotic neurons) and quiescent stem cells, loss of the end-joining function of cNHEJ, in the case of XRCC4 or Lig4 deficiency, causes embryonic lethality with severe post-mitotic neuronal apoptosis (Barnes et al. 1998; Frank et al. 1998; Gao et al. 1998b), very similar to patients with hypomorphic Lig4 mutations. KU-deficient mice also have neuronal apoptosis, albeit less severe, and are born at expected ratio (Gu et al. 1997; Nussenzweig et al. 1997). This is likely due to the end-protection role of KU independent of cNHEJ. In yeast, KU prevents end-resection by Exo1 (Mimitou and Symington 2010; Symington and Gautier 2011). Thus, it was speculated that KU deficiency allows other repair pathway, such as the A-EJ to repair a subset of breaks. Indeed, KU deficiency rescues the embryonic lethality of Ligase 4-deficient mice (Boboila et al. 2012; Karanjawala et al. 2002). During lymphocyte development, the absence of KU, Lig4 or XRCC4 abrogates V(D)J recombination and severely affects CSR (50% or more reduction), with the residual joining mediated by the A-EJ pathway that preferentially uses MH at the junctions (Boboila et al. 2010; Yan et al. 2007).
DNA-PKcs is mutated in a spontaneous severe combined immunodeficient (SCID) mouse strain, which carries a homozygous nonsense mutation that leads to the loss of C-terminal 83aa. Although the truncation itself does not affect the integrity of the kinase domain (Blunt et al. 1996; Araki et al. 1997), the truncated DNA-PKcs protein is present at a very low level (Beamish et al. 2000). Thus, the phenotype of the SCID mice is very similar to the other mouse models with targeted disruption of DNA-PKcs (official gene name Prkdc) (Gao et al. 1998a; Kurimasa et al. 1999; Priestley et al. 1998). In general, DNA-PKcs−/− mice are viable, obtained at Mendelian ratio, fertile and do not have growth retardation nor neuronal apoptosis. But thymus, spleen and lymph nodes from DNA-PKcs−/− mice are significantly smaller than controls, with a severe developmental blockade at the T and B cell progenitor stage. This SCID phenotype is due to a defect in V(D)J recombination, especially at the step of CE hairpin opening. In this context, Artemis-deficient mice also suffer from isolated SCID due to the inability to open hairpin at the CEs (Rooney et al. 2002). However, it is noted that in XLF-deficient backgrounds, DNA-PKcs, but not Artemis, is critical for end-ligation (Oksenych et al. 2013). Genetically double knockouts of DNA-PKcs with many other genes implicated in DNA repair (e.g., H2AX and ATM) have much more severe phenotype than loss of Artemis, clearly supporting an Artemis-independent function of DNA-PKcs beyond end-processing and NHEJ (see below).
A kinase-dead mouse model reveals a structural function for DNA-PKcs
DNA-PKcs is the best characterized substrate of DNA-PK. To understand how DNA-PKcs kinase activity regulates DNA-PKcs protein function during cNHEJ, we generated a mouse model expressing a kinase-dead DNA-PKcs protein by mutating the conserved aspartate D3922 to alanine (Jiang et al. 2015a). In contrast to the DNA-PKcs−/− mice, DNA-PKcsKD/KD and DNA-PKcsKD/− mice died embryonically with severe neuronal apoptosis, similar to the XRCC4−/− and Lig4−/− mice. Both SJ and CJ formations are blocked in DNA-PKcsKD/KD lymphocytes (Jiang et al. 2015a) and DNA-PKcsKD/KD mature B cells display CSR defects and preferentially usage of MH at the junctions nearly identical from those of XRCC4−/− B cells (Crowe et al. 2018). Moreover, deletion of KU and expression of Ku80 without the C-terminal domain required for DNA-PKcs recruitment rescue the embryonic lethality and partially rescue end-ligation in DNA-PKcsKD/KD cells, indicating that DNA-PKcs protein poses a physical block to cNHEJ in the absence of its kinase activity. It is worth noting that DNA-PKcs+/KD mice have no measurable defects in end-ligation, consistent with the intermolecular phosphorylation of DNA-PKcs, during which the WT DNA-PKcs could phosphorylate the DNA-PKcs-KD to relieve its inhibition on end-ligation.
Although direct phosphorylation of Artemis by DNA-PKcs is essential for Artemis activation in vitro (Ma et al. 2002), Artemis-dependent hairpin opening is normal in DNA-PKcsKD/KD cells and requires the kinase activity of ATM in vivo (Jiang et al. 2015a), suggesting that ATM kinase activity can substitute DNA-PKcs kinase activity in the presence of DNA-PKcs protein to recruit and activate Artemis (Fig. 3a). Together with the lack of hairpin opening in DNA-PKcs−/− cells (Gao et al. 1998a; Kurimasa et al. 1999; Priestley et al. 1998), these findings indicate that DNA-PKcs protein has a structural function during Artemis activation beyond its kinase activity.
DNA-PKcs phosphorylation in cNHEJ and beyond
Together with the effective and ATM-dependent hairpin opening in DNA-PKcsKD/KD cells, characterization of the DNA-PKcsKD/KD mice lead to the proposal that perhaps two different kinds of phosphorylation events might exist—with auto-phosphorylation licensing end-ligation and trans-phosphorylation by ATM regulating end-processing. In this context, several major auto- and trans- phosphorylation clusters have been characterized in DNA-PKcs, including, but not limited to, S2056, T2609 and T3950 clusters (Douglas et al. 2007). While IR induces Ser2056 auto-phosphorylation that is significantly reduced in cells expressing kinase-dead DNA-PKcs (Chen et al. 2005), phosphorylation at the Thr2609 cluster (Thr2609, Thr2638 and Thr2647 in human) mainly depends on ATM and ATR under IR and UV treatment, respectively (Chen et al. 2007; Yajima et al. 2006). Impaired phosphorylation at either these sites increases radio sensitivity and simultaneous ablation of both S2056 and T2609 clusters nearly completely abrogates DNA-PKcs-dependent IR resistance (Chen et al. 2007; Yajima et al. 2006). To understand the physiological function of the T2609 cluster phosphorylation, Chen and colleagues generated a mouse model in which Thr2605, Thr2634 and Thr2643 (the Thr2609 cluster) are mutated to alanine in the endogenous DNA-PKcs mouse locus (3A knockin mouse model) (Chiarle et al. 2011). The 3A mutation does not affect the kinase activity of DNA-PKcs and DNA-PKcs3A/3A lymphocytes undergo V(D)J recombination efficiently in an ATM kinase-dependent manner (Lee et al. 2013). However, homozygous DNA-PKcs3A/3A mice die shortly after birth due to p53-dependent bone marrow failure, which was tentatively attributed to impairments in the Fanconi pathway and potential telomere instability (Zhang et al. 2011, 2016). Loss of TP53 doubled the life span of homozygous DNA-PKcs3A/3A mice and alleviated the stem cell failure and the developmental defects of lymphocytes, indicating p53-mediated apoptotic response as the pathway responsible for the loss of hematopoietic stem cells (HSC). These data provide a physiological role for the phosphorylation of Thr2609 cluster in regulating the replication stress response and tissue homeostasis. However, the divergent phenotypes of DNA-PKcs3A/3A mice from the DNA-PKcs−/− and DNA-PKcsKD/KD mice also suggest that the IR-associated T2609 phosphorylation defect might not be completely due to cNHEJ defects and DNA-PKcs might have structural functions in other pathways.
In this context, several lines of evidence suggest that DNA-PKcs and KU might have end-joining-independent function in maintain genomic stability. Specifically, KU and DNA-PKcs have been implicated in telomere maintenance independent of other cNHEJ factors (Gilley et al. 2001; Hsu et al. 2000). In yeast, KU has a role in telomere length maintenance by recruiting the telomerase to the chromosome ends (Fisher and Zakian 2005). In human cells, loss of KU and DNA-PKcs, but not other cNHEJ factors (e.g., Lig4), is not compatible with cellular viability and causes immediate fusion of telomeres (Lee et al. 1997; Li et al. 2002). With the development of potent and specific DNA-PKcs inhibitors, it would be critical to understand the full spectrum of the structural as well as kinase-dependent functions of DNA-PKcs in cNHEJ, DDR and beyond and understand the physiological significance of other DNA-PKcs phosphorylation sites (S2056 and T3950) in the future.
ATR
The ATR essential kinase in the replication stress response
In addition to be activated by resected DNA DSBs, ATR kinase responds to a wide variety of stimuli that are able to generate RPA coated ssDNA, often in the absence of strand breaks. For example, replication through difficult regions of the genome and fragile sites, generation of R-loops during transcription, deoxyribonucleoside triphosphate (dNTP) depletion, DNA polymerases or topoisomerases inhibitors, base-alkylating agents, all activate the ATR kinase preferentially, sometimes with minimal or no activation of ATM or DNA-PKcs. Once RPA binds to ssDNA, it recruits ATR through direct interactions with the obligatory partner of ATR–ATR interacting protein (ATRIP) (Zou and Elledge 2003). The recruitment is essential, but not sufficient for full ATR activation. The full activation of ATR also requires additional factors, including RAD17, RAD9-Rad1-HUS1 (9-1-1), topoisomerase II binding protein 1 (TOPBP1) (Kumagai et al. 2006), Ewing tumor-associated antigen 1 (ETAA1) and others (Haahr et al. 2016; Bass et al. 2016; Feng et al. 2016; Lee et al. 2016). In particularly, TopBP1 and ETAA1 are two allosteric activators of ATR that both contain ATR-activating domains (AAD) and can activate ATR in parallel in vivo and in vitro. Besides having overlapping substrates with ATM and DNA-PKcs (e.g., the histone H2AX), ATR preferentially phosphorylates several other substrates, including many replication proteins (e.g., MCM2, RFC, RPA, etc.) (Cortez et al. 2004; Vassin et al. 2009) and its main effector kinase CHK1 (Zhao and Piwnica-Worms 2001); (Guo et al. 2000). Together, ATR and CHK1 activate G2/M cell cycle checkpoint through phosphorylation and inactivation of CDC25 phosphatases, prevent dormant origins firing, maintain replication fork stability and regulate nucleotide availability (Saldivar et al. 2017), which together contribute to their essential role during cell proliferation (Fig. 4a). In addition to its well-characterized role in S phase during DNA replication, ATR is also found to be activated at centromeres by R-loops during mitosis (Kabeche et al. 2018). While the physiological consequences of ATR loss at mitosis are yet to be determined, it could also contribute to the essential role of ATR in replicating cells.
Fig. 4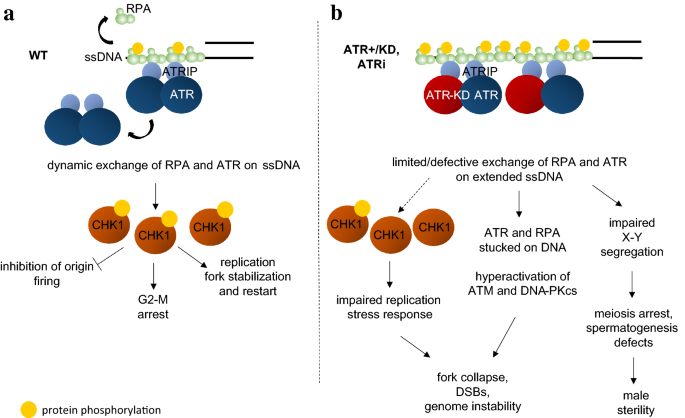
a ATR and RPA dynamically exchange at the sites of ssDNA formation in a manner partially dependent on ATR kinase activity. ATR and its main downstream target, CHK1, are required during the replication stress response for cell cycle arrest, inhibition of origin firing and replication fork stabilization and restart. b In kinase-dead ATR expressing cells, or ATR inhibitor treated cells, the dynamic exchange of ATR and RPA is severely compromised and has several implications, both physiologically (impaired spermatogenesis, male sterility) and damage-induced (hypophosphorylation of CHK1, hyperactivation of ATM and DNA-PKcs, fork stalling, genome instability)
Full size imageATR deficiency in human patients
ATR and CHK1 are essential genes. While complete loss of ATR is not compatible with life, hypomorphic mutations of ATR have been identified in patients with Seckel Syndrome, which is characterized by “bird-headed dwarfism” with craniofacial abnormalities, like beaked nose, receding forehead, microcephaly and radial head dislocation, together with other clinical features (Khetarpal et al. 2016). Notably, mutations at several different genes can cause Seckel Syndrome, including mutations in ATR (Mokrani-Benhelli et al. 2013; O’Driscoll et al. 2003), its obligatory binding partner ATRIP (Ogi et al. 2012) as well as other essential replication genes, such as CtIP (Qvist et al. 2011). In the case of ATR, a specific splicing mutation (A2101G) leads to extremely low ATR protein levels (O’Driscoll et al. 2003).
ATR is essential during embryonic development and tissue homeostasis
Differently from Atm and DNA-PKcs, which are not essential for embryonic development, Atr inactivation is cell lethal and causes early embryonic lethality (< E7.5 or E8.5) in mice (Brown and Baltimore 2000, 2003; de Klein et al. 2000). Atr−/− blastocysts have high frequency of mitotic catastrophe, characterized by severe chromosomal fragmentation, suggesting that ATR prevents premature mitotic entry before the completion of DNA syntheses during normal mitosis (Brown and Baltimore, 2000). The importance of ATR kinase activity for proliferating cells is corroborated by the observation that the loss of its main effector kinase Chk1 also leads to early embryonic lethality, accompanied by aberrant nuclear morphology and defective G2/M checkpoint (Liu et al. 2000; Takai et al. 2000). Moreover, an inactivating point mutation (W1147R) introduced in the AAD of TopBP1, required for full activation of ATR kinase activity, also results in early embryonic lethality in mouse and compromises cell proliferation, inducing premature cellular senescence, in vitro (Zhou et al. 2013).
To circumvent the embryonic lethality of Atr knockout mice and understand the somatic impact of ATR inactivation, Brown and colleagues generated a conditional Atr inactivation allele (Brown and Baltimore 2003). Using the tamoxifen inducible Cre recombinase allele (Cre-ERT2), they tested the effect of somatic deletion of ATR in adult animals (Ruzankina et al. 2007). Deletion of Atr results in a dramatic and progressive loss of stem cells and tissue homeostasis, which eventually leads to premature aging characterized by gray hair, alopecia, osteoporosis, cardiac fibrosis and kidney and thymic involution. While p53 deficiency often rescues development or aging associated phenotypes in mouse models of genome instability, loss of TP53 exacerbates the tissue degeneration in adult mice with somatic Atr inactivation (Ruzankina et al. 2009). Metaphases prepared from Atr and TP53 double-deficient cells, as well as Chk1 and TP53 cells, accumulate high levels of chromosomal fragmentation that are not compatible with cell growth or tissue regeneration. The hypersensitivity of p53-deficient cells to ATR and CHK1 inhibition has been attributed to their partially redundant role in maintaining the G2/M checkpoint (Reinhardt et al. 2007). Thus, in the absence of both p53 and ATR/CHK1, proliferating cells enter mitosis with incompletely replicated DNA and eventually display mitotic catastrophe. Based on this synergistically lethal phenotype, ATR and CHK1 inhibitors have been developed to treat p53-deficient human cancers (Karnitz and Zou 2015a).
An Atr mutation as a model of the human Seckel syndrome (SS)
To understand the Seckel syndrome associated with ATR deficiency, several groups generated the Seckel syndrome model with ATR mutation (Ragland et al. 2009; Murga et al. 2009). In particularly, Capetillo and colleagues replaced the entire mouse genomic fragment encompassing exons 8, 9 and 10 with the human counterpart and then introduced the Seckel splicing mutation (A2101G) (Murga et al. 2009; O’Driscoll et al. 2003). This humanized ATR-Seckel mice (AtrS/S) best recapitulate the clinical presentation of ATR-mutated Seckel syndrome patients. AtrS/S mice were born at sub-Mendelian ratio and have craniofacial abnormalities, growth retardation and microcephaly and die within six months of age with progeroid phenotypes, including hair graying and osteoporosis. Direct targeting of the splicing mutation into mouse Atr does not sufficiently block murine ATR mRNA splicing (Ragland et al. 2009). The retention of the neo resistant cassette in the Atr locus reduced ATR levels by nearly 80% and recapitulated many cellular phenotypes of ATR-Seckel cells. The similarly between the Seckel mouse models and the somatic deletion of ATR in adult tissues clearly indicates that ATR is required for the homeostasis of proliferating tissues. Seckel embryos and derived mouse embryonic fibroblasts (MEFs) experience high levels of replicative stress, highlighted by pan-nuclear γH2AX staining, indicative of a DNA Damage Response activated even without exogenous genotoxic agents. In this scenario, similarly to what has been shown by Brown and colleagues, deletion of p53 does not rescue these phenotypes, but actually aggravates them, accelerating aging and increasing the DNA damage, in both derived embryos and cells. A following report shows increasing dNTP levels, through the increased expression of Rrm2, the catalytic subunit of RNR (ribonucleotide reductase), the enzyme required for the conversion of NTPs in dNTPs, partially rescues the accelerated aging and replication stress phenotype associated with humanized Seckel mice and cells, respectively (Lopez-Contreras et al. 2015). These data indicate how nucleotides are important factors in regulating DNA replication in genetic background with high levels of replicative stress and DNA damage and their potential use in cancer therapy.
Kinase-dead ATR displays a dominant negative function in male meiosis and during DNA damage responses
To determine whether Atr has also a structural function in genomic stability maintenance, like ATM and DNA-PKcs, we generated a knockin mouse harboring kinase-dead ATR protein, in which the conserved aspartate residue D2466 (D2475 in humans) is mutated to alanine (Menolfi et al. 2018). In contrast to the normal development and fertility of Atr+/− mice, male Atr+/KD mice are sterile, preventing us from generating AtrKD/KD mice. Specifically we found that the presence of ATR-KD, but not lack of normal ATR (Atr+/−), dominantly negatively inhibits H2AX phosphorylation on the non-homologous region of X–Y chromosome during pachytene stage. γH2AX is necessary for transcriptional silencing of X–Y chromosomal associated genes. Meiotic sex chromosomal transcriptional silencing is essential for spermatogenesis (Fernandez-Capetillo et al. 2003; Royo et al. 2013). A recent meiotic analysis of the ATR-Seckel mouse model and pharmacological inhibition of ATR also revealed that ATR is crucial for meiotic recombination at meiotic prophase (Pacheco et al. 2018). Cell biology experiments suggest that ATR-KD exhibits the dominant negative function in part by limiting the dynamic exchange of ATR itself and a subset of RPA on ssDNA filaments. As such, Atr+/KD cells display spontaneous genomic instability in regions that expose significant amounts of ssDNA (telomeres, rDNA, fragile sites) and are unable to robustly phosphorylate CHK1 in response to ssDNA generating replication stresses (e.g., Hydroxyurea and Aphidicolin) (Fig. 4b). The Atr+/KD mice present a set of ssDNA-dependent phenotypes during spermatogenesis at the non-homologous region of the X–Y body, mild proliferation defects and lymphocytopenia. In contrast to ATM-KD or DNA-PKcs-KD proteins, ATR-KD displays clearly dominant negative effects, which might be due to the stable hetero-tetramer formed by two ATR and two ATRIP molecules (Wang et al. 2017), which gives the KD protein the possibility to directly interfere with the exchange of WT-ATR. In this regard, ATM and DNA-PKcs are not known to form stable dimers.
Atr mutations in cancer development and treatment
As an essential gene, complete loss of ATR is not found in cancers. But Atr haplo-insufficiency contributes to tumorigenesis in several DNA repair defective backgrounds. Indeed, the loss of a single Atr allele in a Mismatch Repair (MMR)-deficient mouse (Atr+/−Mlh1−/−) favors tumor development, mainly lymphomas and intestinal adenocarcinomas, at an earlier age and higher frequency than the single mutant Mlh1−/− (Fang et al. 2004). In addition, Brown and Baltimore reported a small (3/4-fold) increase in late-life lymphomas in Atr heterozygotes (Atr+/−) and in Atr and Atm double heterozygotes (Atr+/−Atm+/−) compared with Atm heterozygotes (Atm+/−) mice (Brown and Baltimore 2000). On the other hand, humanized ATR-Seckel mutation prevents myc-driven lymphomas (Murga et al. 2011), suggesting that Atr is a dose-dependent tumor suppressor gene. Indeed, oncogenic stress has been linked to premature DNA synthesis (Macheret and Halazonetis 2018), accumulation of ssDNA and a potential increased reliance on the ATR pathway during replication. In mouse models, 90% reduction of ATR protein does not affect normal tissue homeostasis, but inhibits the growth of various cancer, overexpressing Ras or c-Myc, regardless of p53 status (Schoppy et al. 2012). Based on these findings, ATR inhibitors have entered clinical trial, specifically for the treatment of cancers with high replication stress (Karnitz and Zou 2015b) and further corroborate the usefulness of ATR inhibition combined with classical chemotherapy or PARP inhibition to selectively kill cancer cells (Yazinski et al. 2017).
The interplay between ATM, DNA-PKcs and ATR during the DSB response and NHEJ
While mouse model with inactivation in ATM, ATR and DNA-PKcs has unique phenotypes, highlighting their distinct role in DNA damage response, ATM and DNA-PKcs kinases are both recruited to and activated by DSBs, thus sharing overlapping substrates and functions. Following IR, DNA-PKcs is responsible for the residual phosphorylation of a number of ATM substrates in ATM-deficient or ATM inhibitor-treated cells, including, but not limited to, histone H2AX, SMC1, KAP1, p53, which was thought to explain their critical overlapping functions in both cNHEJ and embryonic development. While both Atm−/− and DNA-PKcs−/− mice are viable, disruption of Atm and Prkdc leads to E7.5 embryonic lethality, with excessive DNA damage and p53-dependent apoptosis (Gurley and Kemp 2001; Sekiguchi et al. 2001). Specifically during cNHEJ, the direct ligation of SEs during V(D)J recombination (Gapud and Sleckman 2011; Zha et al. 2011b) or joining during CSR in DNA-PKcs-deficient cells depends on ATM and ATM kinase activity (Callen et al. 2009). While ATM-deficient cells do not have prominent hairpin opening defects, the hairpin opening in DNA-PKcsKD/KD cells completely depends on ATM (Jiang et al. 2015a). Notably, the synergistic function of DNA-PKcs and ATM during chromosomal end-joining is not limited to their kinase activity, as ATM kinase is also essential for end-ligation in DNA-PKcs3A/3A cells with normal DNA-PKcs kinase activity (Lee et al. 2013). In this context, ATM can directly phosphorylate DNA-PKcs at T2609 and potentially other sites and purified DNA-PKcs can phosphorylate ATM and limit ATM activity (Zhou et al. 2017). Yet, the full physiological implications of the intermolecular trans-phosphorylation between ATM and DNA-PKcs are still under investigation.
Despite the partially overlapping substrate portfolio, ATM and DNA-PKcs deficiency leads to distinct cellular and physiological phenotypes. ATM is essential for DNA damage cell cycle checkpoints, while DNA-PKcs deficiency alone seems dispensable for DSB signaling and p53 activation (Jhappan et al. 2000; Jimenez et al. 1999). ATM-deficient mice are infertile, while DNA-PKcs-deficient mice are fertile. DNA-PKcs null mice have SCID but a rather mild defect in CSR (Bosma et al. 2002; Cook et al. 2003; Crowe et al. 2018; Franco et al. 2008; Kiefer et al. 2007; Manis et al. 2002). In contrast, ATM-deficient mice have mild lymphocytopenia, but a nearly 50% reduction in CSR and almost always succumb to early onset T cell lymphomas (Franco et al. 2006; Lumsden et al. 2004; Reina-San-Martin et al. 2004). Several factors contribute to these distinct phenotypes. For one, DNA-PKcs clearly has an essential structural role during cNHEJ that cannot be substituted by ATM. Moreover, DNA-PKcs is activated at the very end of DNA breaks with KU, while ATM activation can occur along a large region, including the flanking chromatin in an MRN-dependent manner. This difference might explain the much more robust and broader impact of ATM on DNA damage responses and by diverse DNA structures. Indeed, cell biology experiments provide strong evidence for the focal localization of KU at the DNA ends and the spreading of MRN into the chromatin surrounding the breaks that are nearly overlapping with γH2AX (Bekker-Jensen et al. 2006; Britton et al. 2013). Further investigation of ATM and DNA-PKcs and the generation of separation of function mutations would provide additional insights into the complex interaction between the two major PI3KKs.
Given the essential role of ATR in cell growth and viability, little is known about the genetic redundancy between ATR and ATM/DNA-PKcs. In this context, ATM-deficient cells are hypersensitive to ATR kinase inhibition (Kwok et al. 2016). The availability of various haploid-insufficient or dominant negative ATR models provides the tools to study functional interactions between ATR and the related ATM and DNA-PK kinases.
Concluding remarks
Mouse models for ATM, DNA-PKcs and ATR have advanced our knowledge in understanding their regulation and their role in embryogenesis, development, physiology and aging. Mutations in these kinases can lead to hereditary diseases (e.g., ataxia–telangiectasia, Seckel syndrome) and are often associated with cancer, rendering the mouse model of vital importance for the study of the genetic basis of diseases and for developing and testing their therapeutic options. Mechanistically, the generation of kinase-dead mouse models has provided unique insights into the structural functions of these proteins beyond their kinase activity, their interaction with co-factors and the developmental and physiological implications. In general, expression of the catalytically inactive kinases is usually associated with more severe genomic instability than loss of the kinases themselves. The presence of the solely ATM-KD and DNA-PK-KD proteins is not compatible with life, while loss of ATM and DNA-PKcs is well tolerated during murine development. Atr+/KD mice have male infertility and lymphocytopenia that are not found in Atr+/− counterparts. By comparing the common and unique phenotypes of the null vs the kinase-dead models, those studies provide strong evidence for a catalysis-dependent structural function of ATM, DNA-PKcs and ATR in addition to their well-characterized signaling role and a clear difference between kinase loss and kinase inhibition.
Outlook for the future perspectives
How the kinase-dead proteins block DNA repair beyond the loss of the kinases themselves? Deletion of corresponding sensor proteins (KU for DNA-PKcs, MRN for ATM) completely or partially relieves the blocks, suggesting that the catalytic inactive protein physically blocks repair at the site of recruitment. The next question is how and where the proteins are stalled. In the case of ATR, the dominant negative phenotype seems to correlate with high levels of RPA coated ssDNA. DNA-PKcs-KD clearly blocks cNHEJ, but notably A-EJ, which also needs access to the DNA ends, is not affected (Crowe et al. 2018), suggesting that the resection mechanism could potentially remove DNA-PKcs-KD. ATM can be activated by several DNA structures, including stalled forks, on which ATM-KD exhibits an inhibitory role. The full spectrum of structures that might trap, if this is the proper word, the catalytically inactive kinases is yet to be discovered and might help us understanding the therapeutic effects and toxicities of specific inhibitors for ATM, ATR and DNA-PKcs that are entering the clinics. Mechanistically, FRAP experiments of ATR (Menolfi et al. 2018) and DNA-PKcs (Uematsu et al. 2007) suggest that the kinase activity promotes dynamic exchange of the kinase at the site of breaks. While auto-phosphorylation seems to be an attractive explanation for these exchange defects, alanine substitution of auto-phosphorylation sites has given either no (in the case of ATM) or different (in the case of DNA-PKcs) phenotype from the kinase-dead model, suggesting that other possibilities might exist. For one, the catalysis might trigger conformation changes of the kinases that are independent of their auto-phosphorylation. Recent structural analyses of ATM, ATR and DNA-PKcs and their yeast orthologs provide evidences for conformation changes upon activation (Wang et al. 2017; Sibanda et al. 2017), which is thought to be regulated by the interaction between the kinases and their respective sensor proteins. If so, it is possible that catalysis could reversely regulate the affinity of the kinase to their sensor proteins, or even the interaction between the sensor protein and the DNA. In this regard, yeast studies suggest that the absence of tel1, the yeast ortholog of Atm, reduces the amount of MRE11 accumulated at the ends of DNA near the breaks. Similar reverse allosteric regulation has been proposed for PARP1, which is also activated upon interaction with DNA ends (Langelier et al. 2018). In this regard, it is important to understand how the FATC and PI3-kinase regulatory domains regulate ATM, DNA-PKcs and ATR activation in the future. Animal models with those mutations will also help us understand the complexity of A–T in human patients and the Achilles’ heel of ATM-mutated human cancers. Moreover, ATM, DNA-PKcs and ATR kinases can phosphorylate each other on different sites, and the mutation of those residues might provide a tool to understand the intricate crosstalk among them and between the kinases and the physical repair. The in vivo interplay between ATR and ATM/DNA-PKcs has not been investigated yet. Since in kinase defective ATR cells, there is a hyperactivation of the other checkpoint kinases, it would be of interest to analyze the in vivo consequences of inactivating ATM and DNA-PKcs in ATR kinase-defective mice with a focus on development, B cells maturation and CSR. Single molecule experiments with defined DNA substrates will also have the unique advantage to provide precise information on protein exchange independent of repair. Moreover, the generation of conditional alleles has allowed the study of otherwise lethal ones and cell-specific conditional Cre alleles can be further exploited to study the function of the kinases in particular cells and tissues and in a specific temporal window (depending on the timing of the deletion), giving an opportunity to investigate biological phenomena like aging and cancer.
References
Aebersold, R., Auffray, C., Baney, E., Barillot, E., Brazma, A., Brett, C., et al. (2009). Report on EU-USA workshop: How systems biology can advance cancer research (27 October 2008). Molecular Oncology,3(1), 9–17. https://doi.org/10.1016/j.molonc.2008.11.003.
Amirifar, P., Ranjouri, M. R., Yazdani, R., Abolhassani, H., & Aghamohammadi, A. (2019). Ataxia-telangiectasia: A review of clinical features and molecular pathology. Pediatric Allergy and Immunology. https://doi.org/10.1111/pai.13020.
Ansel, K. M., Greenwald, R. J., Agarwal, S., Bassing, C. H., Monticelli, S., Interlandi, J., et al. (2004). Deletion of a conserved Il4 silencer impairs T helper type 1-mediated immunity. Nature Immunology,5(12), 1251–1259.
Araki, R., Fujimori, A., Hamatani, K., Mita, K., Saito, T., Mori, M., et al. (1997). Nonsense mutation at Tyr-4046 in the DNA-dependent protein kinase catalytic subunit of severe combined immune deficiency mice. Proceedings of the National Academy of Sciences of the United States of America,94(6), 2438–2443.
Bakkenist, C. J., & Kastan, M. B. (2003). DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature,421(6922), 499–506. https://doi.org/10.1038/nature01368.
Barlow, C., Dennery, P. A., Shigenaga, M. K., Smith, M. A., Morrow, J. D., Roberts, L. J., et al. (1999a). Loss of the ataxia-telangiectasia gene product causes oxidative damage in target organs. Proceedings of the National academy of Sciences of the United States of America,96(17), 9915–9919.
Barlow, C., Eckhaus, M. A., Schaffer, A. A., & Wynshaw-Boris, A. (1999b). Atm haploinsufficiency results in increased sensitivity to sublethal doses of ionizing radiation in mice. Nature Genetics,21(4), 359–360. https://doi.org/10.1038/7684.
Barlow, C., Hirotsune, S., Paylor, R., Liyanage, M., Eckhaus, M., Collins, F., et al. (1996). Atm-deficient mice: A paradigm of ataxia telangiectasia. Cell,86(1), 159–171.
Barnes, D. E., Stamp, G., Rosewell, I., Denzel, A., & Lindahl, T. (1998). Targeted disruption of the gene encoding DNA ligase IV leads to lethality in embryonic mice. Current Biology,8(25), 1395–1398.
Bass, T. E., Luzwick, J. W., Kavanaugh, G., Carroll, C., Dungrawala, H., Glick, G. G., et al. (2016). ETAA1 acts at stalled replication forks to maintain genome integrity. Nature Cell Biology,18(11), 1185–1195. https://doi.org/10.1038/ncb3415.
Bassing, C. H., Suh, H., Ferguson, D. O., Chua, K. F., Manis, J., Eckersdorff, M., et al. (2003). Histone H2AX: A dosage-dependent suppressor of oncogenic translocations and tumors. Cell,114(3), 359–370.
Beamish, H. J., Jessberger, R., Riballo, E., Priestley, A., Blunt, T., Kysela, B., et al. (2000). The C-terminal conserved domain of DNA-PKcs, missing in the SCID mouse, is required for kinase activity. Nucleic Acids Research,28(7), 1506–1513.
Bekker-Jensen, S., Lukas, C., Kitagawa, R., Melander, F., Kastan, M. B., Bartek, J., et al. (2006). Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. Journal of Cell Biology,173(2), 195–206. https://doi.org/10.1083/jcb.200510130.
Blackford, A. N., & Jackson, S. P. (2017). ATM, ATR, and DNA-PK: The trinity at the heart of the DNA damage response. Molecular Cell,66(6), 801–817. https://doi.org/10.1016/j.molcel.2017.05.015.
Block, W. D., & Lees-Miller, S. P. (2005). Putative homologues of the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) and other components of the non-homologous end joining machinery in Dictyostelium discoideum. DNA Repair (Amst),4(10), 1061–1065. https://doi.org/10.1016/j.dnarep.2005.06.008.
Blunt, T., Gell, D., Fox, M., Taccioli, G. E., Lehmann, A. R., Jackson, S. P., et al. (1996). Identification of a nonsense mutation in the carboxyl-terminal region of DNA-dependent protein kinase catalytic subunit in the scid mouse. Proceedings of the National Academy of Sciences of the United States of America,93(19), 10285–10290.
Boboila, C., Oksenych, V., Gostissa, M., Wang, J. H., Zha, S., Zhang, Y., et al. (2012). Robust chromosomal DNA repair via alternative end-joining in the absence of X-ray repair cross-complementing protein 1 (XRCC1). Proceedings of the National Academy of Sciences of the United States of America,109(7), 2473–2478. https://doi.org/10.1073/pnas.1121470109.
Boboila, C., Yan, C., Wesemann, D. R., Jankovic, M., Wang, J. H., Manis, J., et al. (2010). Alternative end-joining catalyzes class switch recombination in the absence of both Ku70 and DNA ligase 4. Journal of Experimental Medicine,207(2), 417–427. https://doi.org/10.1084/jem.20092449.
Boder, E. (1985). Ataxia-telangiectasia: An overview. Kroc Foundation Series,19, 1–63.
Borghesani, P. R., Alt, F. W., Bottaro, A., Davidson, L., Aksoy, S., Rathbun, G. A., et al. (2000). Abnormal development of Purkinje cells and lymphocytes in Atm mutant mice. Proceedings of the National academy of Sciences of the United States of America,97(7), 3336–3341.
Bosma, G. C., Custer, R. P., & Bosma, M. J. (1983). A severe combined immunodeficiency mutation in the mouse. Nature, 301(5900), 527–530. https://doi.org/10.1038/301527a0.
Bosma, G. C., Kim, J., Urich, T., Fath, D. M., Cotticelli, M. G., Ruetsch, N. R., et al. (2002). DNA-dependent protein kinase activity is not required for immunoglobulin class switching. Journal of Experimental Medicine,196(11), 1483–1495.
Boulton, S. J., & Jackson, S. P. (1996). Saccharomyces cerevisiae Ku70 potentiates illegitimate DNA double-strand break repair and serves as a barrier to error-prone DNA repair pathways. EMBO Journal,15(18), 5093–5103.
Britton, S., Coates, J., & Jackson, S. P. (2013). A new method for high-resolution imaging of Ku foci to decipher mechanisms of DNA double-strand break repair. Journal of Cell Biology,202(3), 579–595. https://doi.org/10.1083/jcb.201303073.
Brown, E. J., & Baltimore, D. (2000). ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes & Development,14(4), 397–402.
Brown, E. J., & Baltimore, D. (2003). Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes & Development,17(5), 615–628. https://doi.org/10.1101/gad.1067403.
Buis, J., Wu, Y., Deng, Y., Leddon, J., Westfield, G., Eckersdorff, M., et al. (2008). Mre11 nuclease activity has essential roles in DNA repair and genomic stability distinct from ATM activation. Cell,135(1), 85–96. https://doi.org/10.1016/j.cell.2008.08.015.
Callen, E., Jankovic, M., Wong, N., Zha, S., Chen, H. T., Difilippantonio, S., et al. (2009). Essential role for DNA-PKcs in DNA double-strand break repair and apoptosis in ATM-deficient lymphocytes. Molecular Cell,34(3), 285–297. https://doi.org/10.1016/j.molcel.2009.04.025.
Campbell, A., Krupp, B., Bushman, J., Noble, M., Proschel, C., & Mayer-Proschel, M. (2015). A novel mouse model for ataxia-telangiectasia with a N-terminal mutation displays a behavioral defect and a low incidence of lymphoma but no increased oxidative burden. Human Molecular Genetics,24(22), 6331–6349. https://doi.org/10.1093/hmg/ddv342.
Canman, C. E., Lim, D. S., Cimprich, K. A., Taya, Y., Tamai, K., Sakaguchi, K., et al. (1998). Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science,281(5383), 1677–1679.
Chen, B. P., Chan, D. W., Kobayashi, J., Burma, S., Asaithamby, A., Morotomi-Yano, K., et al. (2005). Cell cycle dependence of DNA-dependent protein kinase phosphorylation in response to DNA double strand breaks. Journal of Biological Chemistry,280(15), 14709–14715. https://doi.org/10.1074/jbc.M408827200.
Chen, C. C., Kass, E. M., Yen, W. F., Ludwig, T., Moynahan, M. E., Chaudhuri, J., et al. (2017). ATM loss leads to synthetic lethality in BRCA1 BRCT mutant mice associated with exacerbated defects in homology-directed repair. Proceedings of the National Academy of Sciences of the United States of America,114(29), 7665–7670. https://doi.org/10.1073/pnas.1706392114.
Chen, B. P., Uematsu, N., Kobayashi, J., Lerenthal, Y., Krempler, A., Yajima, H., et al. (2007). Ataxia telangiectasia mutated (ATM) is essential for DNA-PKcs phosphorylations at the Thr-2609 cluster upon DNA double strand break. Journal of Biological Chemistry,282(9), 6582–6587. https://doi.org/10.1074/jbc.M611605200.
Chiarle, R., Zhang, Y., Frock, R. L., Lewis, S. M., Molinie, B., Ho, Y. J., et al. (2011). Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell,147(1), 107–119. https://doi.org/10.1016/j.cell.2011.07.049.
Choi, M., Kipps, T., & Kurzrock, R. (2016). ATM mutations in cancer: Therapeutic implications. Molecular Cancer Therapeutics,15(8), 1781–1791. https://doi.org/10.1158/1535-7163.MCT-15-0945.
Cook, A. J., Oganesian, L., Harumal, P., Basten, A., Brink, R., & Jolly, C. J. (2003). Reduced switching in SCID B cells is associated with altered somatic mutation of recombined S regions. Journal of Immunology,171(12), 6556–6564.
Cortez, D., Glick, G., & Elledge, S. J. (2004). Minichromosome maintenance proteins are direct targets of the ATM and ATR checkpoint kinases. Proceedings of the National Academy of Sciences of the United States of America,101(27), 10078–10083. https://doi.org/10.1073/pnas.0403410101.
Cosentino, C., Grieco, D., & Costanzo, V. (2011). ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO Journal,30(3), 546–555. https://doi.org/10.1038/emboj.2010.330.
Crowe, J. L., Shao, Z., Wang, X. S., Wei, P. C., Jiang, W., Lee, B. J., et al. (2018). Kinase-dependent structural role of DNA-PKcs during immunoglobulin class switch recombination. Proceedings of the National academy of Sciences of the United States of America. https://doi.org/10.1073/pnas.1808490115.
Daniel, J. A., Pellegrini, M., Lee, B. S., Guo, Z., Filsuf, D., Belkina, N. V., et al. (2012). Loss of ATM kinase activity leads to embryonic lethality in mice. Journal of Cell Biology,198(3), 295–304. https://doi.org/10.1083/jcb.201204035.
Daniel, J. A., Pellegrini, M., Lee, J. H., Paull, T. T., Feigenbaum, L., & Nussenzweig, A. (2008). Multiple autophosphorylation sites are dispensable for murine ATM activation in vivo. Journal of Cell Biology,183(5), 777–783. https://doi.org/10.1083/jcb.200805154.
de Klein, A., Muijtjens, M., van Os, R., Verhoeven, Y., Smit, B., Carr, A. M., et al. (2000). Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Current Biology,10(8), 479–482.
Di Giacomo, M., Barchi, M., Baudat, F., Edelmann, W., Keeney, S., & Jasin, M. (2005). Distinct DNA-damage-dependent and -independent responses drive the loss of oocytes in recombination-defective mouse mutants. Proceedings of the National Academy of Sciences of the United States of America,102(3), 737–742. https://doi.org/10.1073/pnas.0406212102.
Difilippantonio, S., Celeste, A., Fernandez-Capetillo, O., Chen, H. T., Reina San Martin, B., Van Laethem, F., et al. (2005). Role of Nbs1 in the activation of the Atm kinase revealed in humanized mouse models. Nature Cell Biology,7(7), 675–685. https://doi.org/10.1038/ncb1270.
Douglas, P., Cui, X., Block, W. D., Yu, Y., Gupta, S., Ding, Q., et al. (2007). The DNA-dependent protein kinase catalytic subunit is phosphorylated in vivo on threonine 3950, a highly conserved amino acid in the protein kinase domain. Molecular and Cellular Biology,27(5), 1581–1591. https://doi.org/10.1128/mcb.01962-06.
Duecker, R., Baer, P., Eickmeier, O., Strecker, M., Kurz, J., Schaible, A., et al. (2018). Oxidative stress-driven pulmonary inflammation and fibrosis in a mouse model of human ataxia-telangiectasia. Redox Biology,14, 645–655. https://doi.org/10.1016/j.redox.2017.11.006.
Dupré, A., Boyer-Chatenet, L., & Gautier, J. (2006). Two-step activation of ATM by DNA and the Mre11–Rad50–Nbs1 complex. Nature Structural & Molecular Biology, 13(5), 451–457. https://doi.org/10.1038/nsmb1090.
Elson, A., Wang, Y., Daugherty, C. J., Morton, C. C., Zhou, F., Campos-Torres, J., et al. (1996). Pleiotropic defects in ataxia-telangiectasia protein-deficient mice. Proceedings of the National academy of Sciences of the United States of America,93(23), 13084–13089.
Enders, A., Fisch, P., Schwarz, K., Duffner, U., Pannicke, U., Nikolopoulos, E., et al. (2006). A severe form of human combined immunodeficiency due to mutations in DNA ligase IV. Journal of Immunology,176(8), 5060–5068.
Fang, L., Choudhary, S., Zhao, Y., Edeh, C. B., Yang, C., Boldogh, I., et al. (2014). ATM regulates NF-kappaB-dependent immediate-early genes via RelA Ser 276 phosphorylation coupled to CDK9 promoter recruitment. Nucleic Acids Research,42(13), 8416–8432. https://doi.org/10.1093/nar/gku529.
Fang, Y., Tsao, C. C., Goodman, B. K., Furumai, R., Tirado, C. A., Abraham, R. T., et al. (2004). ATR functions as a gene dosage-dependent tumor suppressor on a mismatch repair-deficient background. EMBO Journal,23(15), 3164–3174. https://doi.org/10.1038/sj.emboj.7600315.
Feng, S., Zhao, Y., Xu, Y., Ning, S., Huo, W., Hou, M., et al. (2016). Ewing tumor-associated antigen 1 interacts with replication protein A to promote restart of stalled replication forks. Journal of Biological Chemistry,291(42), 21956–21962. https://doi.org/10.1074/jbc.C116.747758.
Fernandez-Capetillo, O., Mahadevaiah, S. K., Celeste, A., Romanienko, P. J., Camerini-Otero, R. D., Bonner, W. M., et al. (2003). H2AX is required for chromatin remodeling and inactivation of sex chromosomes in male mouse meiosis. Developmental Cell,4(4), 497–508.
Fisher, T. S., & Zakian, V. A. (2005). Ku: A multifunctional protein involved in telomere maintenance. DNA Repair (Amst),4(11), 1215–1226. https://doi.org/10.1016/j.dnarep.2005.04.021.
Franco, S., Gostissa, M., Zha, S., Lombard, D. B., Murphy, M. M., Zarrin, A. A., et al. (2006). H2AX prevents DNA breaks from progressing to chromosome breaks and translocations. Molecular Cell,21(2), 201–214.
Franco, S., Murphy, M. M., Li, G., Borjeson, T., Boboila, C., & Alt, F. W. (2008). DNA-PKcs and Artemis function in the end-joining phase of immunoglobulin heavy chain class switch recombination. Journal of Experimental Medicine,205(3), 557–564.
Frank, K. M., Sekiguchi, J. M., Seidl, K. J., Swat, W., Rathbun, G. A., Cheng, H. L., et al. (1998). Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. Nature,396(6707), 173–177. https://doi.org/10.1038/24172.
Gao, Y., Chaudhuri, J., Zhu, C., Davidson, L., Weaver, D. T., & Alt, F. W. (1998a). A targeted DNA-PKcs-null mutation reveals DNA-PK-independent functions for KU in V(D)J recombination. Immunity,9(3), 367–376.
Gao, Y., Sun, Y., Frank, K. M., Dikkes, P., Fujiwara, Y., Seidl, K. J., et al. (1998b). A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell,95(7), 891–902.
Gapud, E. J., & Sleckman, B. P. (2011). Unique and redundant functions of ATM and DNA-PKcs during V(D)J recombination. Cell Cycle,10(12), 1928–1935. (pii:16011).
Gilley, D., Tanaka, H., Hande, M. P., Kurimasa, A., Li, G. C., Oshimura, M., et al. (2001). DNA-PKcs is critical for telomere capping. Proceedings of the National Academy of Sciences of the United States of America,98(26), 15084–15088. https://doi.org/10.1073/pnas.261574698.
Graham, T. G., Walter, J. C., & Loparo, J. J. (2016). Two-stage synapsis of DNA ends during non-homologous end joining. Molecular Cell,61(6), 850–858. https://doi.org/10.1016/j.molcel.2016.02.010.
Grundy, G. J., Rulten, S. L., Arribas-Bosacoma, R., Davidson, K., Kozik, Z., Oliver, A. W., et al. (2016). The Ku-binding motif is a conserved module for recruitment and stimulation of non-homologous end-joining proteins. Nature Communications,7, 11242. https://doi.org/10.1038/ncomms11242.
Gu, Y., Jin, S., Gao, Y., Weaver, D. T., & Alt, F. W. (1997). Ku70-deficient embryonic stem cells have increased ionizing radiosensitivity, defective DNA end-binding activity, and inability to support V(D)J recombination. Proceedings of the National academy of Sciences of the United States of America,94(15), 8076–8081.
Guo, Z., Kozlov, S., Lavin, M. F., Person, M. D., & Paull, T. T. (2010). ATM activation by oxidative stress. Science,330(6003), 517–521. https://doi.org/10.1126/science.1192912.
Guo, Z., Kumagai, A., Wang, S. X., & Dunphy, W. G. (2000). Requirement for Atr in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes & Development,14(21), 2745–2756.
Gurley, K. E., & Kemp, C. J. (2001). Synthetic lethality between mutation in Atm and DNA-PK(cs) during murine embryogenesis. Current Biology,11(3), 191–194.
Haahr, P., Hoffmann, S., Tollenaere, M. A., Ho, T., Toledo, L. I., Mann, M., et al. (2016). Activation of the ATR kinase by the RPA-binding protein ETAA1. Nature Cell Biology,18(11), 1196–1207. https://doi.org/10.1038/ncb3422.
Herzog, K. H., Chong, M. J., Kapsetaki, M., Morgan, J. I., & McKinnon, P. J. (1998). Requirement for Atm in ionizing radiation-induced cell death in the developing central nervous system. Science,280(5366), 1089–1091.
Hsu, H. L., Gilley, D., Galande, S. A., Hande, M. P., Allen, B., Kim, S. H., et al. (2000). Ku acts in a unique way at the mammalian telomere to prevent end joining. Genes & Development,14(22), 2807–2812.
Hung, P. J., Johnson, B., Chen, B. R., Byrum, A. K., Bredemeyer, A. L., Yewdell, W. T., et al. (2018). MRI is a DNA damage response adaptor during classical non-homologous end joining. Molecular Cell,71(2), 332–342 e338. https://doi.org/10.1016/j.molcel.2018.06.018.
Ito, K., Hirao, A., Arai, F., Matsuoka, S., Takubo, K., Hamaguchi, I., et al. (2004). Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature,431(7011), 997–1002. https://doi.org/10.1038/nature02989.
Jhappan, C., Yusufzai, T. M., Anderson, S., Anver, M. R., & Merlino, G. (2000). The p53 response to DNA damage in vivo is independent of DNA-dependent protein kinase. Molecular and Cellular Biology,20(11), 4075–4083.
Jiang, H., Chang, F. C., Ross, A. E., Lee, J., Nakayama, K., Nakayama, K., et al. (2005). Ubiquitylation of RAG-2 by Skp2-SCF links destruction of the V(D)J recombinase to the cell cycle. Molecular Cell,18(6), 699–709. https://doi.org/10.1016/j.molcel.2005.05.011.
Jiang, W., Crowe, J. L., Liu, X., Nakajima, S., Wang, Y., Li, C., et al. (2015a). Differential phosphorylation of DNA-PKcs regulates the interplay between end-processing and end-ligation during nonhomologous end-joining. Molecular Cell,58(1), 172–185. https://doi.org/10.1016/j.molcel.2015.02.024.
Jiang, W., Lee, B. J., Li, C., Dubois, R. L., Gostissa, M., Alt, F. W., et al. (2015b). Aberrant TCRdelta rearrangement underlies the T-cell lymphocytopenia and t(12;14) translocation associated with ATM deficiency. Blood,125(17), 2665–2668. https://doi.org/10.1182/blood-2015-01-622621.
Jimenez, G. S., Bryntesson, F., Torres-Arzayus, M. I., Priestley, A., Beeche, M., Saito, S., et al. (1999). DNA-dependent protein kinase is not required for the p53-dependent response to DNA damage. Nature,400(6739), 81–83. https://doi.org/10.1038/21913.
Kabeche, L., Nguyen, H. D., Buisson, R., & Zou, L. (2018). A mitosis-specific and R loop-driven ATR pathway promotes faithful chromosome segregation. Science,359(6371), 108–114. https://doi.org/10.1126/science.aan6490.
Kabotyanski, E. B., Gomelsky, L., Han, J. O., Stamato, T. D., & Roth, D. B. (1998). Double-strand break repair in Ku86- and XRCC4-deficient cells. Nucleic Acids Research,26(23), 5333–5342.
Kamsler, A., Daily, D., Hochman, A., Stern, N., Shiloh, Y., Rotman, G., et al. (2001). Increased oxidative stress in ataxia telangiectasia evidenced by alterations in redox state of brains from Atm-deficient mice. Cancer Research,61(5), 1849–1854.
Karanjawala, Z. E., Adachi, N., Irvine, R. A., Oh, E. K., Shibata, D., Schwarz, K., et al. (2002). The embryonic lethality in DNA ligase IV-deficient mice is rescued by deletion of Ku: Implications for unifying the heterogeneous phenotypes of NHEJ mutants. DNA Repair (Amst),1(12), 1017–1026. (pii:S1568786402001519).
Karnitz, L. M., & Zou, L. (2015a). Molecular pathways: Targeting ATR in cancer therapy. Clinical Cancer Research,5, 10. https://doi.org/10.1158/1078-0432.ccr-15-0479.
Karnitz, L. M., & Zou, L. (2015b). Molecular pathways: Targeting ATR in cancer therapy. Clinical Cancer Research,21(21), 4780–4785. https://doi.org/10.1158/1078-0432.CCR-15-0479.
Katyal, S., Lee, Y., Nitiss, K. C., Downing, S. M., Li, Y., Shimada, M., et al. (2014). Aberrant topoisomerase-1 DNA lesions are pathogenic in neurodegenerative genome instability syndromes. Nature Neuroscience,17(6), 813–821. https://doi.org/10.1038/nn.3715.
Khetarpal, P., Das, S., Panigrahi, I., & Munshi, A. (2016). Primordial dwarfism: Overview of clinical and genetic aspects. Molecular Genetics and Genomics,291(1), 1–15. https://doi.org/10.1007/s00438-015-1110-y.
Kiefer, K., Oshinsky, J., Kim, J., Nakajima, P. B., Bosma, G. C., & Bosma, M. J. (2007). The catalytic subunit of DNA-protein kinase (DNA-PKcs) is not required for Ig class-switch recombination. Proceedings of the National Academy of Sciences of the United States of America,104(8), 2843–2848.
Kim, S. T., Lim, D. S., Canman, C. E., & Kastan, M. B. (1999). Substrate specificities and identification of putative substrates of ATM kinase family members. Journal of Biological Chemistry,274(53), 37538–37543.
Kojis, T. L., Gatti, R. A., & Sparkes, R. S. (1991). The cytogenetics of ataxia telangiectasia. Cancer Genetics and Cytogenetics,56(2), 143–156.
Kozlov, S. V., Graham, M. E., Jakob, B., Tobias, F., Kijas, A. W., Tanuji, M., et al. (2011). Autophosphorylation and ATM activation: Additional sites add to the complexity. Journal of Biological Chemistry,286(11), 9107–9119. https://doi.org/10.1074/jbc.M110.204065.
Kozlov, S., Gueven, N., Keating, K., Ramsay, J., & Lavin, M. F. (2003). ATP activates ataxia-telangiectasia mutated (ATM) in vitro. Importance of autophosphorylation. Journal of Biological Chemistry,278(11), 9309–9317.
Kozlov, S. V., Waardenberg, A. J., Engholm-Keller, K., Arthur, J. W., Graham, M. E., & Lavin, M. (2016). Reactive oxygen species (ROS)-activated ATM-dependent phosphorylation of cytoplasmic substrates identified by large-scale phosphoproteomics screen. Molecular and Cellular Proteomics,15(3), 1032–1047. https://doi.org/10.1074/mcp.M115.055723.
Kumagai, A., Lee, J., Yoo, H. Y., & Dunphy, W. G. (2006). TopBP1 activates the ATR-ATRIP complex. Cell,124(5), 943–955. https://doi.org/10.1016/j.cell.2005.12.041.
Kumar, V., Alt, F. W., & Frock, R. L. (2016). PAXX and XLF DNA repair factors are functionally redundant in joining DNA breaks in a G1-arrested progenitor B-cell line. Proceedings of the National Academy of Sciences of the United States of America,113(38), 10619–10624. https://doi.org/10.1073/pnas.1611882113.
Kurimasa, A., Ouyang, H., Dong, L. J., Wang, S., Li, X., Cordon-Cardo, C., et al. (1999). Catalytic subunit of DNA-dependent protein kinase: Impact on lymphocyte development and tumorigenesis. Proceedings of the National Academy of Sciences of the United States of America,96(4), 1403–1408.
Kwok, M., Davies, N., Agathanggelou, A., Smith, E., Oldreive, C., Petermann, E., et al. (2016). ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood,127(5), 582–595. https://doi.org/10.1182/blood-2015-05-644872.
Langelier, M. F., Zandarashvili, L., Aguiar, P. M., Black, B. E., & Pascal, J. M. (2018). NAD(+) analog reveals PARP-1 substrate-blocking mechanism and allosteric communication from catalytic center to DNA-binding domains. Nature Communications,9(1), 844. https://doi.org/10.1038/s41467-018-03234-8.
Lee, Y., Brown, E. J., Chang, S., & McKinnon, P. J. (2014). Pot1a prevents telomere dysfunction and ATM-dependent neuronal loss. Journal of Neuroscience,34(23), 7836–7844. https://doi.org/10.1523/JNEUROSCI.4245-13.2014.
Lee, B. S., Gapud, E. J., Zhang, S., Dorsett, Y., Bredemeyer, A., George, R., et al. (2013). Functional intersection of ATM and DNA-PKcs in coding end joining during V(D)J recombination. Molecular and Cellular Biology,5, 10. https://doi.org/10.1128/mcb.00308-13.
Lee, S. E., Mitchell, R. A., Cheng, A., & Hendrickson, E. A. (1997). Evidence for DNA-PK-dependent and -independent DNA double-strand break repair pathways in mammalian cells as a function of the cell cycle. Molecular and Cellular Biology,17(3), 1425–1433.
Lee, J. H., & Paull, T. T. (2004). Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science,304(5667), 93–96.
Lee, J. H., & Paull, T. T. (2005). ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science,308(5721), 551–554.
Lee, Y. C., Zhou, Q., Chen, J., & Yuan, J. (2016). RPA-binding protein ETAA1 is an ATR activator involved in DNA replication stress response. Current Biology,26(24), 3257–3268. https://doi.org/10.1016/j.cub.2016.10.030.
Lescale, C., Lenden Hasse, H., Blackford, A. N., Balmus, G., Bianchi, J. J., Yu, W., et al. (2016). Specific roles of XRCC4 paralogs PAXX and XLF during V(D)J recombination. Cell Reports. https://doi.org/10.1016/j.celrep.2016.08.069.
Li, N., Banin, S., Ouyang, H., Li, G. C., Courtois, G., Shiloh, Y., et al. (2001). ATM is required for IkappaB kinase (IKKk) activation in response to DNA double strand breaks. Journal of Biological Chemistry,276(12), 8898–8903. https://doi.org/10.1074/jbc.M009809200.
Li, G., Nelsen, C., & Hendrickson, E. A. (2002). Ku86 is essential in human somatic cells. Proceedings of the National academy of Sciences of the United States of America,99(2), 832–837.
Lieber, M. R. (2010). The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annual Review of Biochemistry,79, 181–211. https://doi.org/10.1146/annurev.biochem.052308.093131.
Liu, Q., Guntuku, S., Cui, X. S., Matsuoka, S., Cortez, D., Tamai, K., et al. (2000). Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes & Development,14(12), 1448–1459.
Liu, X., Jiang, W., Dubois, R. L., Yamamoto, K., Wolner, Z., & Zha, S. (2012). Overlapping functions between XLF repair protein and 53BP1 DNA damage response factor in end joining and lymphocyte development. Proceedings of the National Academy of Sciences of the United States of America,109(10), 3903–3908. https://doi.org/10.1073/pnas.1120160109.
Liu, X., Shao, Z., Jiang, W., Lee, B. J., & Zha, S. (2017). PAXX promotes KU accumulation at DNA breaks and is essential for end-joining in XLF-deficient mice. Nature Communications,8, 13816. https://doi.org/10.1038/ncomms13816.
Liyanage, M., Weaver, Z., Barlow, C., Coleman, A., Pankratz, D. G., Anderson, S., et al. (2000). Abnormal rearrangement within the alpha/delta T-cell receptor locus in lymphomas from Atm-deficient mice. Blood,96(5), 1940–1946.
Lopez-Contreras, A. J., Specks, J., Barlow, J. H., Ambrogio, C., Desler, C., Vikingsson, S., et al. (2015). Increased Rrm2 gene dosage reduces fragile site breakage and prolongs survival of ATR mutant mice. Genes & Development,29(7), 690–695. https://doi.org/10.1101/gad.256958.114.
Lumsden, J. M., McCarty, T., Petiniot, L. K., Shen, R., Barlow, C., Wynn, T. A., et al. (2004). Immunoglobulin class switch recombination is impaired in Atm-deficient mice. Journal of Experimental Medicine,200(9), 1111–1121.
Luo, G., Yao, M. S., Bender, C. F., Mills, M., Bladl, A. R., Bradley, A., et al. (1999). Disruption of mRad50 causes embryonic stem cell lethality, abnormal embryonic development, and sensitivity to ionizing radiation. Proceedings of the National Academy of Sciences of the United States of America,96(13), 7376–7381.
Ma, Y., Pannicke, U., Schwarz, K., & Lieber, M. R. (2002). Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell,108(6), 781–794.
Macheret, M., & Halazonetis, T. D. (2018). Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature,555(7694), 112–116. https://doi.org/10.1038/nature25507.
Manis, J. P., Dudley, D., Kaylor, L., & Alt, F. W. (2002). IgH class switch recombination to IgG1 in DNA-PKcs-deficient B cells. Immunity,16(4), 607–617.
Menolfi, D., Jiang, W., Lee, B. J., Moiseeva, T., Shao, Z., Estes, V., et al. (2018). Kinase-dead ATR differs from ATR loss by limiting the dynamic exchange of ATR and RPA. Nature Communications,9(1), 5351. https://doi.org/10.1038/s41467-018-07798-3.
Meyn, M. S. (1993). High spontaneous intrachromosomal recombination rates in ataxia-telangiectasia. Science,260(5112), 1327–1330.
Mimitou, E. P., & Symington, L. S. (2010). Ku prevents Exo1 and Sgs1-dependent resection of DNA ends in the absence of a functional MRX complex or Sae2. EMBO Journal,29(19), 3358–3369. https://doi.org/10.1038/emboj.2010.193.
Mokrani-Benhelli, H., Gaillard, L., Biasutto, P., Le Guen, T., Touzot, F., Vasquez, N., et al. (2013). Primary microcephaly, impaired DNA replication, and genomic instability caused by compound heterozygous ATR mutations. Human Mutation,34(2), 374–384. https://doi.org/10.1002/humu.22245.
Murga, M., Bunting, S., Montana, M. F., Soria, R., Mulero, F., Canamero, M., et al. (2009). A mouse model of ATR-Seckel shows embryonic replicative stress and accelerated aging. Nature Genetics,41(8), 891–898. https://doi.org/10.1038/ng.420.
Murga, M., Campaner, S., Lopez-Contreras, A. J., Toledo, L. I., Soria, R., Montana, M. F., et al. (2011). Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nature Structural & Molecular Biology,18(12), 1331–1335. https://doi.org/10.1038/nsmb.2189.
Murray, J. E., Bicknell, L. S., Yigit, G., Duker, A. L., van Kogelenberg, M., Haghayegh, S., et al. (2014). Extreme growth failure is a common presentation of ligase IV deficiency. Human Mutation,35(1), 76–85. https://doi.org/10.1002/humu.22461.
Nussenzweig, A., Sokol, K., Burgman, P., Li, L., & Li, G. C. (1997). Hypersensitivity of Ku80-deficient cell lines and mice to DNA damage: The effects of ionizing radiation on growth, survival, and development. Proceedings of the National academy of Sciences of the United States of America,94(25), 13588–13593.
Ochi, T., Blackford, A. N., Coates, J., Jhujh, S., Mehmood, S., Tamura, N., et al. (2015). DNA repair. PAXX, a paralog of XRCC4 and XLF, interacts with Ku to promote DNA double-strand break repair. Science,347(6218), 185–188. https://doi.org/10.1126/science.1261971.
O’Driscoll, M., Ruiz-Perez, V. L., Woods, C. G., Jeggo, P. A., & Goodship, J. A. (2003). A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nature Genetics,33(4), 497–501. https://doi.org/10.1038/ng1129.
Ogi, T., Walker, S., Stiff, T., Hobson, E., Limsirichaikul, S., Carpenter, G., et al. (2012). Identification of the first ATRIP-deficient patient and novel mutations in ATR define a clinical spectrum for ATR-ATRIP Seckel Syndrome. PLoS Genetics,8(11), e1002945. https://doi.org/10.1371/journal.pgen.1002945.
Oksenych, V., Kumar, V., Liu, X., Guo, C., Schwer, B., Zha, S., et al. (2013). Functional redundancy between the XLF and DNA-PKcs DNA repair factors in V(D)J recombination and nonhomologous DNA end joining. Proceedings of the National Academy of Sciences of the United States of America,110(6), 2234–2239. https://doi.org/10.1073/pnas.1222573110.
Pacheco, S., Maldonado-Linares, A., Marcet-Ortega, M., Rojas, C., Martinez-Marchal, A., Fuentes-Lazaro, J., et al. (2018). ATR is required to complete meiotic recombination in mice. Nature Communications,9(1), 2622. https://doi.org/10.1038/s41467-018-04851-z.
Pellegrini, M., Celeste, A., Difilippantonio, S., Guo, R., Wang, W., Feigenbaum, L., et al. (2006). Autophosphorylation at serine 1987 is dispensable for murine Atm activation in vivo. Nature,443(7108), 222–225. https://doi.org/10.1038/nature05112.
Perry, J., & Kleckner, N. (2003). The ATRs, ATMs, and TORs are giant HEAT repeat proteins. Cell,112(2), 151–155.
Petiniot, L. K., Weaver, Z., Barlow, C., Shen, R., Eckhaus, M., Steinberg, S. M., et al. (2000). Recombinase-activating gene (RAG) 2-mediated V(D)J recombination is not essential for tumorigenesis in Atm-deficient mice. Proceedings of the National academy of Sciences of the United States of America,97(12), 6664–6669.
Petiniot, L. K., Weaver, Z., Vacchio, M., Shen, R., Wangsa, D., Barlow, C., et al. (2002). RAG-mediated V(D)J recombination is not essential for tumorigenesis in Atm-deficient mice. Molecular and Cellular Biology,22(9), 3174–3177.
Plowman, P. N., Bridges, B. A., Arlett, C. F., Hinney, A., & Kingston, J. E. (1990). An instance of clinical radiation morbidity and cellular radiosensitivity, not associated with ataxia-telangiectasia. British Journal of Radiology,63(752), 624–628. https://doi.org/10.1259/0007-1285-63-752-624.
Priestley, A., Beamish, H. J., Gell, D., Amatucci, A. G., Muhlmann-Diaz, M. C., Singleton, B. K., et al. (1998). Molecular and biochemical characterisation of DNA-dependent protein kinase-defective rodent mutant irs-20. Nucleic Acids Research,26(8), 1965–1973.
Quick, K. L., & Dugan, L. L. (2001). Superoxide stress identifies neurons at risk in a model of ataxia-telangiectasia. Annals of Neurology,49(5), 627–635.
Qvist, P., Huertas, P., Jimeno, S., Nyegaard, M., Hassan, M. J., Jackson, S. P., et al. (2011). CtIP mutations cause Seckel and Jawad syndromes. PLoS Genetics,7(10), e1002310. https://doi.org/10.1371/journal.pgen.1002310.
Ragland, R. L., Arlt, M. F., Hughes, E. D., Saunders, T. L., & Glover, T. W. (2009). Mice hypomorphic for Atr have increased DNA damage and abnormal checkpoint response. Mammalian Genome,20(6), 375–385. https://doi.org/10.1007/s00335-009-9195-4.
Rass, E., Chandramouly, G., Zha, S., Alt, F. W., & Xie, A. (2013). Ataxia telangiectasia mutated (ATM) is dispensable for endonuclease I-SceI-induced homologous recombination in mouse embryonic stem cells. Journal of Biological Chemistry,288(10), 7086–7095. https://doi.org/10.1074/jbc.M112.445825.
Reichenbach, J., Schubert, R., Schindler, D., Muller, K., Bohles, H., & Zielen, S. (2002). Elevated oxidative stress in patients with ataxia telangiectasia. Antioxidants & Redox Signaling,4(3), 465–469. https://doi.org/10.1089/15230860260196254.
Reid, D. A., Keegan, S., Leo-Macias, A., Watanabe, G., Strande, N. T., Chang, H. H., et al. (2015). Organization and dynamics of the nonhomologous end-joining machinery during DNA double-strand break repair. Proceedings of the National Academy of Sciences of the United States of America,112(20), E2575–E2584. https://doi.org/10.1073/pnas.1420115112.
Reina-San-Martin, B., Chen, H. T., Nussenzweig, A., & Nussenzweig, M. C. (2004). ATM is required for efficient recombination between immunoglobulin switch regions. Journal of Experimental Medicine,200(9), 1103–1110.
Reinhardt, H. C., Aslanian, A. S., Lees, J. A., & Yaffe, M. B. (2007). p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell,11(2), 175–189.
Reliene, R., & Schiestl, R. H. (2006). Antioxidant N-acetyl cysteine reduces incidence and multiplicity of lymphoma in Atm deficient mice. DNA Repair (Amst).,5(7), 852–859.
Renwick, A., Thompson, D., Seal, S., Kelly, P., Chagtai, T., Ahmed, M., et al. (2006). ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nature Genetics,38(8), 873–875. https://doi.org/10.1038/ng1837.
Rooney, S., Sekiguchi, J., Zhu, C., Cheng, H. L., Manis, J., Whitlow, S., et al. (2002). Leaky scid phenotype associated with defective V(D)J coding end processing in Artemis-deficient mice. Molecular Cell,10(6), 1379–1390.
Roy, S., de Melo, A. J., Xu, Y., Tadi, S. K., Negrel, A., Hendrickson, E., et al. (2015). XRCC4/XLF interaction is variably required for DNA repair and is not required for ligase IV stimulation. Molecular and Cellular Biology,35(17), 3017–3028. https://doi.org/10.1128/MCB.01503-14.
Royo, H., Prosser, H., Ruzankina, Y., Mahadevaiah, S. K., Cloutier, J. M., Baumann, M., et al. (2013). ATR acts stage specifically to regulate multiple aspects of mammalian meiotic silencing. Genes & Development,27(13), 1484–1494. https://doi.org/10.1101/gad.219477.113.
Ruzankina, Y., Pinzon-Guzman, C., Asare, A., Ong, T., Pontano, L., Cotsarelis, G., et al. (2007). Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell,1(1), 113–126. https://doi.org/10.1016/j.stem.2007.03.002.
Ruzankina, Y., Schoppy, D. W., Asare, A., Clark, C. E., Vonderheide, R. H., & Brown, E. J. (2009). Tissue regenerative delays and synthetic lethality in adult mice after combined deletion of Atr and Trp53. Nature Genetics,41(10), 1144–1149. https://doi.org/10.1038/ng.441.
Saldivar, J. C., Cortez, D., & Cimprich, K. A. (2017). The essential kinase ATR: Ensuring faithful duplication of a challenging genome. Nature Reviews Molecular Cell Biology,18(10), 622–636. https://doi.org/10.1038/nrm.2017.67.
Savitsky, K., Bar-Shira, A., Gilad, S., Rotman, G., Ziv, Y., Vanagaite, L., et al. (1995). A single ataxia telangiectasia gene with a product similar to PI-3Kinase. Science,268(5218), 1749–1753.
Schoppy, D. W., Ragland, R. L., Gilad, O., Shastri, N., Peters, A. A., Murga, M., et al. (2012). Oncogenic stress sensitizes murine cancers to hypomorphic suppression of ATR. Journal of Clinical Investigation,122(1), 241–252. https://doi.org/10.1172/JCI58928.
Schroeder, S. A., Swift, M., Sandoval, C., & Langston, C. (2005). Interstitial lung disease in patients with ataxia-telangiectasia. Pediatric Pulmonology,39(6), 537–543. https://doi.org/10.1002/ppul.20209.
Schroeder, S. A., & Zielen, S. (2014). Infections of the respiratory system in patients with ataxia-telangiectasia. Pediatric Pulmonology,49(4), 389–399. https://doi.org/10.1002/ppul.22817.
Schubert, R., Erker, L., Barlow, C., Yakushiji, H., Larson, D., Russo, A., et al. (2004). Cancer chemoprevention by the antioxidant tempol in Atm-deficient mice. Human Molecular Genetics,13(16), 1793–1802. https://doi.org/10.1093/hmg/ddh189.
Sekiguchi, J., Ferguson, D. O., Chen, H. T., Yang, E. M., Earle, J., Frank, K., et al. (2001). Genetic interactions between ATM and the nonhomologous end-joining factors in genomic stability and development. Proceedings of the National academy of Sciences of the United States of America,98(6), 3243–3248.
Shackelford, R. E., Innes, C. L., Sieber, S. O., Heinloth, A. N., Leadon, S. A., & Paules, R. S. (2001). The Ataxia telangiectasia gene product is required for oxidative stress-induced G1 and G2 checkpoint function in human fibroblasts. Journal of Biological Chemistry,276(24), 21951–21959. https://doi.org/10.1074/jbc.M011303200.
Shiloh, Y., & Ziv, Y. (2013). The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nature Reviews Molecular Cell Biology,14(4), 197–210.
Sibanda, B. L., Chirgadze, D. Y., Ascher, D. B., & Blundell, T. L. (2017). DNA-PKcs structure suggests an allosteric mechanism modulating DNA double-strand break repair. Science,355(6324), 520–524. https://doi.org/10.1126/science.aak9654.
Singleton, B. K., Torres-Arzayus, M. I., Rottinghaus, S. T., Taccioli, G. E., & Jeggo, P. A. (1999). The C terminus of Ku80 activates the DNA-dependent protein kinase catalytic subunit. Molecular and Cellular Biology,19(5), 3267–3277.
Spring, K., Ahangari, F., Scott, S. P., Waring, P., Purdie, D. M., Chen, P. C., et al. (2002). Mice heterozygous for mutation in Atm, the gene involved in ataxia-telangiectasia, have heightened susceptibility to cancer. Nature Genetics,32(1), 185–190. https://doi.org/10.1038/ng958.
Swift, M., Morrell, D., Cromartie, E., Chamberlin, A. R., Skolnick, M. H., & Bishop, D. T. (1986). The incidence and gene frequency of ataxia-telangiectasia in the United States. American Journal of Human Genetics,39(5), 573–583.
Symington, L. S., & Gautier, J. (2011). Double-strand break end resection and repair pathway choice. Annual Review of Genetics,45, 247–271. https://doi.org/10.1146/annurev-genet-110410-132435.
Taccioli, G. E., Amatucci, A. G., Beamish, H. J., Gell, D., Xiang, X. H., Torres Arzayus, M. I., Priestley, A., Jackson, S. P., Marshak Rothstein, A., Jeggo, P. A., & Herrera, V. L. (1998). Targeted disruption of the catalytic subunit of the DNA-PK gene in mice confers severe combined immunodeficiency and radiosensitivity. Immunity, 9(3), 355–366. https://doi.org/10.1016/S1074-7613(00)80618-4.
Takai, H., Tominaga, K., Motoyama, N., Minamishima, Y. A., Nagahama, H., Tsukiyama, T., et al. (2000). Aberrant cell cycle checkpoint function and early embryonic death in Chk1(−/−) mice. Genes & Development,14(12), 1439–1447.
Theunissen, J. W., Kaplan, M. I., Hunt, P. A., Williams, B. R., Ferguson, D. O., Alt, F. W., et al. (2003). Checkpoint failure and chromosomal instability without lymphomagenesis in Mre11(ATLD1/ATLD1) mice. Molecular Cell,12(6), 1511–1523.
Tripathi, D. N., Chowdhury, R., Trudel, L. J., Tee, A. R., Slack, R. S., Walker, C. L., et al. (2013). Reactive nitrogen species regulate autophagy through ATM-AMPK-TSC2-mediated suppression of mTORC1. Proceedings of the National Academy of Sciences of the United States of America,110(32), E2950–E2957. https://doi.org/10.1073/pnas.1307736110.
Uematsu, N., Weterings, E., Yano, K., Morotomi-Yano, K., Jakob, B., Taucher-Scholz, G., et al. (2007). Autophosphorylation of DNA-PKCS regulates its dynamics at DNA double-strand breaks. Journal of Cell Biology,177(2), 219–229. https://doi.org/10.1083/jcb.200608077.
Uziel, T., Lerenthal, Y., Moyal, L., Andegeko, Y., Mittelman, L., & Shiloh, Y. (2003). Requirement of the MRN complex for ATM activation by DNA damage. EMBO Journal,22(20), 5612–5621. https://doi.org/10.1093/emboj/cdg541.
van der Burg, M., Ijspeert, H., Verkaik, N. S., Turul, T., Wiegant, W. W., Morotomi-Yano, K., et al. (2009). A DNA-PKcs mutation in a radiosensitive T-B-SCID patient inhibits Artemis activation and nonhomologous end-joining. Journal of Clinical Investigation,119(1), 91–98. https://doi.org/10.1172/JCI37141.
Vassin, V. M., Anantha, R. W., Sokolova, E., Kanner, S., & Borowiec, J. A. (2009). Human RPA phosphorylation by ATR stimulates DNA synthesis and prevents ssDNA accumulation during DNA-replication stress. Journal of Cell Science,122(Pt 22), 4070–4080. https://doi.org/10.1242/jcs.053702.
Viniegra, J. G., Martinez, N., Modirassari, P., Hernandez Losa, J., Parada Cobo, C., Sanchez-Arevalo Lobo, V. J., et al. (2005). Full activation of PKB/Akt in response to insulin or ionizing radiation is mediated through ATM. Journal of Biological Chemistry,280(6), 4029–4036. https://doi.org/10.1074/jbc.M410344200.
Wang, H., Perrault, A. R., Takeda, Y., Qin, W., & Iliakis, G. (2003). Biochemical evidence for Ku-independent backup pathways of NHEJ. Nucleic Acids Research,31(18), 5377–5388.
Wang, X., Ran, T., Zhang, X., Xin, J., Zhang, Z., Wu, T., et al. (2017). 3.9 Å structure of the yeast Mec1-Ddc2 complex, a homolog of human ATR-ATRIP. Science,358(6367), 1206–1209. https://doi.org/10.1126/science.aan8414.
Williams, B. R., Mirzoeva, O. K., Morgan, W. F., Lin, J., Dunnick, W., & Petrini, J. H. (2002). A murine model of Nijmegen breakage syndrome. Current Biology,12(8), 648–653.
Woodbine, L., Gennery, A. R., & Jeggo, P. A. (2014). The clinical impact of deficiency in DNA non-homologous end-joining. DNA Repair (Amst),16, 84–96. https://doi.org/10.1016/j.dnarep.2014.02.011.
Woodbine, L., Neal, J. A., Sasi, N. K., Shimada, M., Deem, K., Coleman, H., et al. (2013). PRKDC mutations in a SCID patient with profound neurological abnormalities. Journal of Clinical Investigation,123(7), 2969–2980. https://doi.org/10.1172/JCI67349.
Wu, Z. H., Wong, E. T., Shi, Y., Niu, J., Chen, Z., Miyamoto, S., et al. (2010). ATM- and NEMO-dependent ELKS ubiquitination coordinates TAK1-mediated IKK activation in response to genotoxic stress. Molecular Cell,40(1), 75–86. https://doi.org/10.1016/j.molcel.2010.09.010.
Xing, M., Yang, M., Huo, W., Feng, F., Wei, L., Jiang, W., et al. (2015). Interactome analysis identifies a new paralogue of XRCC4 in non-homologous end joining DNA repair pathway. Nature Communications,6, 6233. https://doi.org/10.1038/ncomms7233.
Xu, Y., Ashley, T., Brainerd, E. E., Bronson, R. T., Meyn, M. S., & Baltimore, D. (1996). Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes & Development,10(19), 2411–2422.
Yajima, H., Lee, K. J., & Chen, B. P. (2006). ATR-dependent phosphorylation of DNA-dependent protein kinase catalytic subunit in response to UV-induced replication stress. Molecular and Cellular Biology,26(20), 7520–7528. https://doi.org/10.1128/MCB.00048-06.
Yamamoto, K., Wang, Y., Jiang, W., Liu, X., Dubois, R. L., Lin, C. S., et al. (2012). Kinase-dead ATM protein causes genomic instability and early embryonic lethality in mice. Journal of Cell Biology,198(3), 305–313. https://doi.org/10.1083/jcb.201204098.
Yamamoto, K., Wang, J., Sprinzen, L., Xu, J., Haddock, C. J., Li, C., et al. (2016). Kinase-dead ATM protein is highly oncogenic and can be preferentially targeted by Topo-isomerase I inhibitors. Elife,5, e14709. https://doi.org/10.7554/elife.14709.
Yan, C. T., Boboila, C., Souza, E. K., Franco, S., Hickernell, T. R., Murphy, M., et al. (2007). IgH class switching and translocations use a robust non-classical end-joining pathway. Nature,449(7161), 478–482.
Yang, D. Q., & Kastan, M. B. (2000). Participation of ATM in insulin signalling through phosphorylation of eIF-4E-binding protein 1. Nature Cell Biology,2(12), 893–898. https://doi.org/10.1038/35046542.
Yang, Y., Xia, F., Hermance, N., Mabb, A., Simonson, S., Morrissey, S., et al. (2011). A cytosolic ATM/NEMO/RIP1 complex recruits TAK1 to mediate the NF-kappaB and p38 mitogen-activated protein kinase (MAPK)/MAPK-activated protein 2 responses to DNA damage. Molecular and Cellular Biology,31(14), 2774–2786. https://doi.org/10.1128/MCB.01139-10.
Yazinski, S. A., Comaills, V., Buisson, R., Genois, M. M., Nguyen, H. D., Ho, C. K., et al. (2017). ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes & Development,31(3), 318–332. https://doi.org/10.1101/gad.290957.116.
Yi, M., Rosin, M. P., & Anderson, C. K. (1990). Response of fibroblast cultures from ataxia-telangiectasia patients to oxidative stress. Cancer Letters,54(1–2), 43–50.
Zha, S., Bassing, C. H., Sanda, T., Brush, J. W., Patel, H., Goff, P. H., et al. (2010a). ATM-deficient thymic lymphoma is associated with aberrant tcrd rearrangement and gene amplification. Journal of Experimental Medicine,207(7), 1369–1380. https://doi.org/10.1084/Jem.20100285.
Zha, S., Bassing, C. H., Sanda, T., Brush, J. W., Patel, H., Goff, P. H., et al. (2010b). ATM-deficient thymic lymphoma is associated with aberrant tcrd rearrangement and gene amplification. Journal of Experimental Medicine,207(7), 1369–1380. https://doi.org/10.1084/jem.20100285.
Zha, S., Guo, C., Boboila, C., Oksenych, V., Cheng, H. L., Zhang, Y., et al. (2011a). ATM damage response and XLF repair factor are functionally redundant in joining DNA breaks. Nature,469(7329), 250–254. https://doi.org/10.1038/nature09604.
Zha, S., Jiang, W., Fujiwara, Y., Patel, H., Goff, P. H., Brush, J. W., et al. (2011b). Ataxia telangiectasia-mutated protein and DNA-dependent protein kinase have complementary V(D)J recombination functions. Proceedings of the National Academy of Sciences of the United States of America,108(5), 2028–2033. https://doi.org/10.1073/pnas.1019293108.
Zhang, S., Matsunaga, S., Lin, Y. F., Sishc, B., Shang, Z., Sui, J., et al. (2016). Spontaneous tumor development in bone marrow-rescued DNA-PKcs(3A/3A) mice due to dysfunction of telomere leading strand deprotection. Oncogene,35(30), 3909–3918. https://doi.org/10.1038/onc.2015.459.
Zhang, J., Tripathi, D. N., Jing, J., Alexander, A., Kim, J., Powell, R. T., et al. (2015). ATM functions at the peroxisome to induce pexophagy in response to ROS. Nature Cell Biology,17(10), 1259–1269. https://doi.org/10.1038/ncb3230.
Zhang, S., Yajima, H., Huynh, H., Zheng, J., Callen, E., Chen, H. T., et al. (2011). Congenital bone marrow failure in DNA-PKcs mutant mice associated with deficiencies in DNA repair. Journal of Cell Biology,193(2), 295–305. https://doi.org/10.1083/jcb.201009074.
Zhao, H., & Piwnica-Worms, H. (2001). ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Molecular and Cellular Biology,21(13), 4129–4139. https://doi.org/10.1128/MCB.21.13.4129-4139.2001.
Zhou, Y., Lee, J. H., Jiang, W., Crowe, J. L., Zha, S., & Paull, T. T. (2017). Regulation of the DNA damage response by DNA-PKcs inhibitory phosphorylation of ATM. Molecular Cell,65(1), 91–104. https://doi.org/10.1016/j.molcel.2016.11.004.
Zhou, Z. W., Liu, C., Li, T. L., Bruhn, C., Krueger, A., Min, W., et al. (2013). An essential function for the ATR-activation-domain (AAD) of TopBP1 in mouse development and cellular senescence. PLoS Genetics,9(8), e1003702. https://doi.org/10.1371/journal.pgen.1003702.
Zhu, J., Petersen, S., Tessarollo, L., & Nussenzweig, A. (2001). Targeted disruption of the Nijmegen breakage syndrome gene NBS1 leads to early embryonic lethality in mice. Current Biology,11(2), 105–109.
Zou, L., & Elledge, S. J. (2003). Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science,300(5625), 1542–1548. https://doi.org/10.1126/science.1083430.
Acknowledgements
We thank all members of Zha lab for useful discussions. We apologize for original papers we were not able to cite but were covered by reviews instead. The authors were in part supported by NIH/NCI 5R01CA158073, 5R01CA215067 and 5R01CA184187 to SZ. SZ was a Leukemia Lymphoma Society Scholar and DM has been supported by an American-Italian Cancer Foundation Post-Doctoral Research Fellowship and a senior fellowship from the Leukemia and Lymphoma Society.
Author information
Affiliations
Institute for Cancer Genetics, Vagelos College of Physicians and Surgeons, Columbia University, New York, NY, 10032, USA
Demis Menolfi & Shan Zha
Department of Pathology and Cell Biology, Vagelos College of Physicians and Surgeons, Columbia University, New York, NY, 10032, USA
Shan Zha
Division of Pediatric Oncology, Hematology and Stem Cell Transplantation, Department of Pediatrics, Vagelos College of Physicians and Surgeons, Columbia University, New York, NY, 10032, USA
Shan Zha
Corresponding author
Correspondence to Shan Zha.
Rights and permissions
About this article
Cite this article
Menolfi, D., Zha, S. ATM, DNA-PKcs and ATR: shaping development through the regulation of the DNA damage responses. GENOME INSTAB. DIS. 1, 47–68 (2020). https://doi.org/10.1007/s42764-019-00003-9
Received30 November 2018
Revised26 March 2019
Accepted26 April 2019
Published20 May 2019
Issue DateMarch 2020
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
Mouse models
DNA double strand breaks
DNA replication
Lymphocyte development
用户登录
还没有账号?
立即注册