






Crosstalk between signaling pathways and DNA damage response
Review Article
Kangjunjie Wang, Long Li, Yuxue Zhang & Daming Gao
Genome Instability & Disease 1, 81–91(2020)
Abstract
Living organisms have developed a complex signaling system to sense diverse growth signals and co-ordinate intracellular bioprocesses, so that they survive in various conditions. Since organisms are often exposed to environmental and genomic threats which cause DNA damages, DNA damage response (DDR) and repair systems are important for maintenance of genome stability and integrity. A line of evidences has demonstrated that there is tight crosstalk between growth signaling pathways and DDR systems. In this review, we give a brief overview of recent reports dissecting the interaction between signaling pathways and DDR at molecular level, which may further expand the knowledge of the signaling network and provide clues for disease therapy.
Introduction
Living organisms are usually exposed to various environmental and endogenous risk factors, which are harmful to genome stability and integrity. Different factors can lead to different types of DNA damages (Table 1). For example, ionizing radiation (IR) and chemotherapy drugs can lead to double-strand DNA breaks (DSBs); ultraviolet light (UV) can result in single-strand breaks (SSBs) and double-strand DNA breaks; reactive oxygen species (ROS) can cause single-strand breaks; replication errors can lead to mismatch damages. If such DNA lesions are not repaired correctly and timely, genome stability and integrity would be impaired and lead to severe consequences including cell death or malignant transformation.
| Threaten factors | Types of damage | Repair pathway |
|---|---|---|
| Ultraviolet light (UV) | Single-strand breaks (SSB) | Nucleotide excision repair (NER) |
| Reactive oxygen species (ROS) | Single-strand breaks (SSB) | Base excision repair (BER) |
| Ionizing radiation (IR) | Double-strand breaks (DSB) | Homologous recombination repair (HR) |
| Chemotherapy drugs | Non-homologous end joining (NHEJ) | |
| Single-strand annealing (SSA) | ||
| Microhomology-mediated end joining (MMEJ) | ||
| Replication errors | Mismatch | Mismatch repair (MMR) |
Table 1 DNA damage-threatening factors, types and repair pathways
Luckily, DNA damages can be quickly detected by cells and then induce various intracellular responses, which are known as DNA damage response (DDR) and include a series of mechanisms to respond and repair damaged DNA. When DNA damages were detected, depending on the types of DNA damage, certain kind of DNA repair pathways can be activated solely or together to fix the damages. For example, in mismatch repair (MMR), the only step to repair the lesions is to replace the wrong nucleotide with the correct one (Reyes et al. 2015). Base excision repair (BER) and nucleotide excision repair (NER) are participating in SSBs repair (Scharer 2013; Whitaker et al. 2017). Homologous recombination repair (HR), non-homologous end joining (NHEJ) and single-strand annealing (SSA) are the three main pathways employed for DSB repair (Bhargava et al. 2016; Chapman et al. 2012). Notably, recent studies suggested that microhomology-mediated end joining (MMEJ) is a new error-prone mechanism in DSB repair system which occurs even when HR and NHEJ are available, and it is particularly important in HR-defective tumors (Chiruvella et al. 2013; Wang and Xu 2017) (Table 1).
To control the DNA damage, once DNA lesions are detected, cell cycle is arrested to provide enough time and energy for DNA repair. Then, a series of cell processes are initiated to repair the damages by coordinating various repair machineries and gene transcription. On the other hand, multicellular organisms are highly organized life systems, which rely on signaling transduction network to co-ordinate substances–energy metabolism and efficient intra-cell communication. For example, amino acids activate mTOR signaling to regulate protein and lipid synthesis, autophagy and glucose metabolism; growth factors such as EGF and insulin activate PI3K–AKT pathway to regulate cell growth, cell fate decision and cell cycle; NF-κB signaling can regulate innate immune response and cell death pathways. Recent studies have provided many clues suggesting that such signaling pathways either activate or inhibit one or multiple factors in DDR process. Moreover, some components of signaling network could function also as DNA damage sensors or transducers to actively participate in DDR or DNA repair process. In current review, we focus on introducing recent studies about how DDR or DNA repair process is influenced by or crosstalk with major signaling pathways at molecular level and discuss about how such knowledge would expand our understanding of genome stability maintenance in physiological and pathological conditions.
mTOR signaling and DDR
Mechanistic target of rapamycin (mTOR) is the central regulator of nutrition sensing and it integrates growth signals to control cell homeostasis, growth, survival and proliferation. mTOR regulates protein and lipid synthesis, energy metabolism and autophagy through forming two multi-subunit complexes known as mTORC1 and mTORC2 (Laplante and Sabatini 2012). Although mTORC1 and mTORC2 share some mutual components, such as mTOR, mammalian lethal with sec-13 protein 8 (mLST8), DEP domain containing mTOR-interacting protein (DEPTOR) and the Tti1/Tel2 complex (Jacinto et al. 2004; Kaizuka et al. 2010; Kim et al. 2003; Peterson et al. 2009), there are several subunits only specifically functioning in either mTORC1 or mTORC2. For example, mTORC1 contains regulatory-associated protein of mammalian target of rapamycin (Raptor) and proline-rich Akt substrate 40 kD (PRAS40) (Kim et al. 2002; Wang et al. 2007), while mTORC2 contains rapamycin-insensitive companion of mTOR (Rictor), mammalian stress-activated map kinase-interacting protein 1 (mSin1) and protein observed with Rictor 1 and 2 (Protor1/2) (Frias et al. 2006; Pearce et al. 2007; Pearce et al. 2011; Sarbassov et al. 2004).
mTORC1 is generally activated by amino acids, but also can respond to growth factors, energy status, oxygen and stress. mTORC1 regulates protein and lipid synthesis, energy metabolism, and inhibits autophagy by phosphorylating downstream substrates, which is consistent with its most important function of sensing nutrients. In low amino acids status, SESN1/2 and CASTOR1 (two amino acid sensors for leucine and arginine, respectively) interact with GATOR2 complex to suppress its inhibitory function to GATOR1 complex; meanwhile, GATOR1 inhibits mTORC1 activity by acting as GTPase-activating proteins (GAPs) for GTP-charged RagA. (Bar-Peled et al. 2013; Chantranupong et al. 2014, 2016). On the contrary, abundant leucine and arginine in cytoplasm block SESN1/2 and CASTOR1 from binding to GATOR2, so that GATOR2 can interact with GATOR1 and result in activation of mTORC1. Another arginine sensor is SLC38A9 which localizes on the lysosome membrane and transports essential amino acids into the cytoplasm (Wyant et al. 2017). There are also other lysosome membrane anchoring proteins involving in activating mTORC1, including v-ATPase, RAGULATOR-Rag complex and Folliculin (FLCN) (Sancak et al. 2010; Zoncu et al. 2011). After the change in amino acids level, these proteins change the GTP/GDP charging status of Rag complex to recruit mTORC1 to the lysosome membrane where mTORC1 can be activated by small GTP-binding protein RHEB (Long et al. 2005) (Fig. 1a). Activated mTORC1 directly phosphorylates downstream substrates including S6K1, 4E-BP1 and so on to regulate protein and lipid synthesis, autophagy and energy metabolism (Fig. 1a). In comparison with mTORC1, the activation mechanism of mTORC2 is much less understood. mTORC2 can be activated by growth factors via phosphoinositide 3-kinase (PI3K), such as insulin, but not by nutrients. mTORC2 regulates autophagy, cell survival, metabolism, and cytoskeleton organization by phosphorylating downstream AKT1, SGK1 and PKCα. The dysfunction of mTOR signaling can induce various diseases, including cancer, obesity, type 2 diabetes and neurodegeneration (ref).
Fig. 1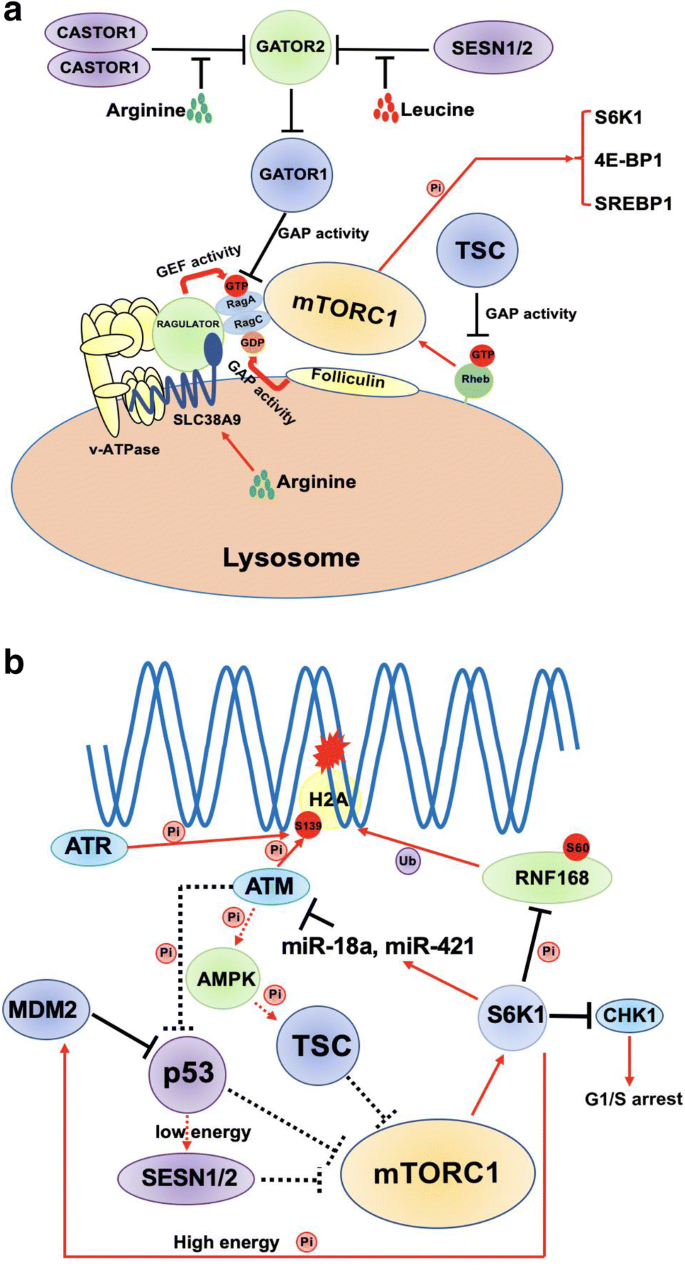
Crosstalk between mTOR signaling and DDR. a A summarized model of mTORC1 activation by amino acids. b mTORC1 interplays with DDR systems. Star indicates DNA damage; black lines indicate inhibitory function; red arrows indicate stimulation or activation; dashed lines indicate feedback regulations
Full size imageRecent studies suggest that mTORC1 pathway plays an important role in one or multiple DDR steps to modulate genome stability, which strongly indicates an underlying association between nutrients/metabolism regulation and genome stability maintenance. Three kinases, ataxia telangiectasia and Rad3-related (ATR), ataxia telangiectasia mutated (ATM) and DNA protein kinase (DNA-PK), are activated to initiate subsequent DDR upon DNA damage. Interestingly, ATR, ATM, DNA-PK and mTORC1 all belong to phosphatidylinositol 3-kinase PI3K-related (PIKKs) family (Ma et al. 2018). For instance, when DSBs occur, activated ATM is recruited to the damage sites rapidly after the loading of MRE11-RAD50-NBS1 (MRN) complex (Paull, 2015). Then, ATM phosphorylates H2A.X at Ser139 (known as γ-H2A.X), which is indispensable for recruiting mediator of DNA damage checkpoint protein 1 (MDC1). Once MDC1 is in place, it acts as a scaffold for E3 ligases RNF8 and RNF168 to ubiquitylate histones for recruiting downstream proteins to initiate NHEJ or HR repair process (Doil et al. 2009; Huen et al. 2007; Kolas et al. 2007; Mailand et al. 2007; Stewart et al. 2009). Our recent study has demonstrated that mTORC1-S6K pathway can inhibit DDR through phosphorylating RNF168 at Ser60 by impairing its E3 ligase activity and promoting TRIP12-dependent RNF168 degradation. Importantly, ectopic expression of phosphorylation-resistant mutant, RNF168-S60A, rescues DNA damage defects and inhibits Lkb1 deletion (causing hyper-activation of mTORC1-S6K pathway)-induced lung tumor development (Xie et al. 2018) (Fig. 1b). These findings provide a new molecular link that connects nutrients/growth signaling to genome stability and tumorigenesis via mTORC1-S6K pathway.
Some other research evidences also suggest that mTORC1 may interfere with the DDR via additional mechanisms. S6K regulates MYCN transcription factor positively, and knockout of S6k1 and S6k2 in MEF leading to MYCN decreases, which controls certain miRNAs transcription. It was further indicated that mTORC1-S6K pathway controls the level of two microRNAs, miR-18a and miR-421, which target ATM mRNA and induce ATM inhibition in childhood sarcoma (Shen and Houghton 2013) (Fig. 1b), adding another layer of evidence that mTORC1-S6K pathway promotes DDR defects and tumorigenesis by impairing ATM checkpoint. As the other side of the feedback loop, mTOR kinase activity is modulated by DDR induced by etoposide via ATM. ATM phosphorylates AMP-activated protein kinase (AMPK) and then AMPK-mediated TSC2 activation cause inhibition of mTORC1 (Alexander et al. 2010) (Fig. 1b).
p53, a transcription factor, is famous for its crucial function in cell cycle control, DDR and cell fate decision. More importantly, p53 is another linker between nutrients sensing and genotoxic stress response. When DNA damage occurs, ATM can phosphorylate p53 at its N-terminal and prevent or promote its degradation mediated by MDM2 (MacLaine and Hupp 2011; Venkata Narayanan et al. 2017) (Fig. 1b). In addition, DNA damage signal activates mTORC1 via p38 MAPK and mTORC1 positively regulates p53 stability via S6K1-mediated MDM2 phosphorylation and inhibition (Lai et al. 2010) (Fig. 1b). On the other hand, p53 also inhibits mTORC1 activity via AMPK/TSC2 axis or activation of SESN2 in low energy condition (Budanov and Karin 2008) (Fig. 1b). Therefore, the regulatory balance between p53 and mTORC1 signaling is bi-directional and very dynamic, and it may have impact on DDR. Moreover, mTORC1 was also shown to modulate DDR at transcription levels. One example is that mTORC1-S6K signaling axis can regulate transcription level of CHK1 (Zhou et al. 2017), a serine/threonine protein kinase required for cell cycle arrest (Fig. 1b). mTORC1-S6K signaling up-regulates Cyclin D and Cyclin E to down-regulate the CHK1 transcription level resulting in S phase arrest induced by DNA damage. Altogether, as the major sensor and modulator of cell metabolism, mTORC1 signaling has complex roles in regulating DDR.
Wnt/β-catenin signaling and DDR
Wnt/β-catenin signaling plays an important role in embryonic development by controlling expression of various genes which are important for cell proliferation, polarity, organogenesis and stem cell renewal. Deregulation of Wnt/β-catenin signaling can lead to diseases including cancer and neurodegeneration (Duchartre et al. 2016). Wnt/β-catenin signaling can be activated and function in β-catenin dependent or independent manners (Fig. 2a). In β-catenin dependent activation, Wnt binds to Frzzled (Fz) and LDL-receptor-related protein (LRP), two receptors on the cell surface, leading to β-catenin dissociation from the Axin-APC-GSK3β complex, which keeps GSK3β from phosphorylating β-catenin. Then, free β-catenin translocates into nucleus to promote gene transcription by binding to co-factors like T cell factor (TCF) and lymphoid enhancer factor (LEF) (Gordon and Nusse 2006) (Fig. 2a). In β-catenin-independent pathway, Wnt binds to Fz to activate Dishevelled (Dsh) resulting in the modulation of cytoskeleton through some small GTPases (Fig. 2a). Another β-catenin-independent signal transduction requires Ca2+ . Once Wnt binds to Fz, Ca2+ is released from endoplasmic reticulum (ER), and increase of Ca2+ in cytoplasm activates Ca2+ -dependent proteins, such as protein kinase C (PKC) and cyclic AMP-responsive element-binding protein 1 (CREB1), which in turn promotes downstream genes transcription (Komiya and Habas 2008) (Fig. 2a).
Fig. 2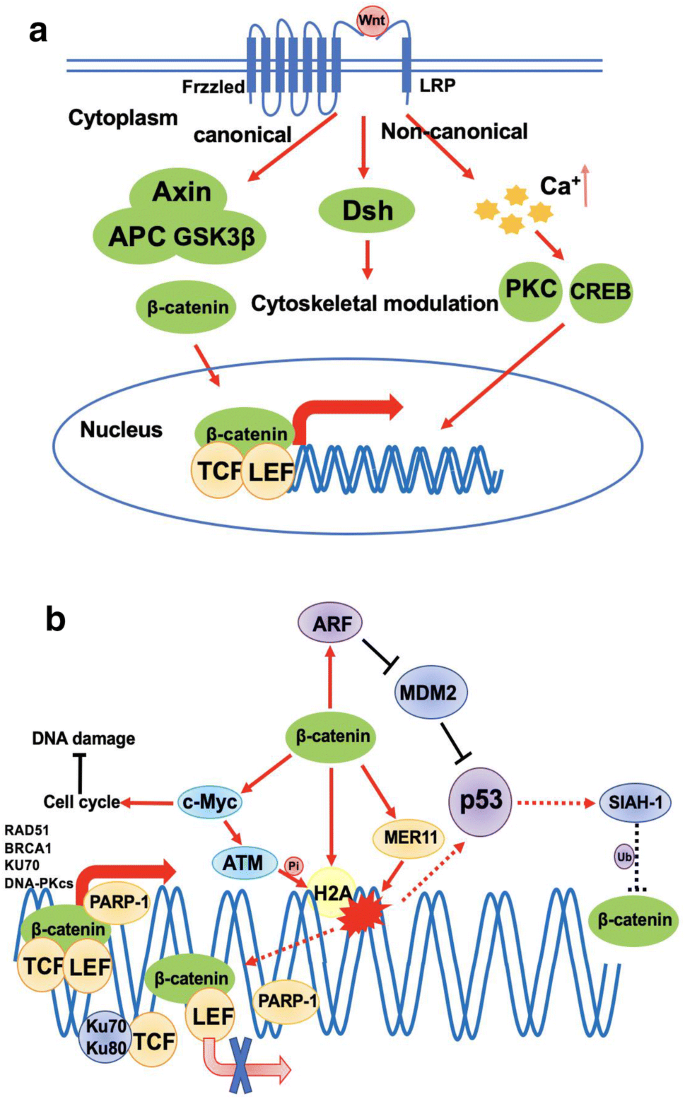
Role of Wnt/β-catenin signaling in DDR. a Simplified mechanisms of canonical and non-canonical Wnt signaling pathway activation. b The crosstalk between Wnt/β-catenin signaling pathway and DDR. Star indicates DNA damage; black lines indicate inhibitory function; red arrows indicate stimulation or activation; dashed lines indicate feedback regulations; thicker red arrows indicate transcription
Full size imageWnt/β-catenin signaling pathway has been indicated in multiple steps of DDR. As a transcription factor, activated β-catenin can promote the expression of MRE11 and H2A.X directly (Deng et al. 2011; Karimaian et al. 2017), which are essential to DDR described above (Fig. 2b). c-Myc, known as a major target gene of β-catenin, contributes to DSBs repair via activation of ATM and transcription of multiple genes in HR and NHEJ pathways, such as RAD51 and BRCA1 (in HR), XRCC4, DNA-PKcs and KU70 (in NHEJ) (Fernandez et al. 2003; Li et al. 2003; Luoto et al. 2010). However, c-Myc also promotes cell cycle progression which is unfavorable for DNA repair as cell cycle arrest is essential for effective DNA repair. Therefore, Wnt/β-catenin signaling may be inhibited temporarily to arrest cell cycle in response to DNA damage. Wnt/β-catenin signaling can also regulate p53 stability through up-regulating the expression of ARF (Inoue and Fry 2018), a tumor suppressor protein that functions along with p53. ARF binds to and inactivates MDM2 to stabilize p53 and promotes p53-dependent responses, such as cell cycle regulation and apoptosis (Fig. 2b). On the contrary, when p53 is activated by DNA damage signals, p53 induces the expression of seven in absentia homolog 1 (SIAH-1) that ubiquitylates β-catenin to promote its degradation (Matsuzawa and Reed 2001) (Fig. 2b). Therefore, in this loop, p53 could be activated by DNA damage and it may restrict Wnt/β-catenin signaling so that cells could fix DNA damage and survive.
As the other side of the feedback loop, DDR can regulate Wnt/β-catenin signaling pathway by controlling β-catenin transcription activity. As described above, β-catenin mainly binds to TCF and LEF to promote target genes transcription (Fig. 2b). Recent research reported that poly (ADP-ribose) polymerase-1 (PARP-1) can bind to β-catenin-TCF complex to enhance its transcription activity (Idogawa et al. 2007). When DNA damage occurs, PARP-1 catalyzes auto-poly (ADP-ribose) modification of itself and dissociate from β-catenin-TCF complex, which decreases the transcription level of downstream genes involved in DNA repair. Ku proteins (Ku70/Ku80) are vital factors in DDR by recognizing DSBs and recruiting DNA-PK to damage sites for further function. It was reported that Ku70/Ku80 can bind to TCF to cause the dissociation of TCF from β-catenin-TCF complex, which decreases the transcription activity of β-catenin (Idogawa et al. 2007). Mechanistically, the dissociation of PARP-1 may allow Ku70 to bind to TCF-4, disrupting the interaction between TCF-4 and β-catenin. In summary, Wnt/β-catenin signaling is delicately coordinated with DDR systems. Once DNA damage occurs, two mechanisms described above contribute to cell cycle arrest through down-regulating β-catenin transcription activity, and Wnt/β-catenin signaling is reactivated to drive cell proliferation after DNA damage fixed, which may induce radioresistance in cancer cells.
PI3K/AKT signaling pathway
Phosphoinositide 3-kinase (PI3K), a lipid kinase responding to growth factors or insulin, catalyzes the phosphorylation and conversion of PtdIns-3,4-P2 (PI3,4P2) to PtdIns-3,4,5-P3 (PIP3) which results in recruiting AKT kinase to the cell membrane. As a consequence of PI3K activation, AKT is phosphorylated at Thr308 and Ser473 by PDK1 and mTORC2, respectively, resulting in its full activation. There are three AKT genes encoded in human genome, known as AKT1, AKT2 and AKT3, but only AKT1 will be discussed in this review because the mechanisms of AKT2 and AKT3 in DNA damage response are less explored. Activated AKT1 can phosphorylate downstream proteins to regulate various cell processes, including cell growth, metabolism, cell cycle, apoptosis and tumorigenesis (Fig. 3a). Recent studies have suggested that dysfunction of PI3K/AKT signaling can lead to diverse pathological diseases, such as cancers, cardiovascular disease, type 2 diabetes, inflammatory and autoimmune disorders, and neurological disorders (Manning and Toker 2017).Some downstream targets of AKT1 are related to cell cycle and DNA repair (Fig. 3b), which means PI3K/AKT signaling may play a role in DDR. AKT1 can regulate transcription level of c-Myc to indirectly promote cell cycle and, meanwhile, inhibits apoptosis (Gartel and Shchors 2003). AKT1 can also regulate c-Myc stability by phosphorylating GSK3β to inhibit its kinase activity, which phosphorylates c-Myc at Thr58 to promote its degradation (Sears et al. 2000). Furthermore, AKT1 can phosphorylate p21 and p27 to inhibit their suppressive function in G1/S transition (Viglietto et al. 2002; Zhou et al. 2001). Similarly, AKT1-mediated phosphorylation and activation of Cdc25B and Cdk2 are important for S/G2 transition (Baldin et al. 2003; Maddika et al. 2008). It was reported that AKT1 phosphorylates MDM2 at Ser166 and Ser186 to stabilize MDM2 and promote its translocation into nucleus for degradation of p53 (Ogawara et al. 2002). Therefore, PI3K–AKT signaling modulates cell cycle via various mechanisms which may indirectly affect DDR.
Fig. 3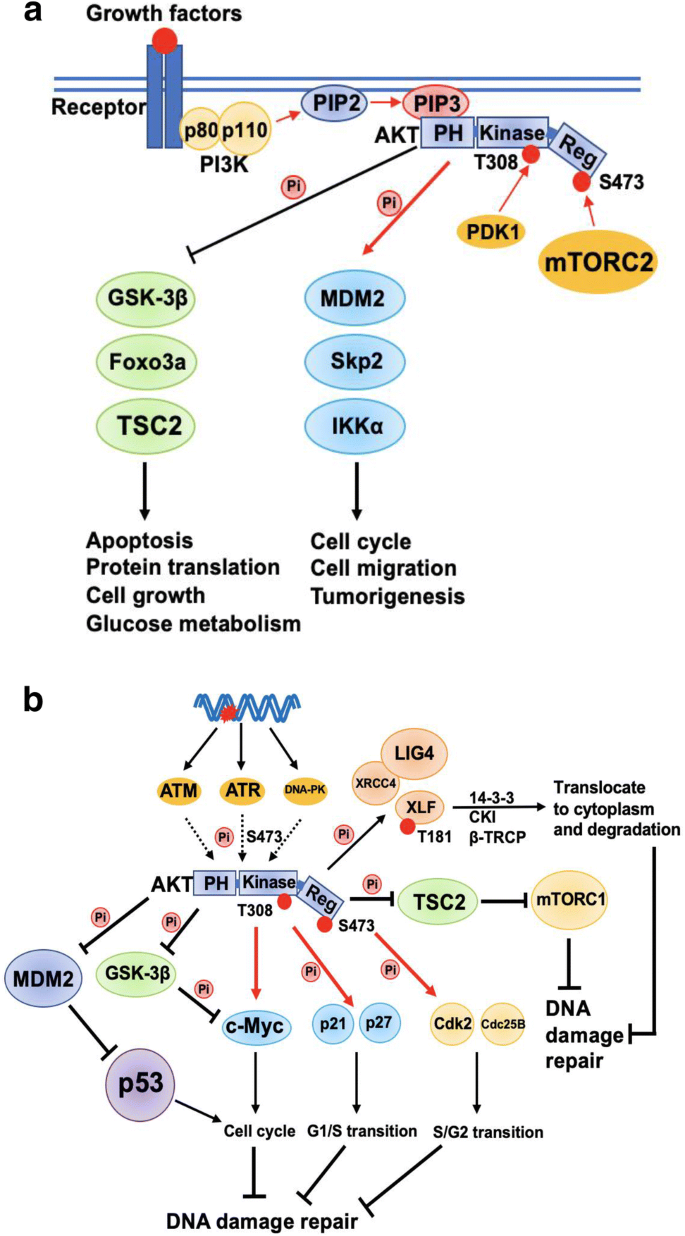
Interaction of PI3K/AKT signaling pathway and DDR. a Model of AKT activation and function via phosphorylating downstream substrates. b AKT1 modulates DDR directly via phosphorylating XLF or indirectly via regulating cell cycle. Star indicates DNA damage; black lines indicate inhibitory function; red arrows indicate stimulation or activation; dashed lines indicate feedback regulations
Full size imageRecent studies also suggest that AKT1 may phosphorylate repair factors directly, such as XLF, BRCA1 and RPS3 (Liu et al. 2015; Plo et al. 2008), to regulate different types of DNA repair (Fig. 3b). AKT1 can also participate in DBSs repair through recruiting DNA-PKcs to the damage sites. Some experimental evidences showed that reduction of AKT1 abundance and activity can decrease the DNA damage-induced phosphorylation of DNA-PKcs, which is essential for activation of DNA-PKcs (Chen et al. 2007). On the contrary, AKT1 activity is also affected by DNA damage. Three PIKK family members, ATM, ATR and DNA-PK, all contribute to AKT1 Ser473 phosphorylation in DDR (Fig. 3b), which indicates that AKT1 is activated not only by growth factors but also by genotoxic stress (Caporali et al. 2008). Moreover, since there is crosstalk between PI3K/AKT, mTOR, and Wnt/β-catenin pathways, PI3K/AKT signaling could also modulate DDR via other signaling pathways. For example, AKT1 activates mTORC1 via phosphorylating and inhibiting TSC2 (Fig. 3b). Similarly, Wnt/β-catenin signaling could also be regulated by AKT1 via phosphorylation of GSK3β (Fig. 3b), which leads to inhibition of GSK3β and stabilization of β-catenin. Taken together, these reports support a complex role of PI3K/AKT pathway in regulating DDR.
cAMP/PKA signaling and DDR
GPCRs are the biggest family of cell membrane receptors, and they can be activated by hormones, cytokines and neurotransmitters. When hormones bind to GPCRs, α subunit of G protein (Gα) changes its structure to bind to GTP and dissociate from β and γ subunits of G protein (Gβ and Gγ). Then, Gα further activates adenylate cyclase (AC) to produce cAMP, which subsequently activates Protein kinase A (PKA). PKA is composed of two regulatory subunits and two catalytic subunits. cAMP binds to PKA regulatory subunits to release and activate catalytic subunits. Free catalytic subunits then phosphorylate downstream substrates, such as cAMP responsive element binding protein (CREB) and glycogen phosphorylase kinase (GPK) (Torres-Quesada et al. 2017) (Fig. 4a). cAMP signaling pathway plays an important role in regulating various cell processes including secretion, cardiac contraction, metabolism, cell differentiation, cell cycle and synaptic plasticity. The disorder of cAMP/PKA signaling pathway causes various diseases including cancer, neurodegeneration and endocrine diseases (ref).
Fig. 4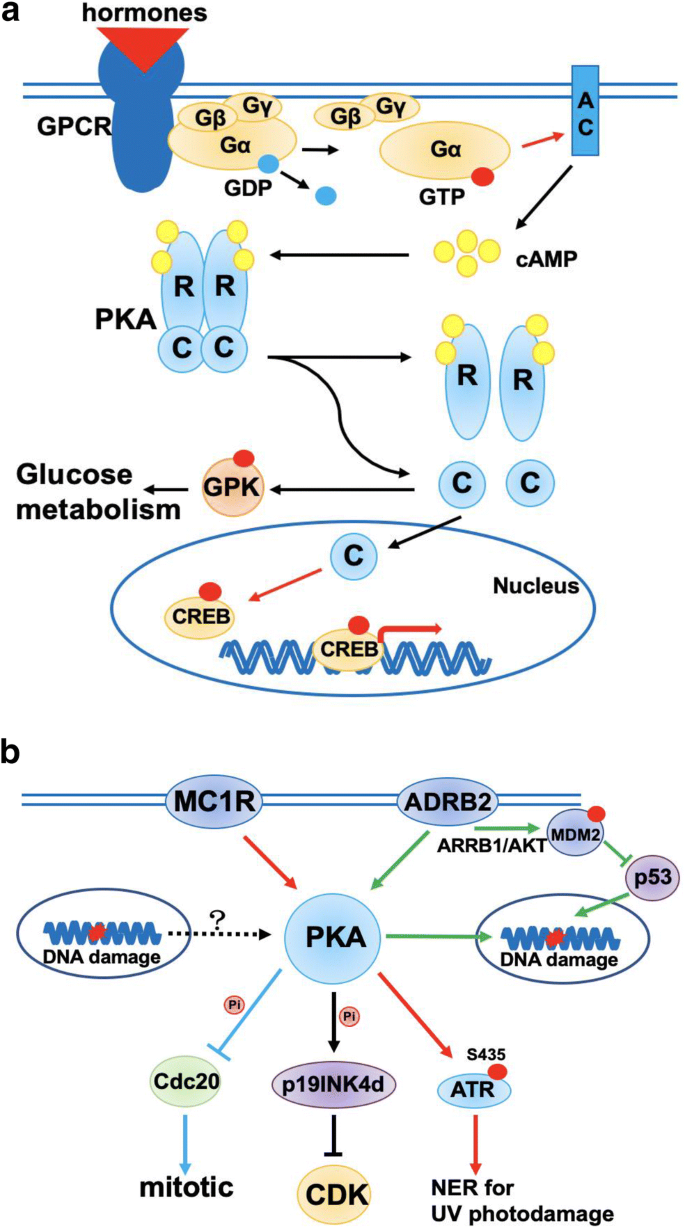
Chronic stress is related to DDR. a Mechanism of GPCRs-mediated cAMP/PKA signaling pathway activation. GPCRs binding to hormones lead to Gα allosteric activation which results in AC activation. Activated AC transfers ATP to cAMP which is essential to PKA activation. R represents regulator subunit of PKA; C represents catalytic subunit of PKA. b PKA-mediated chronic stress cell response in DDR. The lines and arrows of each color represent a chronic stress regulation mechanism in DDR, respectively
Full size imageRecent studies indicated a functional link between cAMP/PKA signaling and DDR. p19INK4d, a member of INK4 family and an inhibitor of cycle-dependent kinase (CDKs), is dependent on PKA to promote its nuclear location and exert its function in DNA repair (Marazita et al. 2012) (Fig. 4b). Moreover, PKA phosphorylates Cdc20 to inhibit its function of inducing mitosis in budding yeast after DNA damage, although the mechanism underlying PKA activation after DNA damage remains unclear (Searle et al. 2004). Melanocortin 1 receptor (MC1R), known as a stimulatory GPCR and an important factor in melanoma, is another bridge connecting cAMP/PKA signaling and DDR, particularly by PKA-dependent ATR-Ser435 phosphorylation and sequential recruitment of XPA to initiate nucleoside excision repair (NER) induced by UV (Jarrett et al. 2014) (Fig. 4b).
Human body and mind are often under various stresses, and chronic stress can stimulate our sympathetic nervous system to persistently secrete catecholamine, adrenaline and noradrenaline. Those hormones may lead to the activation of β2-adrenoreceptors (ADRB2) and the accumulation of DNA damages. An interesting study showed that the activation of ADRB2 can inhibit DNA damage repair by destabilizing p53 protein through a PKA and β-arrestin-1 (ARRB1)-mediated signaling pathway (Hara et al. 2011). Mechanistically chronic stress can activate ARRB1 to promote AKT-mediated activation of MDM2 which leads to enhanced p53 degradation (Fig. 4b). A mouse model of ovarian carcinoma shows that ADRB2-mediated activation of cAMP/PKA signaling pathway can promote tumor growth and angiogenesis (Thaker et al. 2006). Therefore, these evidences support a connection between cAMP/PKA signaling and DDR, which may provide a new explanation for the role of physiologic and psychosocial stress in cancer development.
NF-κB signaling and DDR
Nuclear factor (NF)-κB transcription factors bind to specific DNA senquences directly, known as κB elements, to regulate the expression of genes in immune and inflammatory responses. NF-κB signaling can be activated by canonical and non-canonical pathways. In canonical pathway, extracellular signal proteins, such as tumor necrosis factor-α (TNF-α) and interleukin-1 (IL-1), stimulate TNF receptor1 (TNFR1) to engage receptor adaptor proteins (TRADD and RIP1) and recruit a series of E2 and E3 ubiquitin enzymes to promote mixed poly-ubiquitination of factors that are required for the activation of IKK kinase. Then, IKK phosphorylates inhibitor of κB (IκB) to trigger its ubiquitination and degradation via 26S proteasome, which releases NF-κB from the NF-κB-IκBs complex to translocate into nucleus to regulate gene expression (Fig. 5a). This IKK-dependent canonical NF-κB pathway could also be activated by genotoxic stress. It was reported that NF-κB essential modifier (NEMO) is SUMOylated and ubiquitylated in nucleus after DNA damage activates ATM. Then, modified NEMO transfers to cytoplasm with ATM to activate IKK, which is similar to the mechanism induced by TNF-α (Hayden and Ghosh 2008; Wu and Miyamoto 2007) (Fig. 5a). In non-canonical pathway, after stimulated by lipopolysaccharide (LPS) or CD40, NF-κB-inducing kinase (NIK) activates IKK and then IKK phosphorylates NF-κB subunit p100 and causes p100 ubiquitination and proteasome-dependent processing to produce p52, which translocates into nucleus to regulate gene expression (Perkins 2007).
Fig. 5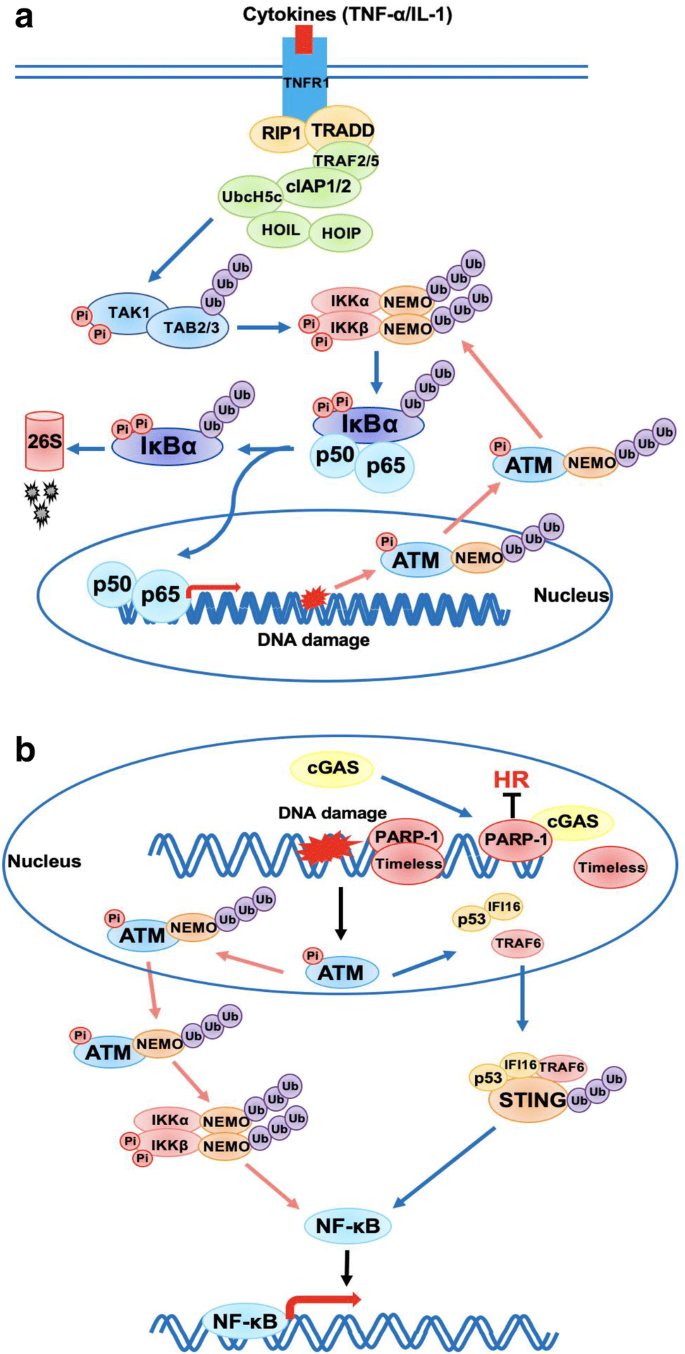
NF-κB signaling pathway interacts with DDR. a Schematic diagram of canonical NF-κB signaling pathway. b A model of innate immune response induced by DNA damage. DNA damage induced by etoposide activates p53, then IFI16 and TRAF6 relocate into cytoplasm to form a complex with and activate STING. This non-canonical activation of STING can activate NF-κB transcription rather than IRF-3
Full size imageRecent experiment evidences suggest that DNA damage can activate innate immune response by activating NF-κB signaling pathway. When DSBs occur, ATM plays an essential role in DNA damage repair and also functions as a key factor in IR-induced activation of NEMO-NF-κB pathway as described above. In addition, one study showed that IKK can be activated through canonical pathway in wild-type mice but not in the Atm−/− mice after exposure to radiation (Li et al. 2001), which demonstrates that ATM is required for canonical activation of NF-κB signaling in physiological condition. Moreover, a recent study reported that etoposide-induced DNA damage can active innate immune response via Cyclic guanosine monophosphate (GMP)-AMP synthase (cGAS), the cytosolic DNA sensor. Once cGAS is activated, it binds to STING at ER leading to the activation of STING and subsequent activation of TBK1 and IRF-3, which contributes to the transcription of interferon-β (IFN-β). But a study by Dunphy et al., suggested that ATM and interferon-γ-inducible factor 16 (IFI16) can activate STING via a cGAS independent pathway. In that pathway, NF-κB, rather than IRF-3, is activated predominantly by STING after nuclear DNA damage occurs. Mechanistically, etoposide-induced activation of ATM and PARP-1 promotes p53-IFI16-TRAF6-STING complex formation in the cytoplasm, where TRAF6 ubiquitylates STING to promote NF-κB-mediated transcription program and downstream innate immune response (Dunphy et al. 2018) (Fig. 5b). Moreover, a latest study shows that DNA damage induces cGAS translocation into nucleus to suppress homologous recombination-mediated DNA repair by abrogating the formation of the PARP1-timeless complex and inhibit tumor growth (Liu et al. 2018). These evidences demonstrate that not only the damaged DNA caused by virus infection but also IR or chemo drugs-induced DNA damage in nucleus can active innate immune response via NF-κB pathway.
Discussion and perspectives
Extracellular factors, such as amino acids, growth factors, hormones and secretory cytokines can regulate cell growth, proliferation, survival under stress, cell cycle control, cell fate decision and metabolism through various signaling pathways. Deregulated signaling pathways may contribute to multiple diseases including cancer. During tumorigenesis, loss-of-function mutation of tumor suppressors and gain-of-function mutation of oncogenes are often observed to cause subsequent activation of signaling pathways. For example, frequently observed p53 loss in cancer contributes to mTORC1 activation (Feng et al. 2005). In mice study, loss of PTEN, a tumor suppressor suppressing mTORC2-AKT pathway, may promote the development of prostate cancer (Guertin et al. 2009). Loss of APC, a famous tumor suppressor, frequently mutated in human cancers, is the main cause of familial adenomatous polyposis which may develop to colorectal cancer (Polakis 2012). Therefore, the key components of signaling pathways are potentially targets for cancer therapy.
Signaling pathways not only play important roles in cancer development but also participate in genome instability regulation by interfering with DDR. Radiotherapy and chemotherapy are common and powerful treatments for cancers, which lead to a large amount of DNA damages and result in cell death. Therefore, dissection of the crosstalk between signaling pathways and DDR systems is important for exploring new targets and strategies to overcome radio resistance and tumor recurrence. For example, it is reported that Wnt/β-catenin signaling is active in many radio-resistant cancers including breast cancer, colon cancer and head and neck cancer (Zhao et al. 2018). Therefore, it is possible to enhance the efficiency of DNA damage therapy by suppressing Wnt/β-catenin signaling components in such cases. In keeping with this notion, iCRT14 and K-756, two inhibitors of Wnt/β-catenin signaling, were reported to promote colorectal cancer radio-sensitivity by targeting TCF/β-catenin and Axin (Jun et al. 2016; Okada-Iwasaki et al. 2016). Moreover, rapamycin analogs and AKT inhibitors are used to inhibit mTOR and PI3K/AKT signaling activity, which increased radiation sensitivity of human breast cancer and non-small cell lung cancer cell lines (Holler et al. 2016). These studies may provide a general strategy to use combination therapies by applying signaling pathways inhibitors and radio/chemotherapy together to enhance the efficiency of cancer therapy.
Overall, accumulating evidences have suggested tight interactions between signaling pathways and DDR systems at molecular level, which provides possibility of identifying new feedback loops within the regulatory network. Further progress of basic research in this direction may expand our knowledge to design better and more efficient therapeutic plans to treat human diseases including cancer.
References
Alexander, A., Cai, S. L., Kim, J., Nanez, A., Sahin, M., MacLean, K. H., et al. (2010). ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proceedings of the National Academy of Sciences,107(9), 4153–4158. https://doi.org/10.1073/pnas.0913860107.
Baldin, V., Theis-Febvre, N., Benne, C., Froment, C., Cazales, M., Burlet-Schiltz, O., et al. (2003). PKB/Akt phosphorylates the CDC25B phosphatase and regulates its intracellular localisation. Biology of the Cell,95(8), 547–554.
Bar-Peled, L., Chantranupong, L., Cherniack, A. D., Chen, W. W., Ottina, K. A., Grabiner, B. C., et al. (2013). A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science,340(6136), 1100–1106. https://doi.org/10.1126/science.1232044.
Bhargava, R., Onyango, D. O., & Stark, J. M. (2016). Regulation of single-strand annealing and its role in genome maintenance. Trends in Genetics,32(9), 566–575. https://doi.org/10.1016/j.tig.2016.06.007.
Budanov, A. V., & Karin, M. (2008). p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell,134(3), 451–460. https://doi.org/10.1016/j.cell.2008.06.028.
Caporali, S., Levati, L., Starace, G., Ragone, G., Bonmassar, E., Alvino, E., et al. (2008). AKT is activated in an ataxia-telangiectasia and Rad3-related-dependent manner in response to temozolomide and confers protection against drug-induced cell growth inhibition. Molecular Pharmacology,74(1), 173–183. https://doi.org/10.1124/mol.107.044743.
Chantranupong, L., Scaria, S. M., Saxton, R. A., Gygi, M. P., Shen, K., Wyant, G. A., et al. (2016). The CASTOR proteins are arginine sensors for the mTORC1 Pathway. Cell,165(1), 153–164. https://doi.org/10.1016/j.cell.2016.02.035.
Chantranupong, L., Wolfson, R. L., Orozco, J. M., Saxton, R. A., Scaria, S. M., Bar-Peled, L., et al. (2014). The Sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Reports,9(1), 1–8. https://doi.org/10.1016/j.celrep.2014.09.014.
Chapman, J. R., Taylor, M. R., & Boulton, S. J. (2012). Playing the end game: DNA double-strand break repair pathway choice. Molecular Cell,47(4), 497–510. https://doi.org/10.1016/j.molcel.2012.07.029.
Chen, B. P., Uematsu, N., Kobayashi, J., Lerenthal, Y., Krempler, A., Yajima, H., et al. (2007). Ataxia telangiectasia mutated (ATM) is essential for DNA-PKcs phosphorylations at the Thr-2609 cluster upon DNA double strand break. Journal of Biological Chemistry,282(9), 6582–6587. https://doi.org/10.1074/jbc.M611605200.
Chiruvella, K. K., Liang, Z., & Wilson, T. E. (2013). Repair of double-strand breaks by end joining. Cold Spring Harbor Perspectives in Biology,5(5), a012757. https://doi.org/10.1101/cshperspect.a012757.
Deng, R., Tang, J., Ma, J. G., Chen, S. P., Xia, L. P., Zhou, W. J., et al. (2011). PKB/Akt promotes DSB repair in cancer cells through upregulating Mre11 expression following ionizing radiation. Oncogene,30(8), 944–955. https://doi.org/10.1038/onc.2010.467.
Doil, C., Mailand, N., Bekker-Jensen, S., Menard, P., Larsen, D. H., Pepperkok, R., et al. (2009). RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell,136(3), 435–446. https://doi.org/10.1016/j.cell.2008.12.041.
Duchartre, Y., Kim, Y. M., & Kahn, M. (2016). The Wnt signaling pathway in cancer. Critical Reviews in Oncology Hematology,99, 141–149. https://doi.org/10.1016/j.critrevonc.2015.12.005.
Dunphy, G., Flannery, S. M., Almine, J. F., Connolly, D. J., Paulus, C., Jonsson, K. L., et al. (2018). Non-canonical activation of the DNA sensing adaptor STING by ATM and IFI16 mediates NF-kappaB signaling after nuclear DNA damage. Molecular Cell,71(5), 745–760e745. https://doi.org/10.1016/j.molcel.2018.07.034.
Feng, Z., Zhang, H., Levine, A. J., & Jin, S. (2005). The coordinate regulation of the p53 and mTOR pathways in cells. Proceedings of the National Academy of Sciences,102(23), 8204–8209. https://doi.org/10.1073/pnas.0502857102.
Fernandez, P. C., Frank, S. R., Wang, L., Schroeder, M., Liu, S., Greene, J., et al. (2003). Genomic targets of the human c-Myc protein. Genes & Development,17(9), 1115–1129. https://doi.org/10.1101/gad.1067003.
Frias, M. A., Thoreen, C. C., Jaffe, J. D., Schroder, W., Sculley, T., Carr, S. A., et al. (2006). mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Current Biology,16(18), 1865–1870. https://doi.org/10.1016/j.cub.2006.08.001.
Gartel, A. L., & Shchors, K. (2003). Mechanisms of c-myc-mediated transcriptional repression of growth arrest genes. Experimental Cell Research,283(1), 17–21.
Gordon, M. D., & Nusse, R. (2006). Wnt signaling: multiple pathways, multiple receptors, and multiple transcription factors. Journal of Biological Chemistry,281(32), 22429–22433. https://doi.org/10.1074/jbc.R600015200.
Guertin, D. A., Stevens, D. M., Saitoh, M., Kinkel, S., Crosby, K., Sheen, J. H., et al. (2009). mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell,15(2), 148–159. https://doi.org/10.1016/j.ccr.2008.12.017.
Hara, M. R., Kovacs, J. J., Whalen, E. J., Rajagopal, S., Strachan, R. T., Grant, W., et al. (2011). A stress response pathway regulates DNA damage through beta2-adrenoreceptors and beta-arrestin-1. Nature,477(7364), 349–353. https://doi.org/10.1038/nature10368.
Hayden, M. S., & Ghosh, S. (2008). Shared principles in NF-kappaB signaling. Cell,132(3), 344–362. https://doi.org/10.1016/j.cell.2008.01.020.
Holler, M., Grottke, A., Mueck, K., Manes, J., Jucker, M., Rodemann, H. P., et al. (2016). Dual targeting of Akt and mTORC1 impairs repair of DNA double-strand breaks and increases radiation sensitivity of human tumor cells. PLoS One,11(5), e0154745. https://doi.org/10.1371/journal.pone.0154745.
Huen, M. S., Grant, R., Manke, I., Minn, K., Yu, X., Yaffe, M. B., et al. (2007). RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell,131(5), 901–914. https://doi.org/10.1016/j.cell.2007.09.041.
Idogawa, M., Masutani, M., Shitashige, M., Honda, K., Tokino, T., Shinomura, Y., et al. (2007). Ku70 and poly(ADP-ribose) polymerase-1 competitively regulate beta-catenin and T-cell factor-4-mediated gene transactivation: possible linkage of DNA damage recognition and Wnt signaling. Cancer Research,67(3), 911–918. https://doi.org/10.1158/0008-5472.CAN-06-2360.
Inoue, K., & Fry, E. A. (2018). Tumor suppression by the EGR1, DMP1, ARF, p53, and PTEN Network. Cancer Investigation,2018, 1–17. https://doi.org/10.1080/07357907.2018.1533965.
Jacinto, E., Loewith, R., Schmidt, A., Lin, S., Ruegg, M. A., Hall, A., et al. (2004). Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nature Cell Biology,6(11), 1122–1128. https://doi.org/10.1038/ncb1183.
Jarrett, S. G., Wolf Horrell, E. M., Christian, P. A., Vanover, J. C., Boulanger, M. C., Zou, Y., et al. (2014). PKA-mediated phosphorylation of ATR promotes recruitment of XPA to UV-induced DNA damage. Molecular Cell,54(6), 999–1011. https://doi.org/10.1016/j.molcel.2014.05.030.
Jun, S., Jung, Y. S., Suh, H. N., Wang, W., Kim, M. J., Oh, Y. S., et al. (2016). LIG4 mediates Wnt signalling-induced radioresistance. Nature Communications,7, 10994. https://doi.org/10.1038/ncomms10994.
Kaizuka, T., Hara, T., Oshiro, N., Kikkawa, U., Yonezawa, K., Takehana, K., et al. (2010). Tti1 and Tel2 are critical factors in mammalian target of rapamycin complex assembly. Journal of Biological Chemistry,285(26), 20109–20116. https://doi.org/10.1074/jbc.M110.121699.
Karimaian, A., Majidinia, M., Bannazadeh Baghi, H., & Yousefi, B. (2017). The crosstalk between Wnt/beta-catenin signaling pathway with DNA damage response and oxidative stress: Implications in cancer therapy. DNA Repair (Amsterdam),51, 14–19. https://doi.org/10.1016/j.dnarep.2017.01.003.
Kim, D. H., Sarbassov, D. D., Ali, S. M., King, J. E., Latek, R. R., Erdjument-Bromage, H., et al. (2002). mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell,110(2), 163–175.
Kim, D. H., Sarbassov, D. D., Ali, S. M., Latek, R. R., Guntur, K. V., Erdjument-Bromage, H., et al. (2003). GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Molecular Cell,11(4), 895–904.
Kolas, N. K., Chapman, J. R., Nakada, S., Ylanko, J., Chahwan, R., Sweeney, F. D., et al. (2007). Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science,318(5856), 1637–1640. https://doi.org/10.1126/science.1150034.
Komiya, Y., & Habas, R. (2008). Wnt signal transduction pathways. Organogenesis,4(2), 68–75.
Lai, K. P., Leong, W. F., Chau, J. F., Jia, D., Zeng, L., Liu, H., et al. (2010). S6K1 is a multifaceted regulator of Mdm2 that connects nutrient status and DNA damage response. EMBO Journal,29(17), 2994–3006. https://doi.org/10.1038/emboj.2010.166.
Laplante, M., & Sabatini, D. M. (2012). mTOR signaling in growth control and disease. Cell,149(2), 274–293. https://doi.org/10.1016/j.cell.2012.03.017.
Li, N., Banin, S., Ouyang, H., Li, G. C., Courtois, G., Shiloh, Y., et al. (2001). ATM is required for IkappaB kinase (IKKk) activation in response to DNA double strand breaks. Journal of Biological Chemistry,276(12), 8898–8903. https://doi.org/10.1074/jbc.M009809200.
Li, Z., Van Calcar, S., Qu, C., Cavenee, W. K., Zhang, M. Q., & Ren, B. (2003). A global transcriptional regulatory role for c-Myc in Burkitt’s lymphoma cells. Proceedings of the National Academy of Sciences,100(14), 8164–8169. https://doi.org/10.1073/pnas.1332764100.
Liu, P., Gan, W., Guo, C., Xie, A., Gao, D., Guo, J., et al. (2015). Akt-mediated phosphorylation of XLF impairs non-homologous end-joining DNA repair. Molecular Cell,57(4), 648–661. https://doi.org/10.1016/j.molcel.2015.01.005.
Liu, H., Zhang, H., Wu, X., Ma, D., Wu, J., Wang, L., et al. (2018). Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature,563(7729), 131–136. https://doi.org/10.1038/s41586-018-0629-6.
Long, X., Lin, Y., Ortiz-Vega, S., Yonezawa, K., & Avruch, J. (2005). Rheb binds and regulates the mTOR kinase. Current Biology,15(8), 702–713. https://doi.org/10.1016/j.cub.2005.02.053.
Luoto, K. R., Meng, A. X., Wasylishen, A. R., Zhao, H., Coackley, C. L., Penn, L. Z., et al. (2010). Tumor cell kill by c-MYC depletion: role of MYC-regulated genes that control DNA double-strand break repair. Cancer Research,70(21), 8748–8759. https://doi.org/10.1158/0008-5472.CAN-10-0944.
Ma, Y., Vassetzky, Y., & Dokudovskaya, S. (2018). mTORC1 pathway in DNA damage response. Biochimica Biophysica Acta Molecular Cell Research,1865(9), 1293–1311. https://doi.org/10.1016/j.bbamcr.2018.06.011.
MacLaine, N. J., & Hupp, T. R. (2011). How phosphorylation controls p53. Cell Cycle,10(6), 916–921. https://doi.org/10.4161/cc.10.6.15076.
Maddika, S., Ande, S. R., Wiechec, E., Hansen, L. L., Wesselborg, S., & Los, M. (2008). Akt-mediated phosphorylation of CDK2 regulates its dual role in cell cycle progression and apoptosis. Journal of Cell Science,121(Pt 7), 979–988. https://doi.org/10.1242/jcs.009530.
Mailand, N., Bekker-Jensen, S., Faustrup, H., Melander, F., Bartek, J., Lukas, C., et al. (2007). RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell,131(5), 887–900. https://doi.org/10.1016/j.cell.2007.09.040.
Manning, B. D., & Toker, A. (2017). AKT/PKB signaling: navigating the network. Cell,169(3), 381–405. https://doi.org/10.1016/j.cell.2017.04.001.
Marazita, M. C., Ogara, M. F., Sonzogni, S. V., Marti, M., Dusetti, N. J., Pignataro, O. P., et al. (2012). CDK2 and PKA mediated-sequential phosphorylation is critical for p19INK4d function in the DNA damage response. PLoS One,7(4), e35638. https://doi.org/10.1371/journal.pone.0035638.
Matsuzawa, S. I., & Reed, J. C. (2001). Siah-1, SIP, and Ebi collaborate in a novel pathway for beta-catenin degradation linked to p53 responses. Molecular Cell,7(5), 915–926.
Ogawara, Y., Kishishita, S., Obata, T., Isazawa, Y., Suzuki, T., Tanaka, K., et al. (2002). Akt enhances Mdm2-mediated ubiquitination and degradation of p53. Journal of Biological Chemistry,277(24), 21843–21850. https://doi.org/10.1074/jbc.M109745200.
Okada-Iwasaki, R., Takahashi, Y., Watanabe, Y., Ishida, H., Saito, J., Nakai, R., et al. (2016). The discovery and characterization of K-756, a novel Wnt/beta-catenin pathway inhibitor targeting tankyrase. Molecular Cancer Therapeutics,15(7), 1525–1534. https://doi.org/10.1158/1535-7163.MCT-15-0938.
Paull, T. T. (2015). Mechanisms of ATM activation. Annual Review of Biochemistry,84, 711–738. https://doi.org/10.1146/annurev-biochem-060614-034335.
Pearce, L. R., Huang, X., Boudeau, J., Pawlowski, R., Wullschleger, S., Deak, M., et al. (2007). Identification of protor as a novel Rictor-binding component of mTOR complex-2. Biochemical Journal,405(3), 513–522. https://doi.org/10.1042/BJ20070540.
Pearce, L. R., Sommer, E. M., Sakamoto, K., Wullschleger, S., & Alessi, D. R. (2011). Protor-1 is required for efficient mTORC2-mediated activation of SGK1 in the kidney. Biochemical Journal,436(1), 169–179. https://doi.org/10.1042/BJ20102103.
Perkins, N. D. (2007). Integrating cell-signalling pathways with NF-kappaB and IKK function. Nature Reviews Molecular Cell Biology,8(1), 49–62. https://doi.org/10.1038/nrm2083.
Peterson, T. R., Laplante, M., Thoreen, C. C., Sancak, Y., Kang, S. A., Kuehl, W. M., et al. (2009). DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell,137(5), 873–886. https://doi.org/10.1016/j.cell.2009.03.046.
Plo, I., Laulier, C., Gauthier, L., Lebrun, F., Calvo, F., & Lopez, B. S. (2008). AKT1 inhibits homologous recombination by inducing cytoplasmic retention of BRCA1 and RAD51. Cancer Research,68(22), 9404–9412. https://doi.org/10.1158/0008-5472.CAN-08-0861.
Polakis, P. (2012). Wnt signaling in cancer. Cold Spring Harbor Perspectives in Biology,4(5), e00852. https://doi.org/10.1101/cshperspect.a008052.
Reyes, G. X., Schmidt, T. T., Kolodner, R. D., & Hombauer, H. (2015). New insights into the mechanism of DNA mismatch repair. Chromosoma,124(4), 443–462. https://doi.org/10.1007/s00412-015-0514-0.
Sancak, Y., Bar-Peled, L., Zoncu, R., Markhard, A. L., Nada, S., & Sabatini, D. M. (2010). Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell,141(2), 290–303. https://doi.org/10.1016/j.cell.2010.02.024.
Sarbassov, D. D., Ali, S. M., Kim, D. H., Guertin, D. A., Latek, R. R., Erdjument-Bromage, H., et al. (2004). Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Current Biology,14(14), 1296–1302. https://doi.org/10.1016/j.cub.2004.06.054.
Scharer, O. D. (2013). Nucleotide excision repair in eukaryotes. Cold Spring Harbor Perspectives in Biology,5(10), a012609. https://doi.org/10.1101/cshperspect.a012609.
Searle, J. S., Schollaert, K. L., Wilkins, B. J., & Sanchez, Y. (2004). The DNA damage checkpoint and PKA pathways converge on APC substrates and Cdc20 to regulate mitotic progression. Nature Cell Biology,6(2), 138–145. https://doi.org/10.1038/ncb1092.
Sears, R., Nuckolls, F., Haura, E., Taya, Y., Tamai, K., & Nevins, J. R. (2000). Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes & Development,14(19), 2501–2514.
Shen, C., & Houghton, P. J. (2013). The mTOR pathway negatively controls ATM by up-regulating miRNAs. Proceedings of the National Academy of Sciences,110(29), 11869–11874. https://doi.org/10.1073/pnas.1220898110.
Stewart, G. S., Panier, S., Townsend, K., Al-Hakim, A. K., Kolas, N. K., Miller, E. S., et al. (2009). The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell,136(3), 420–434. https://doi.org/10.1016/j.cell.2008.12.042.
Thaker, P. H., Han, L. Y., Kamat, A. A., Arevalo, J. M., Takahashi, R., Lu, C., et al. (2006). Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nature Medicine,12(8), 939–944. https://doi.org/10.1038/nm1447.
Torres-Quesada, O., Mayrhofer, J. E., & Stefan, E. (2017). The many faces of compartmentalized PKA signalosomes. Cellular Signalling,37, 1–11. https://doi.org/10.1016/j.cellsig.2017.05.012.
Venkata Narayanan, I., Paulsen, M. T., Bedi, K., Berg, N., Ljungman, E. A., Francia, S., et al. (2017). Transcriptional and post-transcriptional regulation of the ionizing radiation response by ATM and p53. Scientific Reports,7, 43598. https://doi.org/10.1038/srep43598.
Viglietto, G., Motti, M. L., Bruni, P., Melillo, R. M., D’Alessio, A., Califano, D., et al. (2002). Cytoplasmic relocalization and inhibition of the cyclin-dependent kinase inhibitor p27(Kip1) by PKB/Akt-mediated phosphorylation in breast cancer. Nature Medicine,8(10), 1136–1144. https://doi.org/10.1038/nm762.
Wang, L., Harris, T. E., Roth, R. A., & Lawrence, J. C., Jr. (2007). PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. Journal of Biological Chemistry,282(27), 20036–20044. https://doi.org/10.1074/jbc.M702376200.
Wang, H., & Xu, X. (2017). Microhomology-mediated end joining: new players join the team. Cell Bioscience,7, 6. https://doi.org/10.1186/s13578-017-0136-8.
Whitaker, A. M., Schaich, M. A., Smith, M. R., Flynn, T. S., & Freudenthal, B. D. (2017). Base excision repair of oxidative DNA damage: from mechanism to disease. Frontiers in Bioscience (Landmark edition),22, 1493–1522.
Wu, Z. H., & Miyamoto, S. (2007). Many faces of NF-kappaB signaling induced by genotoxic stress. Journal of Molecular Medicine,85(11), 1187–1202. https://doi.org/10.1007/s00109-007-0227-9.
Wyant, G. A., Abu-Remaileh, M., Wolfson, R. L., Chen, W. W., Freinkman, E., Danai, L. V., et al. (2017). mTORC1 Activator SLC38A9 is required to efflux essential amino acids from lysosomes and use protein as a nutrient. Cell,171(3), 642e612–642e654. https://doi.org/10.1016/j.cell.2017.09.046.
Xie, X., Hu, H., Tong, X., Li, L., Liu, X., Chen, M., et al. (2018). The mTOR-S6K pathway links growth signalling to DNA damage response by targeting RNF168. Nature Cell Biology,20(3), 320–331. https://doi.org/10.1038/s41556-017-0033-8.
Zhao, Y., Tao, L., Yi, J., Song, H., & Chen, L. (2018). The role of canonical Wnt signaling in regulating radioresistance. Cellular Physiology and Biochemistry,48(2), 419–432. https://doi.org/10.1159/000491774.
Zhou, B. P., Liao, Y., Xia, W., Spohn, B., Lee, M. H., & Hung, M. C. (2001). Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nature Cell Biology,3(3), 245–252. https://doi.org/10.1038/35060032.
Zhou, X., Liu, W., Hu, X., Dorrance, A., Garzon, R., Houghton, P. J., et al. (2017). Regulation of CHK1 by mTOR contributes to the evasion of DNA damage barrier of cancer cells. Scientific Reports,7(1), 1535. https://doi.org/10.1038/s41598-017-01729-w.
Zoncu, R., Bar-Peled, L., Efeyan, A., Wang, S., Sancak, Y., & Sabatini, D. M. (2011). mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science,334(6056), 678–683. https://doi.org/10.1126/science.1207056.
Acknowledgments
This work was supported by the National Key Research and Development Program from the Ministry of Science and Technology of China (Grant 2015CB964502), National Natural Science Foundation of China (Grants 81790253, 91853130), Strategic Priority Research Program of the Chinese Academy of Sciences (Grants XDB19020203, XDA12020364).
Author information
Affiliations
State Key Laboratory of Cell Biology, CAS Key Laboratory of Systems Biology, CAS Center for Excellence in Molecular Cell Science, Innovation Center for Cell Signaling Network, Shanghai Institute of Biochemistry and Cell Biology, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 320 Yueyang Road, Shanghai, 200031, China
Kangjunjie Wang, Long Li, Yuxue Zhang & Daming Gao
Corresponding author
Correspondence to Daming Gao.
Rights and permissions
About this article
Cite this article
Wang, K., Li, L., Zhang, Y. et al. Crosstalk between signaling pathways and DNA damage response. GENOME INSTAB. DIS. 1, 81–91 (2020). https://doi.org/10.1007/s42764-019-00005-7
Received14 February 2019
Revised05 June 2019
Accepted30 July 2019
Published08 August 2019
Issue DateMarch 2020
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
Growth signaling pathways
DDR
Crosstalk
用户登录
还没有账号?
立即注册