






Ubiquitin and the DNA double-strand break repair pathway
Review Article
Somaira Nowsheen, Min Deng & Zhenkun Lou
Genome Instability & Disease 1, 69–80(2020)
Abstract
The ubiquitin system plays a central role in diverse cellular processes including DNA damage response. As such, it is not surprising that its dysfunction contributes to various diseases including cancer and neurodegenerative disorders. An understanding of the ubiquitin system is, therefore, important in devising treatments for such diseases. In this review, we discuss the central role of ubiquitin in DNA damage response, specifically DNA double-strand break repair. We focus on recent findings on the role of ubiquitin in the DNA double-strand break repair pathway, possible nodes of modulation, and finally their implications for treatment of various diseases.
Introduction
To ensure the fidelity of the genome, cells have developed a complex network of response pathways. As soon as damaged DNA is detected, a number of signaling molecules are activated to halt cell cycle progression so that the damaged DNA is not passed on to the progeny. In addition, repair factors are activated in an attempt to repair the DNA lesion. This multifaceted response also includes transcriptional changes to generate any protein that may be required. In the case of failed DNA repair, cell death pathways such as apoptosis are triggered (Fig. 1).
Fig. 1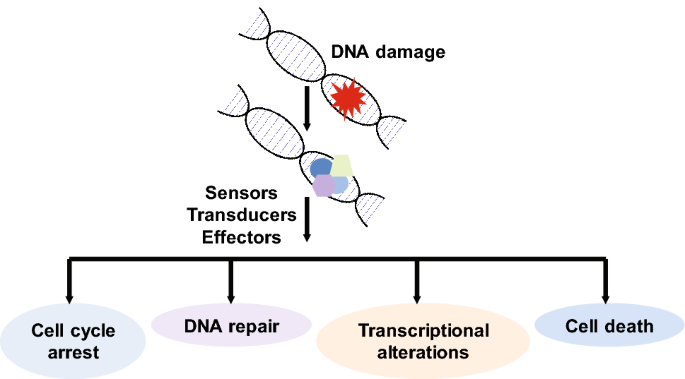
DNA damage leads to the activation of multiple signaling pathways
Full size imageThe apical kinases ATM and ATR initiate the signaling cascade in response to DNA damage or replication stress (Falck et al. 2005; Shiloh 2003; Brown and Baltimore 2000). Detection of DNA damage by complexes such as MRN (Mre11, Rad50, and NBS1) triggers the activation of these protein kinases (Lee and Paull 2004, 2005). Of the various types of DNA damage, double-strand breaks (DSBs) are the most lethal. This is because these lesions can lead to grossly abnormal chromosomal loss or rearrangements which may ultimately result in tumorigenesis. In this review, we will focus our discussion on cellular response to DSBs.
Following DSB, ATM phosphorylates the histone protein H2AX at serine 139 which subsequently acts as a docking site for downstream repair factors (Rogakou et al. 1998). Before phosphorylated serine 139 on H2AX (γH2AX) can operate in this role, H2AX needs to be dephosphorylated at another residue, tyrosine 142 (Y142) (Cook et al. 2009). In unstressed cells, this Y142 residue is basally phosphorylated by the kinase WSTF (Xiao et al. 2009). The protein phosphatase EYA is responsible for dephosphorylation of this amino acid on H2AX (Cook et al. 2009). In addition, the adaptor protein ZNF506 is required for EYA to function in this capacity. Without ZNF506, EYA is unable to recognize and interact with pY142 H2AX, thus, leading to deficient DNA repair (Nowsheen et al. 2018).
Next, the mediator protein MDC1 helps to amplify the signal by binding to γH2AX and recruiting more repair substrates (Lou et al. 2003, 2006). MDC1 recruits the E3 ubiquitin ligase RNF8 (Huen et al. 2007; Mailand et al. 2007). RNF8 is responsible for ubiquitylation of the polycomb group protein L3MBTL2 which subsequently acts as a scaffold for the next E3 ligase in the signaling cascade, RNF168 (Nowsheen et al. 2018). RNF168 is responsible for the modification of histone H2A on lysine (K) 13 (K13) and K15 residues (Mattiroli et al. 2012). These residues act as docking sites for major DNA DSB proteins such as 53BP1 and Rap80/BRCA1 (Fradet-Turcotte et al. 2013; Coleman and Greenberg 2011; Kim et al. 2007; Sobhian et al. 2007; Shao et al. 2009; Wang and Elledge 2007). Together, this signaling cascade thereby helps to repair the DSB (Fig. 2).
Fig. 2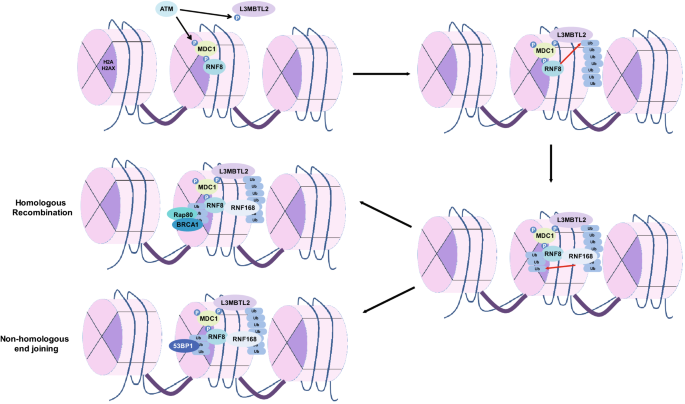
A simplified overview of the DNA double-strand break response pathway
Full size imageUbiquitin
Ubiquitin (Ub) is a highly conserved 76 amino acid polypeptide. Ubiquitylation is an essential reversible post-translational modification that involves covalent attachment of Ub to the substrate. It is ubiquitous (hence the name) and confers specific, quantitative and reversible regulation on biological pathways. The process involves three subsets of proteins: E1 which is an ubiquitin-activating enzyme, E2 which is an ubiquitin-conjugating enzyme, and E3 which is an ubiquitin ligase (Scheffner et al. 1995). The E3 ligases act as writers. In addition, there are ubiquitin-binding effectors which act as readers, and deubiquitylases which are the erasers of the modification (Fig. 3) (Nijman et al. 2005). Ubiquitin can be conjugated to substrates using various amino acids on the protein including methionine and lysine residues (Mattiroli et al. 2012; Kim et al. 2014; Hoege et al. 2002). This process regulates a variety of cellular processes including cell division, differentiation, and DNA damage response including DSB repair and DNA replication (Fig. 4) (Mattiroli et al. 2012; Higashitsuji et al. 2005; Zhong et al. 2003).
Fig. 3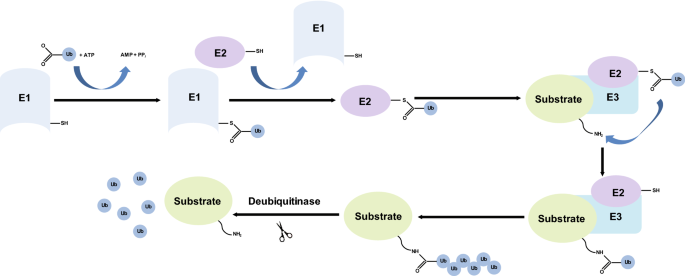
A summary of the ubiquitylation reaction
Fig. 4
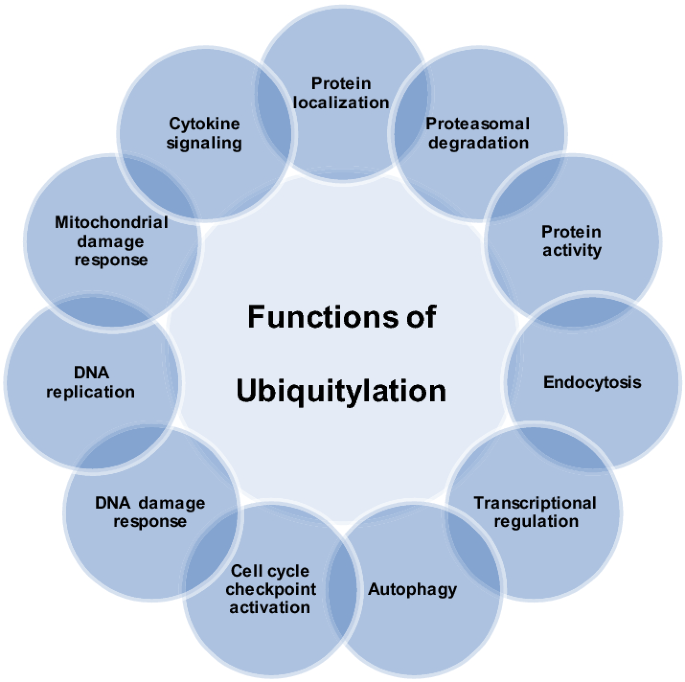
Ubiquitylation regulates various cellular functions
Another unique aspect of this post-translational modification is that it can form various linkages and chains. For instance, transfer of a single ubiquitin molecule to a substrate is known as monoubiquitylation (Andreassen et al. 2004). A substrate may have multiple monoubiquitins conjugated at different amino acid residues. In addition, there can be other conjugations forming chains of various lengths and linkages. For instance, lysine 48 (K48) linkages typically signal proteins for proteasomal degradation (Yau et al. 2017; Chau et al. 1989; Jin et al. 2008). On the other hand, K63 linkages have been shown to play crucial roles in DNA DSB response pathway (Nowsheen et al. 2018; Mattiroli et al. 2012; Fradet-Turcotte et al. 2013). The K63-linked polymers allow for the assembly of large protein complexes during DNA repair. A substrate may undergo a combination of modifications, i.e., both K48- and K63-linked ubiquitylation (Yau et al. 2017), which might have different roles in signal transduction.
Besides K48- and K63-linked polyubiquitin chains, several noncanonical polyubiquitin linkages such as K6, K27 and K33 are also related to DNA repair. For instance, K6- and K33-linked polyubiquitination has been observed to increase significantly in response to DNA damage (Elia et al. 2015). RNF168 has been reported to mediate K27-linked ubiquitylation of histone H2A which is subsequently recognized by DNA double-strand break repair proteins such as Rap80 and 53BP1 to prompt proper activation of the DNA damage response pathway (Gatti et al. 2015). However, the detailed roles of these various modifications are yet to be fully determined.
E3 ligases and deubiquitinases
Assembly and removal of ubiquitin is tightly regulated. E3 ligases are responsible for the addition of this post-translational modification to substrates. These ligases, of which there are over 600+, have characteristic RING, RBR, HECT, or UBOX domains that help to recruit substrates to catalyze the transfer of ubiquitin (Deshaies and Joazeiro 2009; Huang et al. 1999; Hatakeyama et al. 2001; Lechtenberg et al. 2016).
As mentioned above, ubiquitylation is a reversible process. As such, a group of deubiquitinases are responsible for the removal of this modification from substrates. There are about a hundred of these proteins that play critical roles in signal transduction. Deubiquitinases are classified into five families: ubiquitin C-terminal hydrolases (UCHs), ubiquitin-specific proteases (USPs), ovarian tumor proteases (OTUs), Josephins and JAB1/MPN/Mov34 metalloenzymes (JAMMs) that interact with a common hydrophobic patch on ubiquitin. Some deubiquitinases recognize the ubiquitin tag through specific domains and motifs such as UBA, UIM and UEV (Komander et al. 2009; Hurley et al. 2006), while for other deubiquitinases, how they specifically recognize their Ub-conjugated substrates remains unclear. These proteins are critical for maintenance of genomic stability, development and differentiation since mutations in these proteins have been linked to various disorders including cancer and neurodegenerative diseases.
RNF8–RNF168 and DNA double-strand break repair
RNF8 is an important RING E3 ligase which binds and activates the E2 UBC13 to generate the initial K63-linked ubiquitin chains for the amplification of the damage response (Huen et al. 2007; Wang and Elledge 2007). It is generally accepted that RNF8 and RNF168 are sequestered sequentially to the DSB sites. RNF8 localizes to DSBs via its N-terminal forkhead-associated (FHA) domain, which interacts with the ATM-phosphorylated TQXF motifs on MDC1 (Huen et al. 2007; Mailand et al. 2007; Kolas et al. 2007). There is some debate on the exact target of this ubiquitin ligase that is responsible for downstream signal amplification. Histone H1 was proposed to be a target of this ligase. It has been reported that RNF8 polyubiquitylates histone H1 which subsequently acts as a scaffold for proteins such as RNF168 which binds to the modified histones (Thorslund et al. 2015). However, we and others have only been able to detect monoubiquitylated histone H1 upon DNA damage (Nowsheen et al. 2018; Cao and Yan 2012; Liu et al. 2013). The residue on histone H1 that undergoes this modification has not been identified so far. In addition, in a study exploring the histone modifications at DSBs, histone H1 was shown to be removed from these lesions (Clouaire et al. 2018). While there may be other explanations for these observations including the challenge of replacing or knocking out the critical histone to study its affects, a likely explanation may be that a histone-interacting protein may be the target of RNF8. We have shown that the polycomb group-like protein L3MBTL2 is a target of RNF8 and bridges the signaling between RNF8 and RNF168. Upon DNA damage, L3MBTL2 accumulates at DNA DSB sites. It is modified at K659 by RNF8 which then anchors RNF168 to these sites to trigger further signal amplification (Nowsheen et al. 2018). As expected, loss of RNF8 leads to hypersensitivity to DNA damaging agents and impaired cell cycle checkpoint, suggesting that RNF8-dependent signaling is critical for the cellular response to DNA damage.
RNF168 is a RING-type ubiquitin ligase that also works with the E2 enzyme UBC13 (Stewart et al. 2009). Interestingly, the RING domain of RNF168 is not required for recruitment at DNA lesions. Instead, this process is dependent on its two ubiquitin-binding motifs, MIU1 and MIU2 (Doil et al. 2009). Mutations in RNF168 are associated with RIDDLE syndrome (radiosensitivity immunodeficiency dysmorphic features and learning difficulties), which is characterized by cellular defects in repairing DSBs (Stewart et al. 2009).
The DNA DSB response pathway requires tight control to prevent inappropriate activation of the signaling cascade. First, RNF168 is stabilized by the deubiquitylating enzyme USP34 in response to DNA damage (Sy et al. 2013). Second, inhibitory phosphorylation marks on RNF8 at T198 residue by cyclin-dependent kinase 1 (CDK1) during mitosis inhibits the interaction between MDC1 and RNF8 via the MDC1 pT752 site (Orthwein et al. 2014). This is necessary to prevent the fusion of telomeres during mitosis. As yet another layer of regulation, the E3 ligases TRIP12 and UBR5 target RNF168 for degradation, thereby regulating the level of RNF168 and chromatin ubiquitylation (Gudjonsson et al. 2012). Hyper-accumulation of RNF168 alters DNA repair and is prevalent in disease states such as cancer. Finally, other DUBs remove ubiquitylation signals off of histones. One such DUB, OTUB1, also limits the activity of E2-conjugating enzymes. OTUB1 non-catalytically stabilizes the E2 ubiquitin-conjugating enzyme UBE2E1 by preventing its autoubiquitylation. OTUB1 binds to a subset of E2 ubiquitin-conjugating enzymes such as UBE2D, UBE2E, and UBE2N (UBC13), and inhibits their activity by trapping the E2∼ubiquitin thioester and preventing ubiquitin transfer (Nakada et al. 2010).
Role of ubiquitylation in DNA damage response
One of the critical roles of ubiquitylation is stabilization of signaling platforms in the DNA DSB repair pathway, triggering subsequent cell survival and/or cell death pathways. One of the earliest events in the DNA DSB response pathway is the recruitment of the E3 ligase RNF8 to DNA DSB sites by the mediator protein MDC1 (Huen et al. 2007; Mailand et al. 2007; Kolas et al. 2007). MDC1 recognizes γH2AX residues and recruits RNF8. RNF8 subsequently ubiquitylates another adaptor protein, L3MBTL2, adding K63-linked ubiquitin chains to the polycomb group protein. The recruitment of L3MBTL2 to DNA damage sites is dependent on phosphorylation by ATM as well as its interaction with MDC1. Polyubiquitylated L3MBTL2 acts as a docking platform for another E3 ligase, RNF168. RNF168 then ubiquitylates K13 and K15 residues on histone H2A thereby spreading the histone ubiquitylation signal at sites of DNA damage. This positive feedback loop plays a crucial role in the recruitment of key DNA repair proteins (Lou et al. 2003, 2006; Huen et al. 2007; Mailand et al. 2007; Nowsheen et al. 2018; Mattiroli et al. 2012; Fradet-Turcotte et al. 2013; Kim et al. 2007; Sobhian et al. 2007; Shao et al. 2009; Wang and Elledge 2007; Kolas et al. 2007; Thorslund et al. 2015; Stewart et al. 2009; Doil et al. 2009).
Given the critical role of this modification in this pathway, this signaling cascade needs to be tightly regulated. As a result, there are inhibitory proteins present in the cell that prevent inappropriate spread of this ubiquitylation signal. The E3 ligases TRIP12 and UBR5 are responsible for proteasomal degradation of RNF168 (Gudjonsson et al. 2012). In addition, the deubiquitinase OTUB1 limits the activity of E2, adding another layer of regulation to this pathway (Nakada et al. 2010). The deubiquitinase USP26 and USP37 also restrict RNF8/RNF/168-mediated Ub signaling, thereby fine-tuning HR (Typas et al. 2016). Similarly, RNF126, an E3 ligase, can ubiquitylate RNF168 and inhibit RNF168 function (Lee et al. 2018; Zhang et al. 2018).
There are two major DNA DSB repair pathways: homologous recombination (HR) and non-homologous end joining (NHEJ). HR is a high-fidelity repair pathway that uses the sister chromatid as a template. As a result, HR peaks in the S/G2 phases of the cell cycle. In contrast, NHEJ is error prone and ligates the two DNA strands with loss of nucleotides. There is debate on whether RNF168 promotes HR or NHEJ. There is ongoing debate regarding the role of ubiquitylation in shifting the balance between NHEJ and HR.
A number of studies argue in favor of the ubiquitylation by RNF8/RNF168 tipping the balance in favor of NHEJ. For instance, RNF8 regulates the abundance of KU80, a critical player in NHEJ, at sites of DNA damage. Depletion of RNF8 results in prolonged retention of KU80 at damage sites and impairs NHEJ-mediated DSB repair (Feng and Chen 2012). Similarly, RNF168 has been shown to interact with and recruit the NHEJ proteins 53BP1, RIF1 and REV7 to DSB sites, thereby promoting NHEJ-mediated DSB repair over HR (Bohgaki et al. 2013; Chroma et al. 2017). Similar results were obtained in an independent study where the BRCA1–RAP80 complex was found to restrict end resection in S/G2 phases of the cell cycle, thereby limiting HR-mediated DSB repair (Coleman and Greenberg 2011). Another protein, Jumonji Domain-Containing Protein 1C (JMJD1C) that is thought to have multiple roles including acting as a histone demethylase and regulating transcription, has been reported to affix to RNF8 and MDC1 and demethylate MDC1, thereby fostering MDC1–RNF8 interaction, RNF8-dependent MDC1 ubiquitylation, and subsequent recruitment of RAP80–BRCA1 to polyubiquitylated MDC1. Interestingly, in that study, JMJD1C restricted the formation of RAD51 repair foci, arguing in favor of ubiquitylation tipping the balance towards NHEJ over HR (Coleman and Greenberg 2011; Watanabe et al. 2013). Finally, RNF168 depletion has been shown to promote the HR pathways: homology-directed repair and single-strand annealing. Downregulation of RNF168 suppresses defects in HR caused by BRCA1 silencing, but does not suppress HR defects caused by disruption of other HR proteins such as CtIP, RAD50, BRCA2, and RAD51. Interestingly, in that study, RNF168-depleted cells formed ionizing radiation-induced Rad51 foci without forming BRCA1 foci. This suggests that loss of BRCA1 recruitment to DSBs does not necessarily reflect a loss of HR function. Furthermore, the authors in the study also found that RNF168 and 53BP1 have a similar effect on HR (Muñoz et al. 2012). Taken together these studies argue that RNF8/RNF168-mediated ubiquitylation tips the balance from HR to NHEJ.
Yet, there is a body of work supporting ubiquitylation favoring HR over NHEJ. For instance, the NHEJ protein Ku80 is modified by the E3 ligase RNF138 which releases it from DSB sites. In the absence of RNF138, Ku80 persists at these lesions preventing DSB end resection and thereby HR (Ismail et al. 2015). Furthermore, ubiquitylation of CtIP by RNF138 increases the activity of CtIP to promote resection (Schmidt et al. 2015). RNF8, SCF fbxl12, VCP p97, and RNF126 are responsible for targeting Ku for proteasomal degradation (Feng and Chen 2012; Ismail et al. 2015; Rabinovich et al. 2002; Postow and Funabiki 2013). In addition, CtIP is destabilized during G1 by proteasome-mediated degradation. Phosphorylation of CtIP by PLK3/CDK is required for sufficient nuclear accumulation of CtIP for its activity (Barton et al. 2014). The interaction between CtIP and USP4 has also been reported to shift the balance from NHEJ to HR. The interaction between CtIP and the MRN complex is augmented by USP4, thus leading to the recruitment of CtIP to DNA damage sites. Ubiquitylation of USP4 has also been shown to modulate the interaction between CtIP and USP4 (Wijnhoven et al. 2015; Liu et al. 2015). Cullin3–KLHL15 ubiquitin ligase has also been shown to regulate CtIP turnover and fine-tune HR (Ferretti et al. 2016). Thus, these results argue in favor of ubiquitylation swinging the balance between HR and NHEJ.
It is important to note the discrepancies in these studies and lack of consensus on whether RNF8/RNF168 favors one repair pathway over another. The inconsistency may be due to different cell lines used in different studies, experimental conditions and limitations, or regulation by as-yet undiscovered players in this pathway. It should also be noted that ubiquitylation plays other roles in regulation of DSB repair independent of RNF8 and RNF168 function. For instance, as mentioned above, the E3 ligases TRIP12 and UBR5 limit recruitment of RNF168 to DSB lesions (Gudjonsson et al. 2012). In addition, BRCA1 has E3 ligase activity as well that is critical for its function as a tumor suppressor (Wu et al. 2008). In this review, we chose to focus on RNF8/RNF168 since we will not be able to do justice to the numerous groundbreaking studies in the field if we broaden the scope of this review.
Two of the major players in HR and NHEJ are BRCA1 and 53BP1, respectively. 53BP1 restricts resection of DSBs, steering repair away from HR and towards NHEJ (Bunting et al. 2010). In contrast, BRCA1 counteracts this effect by allowing resection to continue even in the presence of 53BP1. 53BP1 recognizes modified histones around DSBs. The Tudor domain of 53BP1 binds to H4K20Me2 while the 53BP1 ubiquitin-dependent recruitment region recognizes RNF168-mediated modified H2A K15ub sites (Fradet-Turcotte et al. 2013; Lee et al. 2014; Jacquet et al. 2016). However, there are additional layers of regulation in place that influence the ability of 53BP1 to bind to these modified histones and promote NHEJ. For instance, TIRR, a protein that binds to the Tudor domain of 53BP1, prevents it from associating with chromatin (Drané et al. 2017). TIP60 also competes with 53BP1 for the H4K20Me2 site (Jacquet et al. 2016). The polycomb group proteins JMJD2A and L3MBTL1 compete with 53BP1 for binding to H4K20Me2 (Mallette et al. 2012; Acs et al. 2011). JMJD2A undergoes proteasomal degradation following DNA damage in an RNF8-dependent manner. JMJD2A has been shown to be ubiquitylated by RNF8 and RNF168, and this modification, therefore, regulates DNA repair by controlling the recruitment of 53BP1 at DNA damage sites (Mallette et al. 2012). There are deubiquitinases in place as well to abrogate this affect and favor HR over NHEJ. The recruitment of JMJD2A is enhanced by the deubiquitinase POH1 while OTUB2 inhibits ubiquitylation-mediated removal of L3MBTL1 (Kato et al. 2014; Butler et al. 2012).
In addition, the E3 ubiquitin ligases RNF169 and RAD18 compete with 53BP1 for binding to H2AK15ub sites independent of E3 ligase activity (Poulsen et al. 2012; Chen et al. 2012; Watanabe et al. 2009). The ability of 53BP1 to bind to H2AK15ub is also limited by phosphorylation and acetylation of key residues within the ubiquitin-dependent recruitment region. PP4C/R3β phosphatase complex dephosphorylates 53BP1 during mitosis to G1 transition (Lee et al. 2014). CBP-mediated acetylation of K1626/1628 in the UDR motif disrupts the interaction between 53BP1 and nucleosomes, thereby blocking the accrual of 53BP1 and its downstream factors PTIP and RIF1 at DSBs (Guo et al. 2017). In addition to its recruitment, the stability of 53BP1 is also regulated via proteasome-mediated degradation by the E2 enzyme, UBCH7 (Han et al. 2014). Replication stress increases the UBCH7 activity, leading to increased degradation of 53BP1 and therefore higher levels of HR.
Similar to 53BP1, the HR protein BRCA1 is also recruited to DNA lesions by the ubiquitin ligases RNF8/RNF168 (Coleman and Greenberg 2011; Kim et al. 2007; Sobhian et al. 2007; Shao et al. 2009; Wang and Elledge 2007). The mechanism by which BRCA1 prevents 53BP1-mediated inhibition of resection and HR is still debated. One mechanism by which BRCA1 relieves the block on DSB resection induced by RIF1 is dependent on the E3 ubiquitin ligase, UHRF1 (Zhang et al. 2016). UHRF1 is recruited to DSBs in S phase by BRCA1. This is dependent on the BRCT domain of BRCA1 and CDK2-phosphorylated Ser674 of UHRF1. Subsequently, UHRF1 mediates K63-linked polyubiquitylation of RIF1 bound to 53BP1 at DNA lesions. This results in the dissociation of RIF1 from 53BP1 thereby facilitating HR initiation (Zhang et al. 2016). Another approach involves PP4C-mediated dephosphorylation of 53BP1 at T1609 and S1618 residues relieving the block on HR (Lee et al. 2014). BRCA1 has been shown to inhibit 53BP1 phosphorylation as well (Feng et al. 2015). Generally, 53BP1 is phosphorylated after DNA damage, leading to association between 53BP1 and its downstream mediators, RIF1 and PTIP, which help promote NHEJ over HR (Callen et al. 2013; Di Virgilio et al. 2013). Since ATM-dependent 53BP1 phosphorylation is restricted to the G1 phase of the cell cycle, RIF1 and PTIP accumulate at DSB sites only in the G1 phase. In other phases of the cell cycle, such as S and G2 phases, both BRCT and RING domains of BRCA1 are necessary for impeding 53BP1 phosphorylation. BRCA1 uses its E3 ligase activity to ubiquitinate 53BP1 and this modification is important for inhibition of 53BP1 phosphorylation (Feng et al. 2015). Another mechanism involves ubiquitylation of H2AK127 by the BRCA1–BARD1 E3 ligase complex, which subsequently triggers SMARCAD1-mediated repositioning of nucleosomes and 53BP1. Thereby, BRCA1 reverses the block on resection imposed by 53BP1 to permit processive DSB resection (Densham et al. 2016).
To promote HR, BRCA1 first recruits Partner and Localizer of BRCA2 (PALB2) and subsequently BRCA2 to the break site, where BRCA2 loads RAD51 (Sy et al. 2009). The interaction between BRCA1 and PALB2 is tightly regulated and represents one of the forks in the choice between HR and NHEJ. For instance, ubiquitylation of key lysine residues in PALB2 by the CRL3–KEAP1 E3 ubiquitin ligase complex blocks association of PALB2 and BRCA1 in G1 phase of the cell cycle, therefore, driving repair towards NHEJ (Ma et al. 2012). The deubiquitinase USP11 puts a check on this process; increased activity of USP11 in the S phase of the cell cycle results in deubiquitylation of PALB2 and formation of the pro-HR BRCA1–PALB2 complex (Orthwein et al. 2015; Buisson et al. 2017). Phosphorylation of PALB2 during specific phases of the cell cycle also adds another layer of adjustment since it dictates the ability of PALB2 to form complexes with BRCA1. ATR activation at resected DSBs leads to phosphorylation of serine 59 in PALB2, which promotes PALB2–BRCA1 complex formation (Buisson et al. 2017). Another critical player in HR, RAD51, is similarly controlled to fine-tune the response to DSB. RFWD3, an E3 ubiquitin ligase ubiquitylates RPA and RAD51 thus promoting their removal by a VCP/p97-dependent pathway. Loss of RFWD3-mediated ubiquitylation results in collapse of HR-mediated DSB repair and manifests as a form of Fanconi anemia (Knies et al. 2017). We have also shown that the deubiquitinases UCHL3 and USP13 regulate HR-mediated DSB repair. Specifically, deubiquitylation of Rad51 facilitates Rad51–BRCA2 binding and Rad51 recruitment. The deubiquitinase UCHL3 is activated by ATM-mediated phosphorylation and in turn deubiquitinates Rad51, stimulating the interaction between Rad51and BRCA2 (Luo et al. 2016). Similarly, the deubiquitinase USP13 is responsible for regulating Rap80–BRCA1 complex formation by deubiquitylating Rap80 in response to DNA damage (Li et al. 2017). Thus, ubiquitylation is pivotal for proper DNA DSB repair.
Ubiquitylation and disease
As can be inferred from the above discussion, ubiquitylation is critical for maintenance of genomic stability. As a result, any aberration in Ub itself or enzymes that catalyze ubiquitylation–deubiquitylation reaction leads to disease. Besides, neurodegenerative disorders such as Parkinson’s disease and Alzheimer’s disease aberrations in ubiquitin pathway are strongly liked with genetic syndromes and cancer. Table 1 lists some of the disorders that are thought to be secondary to mutations in this pathway. Most of these are due to genomic instability secondary to aberrant DNA damage response. Inadequate response to DSBs can lead to genomic rearrangements and loss of genetic material that can result in diseases such as cancer.
| Protein | Disease associated with mutation |
|---|---|
| Ataxin 3 | Spinocerebellar ataxia |
| Machado–Joseph disease | |
| BAP1 | BAP1 tumor predisposition syndrome |
| Cancer | |
| BARD1 | Cancer |
| BRCA1 | Cancer |
| BRCC36 | Cancer |
| Moyamoya disease | |
| Hypergonadotropic hypogonadism | |
| Facial dysmorphism | |
| CHIP/STUB1 | Alzheimer’s disease |
| Parkinson’s disease | |
| Huntington’s disease | |
| Cancer | |
| Spinocerebellar ataxia | |
| Gordon Holmes syndrome | |
| COP1 | Cancer |
| CYLD1 | Cylindromatosis |
| Brooke–Spiegler syndrome | |
| Multiple familial trichoepithelioma | |
| Cancer | |
| FANCD2 | Fanconi anemia |
| FANCL | Fanconi anemia |
| FBXW7 | Cancer |
| Hyperlucent lung | |
| HUWE1 | Mental retardation, X-linked, syndromic, Turner type (MRXST) Angelman syndrome |
| MYSM1 | Skin atrophy |
| Hemorrhagic cystitis | |
| OTUB1 | Cancer |
| Retinitis pigmentosa | |
| Parkin | Alzheimer’s disease |
| Parkinson’s disease | |
| Juvenile recessive Parkinson | |
| Leprosy (Hansen disease) | |
| Cancer | |
| RNF168 | RIDDLE syndrome |
| Cancer | |
| RNF8 | Impaired spermatogenesis |
| Cancer | |
| Morgagni cataract | |
| TRAF6 | Parkinson’s disease |
| Rheumatoid arthritis | |
| Ectodermal dysplasia | |
| Incontinentia pigmenti | |
| UBC13/UBE2N | Cancer |
| Rheumatoid arthritis | |
| UBE3A | Angelman syndrome |
| UCHL1 | Alzheimer’s disease |
| Parkinson’s disease | |
| Spastic paraplegia | |
| UCHL3 | Cancer |
| USP1 | Fanconi anemia |
| USP11 | Cancer |
| USP12 | Cancer |
| USP15 | Microphthalmia |
| Speech and communication disorder | |
| USP16 | Cancer |
| USP2 | Cancer |
| USP21 | Cancer |
| USP36 | Cancer |
| USP7 | Cancer |
| UV sensitivity syndrome | |
| USP8 | Parkinson’s disease |
| Cushing’s disease | |
| Pituitary adenoma | |
| Spastic paraplegia | |
| USP9 | Cancer |
| X-linked mental retardation | |
| VHL | von Hippel–Lindau syndrome |
| Congenital polycythemia | |
| Familial erythrocytosis | |
| Nonsyndromic paraganglioma | |
| Cancers | |
| Hemangioblastoma |
Table 1 Mutations in DNA double-strand break repair proteins which function in the ubiquitin system lead to various genetic disorders
Many of the diseases caused by aberrations in the ubiquitin pathway impose significant challenge and cost to society. As such, a lot of effort has gone into trying to target these proteins and pathways. Since there are multiple points of regulation in the ubiquitin pathway, this presents as an enticing target with regards to therapy. Over the last few decades, a lot of effort has gone into characterizing various cancers and identifying biomarkers that can predict therapeutic response. The goal is to eventually be able to tailor treatment to each mutation present in the tumor. Significant progress has been made in this regard. Immunohistochemistry studies on tumor samples help us assess expression levels and activity of various proteins and, based on the result, choose what therapeutic approach to implement. Considerable effort is going into developing methods that can more consistently predict response to chemotherapy and radiation treatment.
Besides the chemotherapy and radiation treatment that are already heavily used in cancer treatment, another treatment approach involves identifying and developing small molecules and peptides that can alter the function of key players in the DNA damage response pathway. Since the structure of many of these ubiquitin complexes and proteins are known, molecular imaging and prediction models can be used to screen and identify candidate small molecules and peptides. Side effects due to non-selectivity pose the greatest challenge in this regard. Development of innovative approaches that target protein–protein interactions more selectively will allow us to more effectively translate the findings from the bench to the clinic.
Other Ub-like post-translational modifications
A number of ubiquitin-like modifiers have been identified, including small ubiquitin-like modifiers (SUMOs), neural precursor cell expressed, developmentally down-regulated 8 (NEDD8), interferon-stimulated gene 15 (ISG15), human leukocyte antigen (HLA)-F adjacent transcript 10 (FAT10), ubiquitin-fold modifier 1 (UFM1), ubiquitin-related modifier-1 (URM1), autophagy-related protein 12 (ATG12), autophagy-related protein 8 (ATG8), fan ubiquitin-like protein 1 (FUB1) and histone monoubiquitylation 1 (HUB1). The exact roles of these modifications in the DNA damage response pathway, specifically DNA DSB repair, have not been fully elucidated.
What we do know is that neddylation of Histone H2A by RNF168, i.e., addition of NEDD8 to H2A, antagonizes H2A ubiquitylation and thus inhibits DNA repair (Li et al. 2014; Ma et al. 2013). Moreover, RNF168 undergoes neddylation as well and this modification regulates its E3 ubiquitin ligase activity. Inhibition of RNF168 neddylation impairs the interaction between RNF168 and its E2 enzyme Ubc13. The CRL4 E3 ubiquitin ligase, which contains either CUL4A or CUL4B, has additionally been implicated in the removal of Ku via an ubiquitin-dependent mechanism that is dependent on neddylation (Li et al. 2014; Brown et al. 2015).
SUMOylation shares a similar activating pathway with ubiquitin but uses SUMO as a substrate. There are four different variants of SUMO (SUMO 1–4). SAE1/UBA2 is the only E1 and UBC9 is the only SUMO E2 identified thus far. The mediator protein MDC1 is one of the substrates SUMOylated upon DNA damage. SUMOylation of MDC1 triggers ubiquitylation by RNF4 and together these modifications trigger degradation and removal of MDC1 and 53BP1 from sites of DNA damage (Galanty et al. 2012; Luo et al. 2012, 2015; Pfeiffer et al. 2017; Yin et al. 2012).
Similar to ubiquitylation, ISG15 is conjugated to target substrates by an enzymatic cascade: UBE1L (E1), UBCH8 (E2), and E3 ligases. This process can be reversed by deISGylating enzymes such as UBP43 (Skaug and Chen 2010; Ritchie et al. 2004). DNA damage induces ISGylation of p53 which leads to transcriptional alterations (Huang et al. 2014). However, any direct role of this modification in DNA repair has not been reported so far. Similarly, DNA damage induces an upregulation in proteins involved in FATylation. Proliferating cell nuclear antigen (PCNA), which plays an important role in DNA replication and repair, is FATylated in response to DNA damage. This subsequently leads to the degradation of PCNA via the 26S proteasome (Chen et al. 2018). Other functions of FATylation are yet to be explored.
A similar modification that has not been explored as much is UFMylation. This reversible process is very similar to ubiquitylation. It involves the conjugation of ubiquitin-fold modifier 1 (UFM1) to its target proteins using a three step enzymatic reaction involving an E1 to activate UFM1 (UFM1-activating enzyme) ubiquitin-like modifier-activating enzyme 5 (UBA5); the E2 UFM1-conjugating enzyme 1 (UFC1) to transfer the activated UFM1 to the active site of the E2; and an E3 UFM1-specific ligase 1 (UFL1) to recognize its substrate, transfer, and ligate the UFM1 from E2 to the substrate (DeJesus et al. 2016; Habisov et al. 2016). Similar to deubiquitinases, UFM1-specific proteases (UfSPs) cleave UFM1 chains from its target proteins (Wei and Xu 2016). Our lab recently reported that the process of UFMylation is important in the activation of ATM. UFL1 is recruited to DSBs by the MRN complex, and monoufmylates histone H4 following DNA damage. Monoufmylated histone H4 is important for Suv39h1 and Tip60 recruitment. Furthermore, ATM phosphorylates UFL1 at Serine 462, enhancing UFL1 E3 ligase activity and promoting ATM activation in a positive feedback loop (Qin et al. 2019). These findings reveal that UFMylation of histone H4 by UFL1 is an important step for amplification of ATM activation and maintenance of genomic integrity. However, further roles of this Ub-like modification in DNA damage response, specifically DSB repair, remains unclear.
Covalent DNA–protein crosslinks are a by-product of metabolism that needs to be processed effectively to maintain genomic stability. While the MRN complex is responsible for processing the DNA component of the DNA–protein crosslink using its endo- and exonuclease activity, proteases of the Spartan (SPRTN) family target the protein component (Stingele et al. 2014, 2015; Centore et al. 2012; Machida et al. 2012). DNA–protein crosslinks are inherently difficult to repair due to the complex structure and issues with accessibility to the lesion. The first step to resolving these lesions involves proteasomal degradation of the proteins at these junctions. Tyrosyl-DNA phosphodiesterases hydrolyze the phosphotyrosyl bond leading to lesions such as single-strand break (SSB) and DSBs which are then repaired by the appropriate pathway. The MRN complex also helps resolve DNA–protein crosslinks, especially those at the end of DSBs (Connelly et al. 2003; Hoa et al. 2016; Deshpande et al. 2016). An interesting aspect of SPRTN activity is that it is regulated by ubiquitylation and DNA. SPRTN can be monoubiquitylated, which blocks its chromatin localization. The protease is active only towards DNA binding proteins. DNA binding and the subsequent conformational change appear to be essential steps for SPRTN activation. SPRTN recruitment to sites of replication stress requires its ubiquitin-binding UBZ domain and PCNA-binding PIP box motif but is independent of RAD18-mediated PCNA monoubiquitylation. Upon DNA–protein crosslink formation, SPRTN is deubiquitylated and this results in its relocalization to the chromatin (Mosbech et al. 2012; Stingele et al. 2016). The enzymes responsible for these alterations remain unknown.
Unanswered questions
In spite of the years of work in the field, there are questions that linger. First, is ubiquitin itself modified during DNA damage response? It has been reported that PTEN-induced putative kinase 1 (PINK 1) phosphorylates ubiquitin residues on ubiquitylated Parkin to dictate mitophagy (Koyano et al. 2014). Ubiquitylation and phosphorylation are the major regulatory post-translational protein modifications. Individually, phosphorylation is a binary signal (on and off switch if you will), while ubiquitylation provides a more adjustable signal. Combining these two modifications provides a far greater complexity and regulation. How does the cell utilize this potential? What are the kinases and phosphatases responsible for these alterations? Protein kinases that may modify ubiquitin in response to nuclear DNA damage are yet to be identified.
Second, does the cellular microenvironment dictate the choice of repair pathway and the chromatin changes that occur following detection of a DNA lesion? Signaling from the extracellular matrix is a fundamental cellular input that influences the choice between cell death and cell proliferation (Meredith et al. 1993; Ilić et al. 1998). Using the integrin family of proteins, cells sense the chemical composition and physical properties of the microenvironment (Zhang et al. 1995). It stands to reason that these may dictate DNA damage response. It was recently reported that ATR mediates a checkpoint at the nuclear envelope in response to mechanical stress (Kumar et al. 2014). However, the role of the extracellular matrix in ubiquitin-mediated response remains to be elucidated.
Third, there are a number of modifications that are similar to ubiquitylation. Can they act as decoys when the process of ubiquitylation is threatened? Are the processes promiscuous? The roles of these processes with regard to DNA DSB repair remain to be fully explored and as such, there are still a lot of unanswered questions lingering in the field.
References
Acs, K., et al. (2011). The AAA-ATPase VCP/p97 promotes 53BP1 recruitment by removing L3MBTL1 from DNA double-strand breaks. Nature Structural & Molecular Biology,18, 1345.
Andreassen, P. R., D’Andrea, A. D., & Taniguchi, T. (2004). ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes & Development,18, 1958–1963.
Barton, O., et al. (2014). Polo-like kinase 3 regulates CtIP during DNA double-strand break repair in G1. Journal of Cell Biology,206, 877–894.
Bohgaki, M., et al. (2013). RNF168 ubiquitylates 53BP1 and controls its response to DNA double-strand breaks. Proceedings of the National Academy of Sciences,110, 20982–20987.
Brown, E. J., & Baltimore, D. (2000). ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes & Development,14, 397–402.
Brown, J. S., et al. (2015). Neddylation promotes ubiquitylation and release of Ku from DNA-damage sites. Cell Reports,11, 704–714.
Buisson, R., et al. (2017). Coupling of homologous recombination and the checkpoint by ATR. Molecular Cell,65, 336–346.
Bunting, S. F., et al. (2010). 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell,141, 243–254.
Butler, L. R., et al. (2012). The proteasomal de-ubiquitinating enzyme POH1 promotes the double-strand DNA break response. The EMBO Journal,31, 3918–3934.
Callen, E., et al. (2013). 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell,153, 1266–1280.
Cao, J., & Yan, Q. (2012). Histone ubiquitination and deubiquitination in transcription, DNA damage response, and cancer. Frontiers in Oncology,2, 26.
Centore, R. C., Yazinski, S. A., Tse, A., & Zou, L. (2012). Spartan/C1orf124, a reader of PCNA ubiquitylation and a regulator of UV-induced DNA damage response. Molecular Cell,46, 625–635.
Chau, V., et al. (1989). A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science,243, 1576–1583.
Chen, J., Feng, W., Jiang, J., Deng, Y., & Huen, M. S. (2012). Ring finger protein RNF169 antagonizes the ubiquitin-dependent signaling cascade at sites of DNA damage. Journal of Biological Chemistry,287, 27715–27722.
Chen, Z., et al. (2018). Ubiquitin-like protein FAT10 regulates DNA damage repair via modification of proliferating cell nuclear antigen. Molecular Medicine Reports,17, 7487–7496.
Chroma, K., et al. (2017). Tumors overexpressing RNF168 show altered DNA repair and responses to genotoxic treatments, genomic instability and resistance to proteotoxic stress. Oncogene,36, 2405–2422.
Clouaire, T., et al. (2018). Comprehensive mapping of histone modifications at DNA double-strand breaks deciphers repair pathway chromatin signatures. Molecular Cell,72, 250–262.e256.
Coleman, K. A., & Greenberg, R. A. (2011a). The BRCA1–RAP80 complex regulates DNA repair mechanism utilization by restricting end resection. Journal of Biological Chemistry,286, 13669–13680.
Coleman, K. A., & Greenberg, R. A. (2011b). The BRCA1–RAP80 complex regulates DNA repair mechanism utilization by restricting end resection. The Journal of biological chemistry,286, 13669–13680.
Connelly, J. C., de Leau, E. S., & Leach, D. R. (2003). Nucleolytic processing of a protein-bound DNA end by the E. coli SbcCD (MR) complex. DNA Repair,2, 795–807.
Cook, P. J., et al. (2009). Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature,458, 591.
DeJesus, R., et al. (2016). Functional CRISPR screening identifies the ufmylation pathway as a regulator of SQSTM1/p62. Elife,5, e17290.
Densham, R. M., et al. (2016). Human BRCA1–BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nature Structural & Molecular Biology,23, 647.
Deshaies, R. J., & Joazeiro, C. A. (2009). RING domain E3 ubiquitin ligases. Annual Review of Biochemistry,78, 399–434.
Deshpande, R. A., Lee, J.-H., Arora, S., & Paull, T. T. (2016). Nbs1 converts the human Mre11/Rad50 nuclease complex into an endo/exonuclease machine specific for protein–DNA adducts. Molecular Cell,64, 593–606.
Di Virgilio, M., et al. (2013). Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science,339, 711–715.
Doil, C., et al. (2009). RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell,136, 435–446.
Drané, P., et al. (2017). TIRR regulates 53BP1 by masking its histone methyl-lysine binding function. Nature,543, 211.
Elia, A. E. H., et al. (2015). Quantitative proteomic atlas of ubiquitination and acetylation in the DNA damage response. Molecular Cell,59, 867–881.
Falck, J., Coates, J., & Jackson, S. P. (2005). Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature,434, 605.
Feng, L., & Chen, J. (2012). The E3 ligase RNF8 regulates KU80 removal and NHEJ repair. Nature Structural & Molecular Biology,19, 201.
Feng, L., et al. (2015). Cell cycle-dependent inhibition of 53BP1 signaling by BRCA1. Cell Discovery,1, 15019.
Ferretti, L. P., et al. (2016). Cullin3-KLHL15 ubiquitin ligase mediates CtIP protein turnover to fine-tune DNA-end resection. Nature Communications,7, 12628.
Fradet-Turcotte, A., et al. (2013). 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature,499, 50.
Galanty, Y., Belotserkovskaya, R., Coates, J., & Jackson, S. P. (2012). RNF4, a SUMO-targeted ubiquitin E3 ligase, promotes DNA double-strand break repair. Genes & Development,26, 1179–1195.
Gatti, M., et al. (2015). RNF168 promotes noncanonical K27 ubiquitination to signal DNA damage. Cell Reports,10, 226–238.
Gudjonsson, T., et al. (2012). TRIP12 and UBR5 suppress spreading of chromatin ubiquitylation at damaged chromosomes. Cell,150, 697–709.
Guo, X., et al. (2017). Acetylation of 53BP1 dictates the DNA double strand break repair pathway. Nucleic Acids Research,46, 689–703.
Habisov, S., et al. (2016). Structural and functional analysis of a novel interaction motif within UFM1-activating enzyme 5 (UBA5) required for binding to ubiquitin-like proteins and ufmylation. Journal of Biological Chemistry,291, 9025–9041.
Han, X., et al. (2014). UbcH7 regulates 53BP1 stability and DSB repair. Proceedings of the National Academy of Sciences,111, 17456–17461.
Hatakeyama, S., Yada, M., Matsumoto, M., Ishida, N., & Nakayama, K.-I. (2001). U box proteins as a new family of ubiquitin-protein ligases. Journal of Biological Chemistry,276, 33111–33120.
Higashitsuji, H., et al. (2005). The oncoprotein gankyrin binds to MDM2/HDM2, enhancing ubiquitylation and degradation of p53. Cancer Cell,8, 75–87.
Hoa, N. N., et al. (2016). Mre11 is essential for the removal of lethal topoisomerase 2 covalent cleavage complexes. Molecular Cell,64, 580–592.
Hoege, C., Pfander, B., Moldovan, G.-L., Pyrowolakis, G., & Jentsch, S. (2002). RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature,419, 135.
Huang, Y.-F., Wee, S., Gunaratne, J., Lane, D. P., & Bulavin, D. V. (2014). Isg15 controls p53 stability and functions. Cell Cycle,13, 2199–2209.
Huang, L., et al. (1999). Structure of an E6AP–UbcH7 complex: Insights into ubiquitination by the E2–E3 enzyme cascade. Science,286, 1321–1326.
Huen, M. S., et al. (2007). RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell,131, 901–914.
Hurley, J. H., Lee, S., & Prag, G. (2006). Ubiquitin-binding domains. Biochemical Journal,399, 361–372.
Ilić, D., et al. (1998). Extracellular matrix survival signals transduced by focal adhesion kinase suppress p53-mediated apoptosis. The Journal of Cell Biology,143, 547–560.
Ismail, I. H., et al. (2015). The RNF138 E3 ligase displaces Ku to promote DNA end resection and regulate DNA repair pathway choice. Nature Cell Biology,17, 1446.
Jacquet, K., et al. (2016). The TIP60 complex regulates bivalent chromatin recognition by 53BP1 through direct H4K20me binding and H2AK15 acetylation. Molecular Cell,62, 409–421.
Jin, L., Williamson, A., Banerjee, S., Philipp, I., & Rape, M. (2008). Mechanism of ubiquitin-chain formation by the human anaphase-promoting complex. Cell,133, 653–665.
Kato, K., et al. (2014). Fine-tuning of DNA damage-dependent ubiquitination by OTUB2 supports the DNA repair pathway choice. Molecular Cell,53, 617–630.
Kim, H., Chen, J., & Yu, X. (2007). Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science,316, 1202–1205.
Kim, H.-K., et al. (2014). The N-terminal methionine of cellular proteins as a degradation signal. Cell,156, 158–169.
Knies, K., et al. (2017). Biallelic mutations in the ubiquitin ligase RFWD3 cause Fanconi anemia. The Journal of Clinical Investigation,127, 3013–3027.
Kolas, N. K., et al. (2007). Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science,318, 1637–1640.
Komander, D., Clague, M. J., & Urbé, S. (2009). Breaking the chains: Structure and function of the deubiquitinases. Nature Reviews Molecular Cell Biology,10, 550.
Koyano, F., et al. (2014). Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature,510, 162.
Kumar, A., et al. (2014). ATR mediates a checkpoint at the nuclear envelope in response to mechanical stress. Cell,158, 633–646.
Lechtenberg, B. C., et al. (2016). Structure of a HOIP/E2~ ubiquitin complex reveals RBR E3 ligase mechanism and regulation. Nature,529, 546.
Lee, J.-H., & Paull, T. T. (2004). Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science,304, 93–96.
Lee, J.-H., & Paull, T. T. (2005). ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science,308, 551–554.
Lee, D.-H., et al. (2014). Dephosphorylation enables the recruitment of 53BP1 to double-strand DNA breaks. Molecular Cell,54, 512–525.
Lee, N. S., et al. (2018). Ring finger protein 126 (RNF126) suppresses ionizing radiation-induced p53-binding protein 1 (53BP1) focus formation. The Journal of Biological Chemistry,293, 588–598.
Li, T., Guan, J., Huang, Z., Hu, X., & Zheng, X. (2014). RNF168-mediated H2A neddylation antagonizes ubiquitylation of H2A and regulates DNA damage repair. Journal of Cell Science,127, 2238–2248.
Li, Y., et al. (2017). USP13 regulates the RAP80–BRCA1 complex dependent DNA damage response. Nature Communications,8, 15752.
Liu, C., et al. (2013). RNF168 forms a functional complex with RAD6 during the DNA damage response. Journal of Cell Science,126, 2042–2051.
Liu, H., et al. (2015). The deubiquitylating enzyme USP4 cooperates with CtIP in DNA double-strand break end resection. Cell Reports,13, 93–107.
Lou, Z., Minter-Dykhouse, K., Wu, X., & Chen, J. (2003). MDC1 is coupled to activated CHK2 in mammalian DNA damage response pathways. Nature,421, 957.
Lou, Z., et al. (2006). MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Molecular Cell,21, 187–200.
Luo, K., Zhang, H., Wang, L., Yuan, J., & Lou, Z. (2012). Sumoylation of MDC1 is important for proper DNA damage response. EMBO Journal,31, 3008–3019.
Luo, K., et al. (2015). CDK-mediated RNF4 phosphorylation regulates homologous recombination in S-phase. Nucleic Acids Research,43, 5465–5475.
Luo, K., et al. (2016). A phosphorylation–deubiquitination cascade regulates the BRCA2–RAD51 axis in homologous recombination. Genes & Development,30, 2581–2595.
Ma, J., et al. (2012). PALB2 interacts with KEAP1 to promote NRF2 nuclear accumulation and function. Molecular and Cellular Biology,32, 1506–1517.
Ma, T., et al. (2013). RNF111-dependent neddylation activates DNA damage-induced ubiquitination. Molecular Cell,49, 897–907.
Machida, Y., Kim, M. S., & Machida, Y. J. (2012). Spartan/C1orf124 is important to prevent UV-induced mutagenesis. Cell Cycle,11, 3395–3402.
Mailand, N., et al. (2007). RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell,131, 887–900.
Mallette, F. A., et al. (2012). RNF8-and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. The EMBO Journal,31, 1865–1878.
Mattiroli, F., et al. (2012). RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell,150, 1182–1195.
Meredith, J., Jr., Fazeli, B., & Schwartz, M. (1993). The extracellular matrix as a cell survival factor. Molecular Biology of the Cell,4, 953–961.
Mosbech, A., et al. (2012). DVC1 (C1orf124) is a DNA damage–targeting p97 adaptor that promotes ubiquitin-dependent responses to replication blocks. Nature Structural & Molecular Biology,19, 1084.
Muñoz, M. C., et al. (2012). Ring finger nuclear factor RNF168 Is important for defects in homologous recombination caused by loss of the breast cancer susceptibility factor BRCA1. Journal of Biological Chemistry,287, 40618–40628.
Nakada, S., et al. (2010). Non-canonical inhibition of DNA damage-dependent ubiquitination by OTUB1. Nature,466, 941.
Nijman, S. M., et al. (2005). A genomic and functional inventory of deubiquitinating enzymes. Cell,123, 773–786.
Nowsheen, S., et al. (2018a). ZNF506-dependent positive feedback loop regulates H2AX signaling after DNA damage. Nature Communications,9, 2736.
Nowsheen, S., et al. (2018b). L3MBTL2 orchestrates ubiquitin signalling by dictating the sequential recruitment of RNF8 and RNF168 after DNA damage. Nature Cell Biology,20, 455.
Orthwein, A., et al. (2014). Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science,344, 189–193.
Orthwein, A., et al. (2015). A mechanism for the suppression of homologous recombination in G1 cells. Nature,528, 422.
Pfeiffer, A., et al. (2017). Ataxin-3 consolidates the MDC1-dependent DNA double-strand break response by counteracting the SUMO-targeted ubiquitin ligase RNF4. EMBO Journal,36, 1066–1083.
Postow, L., & Funabiki, H. (2013). An SCF complex containing Fbxl12 mediates DNA damage-induced Ku80 ubiquitylation. Cell Cycle,12, 587–595.
Poulsen, M., Lukas, C., Lukas, J., Bekker-Jensen, S., & Mailand, N. (2012). Human RNF169 is a negative regulator of the ubiquitin-dependent response to DNA double-strand breaks. Journal of Cell Biology,197, 189–199.
Qin, B., et al. (2019). UFL1 promotes histone H4 ufmylation and ATM activation. Nature Communications,10, 1242.
Rabinovich, E., Kerem, A., Fröhlich, K.-U., Diamant, N., & Bar-Nun, S. (2002). AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Molecular and Cellular Biology,22, 626–634.
Ritchie, K. J., et al. (2004). Role of ISG15 protease UBP43 (USP18) in innate immunity to viral infection. Nature Medicine,10, 1374.
Rogakou, E. P., Pilch, D. R., Orr, A. H., Ivanova, V. S., & Bonner, W. M. (1998). DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. Journal of Biological Chemistry,273, 5858–5868.
Scheffner, M., Nuber, U., & Huibregtse, J. M. (1995). Protein ubiquitination involving an E1–E2–E3 enzyme ubiquitin thioester cascade. Nature,373, 81.
Schmidt, C. K., et al. (2015). Systematic E2 screening reveals a UBE2D–RNF138–CtIP axis promoting DNA repair. Nature Cell Biology,17, 1458.
Shao, G., et al. (2009). The Rap80-BRCC36 de-ubiquitinating enzyme complex antagonizes RNF8-Ubc13-dependent ubiquitination events at DNA double strand breaks. Proceedings of the National Academy of Sciences,106, 3166–3171.
Shiloh, Y. (2003). ATM and related protein kinases: Safeguarding genome integrity. Nature Reviews Cancer,3, 155.
Skaug, B., & Chen, Z. J. (2010). Emerging role of ISG15 in antiviral immunity. Cell,143, 187–190.
Sobhian, B., et al. (2007). RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science,316, 1198–1202.
Stewart, G. S., et al. (2009). The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell,136, 420–434.
Stingele, J., Habermann, B., & Jentsch, S. (2015). DNA–protein crosslink repair: Proteases as DNA repair enzymes. Trends in Biochemical Sciences,40, 67–71.
Stingele, J., Schwarz, M. S., Bloemeke, N., Wolf, P. G., & Jentsch, S. (2014). A DNA-dependent protease involved in DNA–protein crosslink repair. Cell,158, 327–338.
Stingele, J., et al. (2016). Mechanism and regulation of DNA-protein crosslink repair by the DNA-dependent metalloprotease SPRTN. Molecular Cell,64, 688–703.
Sy, S. M., Huen, M. S., & Chen, J. (2009). PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proceedings of the National Academy of Sciences,106, 7155–7160.
Sy, S. M. H., Jiang, J., O, W. S., Deng, Y., & Huen, M. S. Y. (2013). The ubiquitin specific protease USP34 promotes ubiquitin signaling at DNA double-strand breaks. Nucleic Acids Research,41, 8572–8580.
Thorslund, T., et al. (2015). Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature,527, 389.
Typas, D., et al. (2016). The de-ubiquitylating enzymes USP26 and USP37 regulate homologous recombination by counteracting RAP80. Nucleic Acids Research,44, 2976.
Wang, B., & Elledge, S. J. (2007). Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proceedings of the National Academy of Sciences,104, 20759–20763.
Watanabe, K., et al. (2009). RAD18 promotes DNA double-strand break repair during G1 phase through chromatin retention of 53BP1. Nucleic Acids Research,37, 2176–2193.
Watanabe, S., et al. (2013). JMJD1C demethylates MDC1 to regulate the RNF8 and BRCA1–mediated chromatin response to DNA breaks. Nature Structural & Molecular Biology,20, 1425–1433.
Wei, Y., & Xu, X. (2016). UFMylation: A unique & fashionable modification for life. Genomics, Proteomics & Bioinformatics,14, 140–146.
Wijnhoven, P., et al. (2015). USP4 auto-deubiquitylation promotes homologous recombination. Molecular Cell,60, 362–373.
Wu, W., Koike, A., Takeshita, T., & Ohta, T. (2008). The ubiquitin E3 ligase activity of BRCA1 and its biological functions. Cell Division,3, 1–1.
Xiao, A., et al. (2009). WSTF regulates the H2A. X DNA damage response via a novel tyrosine kinase activity. Nature,457, 57.
Yau, R. G., et al. (2017). Assembly and function of heterotypic ubiquitin chains in cell-cycle and protein quality control. Cell,171, 918–933.e920.
Yin, Y., et al. (2012). SUMO-targeted ubiquitin E3 ligase RNF4 is required for the response of human cells to DNA damage. Genes & Development,26, 1196–1208.
Zhang, Z., Vuori, K., Reed, J. C., & Ruoslahti, E. (1995). The alpha 5 beta 1 integrin supports survival of cells on fibronectin and up-regulates Bcl-2 expression. Proceedings of the National Academy of Sciences,92, 6161–6165.
Zhang, H., et al. (2016). A cell cycle-dependent BRCA1–UHRF1 cascade regulates DNA double-strand break repair pathway choice. Nature Communications,7, 10201.
Zhang, L., et al. (2018). RNF126 quenches RNF168 function in the DNA Damage response. Genomics, Proteomics & Bioinformatics,16, 428–438.
Zhong, W., Feng, H., Santiago, F. E., & Kipreos, E. T. (2003). CUL-4 ubiquitin ligase maintains genome stability by restraining DNA-replication licensing. Nature,423, 885.
Acknowledgements
This work was supported by funding from the National Institutes of Health (CA217183 and CA203561) to Zhenkun Lou.
Author information
Affiliations
Mayo Clinic Medical Scientist Training Program, Mayo Clinic Alix School of Medicine and Mayo Clinic Graduate School of Biomedical Sciences, Mayo Clinic, Rochester, MN, 55905, USA
Somaira Nowsheen
Division of Oncology, Mayo Clinic, Rochester, MN, 55905, USA
Min Deng & Zhenkun Lou
Corresponding author
Correspondence to Zhenkun Lou.
Rights and permissions
About this article
Cite this article
Nowsheen, S., Deng, M. & Lou, Z. Ubiquitin and the DNA double-strand break repair pathway. GENOME INSTAB. DIS. 1, 69–80 (2020). https://doi.org/10.1007/s42764-019-00007-5
Received29 January 2019
Revised11 July 2019
Accepted17 August 2019
Published19 September 2019
Issue DateMarch 2020
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
DNA double-strand break repair
DNA damage response
Ubiquitin
Histone
Radiation
RNF168
RNF8
Ligase
BRCA1
53BP1
用户登录
还没有账号?
立即注册