






Helicobacter pylori infection induced genome instability and gastric cancer
Review Article Open Access
Xiangyu Liu, Muhammad Irfan, Xingzhi Xu, Chi-Yen Tay & Barry J. Marshall
Genome Instability & Disease 1, 129–142(2020)
Abstract
Genome stability and integrity are constantly challenged by exogenous insults such as bacterial infections. When genome stability is perturbed, oncogenic transformation can ensue. Helicobacter pylori (H. pylori) infection is a driving factor of gastric cancer, which is the third leading cause of cancer-related mortality worldwide. Mechanistically, H. pylori infection drives inflammation and directly or indirectly induces DNA damage such as oxidative damage and double-strand breaks (DSBs) in host cells. In addition, the resulting genetic and/or epigenetic perturbations alter the choice of DNA repair pathways. These changes result in imprecise DNA repair, genomic instability as well as chromosomal aberrations that eventually lead to gastric carcinogenesis. In this review, we summarize the mechanisms how H. pylori infection cause DNA damage and alter the DNA damage response pathways in host cells. We highlight the relationship between H. pylori infection and genomic instability that can lead to gastric cancer and propose a potential strategy to interrupt gastric carcinogenesis.
Introduction
Helicobacter pylori (H. pylori) is a Gram-negative, microaerophilic, spiral-shaped bacterium that lives deep in the innermost mucus layer of the gastric mucosa in humans. To colonize the human stomach, H. pylori produces urease, an enzyme that converts urea into ammonia, to survive in the harsh, acidic environment of the stomach. In addition, H. pylori moves through mucus by reducing mucin viscoelasticity (Celli et al. 2009). Interestingly, H. pylori not only infects the surface cells of the stomach but also invades deep into the glands and reach the stem cell compartments. H. pylori can activate gastric stem cells and increase stem cell turnover (Sigal, 2015 #703; Sigal, 2017 #704). These discoveries strongly suggest that chronic H. pylori infections are promoters of gastric cancer. Moreover, H. pylori has evolved various mechanisms to evade local immune responses and prevent elimination, such as avoiding pattern recognition receptor (PRRs) detection (Monack et al. 2004). Infection with H. pylori is extremely common: the Centers for Disease Control and Prevention (CDC) of the United States estimate that approximately half of the world’s population harbors the bacterium, with prevalence much higher in developing countries than in developed nations. Although H. pylori has coexisted with humans perhaps even before the migration out of Africa, it was only discovered 3 decades ago (Marshall and Warren 1984). Since then, numerous studies have shown that H. pylori infection is associated with an increased risk of gastric diseases (Parsonnet et al. 1991; Uemura et al. 2001; Eslick et al. 1999). H. pylori-induced gastric cancer is the fifth most common neoplasm and third most deadly cancer (Guggenheim and Shah 2013; Kelley and Duggan 2003; Karimi et al. 2014; de Martel et al. 2013; Krejs 2010; Amieva and Peek 2016; Rawla and Barsouk 2019), killing approximately 783,000 people in 2018 (Bray et al. 2018). Thus, the International Agency for Research on Cancer (IARC) classified H. pylori as a Class I carcinogen as the cause of mucous associated lymphoid tissue (MALT) of the stomach. Besides, H. pylori infection is a risk factor for the development of stomach adenocarcinoma (STAD), which is irreversible even after eradiation of the bacteria.
Gastric cancer is usually found in association with chronic gastritis that is caused by inflammation (Fox and Wang 2007). Gastric inflammation after H. pylori infection can be induced by two different, but related mechanisms: (1) direct epithelial cell damage or enhancement of released pro-inflammatory mediators by physical contact between the pathogen and the host epithelial cells; or (2) altering cell signaling pathways in the host cells by the influence of virulence factors. Virulence factors are bacterial toxins that are produced by most H. pylori strains, including but not limited to the vacuolating cytotoxin A (VacA), the effector cytotoxin-associated gene A (CagA) and neutrophil-activating protein (NapA). H. pylori harbor type IV secretion systems (T4SS) that translocate virulence factors into host cells thereby to modulate the bacterium–host interaction, determine bacterial pathogenicity and influence clinical outcomes. In addition, the presence of these virulence factors facilitates the bacteria to colonize in the host’s gastric mucosa, causing long-lasting chronic inflammation (Naumann et al. 2017), a tissue response which is estimated to contribute to ~ 25% of all human cancers (Sokolova and Naumann 2019; Kawanishi et al. 2017) (Fig. 1).
Fig. 1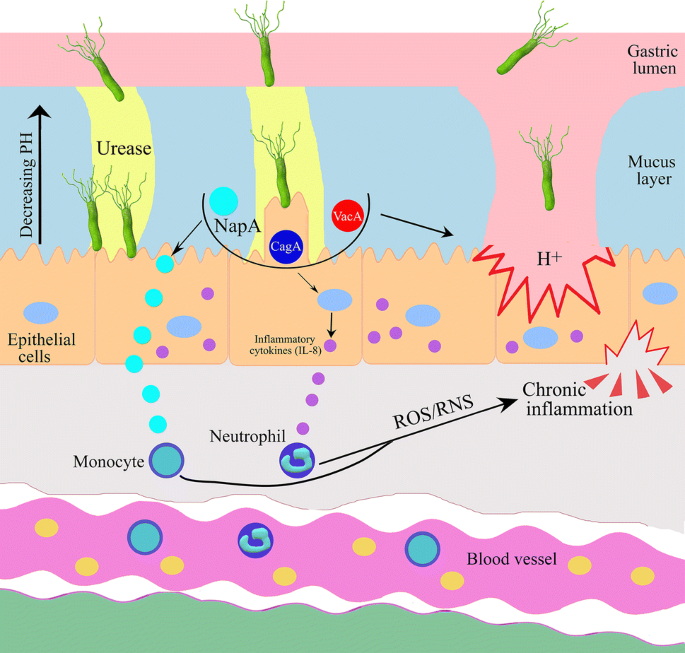
Helicobacter pylori secretes urease and virulence factors (CagA, VacA and NapA) that help to survive in harsh gastric environment and on the stomach mucous membrane. H. pylori induces secretion of cytokines and enhances ROS/RNS which leads to chronic inflammation in host cells
Full size imageMost carcinogens work by inducing DNA damage and/or mutations (Jackson and Bartek 2009); similarly, H. pylori infection causes DNA damage and triggers DNA damage responses (DDR), leading to genomic instability in host cells, which is a hallmark for all types of cancer including gastric cancer. The majority of DNA lesions (75%) are single-stranded DNA (ssDNA) breaks (SSBs), which can arise from oxidative damage during inflammation or base hydrolysis. DNA double-strand breaks (DSBs) are less frequent but more dangerous: they are generated by ssDNA conversion or by direct exogenous sources such as irradiation. DNA damage repair can be achieved by a huge number of proteins that comprise several major DNA repair pathways, such as mismatch repair (MMR), base excision repair (BER), nucleotide excision repair (NER), homologous recombination (HR), nonhomologous end joining (NHEJ) and alternative end joining (A-EJ) (Jeggo et al. 2016). These DNA repair pathways are completed either alone or together with other pathways via a series of signaling responses that ultimately arrest the cell cycle or promote cell death if the ends are irreparable. The latter outcome is essential, as DNA ends that are not properly repaired can generate dangerous outcomes, such as deletions, amplifications or chromosome translocations that might activate oncogenes and promote tumorigenesis (Helleday et al. 2008). Evidences show that H. pylori infection impairs the coordination of different DNA repair machineries through different mechanisms, including methylation of gene promoters and interfering histone modifications. Imbalance of the DNA repair leads to genome instability and subsequent tumorigenesis.
In this review, we summarize how H. pylori infection causes DNA damage and how bacteria interfere the DNA damage repair pathways. In addition, we will discuss how the underlying mechanisms can lead to gastric cancer development. Finally, we will also discuss strategies to target H. pylori-induced genomic instability as possible biomarkers and suggest therapies for gastric cancer.
Helicobacter pylori infection induces DNA damage
The stomach mucosa is continually exposed to environmental exogenous and cellular endogenous mutagens. These mutagens include H. pylori bacteria, nitrates and nitrites, and other contaminants in water and food that can induce DNA damage through different mechanisms (Obst et al. 2000; Farinati et al. 1998; Papa et al. 2002; Hardbower et al. 2014). Here, we discuss different types of DNA damage caused by H. pylori infection through direct or indirect mechanisms.
Helicobacter pylori infection induces inflammation and oxidative damage
Inflammation is the body’s natural response to bacterial infection (Medzhitov and Horng 2009); persistent inflammation, however, can lead to DNA damage and genome instability. H. pylori interacts with PRRs of gastric epithelial cells and activates inflammatory gene expression, including toll-like receptors (TLRs). In addition, virulent H. pylori strains (CagA + , VacA + , NapA +) enhance chemokine activation and stimulate inflammatory pathways in gastric epithelial cells (Eck et al. 2000; Watanabe et al. 1997; Eftang et al. 2012; White et al. 2015). The main inflammatory pathways include the non-canonical and canonical NF-κB, JAK-STAT and MAPK signaling pathways. Activation of each or combined pathways eventually drives abnormal gene transcription that has been implicated in cancer development (Pua et al. 2019). Even though no evidence showed a direct role of NF-κB signaling in gastric cancer stem cells, aberrant activation of the pathway contributes to increased proliferation and evasion of apoptosis, which could contribute to tumorigenesis (Kang et al. 2008; Liu et al. 2004). During chronic inflammation, several factors, such as cytokines, chemokines, growth factors and other immunoregulatory proteins participate in tumor development (Medzhitov and Horng 2009). In addition, chemokines including TNFα, IL-1β and IL-8 that are released from infected gastric epithelial cells can enhance the production of reactive oxygen/nitrogen species (ROS/RNS) (Naito and Yoshikawa 2002; Nozawa et al. 2002). ROS production has been observed in patients and cultured cells infected with H. pylori (Davies et al. 1994; Drake et al. 1998). The sustained release of ROS/RNS together with other risk factors including increased activation-induced cytidine deaminase (AID), inhibited p53 and reactivated TERT expression contribute to DNA damage and genomic instability during chronic inflammation (Pua et al. 2019).
In eukaryotic cells, various proteins involved in ROS production including Spermine Oxidase (SMOX) and Rac1 are activated after H. pylori infection in gastric cancer (Xu et al. 2004; Murray-Stewart et al. 2016; den Hartog et al. 2016). ROS causes changes in the DNA structure by forming oxidized bases [e.g., 8-oxoguanine (8oxoG)], single-strand breaks (SSBs), and/or DNA crosslinks (Panieri and Santoro 2016). ROS-associated DNA damage and genome instability are, thus, considered to be involved in tumor formation and progression (Machado et al. 2013; Cerutti 1994; Dolado et al. 2007; Feig et al. 1994; Frohlich et al. 2008; Gupta et al. 2012).
Moreover, in patients with gastric cancer, H. pylori infection causes oxidative stress-associated DNA damage of Lgr5-positive epithelial stem cells (Uehara et al. 2013). These cells are particularly tolerant to DNA damage and fail to undergo senescence or regulated cell death upon accumulation of genetic lesions (Vitale et al. 2017). Importantly, the Lgr5-positivity was defined as stem-like cells in colorectal cancer (Hirsch et al. 2014); they could also be the cause of gastric carcinogenesis. Together, these findings suggest that H. pylori infection causes inflammation and oxidative damage in host cells which could be a potential reason of tumorigenesis of gastric cancer.
Helicobacter Pylori infection induces double-strand breaks
Double-stranded breaks (DSBs) are generated when the phospho-sugar backbones of both DNA strands are broken at the same position or in sufficient proximity, so that the double helix is cut into two separate parts and the DNA ends are exposed. DSBs are the most dangerous and deleterious forms of DNA damage as the genetic information is lost or altered if the ends are not properly repaired. In addition to loss of genetic information, DSBs lead to fragmentation, loss or rearrangement of chromosomes, which contribute to genomic instability that drives cancer development (Khanna and Jackson 2001). A number of studies have suggested that the DSBs generated in host cells are induced by bacterial infection. For example, Escherichia coli has been linked to DSBs induction and DNA-damage checkpoint activation (Nougayrede et al. 2006). The comet assay is the gold standard to directly measure DNA strand breaks in individual cells (Liao et al. 2009). Using this assay to measure the tail moments, Ladeira et al. found that the levels of DNA damage (tail moment) were significantly higher in H. pylori-infected patients with moderate and severe gastritis than in non-infected patients (Ladeira et al. 2004, 2005). These DSBs, in the long term, escape from or exhaust the repair machinery, and potentially cause gastric carcinogenesis. The induction of DSBs depends on direct contact between living bacteria and mammalian cells.
In addition, using a pulse field gel electrophoresis approach that visualizes fragmented DNA ranging in size from 0.5 to 2.5 Mb, Toller et al. demonstrated that DSBs accumulate in various cell lines and in primary gastric epithelial cells that were incubated with H. pylori in a time-dependent and dose-dependent manner. In their study, DSBs appeared at mitosis and required direct contact between the live bacteria and host cells, but not H. pylori virulence determinants VacA, or ROS-mediated DNA damage (Toller et al. 2011). Using the same approach, Hanada et al. found that although both cag-positive and cag-negative H. pylori strains are capable of inducing DSBs in host cells, the cag-positive strains showed the greater levels of activity (Hanada et al. 2014). Another study found that gastric adenocarcinoma AGS cells infected with several isogenic derivatives of H. pylori lacking the T4SS and associated proteins led to the accumulation of broken DNA, but the increase in damage was partially dependent on the presence of CagA (Koeppel et al. 2015). These evidences showed that the role of virulence factors in DSBs generation is debatable and needs to be further investigated.
Chromatin immunoprecipitation (ChIP)-seq experiments performed by Koeppel et al. using the DSBs biomarker γH2AX showed that H. pylori infection (but not irradiation) induced damage that preferentially accumulated in telomere-proximal regions (~ 10% of all regions) as well as in regions close to the centromeres (~ 5% of all regions) that correlates with the actively transcribed region. This finding is reminiscent of the pattern of structural variations (SVs) obtained from sequencing results of H. pylori-induced gastric cancer (Wang et al. 2014; Koeppel et al. 2015). The accumulation of telomere-proximal DNA damage was most prominent towards the end of chromosome 8, which is a region that is markedly amplified in stomach cancer and close to the MYC oncogene (Koeppel et al. 2015) (Fig. 2). These results support that some of the structural genomic events in gastric cancer could be directly caused by the H. pylori infection and contribute to cellular transformation.
Fig. 2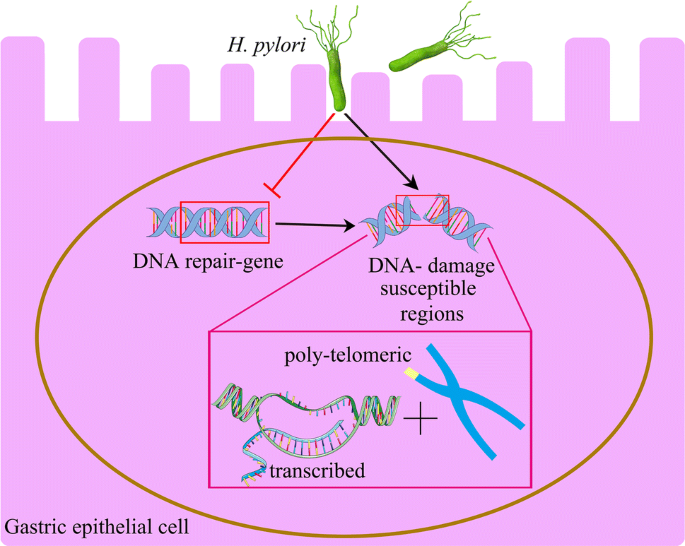
H. pylori mostly influences cellular functioning by targeting the DNA repair gene, transcribed regions and the telomeric region of chromosomes
Full size imageDSBs caused by H. pylori are efficiently repaired if the bacteria were eradicated in a short term, but prolonged infections can lead to residual, unrepaired breaks that negatively affect cell viability (Kidane 2018). The above studies show that H. pylori is able to induce host cellular DSBs damage directly and indirectly, providing a mechanistic explanation for the carcinogenic properties of this bacterial pathogen.
Molecular mechanisms underlying H. pylori-induced DSB formation
As mentioned, H. pylori can lead to chronic gastritis and contributes to genotoxicity by inducing DSBs (Toller et al. 2011; Hartung et al. 2015). However, the molecular mechanism how H. pylori promotes genotoxic activity and how the host response to genotoxic factors drives gastric carcinogenesis is not well understood. Some studies, however, have started to piece together mechanisms how H. pylori functions at the molecular level. For example, a genome-wide screen revealed a critical role for the H. pylori T4SS in XPF/XPG endonucleases-dependent DSB induction (Hartung et al. 2015). T4SS induces NF-κB activation and IL-8 production by consecutively activating integrins. Other studies showed that H. pylori infection on gastric cancer cells activates NF-κB through a signaling pathway including IKKalpha, IKKbeta, NIK, TRAF2, and TRAF6 (Maeda et al. 2000). Enhanced NF-κB activation in host cells upregulates inflammatory mediators and produces inducible nitric oxide synthase (iNOS) (Brandt et al. 2005; Viala et al. 2004; De Luca and Iaquinto 2004). iNOS-mediated nitric oxide (NO) induces DSBs (Baydoun et al. 2015) and enhances the inactivation of DNA repair enzymes, such as MGMT, Fpg, XPA and PARP (O'Connor et al. 1993; Wink and Laval 1994; Tchou et al. 1993; Laval and Wink 1994; Jaiswal et al. 2000; Melino et al. 1997).
Moreover, activated NF-κB translocates to the nucleus where it forms a protein complex with the NER proteins XPG and XPF (Hartung et al. 2015). These endonucleases are critical for excising the damaged DNA strand and removing the DNA lesion. XPG cuts the DNA strand five–six nucleotides downstream of the damage site; while, a ERCC1–XPF protein complex makes an incision in the DNA strand ~ 20–22 nucleotides upstream of the 5′ end of the damage site (Friedberg 2001). XPG/XPF-mediated DSBs amplify NF-κB target inflammatory gene expression and promote host cell survival. Silencing XPG in osteosarcoma patient-derived U2OS cells strongly reduced the DSBs upon H. pylori infection (Hartung et al. 2015). Interestingly, XPG expression level was found to be significantly associated with the presence of H. pylori in human gastric cancer and was related to the progression and prognosis (Deng et al. 2014). These results suggested that the NF-κB as well as the NER pathway plays an important role in H. pylori infection-induced DSBs in host cells.
DSBs can also be caused by the accumulation of unrepaired BER intermediates in DNA replication, when DNA replication meets BER intermediates (Khanna and Jackson 2001; Ensminger et al. 2014). A few studies have shown that the accumulation of abasic site (AP) sites in H. pylori-infected human gastric epithelial cells leads to DSBs (Ding et al. 2004; Bhattacharyya et al. 2009). DNA glycosylase OGG1 that removes 8oxoG and initiates BER in gastric epithelial cells induces the formation of AP sites in host cells after H. pylori infection. These accumulated AP sites arise in replicating DNA and eventually lead to DSB formation (Kidane et al. 2014). However, silencing AP endonuclease 1 (APE1) (part of the BER machinery that process the AP site to generate DSBs) failed to promote H. pylori-induced DSBs in a measurable manner (Hartung et al. 2015). We hypothesize that the protein level of APE1 is not the determinant factor to convert the AP sites into the DSBs. Another possibility is that some other endonucleases could play overlapping roles in this process. The real role of APE1 in the processing of accumulated AP sites into DSBs needs to be further explored.
Helicobacter pylori infection causes mtDNA mutation
Mitochondria are essential organelles that are responsible for energy production: they are involved in apoptosis, calcium homeostasis, lipid and amino acid metabolism. Mitochondria have separate genome DNA (mtDNA) located within the mitochondrion itself. VacA was the first major H. pylori virulence factor identified to target mitochondria (Galmiche and Rassow 2010). Later research discovered that H. pylori uses at least two additional harmful strategies through an alternative VacA-dependent and a VacA-independent mechanism (Chatre et al. 2017). In gastric epithelial cells, VacA localizes to endosomal compartments and reaches the mitochondrial inner membrane where it forms anion-conductive channels (Calore et al. 2010; Domanska et al. 2010). VacA then decreases mitochondrial membrane potential, which ultimately reduces the ATP production efficiency and cytochrome-c release (Galmiche and Rassow 2010). VacA channel activity also disrupts mitochondrial morphological dynamics, resulting in BAX/BAK activation and host cell death (Jain et al. 2011). Mutations in the mtDNA can severely disrupt the mitochondrial oxidative phosphorylation pathway; such mutations are common in many cancers, including gastric cancer with the 53% of the cases have deletion of the mtDNA and 44% of the cases have mtDNA mutation (Carew and Huang 2002). Data from both experimental and population studies have suggested that H. pylori is capable of inducing mutagenesis of both the D-loop region and genes that transcribe subunits of the electron transport chain in mitochondrial DNA (Machado et al. 2009). These findings support that H. pylori infection causes mtDNA mutation.
Helicobacter pylori infection impairs DNA repair pathways
Numerous studies have shown that H. pylori can alter DNA repair gene expression and/or interfere with DNA repair activity (Kidane 2018). In H. pylori-infected cells, a sophisticated network of enzymes coordinate with each other to protect the host cells from staying in the oxidative stress or DNA damage conditions. H. pylori causes differential expression levels of genes implicated in the DNA repair process (Table 1). For example, H. pylori deregulated MMR mechanisms by hypermethylation of related gene promoters (Kim et al. 2002). Failure of the DNA repair mechanisms eventually conduces the DNA breaks into chromosome instability (CIN) and genetic aberrations that favor carcinogenesis (Pino and Chung 2010; Machado et al. 2010).
| Gene | Repair Pathway | Effect of H. pylori infection | References |
|---|---|---|---|
| APE1 | BER | Enhances/decreases in different situations | Hartung et al. (2015); Ding et al. (2004); Machado et al. (2009) |
| ATM | NHEJ and HR | Upregulated | Santos et al. (2018) |
| ATR | HR | Decreased | Koeppel et al. (2015) |
| DNA-PK | NHEJ | Enhances activity and expression | Lee et al. (2007) |
| hMSH2 | MMR | Decreased in mRNA | Kim et al. (2002); Koshiji et al. (2005) |
| hMLH1 | MMR | Decreased in mRNA | Kim et al. (2002; Yao et al. (2006) |
| Ku70/80 | NHEJ | Increase protein level | Bae et al. (2013); Lim et al. (2002) |
| Mre11 | HR | Decreased | Koeppel et al. (2015) |
| NBS1 | HR | Decreased | Koeppel et al. (2015) |
| PARP1 | BER and A-EJ | Activated | Nossa and Blanke (2010) |
| Rad51 | HR | Decreased | Koeppel et al. (2015); Hanada et al. (2014) |
| RPA1 | HR | Decreased | Koeppel et al. (2015) |
| TP53 | Cell cycle arrest | Decreased | Fenoglio-Preiser et al. (2003) |
Helicobacter pylori infection alters the BER pathway
BER is one of the major DNA repair pathways that ultimately removes most of the oxidative and alkylating damage without affecting the DNA double-helix structure. It is initiated by the recognition and excision of the damaged base by specific DNA glycosylases (Wallace et al. 2012; Dianov and Hubscher 2013; Kim and Wilson 2012). Precise orchestration of the different steps of the BER pathway is essential for maintaining genome stability (Allinson et al. 2004). OGG1 glycosylase remains bound to the AP sites and its turnover is stimulated by APE1 (Sidorenko et al. 2007; Mokkapati et al. 2004). After AP site processing and end-remodeling, the single-nucleotide gap is filled by Pol beta, and the nick is sealed by DNA ligase I to complete the repair (Fortini et al. 2003; Robertson et al. 2009).
Helicobacter pylori causes aberrant BER protein function and the processing of H. pylori-induced oxidative DNA damage that leads to SSBs (Cooke et al. 2003) and mutations. Ding et al. reported that H. pylori-induced ROS could upregulate APE1 expression in cultured gastric adenocarcinoma AGS cells and gastric epithelial cells that were isolated from mucosal biopsy samples (Ding et al. 2004). Another study, however, showed that H. pylori inhibits APE1 mRNA expression and impairs the BER pathway (Machado et al. 2009). The opposite results might be dependent on the different Multiplicity of Infection (MOI), infection time or whether live bacteria or protein extracts were used to treat the cells. H. pylori also activates PARP1, an important component of the BER pathway, through a not quite clear mechanism and enhances the inflammatory response, suggesting that the bacterium modulates the host PARP1 status to drive inflammation-associated gastric cancer (Nossa and Blanke 2010). Together, these results suggest that H. pylori infection alters the BER pathway in different situations through different mechanisms, leading to different kinds of DNA damage in the host cells.
Helicobacter pylori impairs DNA mismatch repair
MMR maintains genome stability by correcting errors in the newly synthesized strand during DNA replication. Defects in MMR lead to diseases including hereditary non-polyposis colorectal cancer and brain tumors (Fishel et al. 1993; Modrich and Lahue 1996; Koopman et al. 2009). Several human MMR proteins have been identified, including human homologs of MutS, MutL, EXO1, single-strand DNA-binding protein RPA, proliferating cellular nuclear antigen (PCNA), DNA polymerase δ (pol δ), and DNA ligase I (Li 2008). In human cells, base–base mismatches and small nucleotide insertion/deletion (ID) mispairs of 1 or 2 nucleotides are recognized by the hMSH2–hMSH6 (hMutSα) complex; while the hMSH2–hMSH3 (hMutSβ) complex mainly recognizes larger ID mispairs (Li 2008). Both complexes are required for MMR. In addition, four human MutL homologs (hMLH1, hMLH3, hPMS1, and hPMS2) have been identified. hMLH1 heterodimerizes with hPMS2, hPMS1, or hMLH3 to form hMutLα, hMutLβ, or hMutLγ, respectively, of which hMutLα is required for MMR, while the other two are not absolutely required (Kunkel and Erie 2005).
In eukaryotic cells, defects in DNA MMR can be detected as instability in simple repetitive DNA sequences called microsatellites. Thus, microsatellite instability (MSI) is regarded as a hallmark of MMR deficiency (Modrich and Lahue 1996; Li 2008). Studies have revealed that MSI is a reliable biomarker of gastric cancer (Pietrantonio et al. 2019). Interestingly, individuals with MSI-positive gastric cancers show more active H. pylori infection than those with MSI-negative gastric cancers, suggesting that H. pylori itself might affect the DNA MMR system during gastric carcinogenesis (Leung et al. 2000). Moreover, MSI were found in gastric intestinal metaplasia of both patients with or without gastric carcinoma (Leung et al. 2000), suggesting that the progressive accumulation of MSI in areas of intestinal metaplasia might contribute to gastric cancer development.
Using a co-culture in vitro system in which different gastric cancer cell lines were exposed to H. pylori bacterium or H. pylori products, Kim et al. found that H. pylori infection of gastric epithelial cells leads to a decrease in mRNA levels of the MMR genes hMSH2 and hMLH1 (Kim et al. 2002). Studies on human patients also showed that the percentage of hMLH1-positive epithelial cell nuclei in H. pylori-positive patients was higher than that in H. pylori-negative patients; no significant difference was observed between the two groups regarding the percentage of hMSH2-positive epithelial cell nuclei (Mirzaee et al. 2008). Interestingly, one study found that the expression of DNA MMR proteins increased in the gastric mucosa after H. pylori eradication (Park et al. 2005), further confirming the relationship between H. pylori and MMR.
The underlying mechanism of H. pylori-mediated MMR inhibition has multiple explanations: it might be mediated at least in part by H. pylori-induced hypoxia-inducible factor-1a (HIF1α) expression (Park et al. 2003), which in turn inhibits MMR activity by decreasing both hMSH2 and hMSH6 expression (Koshiji et al. 2005). Interestingly, genome-wide screening showed that H. pylori infection caused aberrant DNA methylation of several CpG islands in gastric epithelial cells through inflammatory processes (Niwa et al. 2010). Yao et al. suggests that accumulation of genomic mutations after H. pylori infection is in part associated with CpG methylation of the hMLH1 promoter (Yao et al. 2006). After comparing the hMLH1 methylation in H. pylori-infected children and adults, Alvarez et al. found that DNA methylation of the genes did not occur in earlier-stage H. pylori infections but was dependent on the duration of infection. The frequency of promoter methylation for hMLH1 was higher in gastric cancer samples than in H. pylori-positive chronic gastritis samples (p < 0.05) (Alvarez et al. 2013).
Also, since both cagA-positive and cagA-negative H. pylori strains can reduce MMR protein levels, it is unlikely that this effect is related to the Cag pathogenicity island proteins. Interestingly, Campylobacter jejuni—a Gram-negative bacterium that is phylogenetically closely related to H. pylori—can also impair MMR protein levels (Kim et al. 2002). This discovery suggests a common molecular pathway affecting MMR after exposure to these bacteria. Taken together, H. pylori infection plays a pivotal role in causing MMR deficiency in gastric epithelial cells and increases the risk of mutation accumulation in gastric mucosa cells and gastric cancer.
Helicobacter pylori triggers DSB repair pathways
The major pathways of DSB repair are classically defined based on whether sequence homology is used to join the DSB ends. In eukaryotic cells, DSBs are mainly repaired through two classical DNA repair pathways: non-homologous end joining (NHEJ), where ends are processed without sequence homology or with few end homology sequences; or homologous recombination (HR), where sequence homology of extensive DNA regions is required in the undamaged sister chromatid or homologous chromosomes. The repair pathway that is chosen depends on the genomic location and cause of the breaks, cell cycle stage and genetic background (Sancar et al. 2004). Interfering with either of these pathways would cause loss of genetic information or induction of abnormal gene expression and caused various human pathologies such as cancer (Smith et al. 2010). We believe that it is informative to uncover the nature of the DSB response and repair after H. pylori infection to prevent infection-related tumorigenesis.
Previous studies showed that H. pylori infection inhibits the G1–S transition in AGS cells (Shirin et al. 1999), suggesting that NHEJ repair might have a central role in H. pylori-induced DSBs repair. Koeppel et al. (2015) compared the responses of host cells to damage caused by H. pylori infection or by different genotoxic treatments; they found that the overall damage during infection was similar to that induced by ionizing radiation, which is considered to be repaired predominantly by the NHEJ pathway (Branzei and Foiani 2008). In the NHEJ pathway, Ku (KU70 and KU80) and DNA-dependent protein kinase (DNA-PKcs) have essential roles in processing DNA ends. Ku proteins bind and protect the break ends, recruiting DNA-PK as a DNA damage sensor and scaffold. DNA-PKcs in turn promotes Artemis endonuclease to the damage site and processes DNA ends. Finally, DNA ligase 4 (Lig4)–XRCC4–XLF–PAXX complex carries out the ligation reaction to complete the repair (X. Liu et al. 2017; Pannunzio et al. 2018).
Ku protein levels are increased in H. pylori-infected gastric mucosal tissues of Mongolian gerbils (Bae et al. 2013), suggesting that H. pylori enhances NHEJ pathways. In addition, Lim et al. showed that H. pylori mediated activation NF-B-Cox2 axis helps to increase the expression of KU70/80 in human gastric cancer cells (Lim et al. 2002). At the same time, DNA-PKcs expression was positive in most H. pylori-associated gastritis, intestinal metaplasia and gastric adenoma tissues, but was negative in foveolar epithelium of normal gastric mucosal tissues (Lee et al. 2007). Moreover, siRNA-mediated knockdown of XRCC4, DNA ligase IV or DNA-PKcs resulted in a significant accumulation of fragmented DNA in H. pylori-infected cells. These findings suggest that abnormalities to the NHEJ pathway participate in the pathological processes in gastric carcinoma after H. pylori infection (Li et al. 2013).
When DSBs are generated in the S or G2 phases of the cell cycle, HR participates and contributes to genome integrity. However, knockdown of the HR protein Rad51, which is involved in homology searching and strand pairing, had no effect on the amount of fragmented DNA generated during H. pylori infection (Hartung et al. 2015), suggesting that H. pylori-induced DSBs are repaired via NHEJ rather than HR. Interestingly, RNA-seq and microarray analyses also showed that H. pylori infection suppresses HR factors, including RPA1, Mre11, NBS1, ATR and RAD51; these data suggest a shift in the choice of the DNA repair pathway away from HR (Koeppel et al. 2015; Hanada et al. 2014). Moreover, reduced RAD51 gene expression is not due to cell cycle arrest in G1 phase, as the cells infected with H. pylori showed a reduced G1 phase and an increased S phase (Hanada et al. 2014). Together, these findings suggest that H. pylori-induced DSBs are repaired majorly through the non-homologous end-joining pathway.
Helicobacter pylori triggers ATM activation
Helicobacter pylori-induced DSBs have been associated with the activation of the serine/threonine kinase ATM (Santos et al. 2018). This activation is considered as one of the most critical events in response to DSBs, as it results in the phosphorylation of several target proteins that are involved in DNA repair. For example, ATM-mediated H2AX phosphorylation at serine 139 (γH2AX) recruits mediator of checkpoint 1 (MDC1), which in turn recruits ATM through a positive feedback loop to amplify ATM signals (Stucki et al. 2005). The γH2AX/MDC1 complex also recruits downstream chromatin modifiers, such as the ubiquitin E3 ligases RNF8/168 to facilitate sequential loading of repair factor 53BP1 to repair damaged DNA (Stucki et al. 2005; Huen et al. 2007; Kolas et al. 2007; Mailand et al. 2007). ATM is also essential for NF-κB activation in response to DSBs (Li et al. 2001).
γH2AX, MDC1, and 53BP1 nuclear foci have all been observed in H. pylori-infected cultured gastric adenocarcinoma cells; pre-treatment with an ATM inhibitor blocked foci formation. Consistently, γH2AX formation was abrogated in ATM-deficient cells infected with H. pylori, indicating that H. pylori induces ATM activation (Toller et al. 2011; Hanada et al. 2014). In a study based on human samples, high γH2AX levels were detected by immunohistochemistry and western blotting in H. pylori-infected gastric biopsies (Xie et al. 2014). Moreover, active (phosphor-S1981) ATM was found in the epithelial cells of gastric biopsy specimens from patients with H. pylori gastritis (Hanada et al. 2014). Further studies have suggested that H. pylori might activate ATM through epigenetic modifications such as histone H3 and H4 hyperacetylation and DNA promoter hypomethylation (Santos et al. 2018), suggesting that H. pylori might affect ATM activation by modulating host cell chromatin. In fact, H. pylori infection causes H3 Ser10 dephosphorylation and other histone modification changes that consequently affect the gastric epithelium and ultimately affect the pathogenicity of the bacterium (Ding et al. 2010; Fehri et al. 2009). Because several studies have shown that chromatin modifications, including certain types of histone modifications, are mechanistically and functionally relevant to ATM activation (Li et al. 2018), it is tempting to speculate that H. pylori can also activate ATM through sophisticated histone modifications. These findings demonstrate that H. pylori infection triggers ATM activation through both activating ATM activity and increasing ATM protein levels.
In most cases, ATM is activated as a result of DNA damage. In certain circumstances, however, ATM is activated independent of DSBs. For example, oxidative stress activates ATM, which is independent of autophosphorylation or MRN (Mre11, Rad50 and Nbs1). (Reliene et al. 2004; Guo et al. 2010). In addition, ATM interacting protein (ATMIN) mediates ATM activation upon hypotonic stress through a MRN-independent pathway (Liu and Zha 2011; Kanu and Behrens 2007; Loizou et al. 2011). Whether H. pylori-induced ATM activation is solely mediated by infection-dependent DSBs or by DSB-independent mechanisms remains to be clarified.
Future perspectives
More than half of the world’s population is infected with H. pylori and about 1–3% of the carriers will develop gastric cancer. H. pylori is normally acquired during childhood and infection can be life-long or until a treatment is provided. H. pylori alone accounts for > 80% of all gastric cancer cases; other factors include the H. pylori genotype, the host genetics, and the complex relationship between host cells and different nutrient conditions (Atherton and Blaser 2009). Due to the complicated multistep process of gastric carcinogenesis (Correa and Piazuelo 2012) and the long time between the initial infection and tumor formation (Amieva and Peek 2016), very little is known about exactly how H. pylori causes gastric cancer. This length of time allows for extensive positive selection for cells with oncogenic growth advantages to colonize. For example, the H. pylori infection increases the phosphorylation of HDM2 and the proteasomal degradation of tumor suppressor p53, which was found to be mutated and similar mutations occur in 0–77% of gastric carcinomas (Fenoglio-Preiser et al. 2003). Epithelial cells with decreased p53 levels will survive with sustained DNA damage and mutations which eventually develop into tumors (Wei et al. 2010). In addition to genetic alterations, studies also have shown that H. pylori infection-related chronic inflammation enhances epigenetic changes as represented by DNA methylation, which is an important mechanism that regulates gene expression (Chiba et al. 2012). Despite these advances, the precise mechanisms that regulate cancer development in response to H. pylori need further exploration.
Manipulation of host signaling cascades by H. pylori is crucial for prolonging chronic infection and the progression to gastric cancer. The strategies by which H. pylori contacts and subverts host cell signal transmission mainly include: (1) adhesion to epithelial cells and T4SS function, (2) regulation of signal transmission in colonized epithelial cells, (3) induced paracrine signaling, and 4) perturbations of the DNA damage response (Naumann et al. 2017) (Fig. 3).
Fig. 3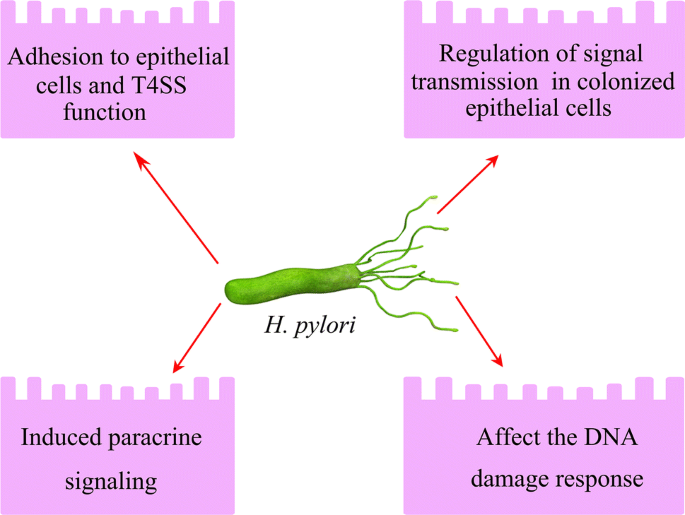
Helicobacter pylori contacts and subverts host cell signal transmission mainly include: adhesion to host cells; regulation of signal transmission; induced paracrine signaling, and affect the DNA damage response
Full size imageMost of the DSBs that are generated by H. pylori infection are repaired by NHEJ pathway, but the ends of the broken DNA can sometimes receive an additional adenine nucleotide. As a result, this could lead to a frameshift mutation and silence a particular gene (Heidenreich et al. 2003; Lieber 2008). Thus, increased expression level of NHEJ factors (KU and DNA-PKcs) by H. pylori could possibly lead to the enhancement of gene mutations and genomic instability in host epithelial cells. Moreover, since H. pylori inhibits components of the HR repair pathway, the alternative repair mechanism, alternative end joining (A-EJ), will be activated to join the ends using microhomology (usually > 4 bp). Such repair mechanism can often lead to insertions or deletions of substantial length (Chang et al. 2017). In leukemia, lymphoma, and many types of solid tumors including gastric cancer, A-EJ can be targeted as a diagnostic marker and a therapeutic target (Simsek et al. 2011). It is tempting to look at the expression level of A-EJ factors such as DNA ligase III or to measure the A-EJ level in H. pylori-infected host cells to determine the relationship between A-EJ and tumorigenesis.
Biomarkers constitute an important tool to detect and diagnose cancers and generate a prognosis in clinic. As DNA damage is a hallmark of carcinogenesis, proteins or products involved in DNA damage might serve as sensitive and faithful biomarkers for gastric cancer (de Sablet et al. 2011). For example, because H. pylori causes an increase in ROS levels and oxidative DNA damage in host epithelial cells, 8-oxoG could be an ideal marker for detecting and assessing the prognosis of gastric carcinoma (Borrego et al. 2013). Ku80 is another potential DNA repair-related biomarker for radiation treatment failure and death, particularly in patients with high-risk HPV-negative head and neck squamous-cell carcinoma (HNSCC) (Moeller et al. 2011). As such, it seems possible and rational to use one of the DNA repair factors as a biomarker for gastric cancer.
Numerous antibiotic-based therapies are available to avoid the occurrence of H. pylori-induced gastric cancer. A 15-year follow-up of a randomized clinical trial found that short-term treatment with antibiotics to eradicate H. pylori reduced the incidence of gastric cancer by almost 40% (Ma et al. 2012). Such therapies, however, have several inherent problems, including the development of resistance to the antibiotics used and associated adverse effects. For patients who have already developed cancer, targeting the bacteria itself would not be sufficient. Because gastric cancer progression is typically accompanied by ATM activation, ATM inhibition with specific inhibitors could be a possible alternative therapeutic strategy. Highly selective small molecule ATM inhibitors such as AZD1390 are currently in the preclinical development stages (Durant et al. 2018). Thus far, this drug has shown promising results in brain tumor models. Whether the inhibitors could be applied in H. pylori-induced gastric cancer would be an interesting question to be explored.
As we learned more about H. pylori, we found that H. pylori has a multitude of effects on various molecular mechanisms that ultimately confer genome instability (Fig. 4). However, many questions remain to be resolved regarding the relationship between H. pylori and genomic instability. How do the host cells choose the DSBs repair pathways under H. pylori infection? Does H. pylori infection increase the tumor latency in patients with DNA repair defects? Future studies also will investigate the development of biomarkers as well as the therapeutic targets for the gastric cancer.
Fig. 4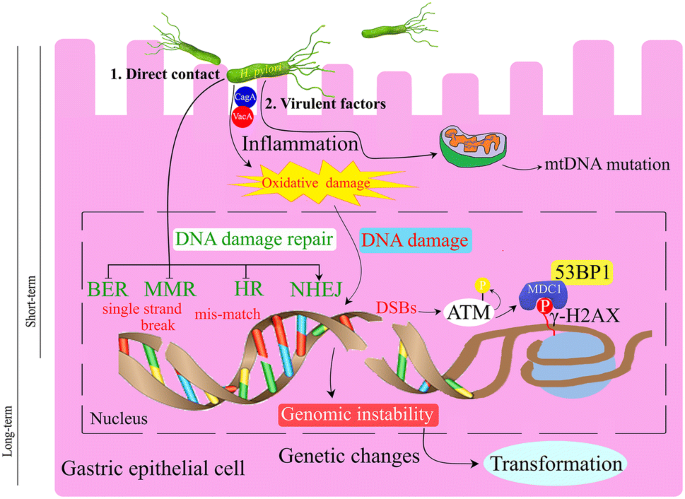
Helicobacter pylori influences the gastric epithelial cells by direct contact and/or virulence factors. The bacteria induces inflammation and oxidative damage to the cells and caused different kind of DNA damage and impairment of the DNA repair pathways. DSBs induced by H. pylori infection caused activation of AMT, phosphorylation of H2AX and recruitment of 53BP1 to facilitate DNA repair. If not properly repaired, the DNA ends will cause genomic instability and the cells will transform into cancerous cells
Full size imageReferences
Allinson, S. L., Sleeth, K. M., Matthewman, G. E., & Dianov, G. L. (2004). Orchestration of base excision repair by controlling the rates of enzymatic activities. DNA Repair (Amsterdam),3(1), 23–31.
Alvarez, M. C., Santos, J. C., Maniezzo, N., Ladeira, M. S., da Silva, A. L., Scaletsky, I. C., et al. (2013). MGMT and MLH1 methylation in Helicobacter pylori-infected children and adults. World Journal of Gastroenterology,19(20), 3043–3051. https://doi.org/10.3748/wjg.v19.i20.3043.
Amieva, M., & Peek, R. M., Jr. (2016). Pathobiology of Helicobacter pylori-induced gastric cancer. Gastroenterology,150(1), 64–78. https://doi.org/10.1053/j.gastro.2015.09.004.
Atherton, J. C., & Blaser, M. J. (2009). Coadaptation of Helicobacter pylori and humans: ancient history, modern implications. Journal of Clinical Investigation,119(9), 2475–2487. https://doi.org/10.1172/JCI38605.
Bae, M., Lim, J. W., & Kim, H. (2013). Oxidative DNA damage response in Helicobacter pylori-infected mongolian gerbils. Journal of Cancer Prevention,18(3), 271–275.
Baydoun, H. H., Cherian, M. A., Green, P., & Ratner, L. (2015). Inducible nitric oxide synthase mediates DNA double strand breaks in Human T-Cell Leukemia Virus Type 1-induced leukemia/lymphoma. Retrovirology,12, 71. https://doi.org/10.1186/s12977-015-0196-y.
Bhattacharyya, A., Chattopadhyay, R., Burnette, B. R., Cross, J. V., Mitra, S., Ernst, P. B., et al. (2009). Acetylation of apurinic/apyrimidinic endonuclease-1 regulates Helicobacter pylori-mediated gastric epithelial cell apoptosis. Gastroenterology,136(7), 2258–2269. https://doi.org/10.1053/j.gastro.2009.02.014.
Borrego, S., Vazquez, A., Dasi, F., Cerda, C., Iradi, A., Tormos, C., et al. (2013). Oxidative stress and DNA damage in human gastric carcinoma: 8-oxo-7'8-dihydro-2'-deoxyguanosine (8-oxo-dG) as a possible tumor marker. International Journal of Molecular Sciences,14(2), 3467–3486. https://doi.org/10.3390/ijms14023467.
Brandt, S., Kwok, T., Hartig, R., Konig, W., & Backert, S. (2005). NF-kappaB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proceedings of National Academy of Science USA,102(26), 9300–9305. https://doi.org/10.1073/pnas.0409873102.
Branzei, D., & Foiani, M. (2008). Regulation of DNA repair throughout the cell cycle. Nature Reviews Molecular Cell Biology,9(4), 297–308. https://doi.org/10.1038/nrm2351.
Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A., & Jemal, A. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer Journal for Clinicians,68(6), 394–424. https://doi.org/10.3322/caac.21492.
Calore, F., Genisset, C., Casellato, A., Rossato, M., Codolo, G., Esposti, M. D., et al. (2010). Endosome-mitochondria juxtaposition during apoptosis induced by H. pylori VacA. Cell Death and Differentiation,17(11), 1707–1716. https://doi.org/10.1038/cdd.2010.42.
Carew, J. S., & Huang, P. (2002). Mitochondrial defects in cancer. Molecular Cancer,1, 9.
Celli, J. P., Turner, B. S., Afdhal, N. H., Keates, S., Ghiran, I., Kelly, C. P., et al. (2009). Helicobacter pylori moves through mucus by reducing mucin viscoelasticity. Proceedings of National Academy of Science USA,106(34), 14321–14326. https://doi.org/10.1073/pnas.0903438106.
Cerutti, P. A. (1994). Oxy-radicals and cancer. The Lancet,344(8926), 862–863. https://doi.org/10.1016/s0140-6736(94)92832-0.
Chang, H. H. Y., Pannunzio, N. R., Adachi, N., & Lieber, M. R. (2017). Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nature Reviews Molecular Cell Biology,18(8), 495–506. https://doi.org/10.1038/nrm.2017.48.
Chatre, L., Fernandes, J., Michel, V., Fiette, L., Ave, P., Arena, G., et al. (2017). Helicobacter pylori targets mitochondrial import and components of mitochondrial DNA replication machinery through an alternative VacA-dependent and a VacA-independent mechanisms. Scientific Reports,7(1), 15901. https://doi.org/10.1038/s41598-017-15567-3.
Chiba, T., Marusawa, H., & Ushijima, T. (2012). Inflammation-associated cancer development in digestive organs: mechanisms and roles for genetic and epigenetic modulation. Gastroenterology,143(3), 550–563. https://doi.org/10.1053/j.gastro.2012.07.009.
Cooke, M. S., Evans, M. D., Dizdaroglu, M., & Lunec, J. (2003). Oxidative DNA damage: mechanisms, mutation, and disease. The FASEB Journal,17(10), 1195–1214. https://doi.org/10.1096/fj.02-0752rev.
Davies, G. R., Simmonds, N. J., Stevens, T. R., Sheaff, M. T., Banatvala, N., Laurenson, I. F., et al. (1994). Helicobacter pylori stimulates antral mucosal reactive oxygen metabolite production in vivo. Gut,35(2), 179–185. https://doi.org/10.1136/gut.35.2.179.
De Luca, A., & Iaquinto, G. (2004). Helicobacter pylori and gastric diseases: a dangerous association. Cancer Letters,213(1), 1–10. https://doi.org/10.1016/j.canlet.2004.06.006.
de Martel, C., Forman, D., & Plummer, M. (2013). Gastric cancer: epidemiology and risk factors. Gastroenterology Clinics of North America,42(2), 219–240. https://doi.org/10.1016/j.gtc.2013.01.003.
Deng, N., Liu, J. W., Sun, L. P., Xu, Q., Duan, Z. P., Dong, N. N., et al. (2014). Expression of XPG protein in the development, progression and prognosis of gastric cancer. PLoS ONE,9(9), e108704. https://doi.org/10.1371/journal.pone.0108704.
den Hartog, G., Chattopadhyay, R., Ablack, A., Hall, E. H., Butcher, L. D., Bhattacharyya, A., et al. (2016). Regulation of Rac1 and reactive oxygen species production in response to infection of gastrointestinal epithelia. PLoS Pathogens,12(1), e1005382. https://doi.org/10.1371/journal.ppat.1005382.
de Sablet, T., Piazuelo, M. B., Shaffer, C. L., Schneider, B. G., Asim, M., Chaturvedi, R., et al. (2011). Phylogeographic origin of Helicobacter pylori is a determinant of gastric cancer risk. Gut,60(9), 1189–1195. https://doi.org/10.1136/gut.2010.234468.
Dianov, G. L., & Hubscher, U. (2013). Mammalian base excision repair: the forgotten archangel. Nucleic Acids Research,41(6), 3483–3490. https://doi.org/10.1093/nar/gkt076.
Ding, S. Z., O'Hara, A. M., Denning, T. L., Dirden-Kramer, B., Mifflin, R. C., Reyes, V. E., et al. (2004). Helicobacter pylori and H2O2 increase AP endonuclease-1/redox factor-1 expression in human gastric epithelial cells. Gastroenterology,127(3), 845–858. https://doi.org/10.1053/j.gastro.2004.06.017.
Ding, S. Z., Fischer, W., Kaparakis-Liaskos, M., Liechti, G., Merrell, D. S., Grant, P. A., et al. (2010). Helicobacter pylori-induced histone modification, associated gene expression in gastric epithelial cells, and its implication in pathogenesis. PLoS ONE,5(4), e9875. https://doi.org/10.1371/journal.pone.0009875.
Dolado, I., Swat, A., Ajenjo, N., De Vita, G., Cuadrado, A., & Nebreda, A. R. (2007). p38alpha MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell,11(2), 191–205. https://doi.org/10.1016/j.ccr.2006.12.013.
Domanska, G., Motz, C., Meinecke, M., Harsman, A., Papatheodorou, P., Reljic, B., et al. (2010). Helicobacter pylori VacA toxin/subunit p34: targeting of an anion channel to the inner mitochondrial membrane. PLoS Pathogens,6(4), e1000878. https://doi.org/10.1371/journal.ppat.1000878.
Drake, I. M., Mapstone, N. P., Schorah, C. J., White, K. L., Chalmers, D. M., Dixon, M. F., et al. (1998). Reactive oxygen species activity and lipid peroxidation in Helicobacter pylori associated gastritis: relation to gastric mucosal ascorbic acid concentrations and effect of H. pylori eradication. Gut,42(6), 768–771. https://doi.org/10.1136/gut.42.6.768.
Durant, S. T., Zheng, L., Wang, Y., Chen, K., Zhang, L., Zhang, T., et al. (2018). The brain-penetrant clinical ATM inhibitor AZD1390 radiosensitizes and improves survival of preclinical brain tumor models. Science Advances,4(6), 1719. https://doi.org/10.1126/sciadv.aat1719.
Eck, M., Schmausser, B., Scheller, K., Toksoy, A., Kraus, M., Menzel, T., et al. (2000). CXC chemokines Gro(alpha)/IL-8 and IP-10/MIG in Helicobacter pylori gastritis. Clinical and Experimental Immunology,122(2), 192–199. https://doi.org/10.1046/j.1365-2249.2000.01374.x.
Eftang, L. L., Esbensen, Y., Tannaes, T. M., Bukholm, I. R., & Bukholm, G. (2012). Interleukin-8 is the single most up-regulated gene in whole genome profiling of H. pylori exposed gastric epithelial cells. BMC Microbiology,12, 9. https://doi.org/10.1186/1471-2180-12-9.
Ensminger, M., Iloff, L., Ebel, C., Nikolova, T., Kaina, B., & Lbrich, M. (2014). DNA breaks and chromosomal aberrations arise when replication meets base excision repair. Journal of Cell Biology,206(1), 29–43. https://doi.org/10.1083/jcb.201312078.
Eslick, G. D., Lim, L. L., Byles, J. E., Xia, H. H., & Talley, N. J. (1999). Association of Helicobacter pylori infection with gastric carcinoma: a meta-analysis. American Journal of Gastroenterology,94(9), 2373–2379. https://doi.org/10.1111/j.1572-0241.1999.01360.x.
Farinati, F., Cardin, R., Degan, P., Rugge, M., Mario, F. D., Bonvicini, P., et al. (1998). Oxidative DNA damage accumulation in gastric carcinogenesis. Gut,42(3), 351–356. https://doi.org/10.1136/gut.42.3.351.
Fehri, L. F., Rechner, C., Janssen, S., Mak, T. N., Holland, C., Bartfeld, S., et al. (2009). Helicobacter pylori-induced modification of the histone H3 phosphorylation status in gastric epithelial cells reflects its impact on cell cycle regulation. Epigenetics,4(8), 577–586. https://doi.org/10.4161/epi.4.8.10217.
Feig, D. I., Reid, T. M., & Loeb, L. A. (1894s). Reactive oxygen species in tumorigenesis. Cancer Research,54(7 Suppl), 1890s–1894s.
Fenoglio-Preiser, C. M., Wang, J., Stemmermann, G. N., & Noffsinger, A. (2003). TP53 and gastric carcinoma: a review. Human Mutation,21(3), 258–270. https://doi.org/10.1002/humu.10180.
Fishel, R., Lescoe, M. K., Rao, M. R., Copeland, N. G., Jenkins, N. A., Garber, J., et al. (1993). The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell,75(5), 1027–1038. https://doi.org/10.1016/0092-8674(93)90546-3.
Fortini, P., Pascucci, B., Parlanti, E., D'Errico, M., Simonelli, V., & Dogliotti, E. (2003). The base excision repair: mechanisms and its relevance for cancer susceptibility. Biochimie,85(11), 1053–1071. https://doi.org/10.1016/j.biochi.2003.11.003.
Fox, J. G., & Wang, T. C. (2007). Inflammation, atrophy, and gastric cancer. The Journal of Clinical Investigation,117(1), 60–69. https://doi.org/10.1172/JCI30111.
Friedberg, E. C. (2001). How nucleotide excision repair protects against cancer. Nature Reviews Cancer,1(1), 22–33. https://doi.org/10.1038/35094000.
Frohlich, D. A., McCabe, M. T., Arnold, R. S., & Day, M. L. (2008). The role of Nrf2 in increased reactive oxygen species and DNA damage in prostate tumorigenesis. Oncogene,27(31), 4353–4362. https://doi.org/10.1038/onc.2008.79.
Galmiche, A., & Rassow, J. (2010). Targeting of Helicobacter pylori VacA to mitochondria. Gut Microbes,1(6), 392–395. https://doi.org/10.4161/gmic.1.6.13894.
Guggenheim, D. E., & Shah, M. A. (2013). Gastric cancer epidemiology and risk factors. Journal of Surgical Oncology,107(3), 230–236. https://doi.org/10.1002/jso.23262.
Guo, Z., Kozlov, S., Lavin, M. F., Person, M. D., & Paull, T. T. (2010). ATM activation by oxidative stress. Science,330(6003), 517–521. https://doi.org/10.1126/science.1192912.
Gupta, S. C., Hevia, D., Patchva, S., Park, B., Koh, W., & Aggarwal, B. B. (2012). Upsides and downsides of reactive oxygen species for cancer: the roles of reactive oxygen species in tumorigenesis, prevention, and therapy. Antioxidants and Redox Signaling,16(11), 1295–1322. https://doi.org/10.1089/ars.2011.4414.
Hanada, K., Uchida, T., Tsukamoto, Y., Watada, M., Yamaguchi, N., Yamamoto, K., et al. (2014). Helicobacter pylori infection introduces DNA double-strand breaks in host cells. Infection and Immunity,82(10), 4182–4189. https://doi.org/10.1128/IAI.02368-14.
Hardbower, D. M., Peek, R. M., Jr., & Wilson, K. T. (2014). At the Bench: Helicobacter pylori, dysregulated host responses, DNA damage, and gastric cancer. Journal of Leukocyte Biology,96(2), 201–212. https://doi.org/10.1189/jlb.4BT0214-099R.
Hartung, M. L., Gruber, D. C., Koch, K. N., Gruter, L., Rehrauer, H., Tegtmeyer, N., et al. (2015). H. pylori-induced DNA strand breaks are introduced by nucleotide excision repair endonucleases and promote NF-kappaB target gene expression. Cell Reports,13(1), 70–79. https://doi.org/10.1016/j.celrep.2015.08.074.
Heidenreich, E., Novotny, R., Kneidinger, B., Holzmann, V., & Wintersberger, U. (2003). Non-homologous end joining as an important mutagenic process in cell cycle-arrested cells. EMBO Journal,22(9), 2274–2283. https://doi.org/10.1093/emboj/cdg203.
Helleday, T., Petermann, E., Lundin, C., Hodgson, B., & Sharma, R. A. (2008). DNA repair pathways as targets for cancer therapy. Nature Reviews Cancer,8(3), 193–204. https://doi.org/10.1038/nrc2342.
Hirsch, D., Barker, N., McNeil, N., Hu, Y., Camps, J., McKinnon, K., et al. (2014). LGR5 positivity defines stem-like cells in colorectal cancer. Carcinogenesis,35(4), 849–858. https://doi.org/10.1093/carcin/bgt377.
Huen, M. S., Grant, R., Manke, I., Minn, K., Yu, X., Yaffe, M. B., et al. (2007). RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell,131(5), 901–914. https://doi.org/10.1016/j.cell.2007.09.041.
Jackson, S. P., & Bartek, J. (2009). The DNA-damage response in human biology and disease. Nature,461(7267), 1071–1078. https://doi.org/10.1038/nature08467.
Jain, P., Luo, Z. Q., & Blanke, S. R. (2011). Helicobacter pylori vacuolating cytotoxin A (VacA) engages the mitochondrial fission machinery to induce host cell death. Proceedings of National Academy of Sciences USA,108(38), 16032–16037. https://doi.org/10.1073/pnas.1105175108.
Jaiswal, M., LaRusso, N. F., Burgart, L. J., & Gores, G. J. (2000). Inflammatory cytokines induce DNA damage and inhibit DNA repair in cholangiocarcinoma cells by a nitric oxide-dependent mechanism. Cancer Research,60(1), 184–190.
Jeggo, P. A., Pearl, L. H., & Carr, A. M. (2016). DNA repair, genome stability and cancer: a historical perspective. Nature Reviews Cancer,16(1), 35–42. https://doi.org/10.1038/nrc.2015.4.
Kang, M. J., Ryu, B. K., Lee, M. G., Han, J., Lee, J. H., Ha, T. K., et al. (2008). NF-kappaB activates transcription of the RNA-binding factor HuR, via PI3K-AKT signaling, to promote gastric tumorigenesis. Gastroenterology,135(6), 2030–2042. https://doi.org/10.1053/j.gastro.2008.08.009.
Kanu, N., & Behrens, A. (2007). ATMIN defines an NBS1-independent pathway of ATM signalling. EMBO Journal,26(12), 2933–2941. https://doi.org/10.1038/sj.emboj.7601733.
Karimi, P., Islami, F., Anandasabapathy, S., Freedman, N. D., & Kamangar, F. (2014). Gastric cancer: descriptive epidemiology, risk factors, screening, and prevention. Cancer Epidemiology, Biomarkers and Prevention,23(5), 700–713. https://doi.org/10.1158/1055-9965.EPI-13-1057.
Kawanishi, S., Ohnishi, S., Ma, N., Hiraku, Y., & Murata, M. (2017). Crosstalk between DNA damage and inflammation in the multiple steps of carcinogenesis. International Journal of Molecular Sciences. https://doi.org/10.3390/ijms18081808.
Kelley, J. R., & Duggan, J. M. (2003). Gastric cancer epidemiology and risk factors. Journal of Clinical Epidemiology,56(1), 1–9. https://doi.org/10.1016/s0895-4356(02)00534-6.
Khanna, K. K., & Jackson, S. P. (2001). DNA double-strand breaks: signaling, repair and the cancer connection. Nature Genetics,27(3), 247–254. https://doi.org/10.1038/85798.
Kidane, D. (2018). Molecular mechanisms of H. pylori-induced DNA double-strand breaks. International Journal of Molecular Sciences. https://doi.org/10.3390/ijms19102891.
Kidane, D., Murphy, D. L., & Sweasy, J. B. (2014). Accumulation of abasic sites induces genomic instability in normal human gastric epithelial cells during Helicobacter pylori infection. Oncogenesis,3, e128. https://doi.org/10.1038/oncsis.2014.42.
Kim, Y. J., & Wilson, D. M., 3rd. (2012). Overview of base excision repair biochemistry. Current Molecular Pharmacology,5(1), 3–13.
Kim, J. J., Tao, H., Carloni, E., Leung, W. K., Graham, D. Y., & Sepulveda, A. R. (2002). Helicobacter pylori impairs DNA mismatch repair in gastric epithelial cells. Gastroenterology,123(2), 542–553. https://doi.org/10.1053/gast.2002.34751.
Koeppel, M., Garcia-Alcalde, F., Glowinski, F., Schlaermann, P., & Meyer, T. F. (2015). Helicobacter pylori infection causes characteristic DNA damage patterns in human cells. Cell Reports,11(11), 1703–1713. https://doi.org/10.1016/j.celrep.2015.05.030.
Kolas, N. K., Chapman, J. R., Nakada, S., Ylanko, J., Chahwan, R., Sweeney, F. D., et al. (2007). Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science,318(5856), 1637–1640. https://doi.org/10.1126/science.1150034.
Koopman, M., Kortman, G. A., Mekenkamp, L., Ligtenberg, M. J., Hoogerbrugge, N., Antonini, N. F., et al. (2009). Deficient mismatch repair system in patients with sporadic advanced colorectal cancer. British Journal of Cancer,100(2), 266–273. https://doi.org/10.1038/sj.bjc.6604867.
Koshiji, M., To, K. K., Hammer, S., Kumamoto, K., Harris, A. L., Modrich, P., et al. (2005). HIF-1alpha induces genetic instability by transcriptionally downregulating MutSalpha expression. Molecular Cell,17(6), 793–803. https://doi.org/10.1016/j.molcel.2005.02.015.
Krejs, G. J. (2010). Gastric cancer: epidemiology and risk factors. Digestive Diseases,28(4–5), 600–603. https://doi.org/10.1159/000320277.
Kunkel, T. A., & Erie, D. A. (2005). DNA mismatch repair. Annual Review of Biochemistry,74, 681–710. https://doi.org/10.1146/annurev.biochem.74.082803.133243.
Ladeira, M. S., Rodrigues, M. A., Salvadori, D. M., Queiroz, D. M., & Freire-Maia, D. V. (2004). DNA damage in patients infected by Helicobacter pylori. Cancer Epidemiology, Biomarkers and Prevention,13(4), 631–637.
Ladeira, M. S., Rodrigues, M. A., Freire-Maia, D. V., & Salvadori, D. M. (2005). Use of comet assay to assess DNA damage in patients infected by Helicobacter pylori: comparisons between visual and image analyses. Mutation Research,586(1), 76–86. https://doi.org/10.1016/j.mrgentox.2005.06.003.
Laval, F., & Wink, D. A. (1994). Inhibition by nitric oxide of the repair protein, O6-methylguanine-DNA-methyltransferase. Carcinogenesis,15(3), 443–447. https://doi.org/10.1093/carcin/15.3.443.
Lee, H. S., Choe, G., Park, K. U., Park, D. J., Yang, H. K., Lee, B. L., et al. (2007). Altered expression of DNA-dependent protein kinase catalytic subunit (DNA-PKcs) during gastric carcinogenesis and its clinical implications on gastric cancer. International Journal of Oncology,31(4), 859–866.
Leung, W. K., Kim, J. J., Kim, J. G., Graham, D. Y., & Sepulveda, A. R. (2000). Microsatellite instability in gastric intestinal metaplasia in patients with and without gastric cancer. American Journal of Pathology,156(2), 537–543. https://doi.org/10.1016/S0002-9440(10)64758-X.
Li, G. M. (2008). Mechanisms and functions of DNA mismatch repair. Cell Research,18(1), 85–98. https://doi.org/10.1038/cr.2007.115.
Liao, W., McNutt, M. A., & Zhu, W. G. (2009). The comet assay: a sensitive method for detecting DNA damage in individual cells. Methods,48(1), 46–53. https://doi.org/10.1016/j.ymeth.2009.02.016.
Lieber, M. R. (2008). The mechanism of human nonhomologous DNA end joining. Journal of Biological Chemistry,283(1), 1–5. https://doi.org/10.1074/jbc.R700039200.
Lim, J. W., Kim, H., & Kim, K. H. (2002). Expression of Ku70 and Ku80 mediated by NF-kappa B and cyclooxygenase-2 is related to proliferation of human gastric cancer cells. Journal of Biological Chemistry,277(48), 46093–46100. https://doi.org/10.1074/jbc.M206603200.
Liu, X., & Zha, S. (2011). ATMIN: a new tumor suppressor in developing B cells. Cancer Cell,19(5), 569–570. https://doi.org/10.1016/j.ccr.2011.05.002.
Liu, C. A., Wang, M. J., Chi, C. W., Wu, C. W., & Chen, J. Y. (2004). Rho/Rhotekin-mediated NF-kappaB activation confers resistance to apoptosis. Oncogene,23(54), 8731–8742. https://doi.org/10.1038/sj.onc.1208106.
Liu, X., Shao, Z., Jiang, W., Lee, B. J., & Zha, S. (2017). PAXX promotes KU accumulation at DNA breaks and is essential for end-joining in XLF-deficient mice. Nature Communications,8, 13816. https://doi.org/10.1038/ncomms13816.
Li, N., Banin, S., Ouyang, H., Li, G. C., Courtois, G., Shiloh, Y., et al. (2001). ATM is required for IkappaB kinase (IKKk) activation in response to DNA double strand breaks. Journal of Biological Chemistry,276(12), 8898–8903. https://doi.org/10.1074/jbc.M009809200.
Li, W., Xie, C., Yang, Z., Chen, J., & Lu, N. H. (2013). Abnormal DNA-PKcs and Ku 70/80 expression may promote malignant pathological processes in gastric carcinoma. World Journal of Gastroenterology,19(40), 6894–6901. https://doi.org/10.3748/wjg.v19.i40.6894.
Li, Z., Li, Y., Tang, M., Peng, B., Lu, X., Yang, Q., et al. (2018). Destabilization of linker histone H1.2 is essential for ATM activation and DNA damage repair. Cell Research,28(7), 756–770. https://doi.org/10.1038/s41422-018-0048-0.
Loizou, J. I., Sancho, R., Kanu, N., Bolland, D. J., Yang, F., Rada, C., et al. (2011). ATMIN is required for maintenance of genomic stability and suppression of B cell lymphoma. Cancer Cell,19(5), 587–600. https://doi.org/10.1016/j.ccr.2011.03.022.
Machado, A. M., Figueiredo, C., Touati, E., Maximo, V., Sousa, S., Michel, V., et al. (2009). Helicobacter pylori infection induces genetic instability of nuclear and mitochondrial DNA in gastric cells. Clinical Cancer Research,15(9), 2995–3002. https://doi.org/10.1158/1078-0432.CCR-08-2686.
Machado, A. M., Figueiredo, C., Seruca, R., & Rasmussen, L. J. (2010). Helicobacter pylori infection generates genetic instability in gastric cells. Biochimica et Biophysica Acta,1806(1), 58–65. https://doi.org/10.1016/j.bbcan.2010.01.007.
Machado, A. M., Desler, C., Boggild, S., Strickertsson, J. A., Friis-Hansen, L., Figueiredo, C., et al. (2013). Helicobacter pylori infection affects mitochondrial function and DNA repair, thus, mediating genetic instability in gastric cells. Mechanisms of Ageing and Development,134(10), 460–466. https://doi.org/10.1016/j.mad.2013.08.004.
Maeda, S., Yoshida, H., Ogura, K., Mitsuno, Y., Hirata, Y., Yamaji, Y., et al. (2000). H. pylori activates NF-kappaB through a signaling pathway involving IkappaB kinases, NF-kappaB-inducing kinase, TRAF2, and TRAF6 in gastric cancer cells. Gastroenterology,119(1), 97–108. https://doi.org/10.1053/gast.2000.8540.
Mailand, N., Bekker-Jensen, S., Faustrup, H., Melander, F., Bartek, J., Lukas, C., et al. (2007). RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell,131(5), 887–900. https://doi.org/10.1016/j.cell.2007.09.040.
Marshall, B. J., & Warren, J. R. (1984). Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. The Lancet,1(8390), 1311–1315. https://doi.org/10.1016/s0140-6736(84)91816-6.
Ma, J. L., Zhang, L., Brown, L. M., Li, J. Y., Shen, L., Pan, K. F., et al. (2012). Fifteen-year effects of Helicobacter pylori, garlic, and vitamin treatments on gastric cancer incidence and mortality. Journal of the National Cancer Institute,104(6), 488–492. https://doi.org/10.1093/jnci/djs003.
Medzhitov, R., & Horng, T. (2009). Transcriptional control of the inflammatory response. Nature Reviews Immunology,9(10), 692–703. https://doi.org/10.1038/nri2634.
Melino, G., Bernassola, F., Knight, R. A., Corasaniti, M. T., Nistico, G., & Finazzi-Agro, A. (1997). S-Nitrosylation regulates apoptosis. Nature,388(6641), 432–433. https://doi.org/10.1038/41237.
Mirzaee, V., Molaei, M., Shalmani, H. M., & Zali, M. R. (2008). Helicobacter pylori infection and expression of DNA mismatch repair proteins. World Journal of Gastroenterology,14(43), 6717–6721. https://doi.org/10.3748/wjg.14.6717.
Modrich, P., & Lahue, R. (1996). Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annual Review of Biochemistry,65, 101–133. https://doi.org/10.1146/annurev.bi.65.070196.000533.
Moeller, B. J., Yordy, J. S., Williams, M. D., Giri, U., Raju, U., Molkentine, D. P., et al. (2011). DNA repair biomarker profiling of head and neck cancer: Ku80 expression predicts locoregional failure and death following radiotherapy. Clinical Cancer Research,17(7), 2035–2043. https://doi.org/10.1158/1078-0432.CCR-10-2641.
Mokkapati, S. K., Wiederhold, L., Hazra, T. K., & Mitra, S. (2004). Stimulation of DNA glycosylase activity of OGG1 by NEIL1: functional collaboration between two human DNA glycosylases. Biochemistry,43(36), 11596–11604. https://doi.org/10.1021/bi049097i.
Monack, D. M., Mueller, A., & Falkow, S. (2004). Persistent bacterial infections: the interface of the pathogen and the host immune system. Nature Reviews Microbiology,2(9), 747–765. https://doi.org/10.1038/nrmicro955.
Murray-Stewart, T., Sierra, J. C., Piazuelo, M. B., Mera, R. M., Chaturvedi, R., Bravo, L. E., et al. (2016). Epigenetic silencing of miR-124 prevents spermine oxidase regulation: implications for Helicobacter pylori-induced gastric cancer. Oncogene,35(42), 5480–5488. https://doi.org/10.1038/onc.2016.91.
Naito, Y., & Yoshikawa, T. (2002). Molecular and cellular mechanisms involved in Helicobacter pylori-induced inflammation and oxidative stress. Free Radical Biology and Medicine,33(3), 323–336. https://doi.org/10.1016/s0891-5849(02)00868-7.
Naumann, M., Sokolova, O., Tegtmeyer, N., & Backert, S. (2017). Helicobacter pylori: a paradigm pathogen for subverting host cell signal transmission. Trends in Microbiology,25(4), 316–328. https://doi.org/10.1016/j.tim.2016.12.004.
Niwa, T., Tsukamoto, T., Toyoda, T., Mori, A., Tanaka, H., Maekita, T., et al. (2010). Inflammatory processes triggered by Helicobacter pylori infection cause aberrant DNA methylation in gastric epithelial cells. Cancer Research,70(4), 1430–1440. https://doi.org/10.1158/0008-5472.CAN-09-2755.
Nossa, C. W., & Blanke, S. R. (2010). Helicobacter pylori activation of PARP-1: usurping a versatile regulator of host cellular health. Gut Microbes,1(6), 373–378. https://doi.org/10.4161/gmic.1.6.13572.
Nougayrede, J. P., Homburg, S., Taieb, F., Boury, M., Brzuszkiewicz, E., Gottschalk, G., et al. (2006). Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science,313(5788), 848–851. https://doi.org/10.1126/science.1127059.
Nozawa, Y., Nishihara, K., Peek, R. M., Nakano, M., Uji, T., Ajioka, H., et al. (2002). Identification of a signaling cascade for interleukin-8 production by Helicobacter pylori in human gastric epithelial cells. Biochemical Pharmacology,64(1), 21–30. https://doi.org/10.1016/s0006-2952(02)01030-4.
Obst, B., Wagner, S., Sewing, K. F., & Beil, W. (2000). Helicobacter pylori causes DNA damage in gastric epithelial cells. Carcinogenesis,21(6), 1111–1115.
O'Connor, T. R., Graves, R. J., de Murcia, G., Castaing, B., & Laval, J. (1993). Fpg protein of Escherichia coli is a zinc finger protein whose cysteine residues have a structural and/or functional role. Journal of Biological Chemistry,268(12), 9063–9070.
Panieri, E., & Santoro, M. M. (2016). ROS homeostasis and metabolism: a dangerous liason in cancer cells. Cell Death Disease,7(6), e2253. https://doi.org/10.1038/cddis.2016.105.
Pannunzio, N. R., Watanabe, G., & Lieber, M. R. (2018). Nonhomologous DNA end-joining for repair of DNA double-strand breaks. Journal of Biological Chemistry,293(27), 10512–10523. https://doi.org/10.1074/jbc.TM117.000374.
Papa, A., Danese, S., Sgambato, A., Ardito, R., Zannoni, G., Rinelli, A., et al. (2002). Role of Helicobacter pylori CagA+ infection in determining oxidative DNA damage in gastric mucosa. Scandinavian Journal of Gastroenterology,37(4), 409–413. https://doi.org/10.1080/003655202317316033.
Park, J. H., Kim, T. Y., Jong, H. S., Kim, T. Y., Chun, Y. S., Park, J. W., et al. (2003). Gastric epithelial reactive oxygen species prevent normoxic degradation of hypoxia-inducible factor-1alpha in gastric cancer cells. Clinical Cancer Research,9(1), 433–440.
Park, D. I., Park, S. H., Kim, S. H., Kim, J. W., Cho, Y. K., Kim, H. J., et al. (2005). Effect of Helicobacter pylori infection on the expression of DNA mismatch repair protein. Helicobacter,10(3), 179–184. https://doi.org/10.1111/j.1523-5378.2005.00309.x.
Parsonnet, J., Friedman, G. D., Vandersteen, D. P., Chang, Y., Vogelman, J. H., Orentreich, N., et al. (1991). Helicobacter pylori infection and the risk of gastric carcinoma. New England Journal of Medicine,325(16), 1127–1131. https://doi.org/10.1056/NEJM199110173251603.
Pietrantonio, F., Miceli, R., Raimondi, A., Kim, Y. W., Kang, W. K., Langley, R. E., et al. (2019). Individual patient data meta-analysis of the value of microsatellite instability as a biomarker in gastric cancer. Journal of Clinical Oncology. https://doi.org/10.1200/JCO.19.01124.
Pino, M. S., & Chung, D. C. (2010). The chromosomal instability pathway in colon cancer. Gastroenterology,138(6), 2059–2072. https://doi.org/10.1053/j.gastro.2009.12.065.
Pua, K. H., Chew, C. L., Lane, D. P., & Tergaonkar, V. (2019). Inflammationassociated genomic instability in cancer. Genome Instability and Disease. https://doi.org/10.1007/s42764-019-00006-6.
Rawla, P., & Barsouk, A. (2019). Epidemiology of gastric cancer: global trends, risk factors and prevention. Przeglad Gastroenterol,14(1), 26–38. https://doi.org/10.5114/pg.2018.80001.
Reliene, R., Fischer, E., & Schiestl, R. H. (2004). Effect of N-acetyl cysteine on oxidative DNA damage and the frequency of DNA deletions in atm-deficient mice. Cancer Research,64(15), 5148–5153. https://doi.org/10.1158/0008-5472.CAN-04-0442.
Robertson, A. B., Klungland, A., Rognes, T., & Leiros, I. (2009). DNA repair in mammalian cells: base excision repair: the long and short of it. Cellular and Molecular Life Sciences,66(6), 981–993. https://doi.org/10.1007/s00018-009-8736-z.
Sancar, A., Lindsey-Boltz, L. A., Unsal-Kacmaz, K., & Linn, S. (2004). Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annual Review of Biochemistry,73, 39–85. https://doi.org/10.1146/annurev.biochem.73.011303.073723.
Santos, J. C., Gambeloni, R. Z., Roque, A. T., Oeck, S., & Ribeiro, M. L. (2018). Epigenetic mechanisms of ATM activation after helicobacter pylori infection. American Journal of Pathology,188(2), 329–335. https://doi.org/10.1016/j.ajpath.2017.10.005.
Shirin, H., Sordillo, E. M., Oh, S. H., Yamamoto, H., Delohery, T., Weinstein, I. B., et al. (1999). Helicobacter pylori inhibits the G1 to S transition in AGS gastric epithelial cells. Cancer Research,59(10), 2277–2281.
Sidorenko, V. S., Nevinsky, G. A., & Zharkov, D. O. (2007). Mechanism of interaction between human 8-oxoguanine-DNA glycosylase and AP endonuclease. DNA Repair (Amsterdam),6(3), 317–328. https://doi.org/10.1016/j.dnarep.2006.10.022.
Simsek, D., Brunet, E., Wong, S. Y., Katyal, S., Gao, Y., McKinnon, P. J., et al. (2011). DNA ligase III promotes alternative nonhomologous end-joining during chromosomal translocation formation. PLoS Genetics,7(6), e1002080. https://doi.org/10.1371/journal.pgen.1002080.
Smith, J., Tho, L. M., Xu, N., & Gillespie, D. A. (2010). The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Advances in Cancer Research,108, 73–112. https://doi.org/10.1016/B978-0-12-380888-2.00003-0.
Sokolova, O., & Naumann, M. (2019). Crosstalk between DNA damage and inflammation in the multiple steps of gastric carcinogenesis. Current Topics in Microbiology and Immunology,421, 107–137. https://doi.org/10.1007/978-3-030-15138-6_5.
Stucki, M., Clapperton, J. A., Mohammad, D., Yaffe, M. B., Smerdon, S. J., & Jackson, S. P. (2005). MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell,123(7), 1213–1226. https://doi.org/10.1016/j.cell.2005.09.038.
Tchou, J., Michaels, M. L., Miller, J. H., & Grollman, A. P. (1993). Function of the zinc finger in Escherichia coli Fpg protein. Journal of Biological Chemistry,268(35), 26738–26744.
Toller, I. M., Neelsen, K. J., Steger, M., Hartung, M. L., Hottiger, M. O., Stucki, M., et al. (2011). Carcinogenic bacterial pathogen Helicobacter pylori triggers DNA double-strand breaks and a DNA damage response in its host cells. Proceedings of National Academy of Sciences USA,108(36), 14944–14949. https://doi.org/10.1073/pnas.1100959108.
Uehara, T., Ma, D., Yao, Y., Lynch, J. P., Morales, K., Ziober, A., et al. (2013). H. pylori infection is associated with DNA damage of Lgr5-positive epithelial stem cells in the stomach of patients with gastric cancer. Digestive Diseases and Sciences,58(1), 140–149. https://doi.org/10.1007/s10620-012-2360-8.
Uemura, N., Okamoto, S., Yamamoto, S., Matsumura, N., Yamaguchi, S., Yamakido, M., et al. (2001). Helicobacter pylori infection and the development of gastric cancer. New England Journal of Medicine,345(11), 784–789. https://doi.org/10.1056/NEJMoa001999.
Viala, J., Chaput, C., Boneca, I. G., Cardona, A., Girardin, S. E., Moran, A. P., et al. (2004). Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nature Immunology,5(11), 1166–1174. https://doi.org/10.1038/ni1131.
Vitale, I., Manic, G., De Maria, R., Kroemer, G., & Galluzzi, L. (2017). DNA damage in stem cells. Molecular Cell,66(3), 306–319. https://doi.org/10.1016/j.molcel.2017.04.006.
Wallace, S. S., Murphy, D. L., & Sweasy, J. B. (2012). Base excision repair and cancer. Cancer Letters,327(1–2), 73–89. https://doi.org/10.1016/j.canlet.2011.12.038.
Wang, K., Yuen, S. T., Xu, J., Lee, S. P., Yan, H. H., Shi, S. T., et al. (2014). Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nature Genetics,46(6), 573–582. https://doi.org/10.1038/ng.2983.
Watanabe, N., Shimada, T., Ohtsuka, Y., Hiraishi, H., & Terano, A. (1997). Proinflammatory cytokines and Helicobacter pylori stimulate CC-chemokine expression in gastric epithelial cells. Journal of Physiology and Pharmacology,48(3), 405–413.
Wei, J., Nagy, T. A., Vilgelm, A., Zaika, E., Ogden, S. R., Romero-Gallo, J., et al. (2010). Regulation of p53 tumor suppressor by Helicobacter pylori in gastric epithelial cells. Gastroenterology,139(4), 1333–1343. https://doi.org/10.1053/j.gastro.2010.06.018.
White, J. R., Winter, J. A., & Robinson, K. (2015). Differential inflammatory response to Helicobacter pylori infection: etiology and clinical outcomes. Journal of Inflammation Research,8, 137–147. https://doi.org/10.2147/JIR.S64888.
Wink, D. A., & Laval, J. (1994). The Fpg protein, a DNA repair enzyme, is inhibited by the biomediator nitric oxide in vitro and in vivo. Carcinogenesis,15(10), 2125–2129. https://doi.org/10.1093/carcin/15.10.2125.
Xie, C., Xu, L. Y., Yang, Z., Cao, X. M., Li, W., & Lu, N. H. (2014). Expression of gammaH2AX in various gastric pathologies and its association with Helicobacter pylori infection. Oncology Letters,7(1), 159–163. https://doi.org/10.3892/ol.2013.1693.
Xu, H., Chaturvedi, R., Cheng, Y., Bussiere, F. I., Asim, M., Yao, M. D., et al. (2004). Spermine oxidation induced by Helicobacter pylori results in apoptosis and DNA damage: implications for gastric carcinogenesis. Cancer Research,64(23), 8521–8525. https://doi.org/10.1158/0008-5472.CAN-04-3511.
Yao, Y., Tao, H., Park, D. I., Sepulveda, J. L., & Sepulveda, A. R. (2006). Demonstration and characterization of mutations induced by Helicobacter pylori organisms in gastric epithelial cells. Helicobacter,11(4), 272–286. https://doi.org/10.1111/j.1523-5378.2006.00408.x.
Acknowledgements
This work was supported by the Guangdong Provincial Key Laboratory of Regional Immunity and Diseases (Grant number: 2019B030301009) and the by the Discipline Construction Funding of Shenzhen (2016-2019). The authors would like to recognize that there are many other valuable papers that were not included in this review because of space limitations.
Author information
Affiliations
Guangdong Provincial Key Laboratory of Regional Immunity and Diseases, Department of Biochemistry and Molecular Biology, School of Medicine, Shenzhen University, Shenzhen, China
Xiangyu Liu & Muhammad Irfan
Guangdong Key Laboratory of Genome Instability and Human Disease Prevention, Shenzhen University Carson Cancer Center, Shenzhen, China
Xiangyu Liu & Xingzhi Xu
Marshall Laboratory of Biomedical Engineering, School of Biomedical Engineering, Shenzhen University Health Science Center, Shenzhen, China
Xiangyu Liu, Xingzhi Xu, Chi-Yen Tay & Barry J. Marshall
The Marshall Centre for Infectious Diseases Research and Training, The University of Western Australia, Perth, 6009, Australia
Chi-Yen Tay & Barry J. Marshall
Corresponding authors
Correspondence to Xiangyu Liu or Barry J. Marshall.
Ethics declarations
Conflict of interest
The authors declare no competing financial interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article
Cite this article
Liu, X., Irfan, M., Xu, X. et al. Helicobacter pylori infection induced genome instability and gastric cancer. GENOME INSTAB. DIS. 1, 129–142 (2020). https://doi.org/10.1007/s42764-020-00010-1
Received04 March 2020
Revised24 March 2020
Accepted25 March 2020
Published21 April 2020
Issue DateMay 2020
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
H. pylori
Genomic instability
Gastric cancer
DNA damage
DNA repair
用户登录
还没有账号?
立即注册