






Suppression of genomic instability by replicative senescence and crisis
Review Article
Genome Instability & Disease 1, 143–150(2020)
Abstract
Replicative senescence and crisis are proliferative barriers, activated as a response to telomere shortening and the resulting loss of chromosome end protection. Cells that enter replicative senescence usually maintain a stable genome, while bypass of senescence through loss of p53 and Rb pathways leads to the accumulation of chromosomal end-to-end fusions. Such fusions activate the spindle assembly checkpoint and a cell death program referred to as replicative crisis. Crisis is remarkably effective in removing cells that have bypassed senescence from the population; however, rarely cells escape, which then are thought to progress to a malignant phenotype. These observations suggest that crisis is the final barrier against cancer formation, yet it is still poorly understood how cells die in crisis, in which molecular cascades play a role in escape from crisis, and whether cells that emerge from crisis can be considered as cancer cells, pinpointing that a better understanding of these pathways is critical to target cancer formation in its earliest stages.
Introduction
Two major barriers prevent the transformation of dividing human cells into cancer cells, replicative senescence and crisis (Fig. 1). In the absence of oncogene-induced stress, short telomeres trigger replicative senescence, also referred to as the Hayflick limit, (Hayflick and Moorhead 1961; Herbig et al. 2004; d’Adda di Fagagna et al. 2003). Telomeres induce senescence through an intermediate-deprotected state, which is recognized by the DNA damage machinery, but still protects chromosomes against end-to-end fusions (Cesare and Karlseder 2012), thereby maintaining a stable diploid genome (Cesare et al. 2013). However, senescence of human cells can be prevented by the impairment of the p53 and pRb controlled cell cycle checkpoints (Fig. 1) (Hara et al. 1996; Beausejour et al. 2003; Shay et al. 1991). Following senescence bypass and continued proliferation with ever shortening telomeres, cells enter a stage referred to as replicative crisis, which is a phase highlighted by substantial telomere deprotection, chromosome end to end fusions and widespread cell death (Wei and Sedivy 1999; Bond et al. 1999). Replicative senescence and crisis are two different cellular responses induced by distinct triggers (as opposed to two different outcomes of the same stressor). Replicative senescence is activated in primary normal cells (with functional checkpoints) in response to a sustained and persistent DDR (DNA Damage Response) pathway emanating from deprotected telomeres (at roughly 3–5 kb in length). In replicative senescence, cells become arrested during the cell cycle (mostly in G1) and remain metabolically active and viable. Cell death is not prominent in replicative senescence and it is generally well accepted that cells in replicative senescence are resistant to cell death.
Fig. 1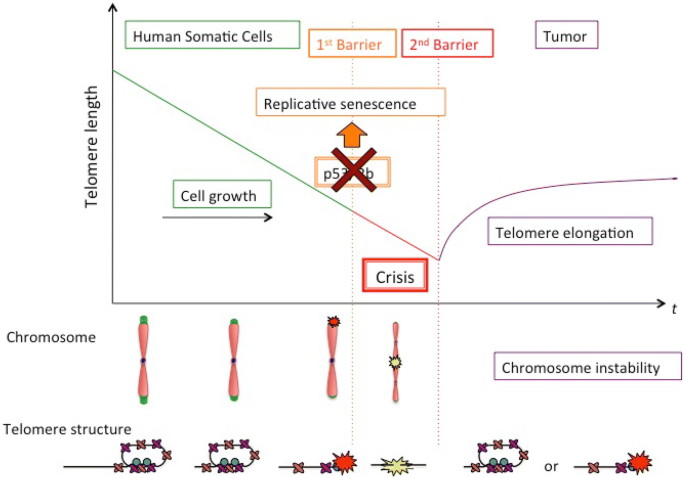
Telomere shortening, senescence, crisis and cancer. Primary human cells lacking telomere maintenance pathways divide exponentially and telomeres shorten until they reach a critical length. At that point, short telomeres activate the p53 and pRb tumor-suppressor pathways, followed by irreversible cell cycle arrest in senescence. Telomeres in senescent cells are recognized as sites of DNA damage, but are still protected from fusion. Suppression of p53 and pRB allows cells to bypass senescence and continue to divide, until chromosome end protection is completely lost, resulting in telomere end to end fusions, which trigger replicative crisis, a state marked by widespread cell death and genome instability. Cells that escape crisis have lost tumor suppressors, have likely activated telomere maintenance mechanisms and harbor an unstable genome, all signs of neoplastic transformation
Full size imageUpon disruption of cell cycle checkpoints, cells become insensitive to DDR-positive telomeres, lose the ability to activate senescence and exit the cell cycle. As a result, cells bypass senescence and continue to divide and to shorten telomeres beyond the length detected in replicative senescence (2–4 kb). At a terminal point, telomeres become critically short, resulting in chromosomal end-to-end fusions, prolonged mitosis and cell death, all characteristics of replicative crisis (Hayashi et al. 2015). Cell death in crisis is extensive and tightly linked to telomere fusions (Maciejowski and de Lange 2017), which stands in contrast to the resistance to cell death of replicative senescence. Crisis is therefore a stringent anti-proliferative barrier, as it removes the vast majority of cells that escape senescence from the population (Fig. 1), independently of their p53/Rb status. Only rarely do cells overcome this barrier; however, these become neoplastic (Shay et al. 1993; Shay and Wright 1989; Halvorsen et al. 1999).
This review focuses on the roles of replicative senescence and crisis in preventing genome instability and thereby cancer formation.
Cellular senescence as response to various types of stress
While signaling from short telomeres triggers replicative senescence, senescence-like arrest can be induced by many different types of cellular stresses. Genomic damage at non-telomeric sites can lead to persistent damage signaling and push cells into senescence (Nakamura et al. 2008; Smogorzewska and de Lange 2002), which is emphasized by the finding that DNA double-stranded breaks are potent senescence inducers (Di Leonardo et al. 1994). Even without DNA breaks, changes in chromatin structure can lead to senescence, demonstrated by the observation that histone deacetylase inhibitors, which relax chromatin without physically damaging DNA, activate the DDR modulators ataxia telangiectasia mutated (ATM) and the p53 tumor suppressor and induce terminal senescence-like differentiation (Bakkenist and Kastan 2003; Munro et al. 2004). It has been proposed that condensin complexes carry out the organization of eu- and heterochromatin chromatin compartments, since condensin localizes to senescence genes and loss of condensing function impaired senescence markers (Iwasaki et al. 2019). In addition to these genomic stressors, cells can enter senescence upon mitotic overstimulation, such as those conferred by oncogene induction (Serrano et al. 1997; Zhu et al. 1998; Dimri et al. 2000). These mitogenic signals, however, can be conferred to genomic damage and persistent DNA damage signaling due to replication fork collapse and misfiring of replication origins (Bartkova et al. 2006; Di Micco et al. 2006; Mallette et al. 2007). Poor tissue culture conditions can also induce senescence-like arrest, termed culture shock, where cells permanently cease to differentiate without signs of genomic damage (Ramirez et al. 2001).
Defining senescent cells is complicated by the fact that no marker that has been identified so far is exclusively applicable to senescence, and in turn, not all senescent cells express all senescence markers. Furthermore, it is likely that replicative senescence, oncogene-induced senescence, chromatin-induced senescence and other senescence-like arrest phenotypes are not perfectly alike, causing confusion in the field and raising the need for better terminology. However, senescent cells are likely best characterized by the expression of a combination of senescence markers:
The cells enter a permanent growth arrest, which cannot be reversed in human cells. While low levels of DNA replication have been observed in senescent cells, where DNA checkpoint kinases have been rendered inactive, it is doubtful that such cells will complete an S phase and mitosis following manipulation (d’Adda di Fagagna et al. 2003).
Senescent cells, as opposed to quiescent or differentiated cells, usually express p16INK4a (Alcorta et al. 1996; Serrano et al. 1997; Hara et al. 1996). This property has been successfully exploited to remove senescent cells from tissues and organisms, by expressing an inducible p16-driven elimination cassette, resulting in the delay of multiple age-associated disorders (Baker et al. 2011).
Senescent cells express senescence-associated b-galactosidase, which is specifically active at pH6 (Dimri et al. 1995).
Senescent cells often harbor senescence-associated heterochromatin foci (SAHF) (Narita et al. 2003), which contain high mobility group A proteins (HMGA) as structural component. These HMGA proteins cooperate with p16 in SAHF formation and in driving the senescence-associated cell cycle arrest (Narita et al. 2006).
Senescent cells are usually much larger in size than their precursors, which was one of the earliest markers for replicative senescence ever described (Hayflick 1965).
Senescent cells often harbor foci that contain activated DNA damage factors such as phosphorylated ATM and ATR (d’Adda di Fagagna et al. 2003; Herbig et al. 2004), and often colocalize with dysfunctional and critically short telomeres (Takai et al. 2003; Kim et al. 2004).
Senescent cells secrete a cocktail of growth factors, cytokines and other factors that can enable the proliferation of surrounding cells, thereby promoting cellular transformation (Coppe et al. 2010a; Acosta et al. 2008; Kuilman et al. 2008).
With the discovery that senescent cells secrete cytokines that stimulate proliferation of the surrounding cells through development of the SASP (Coppe et al. 2010a; Acosta et al. 2008; Kuilman et al. 2008), it became clear that senescence is another process with both beneficial and deleterious properties. Notably, many SASP factors stimulate the proliferation of premalignant epithelial cells or promote invasion of such cells into the basement membrane, such as interleukines 6 and 8, or VEGF (Coppe et al. 2006, 2008, 2010b).
Given these opposing roles of senescence as age-associated tumor-suppressive mechanism as well as growth and invasion promoter through the SASP it was fascinating to observe that targeted removal of senescent cells was associated with overwhelmingly positive consequences in a progeroid murine model (Baker et al. 2011). Deletion of senescent cells marked by elevated p16INK4a expression delayed onset of age-associated deleterious phenotypes in adipose tissue, skeletal muscle and the eye, linking senescence causally to age-associated disease.
Replicative senescence preserves genomic stability
Senescence, in particular replicative senescence, has originally been characterized as a powerful tumor-suppressive pathway (de Lange 1998), underlined by the findings that the stimuli that elicit a senescence response are all potentially carcinogenic, and that the major tumor-suppressive pathways p53 and RB are both required to induce senescence in human cells (Campisi and d’Adda di Fagagna 2007).
An established model proposes that spontaneous telomere deprotection as function of replicative aging progresses through three distinct states that regulate cellular fate (Cesare and Karlseder 2012). In young cells and during the exponential growth phase, closed-state telomeres prevent DDR activation by sequestering chromosome termini within the telomere loop (T-loop), a protective higher-order structure (Griffith et al. 1999). Progressive telomere shortening as a consequence of replicative age, or insufficient coverage of telomeric tracts by shelterin components, renders telomeres partially dysfunctional as intermediate-state telomeres, which are susceptible to a DDR. However, NHEJ (non-homologous end-joining)-dependent fusion of intermediate-state telomeres is still repressed by TRF2 retention on the DDR-positive telomeric chromatin. Quantitative analysis of individual telomeres in cells entering senescence revealed that five or more intermediate-state telomeres in a G1 phase cell are sufficient to activate the senescence response in primary cells, but even more intermediate-state telomeres accumulate in p53 incompetent cells without affecting growth (Kaul et al. 2012). Finally, the uncapped telomere state arises under physiological conditions when chromosome fusions occur as a response to insufficient TRF2 at telomeres, which then fails to inhibit nonhomologous end joining (NHEJ) (van Steensel et al. 1998; Karlseder et al. 1999). This could be expected to occur spontaneously after excessive telomere erosion removes the shelterin-binding sites at chromosome ends and is correlated with cell death at crisis.
Recently, it was discovered that human cells react with a telomere-specific DDR upon prolonged mitotic arrest. This stress response was due to partial dissociation of TRF2 from chromosome ends and resulted in an ATM-dependent telomeric DDR, which activates the prolonged mitotic arrest checkpoint (Hayashi et al. 2012; Hayashi and Karlseder 2013). However, no chromosome fusions were detected, and when cells were released from the mitotic block, p53-competent cells arrest in G1 phase as a consequence of inheritance of deprotected telomeres from the preceding mitosis. Such observations emphasize that human cells can utilize the transition of closed- to intermediate-state telomeres as a powerful mechanism to arrest proliferation in senescence, while avoiding the risk of the genomic instability associated with chromosome fusions.
In primary human fibroblasts, replicative senescence is primarily a response to short telomeres, which signal through an intermediate stage of deprotection to the p53 and RB pathways (Cesare and Karlseder 2012). Importantly, while telomeres are recognized as damage in pre-senescent cells, they still harbor sufficient protective properties to prevent chromosome end-to-end fusion (Cesare and Karlseder 2012). While it was long believed that deprotected telomeres resemble double-stranded breaks, detailed analysis of the signaling pathways revealed that the telomere deprotection response is functionally distinct from the response to intrachromosomal double-stranded breaks (Cesare et al. 2013). Analyzing the final cell cycles prior to senescence entry demonstrated that deprotected telomeres signal differentially from double-stranded breaks during cellular aging and in senescence. While intrachromosomal breaks activate G1 and G2 DNA damage checkpoints, deprotected telomeres fail to activate the CHK2-dependent G2 checkpoint. This enables cells that are about to enter senescence to pass through one last mitosis and stop growth in a stringent G1 arrest with a diploid genome, as a consequence preserving genome integrity (Fig. 2) (Cesare et al. 2013). Progressive telomere erosion during replicative aging dictates that telomeres enter the intermediate state before progressing into the uncapped state. As a result, senescence is constrained to diploid G1 phase cells, preventing genome instability by triggering arrest before genomes become destabilized through telomere fusions.
Fig. 2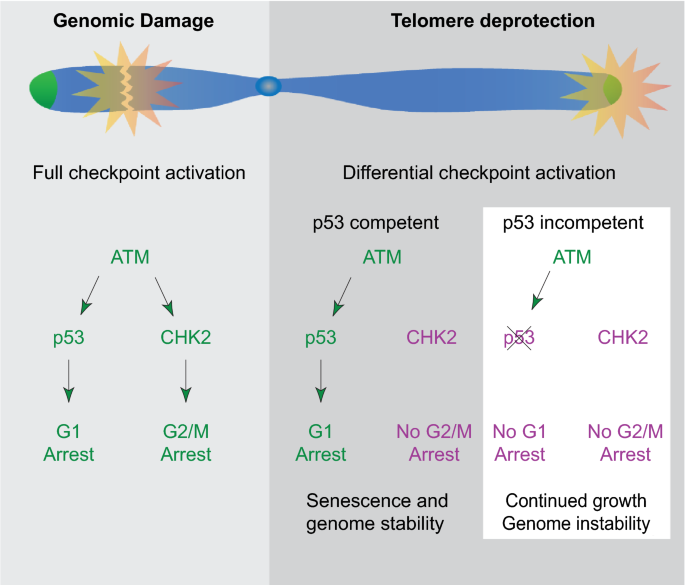
Differential signaling between intrachromosomal breaks and deprotected telomeres. While genomic damage activates cell cycle arrest in G1 and G2 through the p53 and CHK2 pathways, respectively, telomeres in p53 competent cells fail to activate CHK2 signaling. Once p53 has been compromised, neither G1 nor G2 arrest is triggered through telomere dysfunction and cell can continue to divide and accumulate genomic instability
Full size imageReplicative crisis as barrier against cancer formation
Replication-associated telomere shortening is the result of telomere end processing and the inability of conventional polymerases to fully replicate the parental DNA strand through lagging strand synthesis. As a consequence, telomeres shorten by approximately 100–200 base pairs per population doubling (Fig. 1). Once telomeres reach a critical length, they are recognized by the DNA damage machinery, which signals toward cell cycle arrest in response to activation of the p53 and pRb-dependent damage-sensing machineries. In human fibroblasts, this arrest has been characterized as terminal differentiation or replicative senescence. Telomeres in senescent cells are recognized by DNA damage-sensing pathways; however, they are not fully deprotected and retain chromosome end-protective properties. Hence, chromosomes in senescent cells do not fuse, but exist in an intermediate state of telomere deprotection (Cesare and Karlseder 2012; Cesare et al. 2013).
Impairment of p53 and pRb pathways, through viral oncogenes, by targeted deletion or by disruption of the downstream pathways, allows cells to bypass senescence and to keep proliferating until the population reaches replicative crisis, a response quite distinctive to replicative senescence. While senescent cells have active DNA damage checkpoints, crisis cells have bypassed senescence by loss of these checkpoints before the cells ever entered senescence. Therefore, crisis cells are mostly not cells that escaped the senescence-associated arrest, but they are cells that bypassed senescence. Replication-associated telomere shortening during the additional proliferation period leads to further telomere attrition until telomeres lose all protective properties, and chromosome fusions arise as consequence of non-homologous end joining and alternative end joining. As mentioned, a hallmark of crisis is extensive cell death, which has been previously attributed to extensive genome instability resulting from repetitive breakage–fusion–bridge cycles.
Recently, a much more controlled mechanism for cell death in replicative crisis has been proposed instead. Based on experiments with human diploid fibroblasts, a well-controlled cell death mechanism within a single cell cycle has been established. This pathway relies on the chromosome end to end fusions appearing in cells that have bypassed senescence and suffer from extensive replicative telomere shortening. These uncapped telomeres are repaired by NHEJ and in turn the fused telomeres in cells approaching crisis activate the spindle assembly checkpoint, which triggers cell cycle arrest in the following mitosis. During the mitotic arrest, the telomere repeat binding factor TRF2 is removed from telomeres via an Aurora B-dependent pathway, which amplifies telomere deprotection and thereby causes cell death (Hayashi et al. 2012, 2015) (Fig. 3). This model also became interesting for the understanding of cell death upon treatment with mitotic inhibitors, as are frequently used in cancer therapy, such as taxane derivatives, since such drugs also appeared to cause telomere dysfunction (Hayashi et al. 2012). However, this model did not explain the extensive genome instability observed in post-crisis cells and did not offer a molecular explanation of the crisis-associated cell death. It therefore became essential to revisit the cell death response triggered by telomere dysfunction, leading to the unexpected discovery that death in crisis is orchestrated by macroautophagy (Nassour et al. 2019).
Fig. 3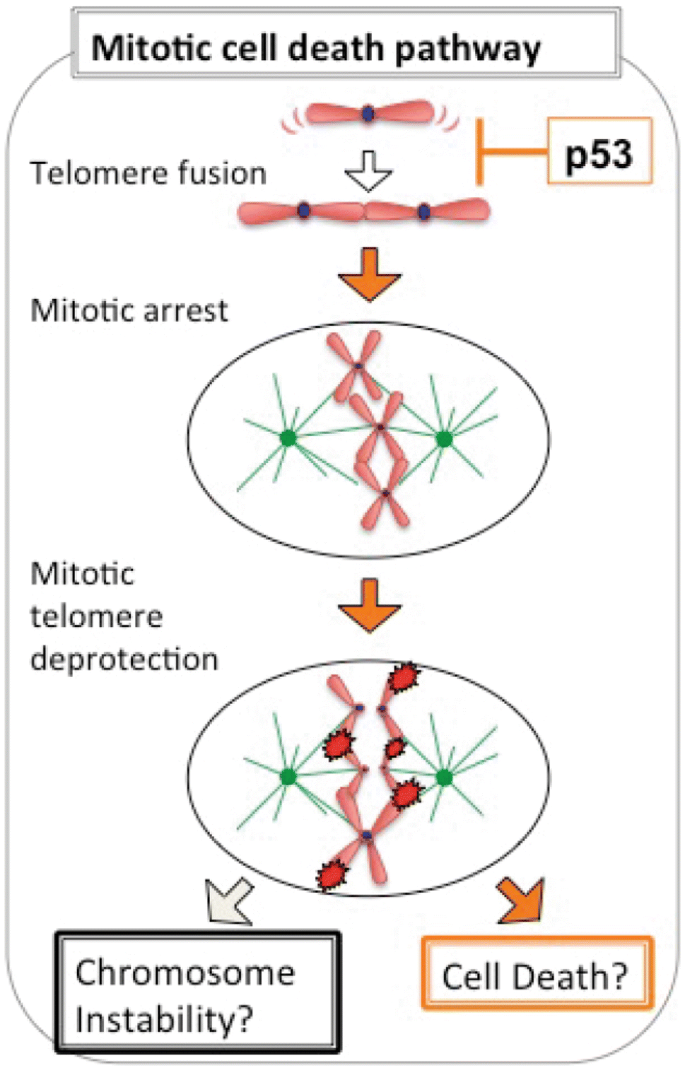
Model for cell death in crisis. Telomere fusions in cells that have bypassed senescence through loss of p53 and RB pathways trigger the spindle assembly checkpoint (SAC). SAC activation leads to mitotic arrest, which amplifies mitotic telomere deprotection via targeted Aurora B kinase-dependent removal of TRF2 from telomeres. The resulting DNA damage responds and kills cells in replicative crisis within a single cell cycle
Full size imageReplicative crisis and genome instability
Autophagy is a “self-digesting” process, whereby damaged or excessive components of the cell are enclosed into double membrane vesicles, called autophagosomes, which then are transformed into single membrane autolysosomes for lysosomal digestion of captured components. Hence, autophagy serves as an adaptive mechanism allowing cells to maintain nutrient and energy homeostasis. Paradoxically, long-term activation of autophagy is deleterious for cells, as it is capable of destroying a large portion of the cytosol, causing an irreversible collapse of cellular functions and consequently cell death (Yonekawa and Thorburn 2013). Apoptosis, on the other hand, is marked by cleavage of caspases and chromatin degradation.
Against expectations, the onset of replicative crisis was associated with extensive cytoplasmic vacuolization in human fibroblasts and epithelial cells, suggestive of macroautophagy (Nassour et al. 2019). The cytoplasm of crisis cells contained numerous vacuoles with features of double-membrane autophagosomes containing intact cytosol or organelles and single-membrane autolysosomes containing digested cellular components. The hallmarks of apoptosis were only detected in staurosporine-treated cells, and not at all in cells approaching crisis. Autophagic flux, depicting the active transition from autophagosomes to autolysosomes, was directly monitored with a tandem fluorescence LC3 reporter (mCherry-GFP-LC3), where activation of autophagic flux leads to an accumulation of both yellow and red LC3 dots (Bampton et al. 2005). Crisis cells exhibited an extensive accumulation of autophagosomes and autolysosomes, where the absolute numbers of both types of vacuoles simultaneously increased. Treatment with bafilomycin A1, an inhibitor of autophagosome–lysosome fusion and lysosomal acidification, in crisis led to an even greater accumulation of autophagosomes, but abolished autolysosomes, corroborating an active autophagic flux (Nassour et al. 2019).
Of course, the question arose what the trigger for autophagy activation in crisis cells was. Currently, the hypothesis is that the fusion of chromsomes with critically short telomeres generates dicentric chromosomes, nuclear bridge formation and breakage of those structures during mitosis. The random breakage releases chromatin fragments into the cytoplasm and this cytoplasmic DNA can trigger the DNA-sensing cGAS (cyclic GMP-AMP synthase)/STING (stimulator of interferon genes) pathway, initiate the production of inflammatory cytokines and stimulate autophagy (Ishikawa et al. 2009; Sun et al. 2013; Comb et al. 2011). Indeed, the cGAS–STING pathway was required for crisis-dependent autophagy activation and cytosolic DNA in crisis cells colocalized with cGAS. Furthermore, cGAS and STING were found essential for crisis, since fibroblasts lacking cGAS or STING proliferated beyond crisis and this crisis bypass was associated with an attenuation of autophagy (Nassour et al. 2019).
It has been hypothesized that escape from replicative crisis results in cells with an unstable genome. However, this was not easily experimentally testable, as escape from crisis is exceedingly rare and an efficient protocol for bypass of crisis did not exist. The discovery that autophagy was essential for the elimination of cells in crisis allowed for the first time to rapidly and efficiently establish cells that bypassed crisis, simply by inhibiting autophagy through suppression of the essential autophagy factors ATG3, 5 or 7 (Nassour et al. 2019). Spectral karyotyping on metaphase chromosomes of cells that bypassed crisis upon autophagy suppression revealed gross chromosomal aberrations, but autophagy suppression alone did not elevate levels of chromosomal aberration. Only when cells bypassed crisis did they accumulate high levels of genome instability and non-reciprocal translocations, deletions and aneuploidy (pseudotetraploidy) were frequent (Nassour et al. 2019), demonstrating that elimination of cells in crisis by autophagy is essential to prevent genome instability (Nassour et al. 2019). A detailed analysis of the impact of telomere-driven crisis on genome instability revealed a role for replication-associated repair and template switching, potentially leading to chromothripsis (Cleal et al. 2019). Chromothripsis was also the result of nuclear envelope rupture driven by chromatin bridges originating from dicentric chromosomes, fully consistent with the requirement of critically short telomeres that fuse during replicative crisis (Maciejowski and de Lange 2017; Maciejowski et al. 2015).
Taken together, these data suggested that cell death in crisis was associated with autophagy, not with apoptosis. It appears that telomere dysfunction in general could be a trigger for autophagy and that telomere dysfunction during replicative crisis induces autophagic cell death. Inhibition of autophagy enables crisis bypass and the establishment of transformed cells with an unstable genome, all together leading to the hypothesis that autophagy activation is an integral component of the final barrier against tumor formation, where cells have to overcome on their pathway to neoplasia.
Conclusions
While senescence and the many pathways leading to this terminal arrest are well understood, replicative crisis is not. Crisis, however, represents a critical barrier against cancer formation by efficiently eliminating cells that have lost the p53 and Rb pathways. The discovery that autophagy activation is required for crisis-associated cell death now allows deeper investigation of the phenomenon, while raising many additional questions: we have only scratched the surface of the pathway between fused telomeres, the cytoplasmic DNA damage response, autophagy activation and cell death. While we know that autophagy is required, we still do not know the exact mode of death. To this point, it is unclear why autophagy appears to be a tumor-suppressive mechanism at the early stages of cancer development, yet switches over to an essential growth-promoting pathway in later stages of cancerogenesis. Furthermore, the role of crisis as tumor suppressor has so far not been investigated in vivo, but the discovery that autophagy allows crisis bypass now allows the generation of complex mouse models with autophagy and telomere dysfunction. Finally, we can now tackle the question whether cells that emerge from crisis truly are cancer cells, or whether there is a proliferative barrier beyond crisis, thereby fundamentally changing the way we look at the early steps of cancer development.
References
Acosta, J. C., O’Loghlen, A., Banito, A., Guijarro, M. V., Augert, A., Raguz, S., et al. (2008). Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell,133(6), 1006–1018.
Alcorta, D. A., Xiong, Y., Phelps, D., Hannon, G., Beach, D., & Barrett, J. C. (1996). Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proceedings of the National Academy of Sciences of the United States of America,93(24), 13742–13747. https://doi.org/10.1073/pnas.93.24.13742.
Baker, D. J., Wijshake, T., Tchkonia, T., LeBrasseur, N. K., Childs, B. G., van de Sluis, B., et al. (2011). Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature,479(7372), 232–236.
Bakkenist, C., & Kastan, M. (2003). DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature,421(6922), 499–506.
Bampton, E. T., Goemans, C. G., Niranjan, D., Mizushima, N., & Tolkovsky, A. M. (2005). The dynamics of autophagy visualized in live cells: from autophagosome formation to fusion with endo/lysosomes. Autophagy,1(1), 23–36.
Bartkova, J., Rezaei, N., Liontos, M., Karakaidos, P., Kletsas, D., Issaeva, N., et al. (2006). Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature,444(7119), 633–637. https://doi.org/10.1038/nature05268.
Beausejour, C. M., Krtolica, A., Galimi, F., Narita, M., Lowe, S. W., Yaswen, P., et al. (2003). Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO Journal,22(16), 4212–4222.
Bond, J. A., Haughton, M. F., Rowson, J. M., Smith, P. J., Gire, V., Wynford-Thomas, D., et al. (1999). Control of replicative life span in human cells: barriers to clonal expansion intermediate between M1 senescence and M2 crisis. Molecular and Cellular Biology,19(4), 3103–3114.
Campisi, J., & d’Adda di Fagagna, F. (2007). Cellular senescence: when bad things happen to good cells. Nature Reviews Molecular Cell Biology,8(9), 729–740.
Cesare, A. J., Hayashi, M. T., Crabbe, L., & Karlseder, J. (2013). The telomere deprotection response is functionally distinct from the genomic DNA damage response. Molecular Cell,51(2), 141–155.
Cesare, A. J., & Karlseder, J. (2012). A three-state model of telomere control over human proliferative boundaries. Current Opinion in Cell Biology,24(6), 731–738.
Cleal, K., Jones, R. E., Grimstead, J. W., Hendrickson, E. A., & Baird, D. M. (2019). Chromothripsis during telomere crisis is independent of NHEJ, and consistent with a replicative origin. Genome Research,29(5), 737–749.
Comb, W. C., Cogswell, P., Sitcheran, R., & Baldwin, A. S. (2011). IKK-dependent, NF-kappaB-independent control of autophagic gene expression. Oncogene,30(14), 1727–1732.
Coppe, J. P., Kauser, K., Campisi, J., & Beausejour, C. M. (2006). Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. Journal of Biological Chemistry,281(40), 29568–29574.
Coppe, J. P., Patil, C. K., Rodier, F., Sun, Y., Munoz, D. P., Goldstein, J., et al. (2008). Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biology,6(12), 2853–2868.
Coppe, J. P., Desprez, P. Y., Krtolica, A., & Campisi, J. (2010a). The senescence-associated secretory phenotype: the dark side of tumor suppression. Annual Review of Pathology: Mechanisms of Disease,5, 99–118. https://doi.org/10.1146/annurev-pathol-121808-102144.
Coppe, J. P., Patil, C. K., Rodier, F., Krtolica, A., Beausejour, C. M., Parrinello, S., et al. (2010b). A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PLoS ONE,5(2), e9188.
d’Adda di Fagagna, F., Reaper, P. M., Clay-Farrace, L., Fiegler, H., Carr, P., Von Zglinicki, T., et al. (2003). A DNA damage checkpoint response in telomere-initiated senescence. Nature,426(6963), 194–198.
de Lange, T. (1998). Telomeres and senescence: ending the debate. Science,279(5349), 334–335.
Di Leonardo, A., Linke, S. P., Clarkin, K., & Wahl, G. M. (1994). DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes & Development,8(21), 2540–2551.
Di Micco, R., Fumagalli, M., Cicalese, A., Piccinin, S., Gasparini, P., Luise, C., et al. (2006). Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature,444(7119), 638–642.
Dimri, G. P., Lee, X., Basile, G., Acosta, M., Scott, G., Roskelley, C., et al. (1995). A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proceedings of the National Academy of Sciences of the United States of America,92(20), 9363–9367.
Dimri, G. P., Itahana, K., Acosta, M., & Campisi, J. (2000). Regulation of a senescence checkpoint response by the E2F1 transcription factor and p14(ARF) tumor suppressor. Molecular and Cellular Biology,20(1), 273–285.
Griffith, J. D., Comeau, L., Rosenfield, S., Stansel, R. M., Bianchi, A., Moss, H., et al. (1999). Mammalian telomeres end in a large duplex loop. Cell,97(4), 503–514.
Halvorsen, T. L., Leibowitz, G., & Levine, F. (1999). Telomerase activity is sufficient to allow transformed cells to escape from crisis. Molecular and Cellular Biology,19(3), 1864–1870.
Hara, E., Smith, R., Parry, D., Tahara, H., Stone, S., & Peters, G. (1996). Regulation of p16CDKN2 expression and its implications for cell immortalization and senescence. Molecular and Cellular Biology,16(3), 859–867.
Hayashi, M. T., Cesare, A. J., Fitzpatrick, J. A., Lazzerini-Denchi, E., & Karlseder, J. (2012). A telomere-dependent DNA damage checkpoint induced by prolonged mitotic arrest. Nature Structural & Molecular Biology,19(4), 387–394.
Hayashi, M. T., Cesare, A. J., Rivera, T., & Karlseder, J. (2015). Cell death during crisis is mediated by mitotic telomere deprotection. Nature,522(7557), 492–496.
Hayashi, M. T., & Karlseder, J. (2013). DNA damage associated with mitosis and cytokinesis failure. Oncogene,32(39), 4593–4601.
Hayflick, L. (1965). The Limited in Vitro Lifetime of Human Diploid Cell Strains. Experimental Cell Research,37, 614–636.
Hayflick, L., & Moorhead, P. S. (1961). The serial cultivation of human diploid cell strains. Experimental Cell Research,25, 585–621.
Herbig, U., Jobling, W. A., Chen, B. P., Chen, D. J., & Sedivy, J. M. (2004). Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Molecular Cell,14(4), 501–513.
Ishikawa, H., Ma, Z., & Barber, G. N. (2009). STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature,461(7265), 788–792.
Iwasaki, O., Tanizawa, H., Kim, K. D., Kossenkov, A., Nacarelli, T., Tashiro, S., et al. (2019). Involvement of condensin in cellular senescence through gene regulation and compartmental reorganization. Nature Communications,10(1), 5688.
Karlseder, J., Broccoli, D., Dai, Y., Hardy, S., & de Lange, T. (1999). p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science,283(5406), 1321–1325.
Kaul, Z., Cesare, A. J., Huschtscha, L. I., Neumann, A. A., & Reddel, R. R. (2012). Five dysfunctional telomeres predict onset of senescence in human cells. EMBO Reports,13(1), 52–59.
Kim, S. H., Beausejour, C., Davalos, A. R., Kaminker, P., Heo, S. J., & Campisi, J. (2004). TIN2 mediates functions of TRF2 at human telomeres. Journal of Biological Chemistry,279(42), 43799–43804.
Kuilman, T., Michaloglou, C., Vredeveld, L. C., Douma, S., van Doorn, R., Desmet, C. J., et al. (2008). Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell,133(6), 1019–1031.
Maciejowski, J., & de Lange, T. (2017). Telomeres in cancer: tumour suppression and genome instability. Nature Reviews Molecular Cell Biology,18(3), 175–186.
Maciejowski, J., Li, Y., Bosco, N., Campbell, P. J., & de Lange, T. (2015). Chromothripsis and kataegis induced by telomere crisis. Cell,163(7), 1641–1654.
Mallette, F. A., Gaumont-Leclerc, M. F., & Ferbeyre, G. (2007). The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes & Development,21(1), 43–48.
Munro, J., Barr, N. I., Ireland, H., Morrison, V., & Parkinson, E. K. (2004). Histone deacetylase inhibitors induce a senescence-like state in human cells by a p16-dependent mechanism that is independent of a mitotic clock. Experimental Cell Research,295(2), 525–538.
Nakamura, A. J., Chiang, Y. J., Hathcock, K. S., Horikawa, I., Sedelnikova, O. A., Hodes, R. J., et al. (2008). Both telomeric and non-telomeric DNA damage are determinants of mammalian cellular senescence. Epigenetics Chromatin,1(1), 6.
Narita, M., Krizhanovsky, V., Nunez, S., Chicas, A., Hearn, S. A., Myers, M. P., et al. (2006). A novel role for high-mobility group a proteins in cellular senescence and heterochromatin formation. Cell,126(3), 503–514.
Narita, M., Nunez, S., Heard, E., Narita, M., Lin, A. W., Hearn, S. A., et al. (2003). Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell,113(6), 703–716.
Nassour, J., Radford, R., Correia, A., Fuste, J. M., Schoell, B., Jauch, A., et al. (2019). Autophagic cell death restricts chromosomal instability during replicative crisis. Nature,565(7741), 659–663.
Ramirez, R. D., Morales, C. P., Herbert, B. S., Rohde, J. M., Passons, C., Shay, J. W., et al. (2001). Putative telomere-independent mechanisms of replicative aging reflect inadequate growth conditions. Genes & Development,15(4), 398–403.
Serrano, M., Lin, A. W., McCurrach, M. E., Beach, D., & Lowe, S. W. (1997). Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell,88(5), 593–602.
Shay, J. W., Pereira-Smith, O. M., & Wright, W. E. (1991). A role for both RB and p53 in the regulation of human cellular senescence. Experimental Cell Research,196(1), 33–39.
Shay, J. W., Van Der Haegen, B. A., Ying, Y., & Wright, W. E. (1993). The frequency of immortalization of human fibroblasts and mammary epithelial cells transfected with SV40 large T-antigen. Experimental Cell Research,209(1), 45–52.
Shay, J. W., & Wright, W. E. (1989). Quantitation of the frequency of immortalization of normal human diploid fibroblasts by SV40 large T-antigen. Experimental Cell Research,184(1), 109–118.
Smogorzewska, A., & de Lange, T. (2002). Different telomere damage signaling pathways in human and mouse cells. EMBO Journal,21(16), 4338–4348.
Sun, L., Wu, J., Du, F., Chen, X., & Chen, Z. J. (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science,339(6121), 786–791.
Takai, H., Smogorzewska, A., & de Lange, T. (2003). DNA damage foci at dysfunctional telomeres. Current Biology,13(17), 1549–1556.
van Steensel, B., Smogorzewska, A., & de Lange, T. (1998). TRF2 protects human telomeres from end-to-end fusions. Cell,92, 401–413.
Wei, W., & Sedivy, J. M. (1999). Differentiation between senescence (M1) and crisis (M2) in human fibroblast cultures. Experimental Cell Research,253(2), 519–522.
Yonekawa, T., & Thorburn, A. (2013). Autophagy and cell death. Essays in Biochemistry,55, 105–117.
Zhu, J., Woods, D., McMahon, M., & Bishop, J. M. (1998). Senescence of human fibroblasts induced by oncogenic Raf. Genes & Development,12(19), 2997–3007.
Author information
Affiliations
Molecular and Cell Biology Laboratory, The Salk Institute for Biological Studies, 10010 N. Torrey Pines Rd., La Jolla, CA, 92037, USA
Jan Karlseder
Corresponding author
Correspondence to Jan Karlseder.
Rights and permissions
About this article
Cite this article
Karlseder, J. Suppression of genomic instability by replicative senescence and crisis. GENOME INSTAB. DIS. 1, 143–150 (2020). https://doi.org/10.1007/s42764-020-00013-y
Received28 March 2020
Revised14 April 2020
Accepted18 April 2020
Published08 May 2020
Issue DateMay 2020
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
Replicative senescence
Replicative crisis
Telomeres
Genome instability
用户登录
还没有账号?
立即注册