






The MRE11 nuclease promotes homologous recombination not only in DNA double-strand break resection but also in post-resection in human TK6 cells
Original Research Paper Open Access
Naoto Shimizu, Remi Akagawa, Shunichi Takeda & Hiroyuki Sasanuma
Genome Instability & Disease 1,184–196(2020)
Abstract
Homologous recombination (HR) repairs double-strand breaks (DSBs) occurring in sister chromatids using the intact sisters as the repair template. HR is initiated by DSB resection, which generates 3′ single-strand DNA (ssDNA). RAD51 recombinase polymerizes on the ssDNA and undergoes strand exchange with intact sister chromatids, generating junction molecules (JMs). The separation of JMs completes HR-dependent DSB repair. Defective resolution of JMs not only leaves DSBs unrepaired but also has the broken sisters remain entangled with the intact sisters, leading to the formation of isochromatid-type breaks, where both sister chromatids are broken at the same sites, in mitotic chromosome spreads. The MRE11 nuclease plays a key role in HR, and it is generally believed that MRE11 does so by initiating DSB resection. We here showed that the loss of MRE11 reduced the efficiency of HR in human TK6 cells without affecting DSB resection, indicating a role for MRE11 in HR also at a post-resection step. MRE11-deficient TK6 cells showed proficient induction of RAD51 foci by ionizing-radiation (IR) and olaparib but significantly delayed their resolution. Although exposure of G2-phase cells to IR cleaves only one of two sister chromatids, the loss of the MRE11-nuclease activity increased the number of isochromosome-type breaks in subsequent M phase. The overexpression of GEN1 resolvase suppressed the formation of IR-induced isochromatid-type breaks in MRE11-nuclease-deficient TK6 cells. These data indicate that MRE11 plays an important role in HR by processing JMs. We propose the dual roles of MRE11 in HR at DSB resection and post-resection steps.
Introduction
DNA double-strand breaks (DSBs) are highly cytotoxic lesions leading to mutations, chromosomal aberrations, and cell death (Symington and Gautier 2011). DSBs are repaired by two main mechanisms: non-homologous end joining (NHEJ) and homologous recombination (HR) (O’Driscoll and Jeggo 2006). While NHEJ is active throughout the entire cell cycle, HR is generally restricted to the S and G2 phases and uses intact sister chromatid sequences as the repair template (Mao et al. 2008; Moynahan and Jasin 2010). While NHEJ-deficient cells are able to proliferate normally, the loss of HR immediately leads to cell death showing very severe genome instability due to an essential role of HR in the repair of lethal DSBs spontaneously arising during unperturbed DNA replication (Sonoda et al. 1998; Mehta and Haber 2014). Olaparib, an inhibitor of poly[ADP ribose]polymerase, suppresses the repair of spontaneously-arising single-strand breaks (SSBs) and increases the number of DSBs during DNA replication (Murai et al. 2012). HR, but not NHEJ, repairs these DSBs, and HR-deficient cells are thus hypersensitive to olaparib (Bryant et al. 2005; Farmer et al. 2005).
HR-mediated DSB repair is carried out by step-wise reactions. In the initial step, the DSB ends are degraded to generate 3′ single-strand DNA (ssDNA) tails, a process called as 5′–3′ DSB resection (Shibata et al. 2014; Chang et al. 2017). DSB resection is done by the DNA2, EXO1, and MRE11 nucleases (Zhu et al. 2008; Mimitou and Symington 2008). The resulting 3′-ssDNA tails are coated with an ssDNA binding protein, replication protein A (RPA), which is subsequently replaced by polymerized RAD51 recombinase (reviewed in Haber (2016)). Polymerized RAD51 then catalyzes homology search and strand invasion of the 3′ ssDNA into homologous intact sister chromatid sequences to form HR intermediates called joint molecules (JMs) with the help of the several auxiliary factors, such as RAD51AP1 and RAD54 (Mazin et al. 2010; Sugawara et al. 2003; Petukhova et al. 1998). JMs include displacement loops (D-loop), single Holliday junctions (sHJs), and double Holliday junctions (dHJs) (Haber 2016). dHJs are separated by dissolution and resolution pathways (Wyatt and West 2014; Bizard and Hickson 2014). Dissolution is carried out by the BTR complex, composed of BLM, topoisomerase III, and RMI1/2, leading to the decatenation of dHJs and the generation of non-crossover products (Wu and Hickson 2003; Yin et al. 2005; Wu et al. 2006). Resolution is performed by structure-specific endonucleases such as GEN1 and MUS81, producing both crossover and non-crossover products (Wyatt and West 2014; Gaillard et al. 2003; Wyatt et al. 2013; Castor et al. 2013). Ionizing-irradiation (IR) treatment at G2 phase followed by the morphological analysis of chromosomes in subsequent M phase makes it possible to distinguish a defect in the formation of JMs from the defective resolution of JMs. γ-irradiation at G2 phase induced DSBs in one of two sister chromatids. In the absence of JM formation, unrepaired DSBs are detectable in the subsequent M phase as chromatid-type breaks, where only one of two sisters is broken, in mitotic chromosome spreads. The defective resolution can cause isochromatid-type breaks, where two sisters are broken at the same sites, through the mechanism that unresolved JMs may interfere with normal chromosome condensation at the sites of the JMs in the G2/M phases (Wechsler et al. 2011; Kikuchi et al. 2013).
MRE11 forms a complex with RAD50 and NBS1 (MRN complex) and plays a major role in DSB repair by HR (Stracker and Petrini 2011). The loss of MRE11 is lethal to cycling vertebrate cells due to severe genome instability, as does the loss of RAD51 (Sonoda et al. 1998; Buis et al. 2008). Genetic studies of Saccharomyces cerevisiae (S. cerevisiae) have established that MRE11 contributes to HR-dependent DSB repair at the initial step of HR, DSB resection, at which step a few hundred nucleotides long 3′ ssDNA tail is formed by a combined action of MRE11 and SAE2, a yeast orthologue of human CTIP endonuclease (Zhu et al. 2008; Mimitou and Symington 2008). It has been believed that this key role of yeast MRE11 in DSB resection is conserved in mammalian cells. Conditionally generated MRE11-null-deficient mice and MRE11-nuclease-deficient mice exhibit a very similar phenotype including a ~ 50% decrease in DSB resection, severe genome instability, hypersensitivity to IR, and cellular mortality, which similarity has been interpreted as the evidence that MRE11 contributes to HR as a nuclease involved in DSB resection (Buis et al. 2008). However, it remains elusive whether only a ~ 50% decrease in DSB resection fully explains the very severe genomic instability of MRE11-deficient TK6 cells. We previously showed that although CTIP depletion by 80% in the human TK6 B lymphoid cell line leads to a ~ 70% decrease in the rate of DSB resection, the HR capacity remains unaffected even in the heteroallelic HR assay, a stringent test for evaluating the capability of HR (Hoa et al. 2015a). This result led us to hypothesize an additional role played by MRE11 in HR at post-resection steps.
In this study, we showed the genetic evidence for the role of MRE11 in the resolution of JMs by examining the MRE11-null (MRE11−/−) mutant cells and nuclease-deficient (MRE11−/H129N) cells derived from the human TK6 B cell line (Hoa et al. 2015a). The conditional loss of MRE11 and inactivation of the MRE11-nuclease activity significantly reduced the efficiency of HR without affecting DSB resection. Thus, these TK6 mutants provided the unique opportunity of addressing the potential role of MRE11 in HR at post-resection steps. These MRE11-deficient TK6 cells exhibited an elevated number of isochromatid-type breaks following γ-irradiation at the G2 phase, and the overexpression of GEN1, a resolvase of dHJs, reduced the number of isochromatid-type breaks. The G2 irradiation induced radial chromosomes in MRE11−/− TK6 cells, but not MRE11−/H129N TK6 cells. These data indicate that MRE11 have catalytic and structural roles, and the latter may maintain JMs for proper resolution. We propose that MRE11 plays a role in HR not only at DSB resection but also at the subsequent step, the processing of JMs in mammalian cells.
Results
Normal induction of RAD51 foci in response to IR in MRE11-deficient TK6 cells
To investigate the impact of MRE11 loss on HR, we utilized human MRE11−/− and MRE11−/H129N TK6 cells (Hoa et al. 2015a). This cell line has been widely used by the governments of developed countries to control hazardous chemicals due to its phenotypic stability and nearly normal karyotype (Fellows and O’Donovan 2010; Lorge 2010; Ibrahim et al. 2020). Table S1 shows the list of the mutant cells analyzed in this study. The addition of 4-hydroxytamoxifen (4-OHT) activates the Cre recombinase and converts the genotype from MRE11loxP/loxP and MRE11loxP/H129N to MRE11−/− and MRE11−/H129N, respectively (Hoa et al. 2015a). We analyzed these cells four days after the addition of 4-OHT, when MRE11-depleted TK6 cells are able to normally proliferate without displaying genome instability. The length of the cell cycle is 13 h, and the amount of MRE11 is decreased by more than 99% at day four after the exposure to 4-OHT (Hoa et al. 2015a).
To monitor the progression of HR-mediated DSB repair, we exposed an asynchronous population of cells to IR, and measured with time the number of RAD51 foci, which represent polymerized RAD51 at resected DSBs, in cyclinA-positive S/G2-phase cells. We did not analyze RPA foci since they do not accurately reflect the extent of DSB resection due to the rapid replacement of RPA by RAD51 on resected DNA (Haber 2016). The number of RAD51 foci peaked (8.7 ± 0.6 per cell) at two hours post-IR, gradually decreased afterwards, and returned to a background level by eight hours (Figs. 1a and S1a). As expected, wild-type, RAD54−/−, and BLM−/− TK6 cells all showed very similar kinetics of RAD51-focus induction at one hour post-IR (Figs. 1b, 1c (1 h) and S1f). Remarkably, MRE11−/− and MRE11−/H129N TK6 cells also exhibited very similar induction of RAD51 foci as did wild-type cells. We previously confirmed proficient DSB resection of these cells by examining bromodeoxyuridine (BrdU), a nucleoside analog, incorporated into 3′-ssDNA of resected DSB ends (Hoa et al. 2015a). These results indicate that other nucleases such as DNA2 may fully compensate for the loss of MRE11 in the DSB resection of TK6 cells (Hoa et al. 2015b) (reviewed in Paull (2018)). Importantly, these data showed that the MRE11−/− and MRE11−/H129N TK6 cells provide a unique opportunity of identifying a potential role played by MRE11 in HR at post-resection steps.
Fig. 1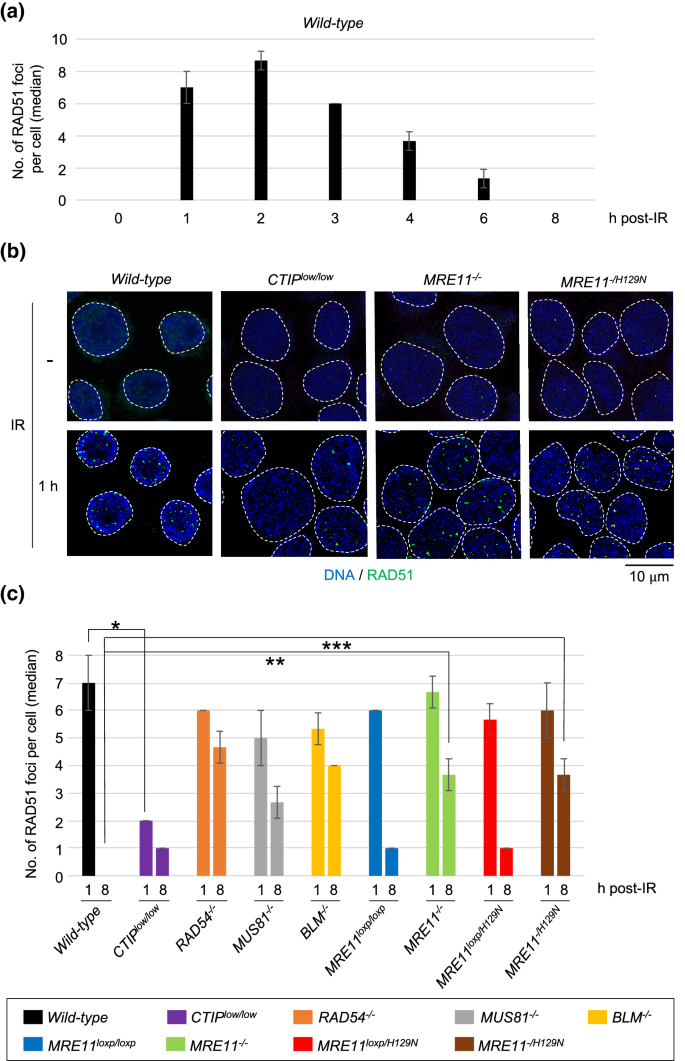
Delayed DSB repair in MRE11-null-deficient and MRE11-nuclease-deficient TK6 cells. a Kinetics of RAD51-focus formation in cyclinA-positive wild-type TK6 cells at the indicated times after 0.5 Gy irradiation. The average of median values with standard deviation (SD) were shown from three independent experiments. More than 100 cells per experiment were counted for each experiment. b Representative images of RAD51 foci in the indicated genotypes without IR exposure and at one hour after IR exposure. Green specks indicate RAD51 signal. The nuclei are outlined. c The median values of RAD51 foci in cyclinA-positive cells for the indicated genotypes at one and eight hours after IR exposure. MRE11−/− and MRE11−/H129N TK6 cells were generated from MRE11loxP/loxP and MRE11loxP/H129N TK6 cells by incubating them with tamoxifen (4-OHT) for 4 days to activate Cre recombinase. Single, double, and triple asterisks indicate p = 2.1 × 10–4, p = 1.7 × 10–4 and p = 1.0 × 10–3, respectively, calculated by a two-tailed unpaired Student’s t test. Error bars indicate the SD calculated from three independent experiments. More than 100 cells per experiment were counted for each experiment
Full size imageA delay in the resolution of RAD51 foci in MRE11-deficient TK6 cells
We previously created CTIPlow/low TK6 cells, where wild-type CTIP cDNA was inserted into the first exon of the endogenous CTIP alleles (Fig. S1b) and their expression levels were only 20% of that in wild-type cells (Fig. S1c). The CTIPlow/low TK6 cells showed a ~ 70% decrease in DSB resection at one hour post-IR (Figs. 1b, c and S1f) (Hoa et al. 2015a). However, CTIPlow/low TK6 cells retain proficient HR capability including the heteroallelic HR (Hoa et al. 2015a). In agreement with this conclusion, wild-type and CTIPlow/low TK6 cells completed HR-dependent DSB repair with the same kinetics as evidenced by the data that wild-type and CTIPlow/low TK6 cells showed a decrease in the number of RAD51 foci to a background level at eight hours post-IR (Figs. 1c and S1f). These data indicate that DSB resection takes place at an excessive level in TK6 cells, and a few times decrease in the length of DSB resection does not affect the efficiency of HR. Thus, even if the MRE11−/− and MRE11−/H129N TK6 cells have a modest defect in DSB resection, this defect could not affect the overall efficiency of HR in these mutants.
BLM−/−, MUS81−/− (Fig. S1d and S1e), and RAD54−/− TK6 cells displayed persistent RAD51 foci at eight hours, which agree with the role of these proteins in HR after the polymerization of RAD51 at resected DSBs (Wyatt and West 2014) (Figs. 1c and S1f). Surprisingly, both the loss of MRE11 and the inactivation of MRE11-nuclease activity caused a significant delay in the resolution of RAD51 foci. In wild-type TK6 cells, virtually 100% of the RAD51 foci that were observed at one hour post-IR were resolved at eight hours, whereas in MRE11−/− and MRE11−/H129N TK6 cells, only 45 and 39% of the foci, respectively, seen at one hour were resolved at eight hours (Fig. 1c). These results indicate that MRE11 may play an important role in HR at steps after DSB resection as do BLM, MUS81, and RAD54.
The important role of MRE11 in HR-mediated repair of olaparib-induced DSBs after DSB resection in TK6 cells
Olaparib, a poly [ADP ribose] polymerase poison, generates the DSBs that occur during DNA replication. These DSBs are repaired exclusively by HR (Murai et al. 2012). We investigated whether or not MRE11 promotes HR-mediated repair of olaparib-induced DSBs in TK6 cells. We pulse-exposed wild-type, RAD54−/−/RAD51AP1−/− (Fig. S2a and S2b), BLM−/−, and MRE11−/− TK6 cells to olaparib for one hour and monitored RAD51 foci present in S/G2-phase (cyclin A-positive) cells immediately and at three hours after the removal of olaparib. The loss of RAD51AP1 in RAD54−/− TK6 cells further increased their sensitivity to olaparib but not to IR (Fig. S2c and S2d). The number of induced RAD51 foci was very similar in these four genotypes immediately after the pulse-exposure to olaparib (Fig. 2a and b), indicating that MRE11 is dispensable for DSB resection at DSBs induced by olaparib. ~ 45% of the DSBs induced at one hour was repaired at three hours of repair time in wild-type TK6 cells (Figs. 2b and S2e). In contrast, RAD54−/−/RAD51AP1−/− and BLM−/− TK6 cells displayed no decrease in the number of RAD51 foci, indicating a significant delay in HR-mediated repair in these cells with olaparib still generating DSBs even after its removal from culture media. The number of olaparib-induced RAD51 foci persisted in MRE11−/− TK6 cells (Figs. 2a, b and S2e) as seen in the repair of IR-induced DSBs (Fig. 1c). These results showed that MRE11 significantly contributes to HR-dependent DSB repair in TK6 cells. Proficient DSB resection and delayed resolution of RAD51 foci not only IR-induced ones but also at at olaparib-induced DSBs imply that MRE11 promotes HR after RAD51 polymerization at resected DSBs.
Fig. 2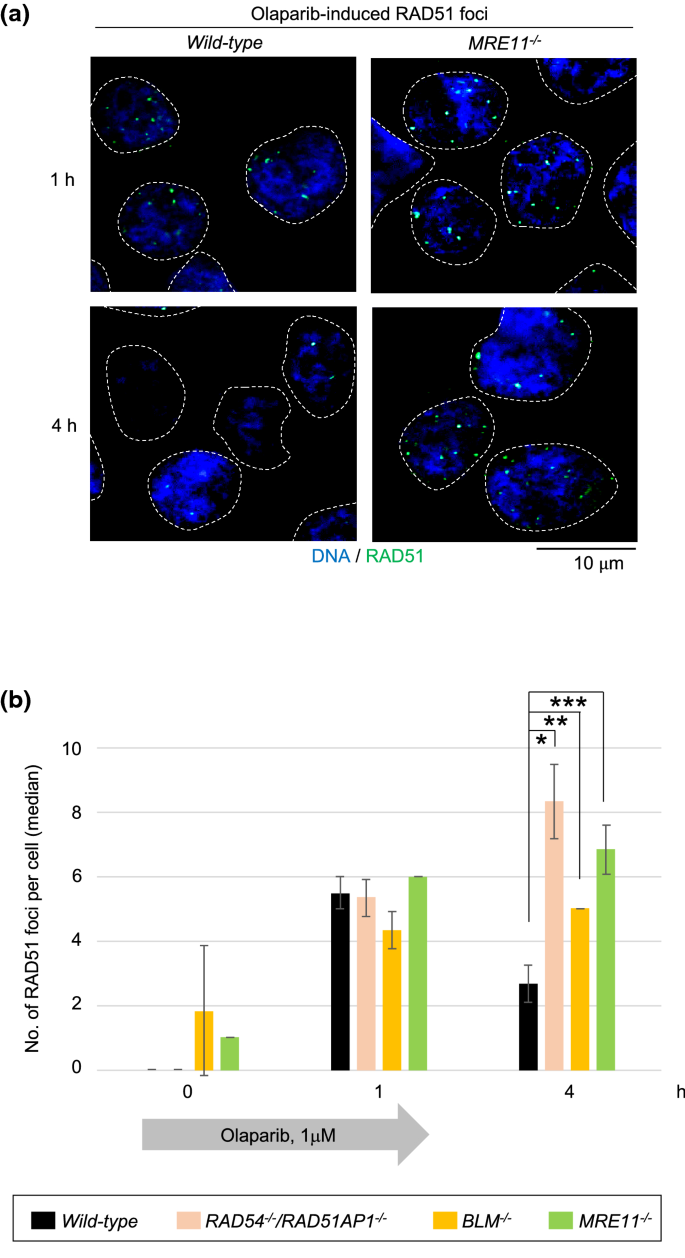
Involvement of MRE11 in the HR repair of olaparib-induced DSBs. a Representative images of RAD51 foci are shown at one and four hours after 1-h pulse exposure to the olaparib (1 μM). TK6 cells were incubated in olaparib-free medium for three hours after olaparib pulse-exposure. Green specks indicate RAD51 signal. The nuclei are outlined. b Kinetics of RAD51-focus formation induced by olaparib in the indicated genotypes. The average of median values of RAD51 foci per cyclinA-positive cells were shown for the indicated genotypes after the olaparib pulse-exposure. Single, double and triple asterisks indicate p = 1.6 × 10–3, p = 2.2 × 10–3, and p = 1.7 × 10–3, respectively, calculated by a two-tailed unpaired Student’s t test. Error bars indicate the SD calculated from three independent experiments. More than 100 cells per experiment were counted for each experiment
Full size imageSpontaneously-arising chromosome aberrations in MRE11−/− and MRE11−/H129N TK6 cells
We re-evaluated spontaneously-arising chromosome aberrations following the conditional inactivation of wild-type MRE11 gene in MRE11loxP/loxP and MRE11loxP/H129N TK6 cells. As previously reported, the conditional inactivation stopped the proliferation of these cells and significantly increased the number of spontaneously-arising chromosome aberrations at day six (Hoa et al. 2016) (Fig. 3a). 31% and 17% of the total chromosome aberrations were isochromatid-type breaks in MRE11−/− and MRE11−/H129N TK6 cells, respectively, at day six (Fig. 3a). This observation is surprising, because HR is carried out between broken sister chromatids and intact ones, and no initiation of HR is supposed to generate chromatid-type breaks, where only one of two sister chromatids is broken, in mitotic chromosome spreads. The presence of isochromatid-type breaks, therefore, suggests that defects in MRE11 may interfere with HR after the formation of JMs (Wechsler et al. 2011; Kikuchi et al. 2013).
Fig. 3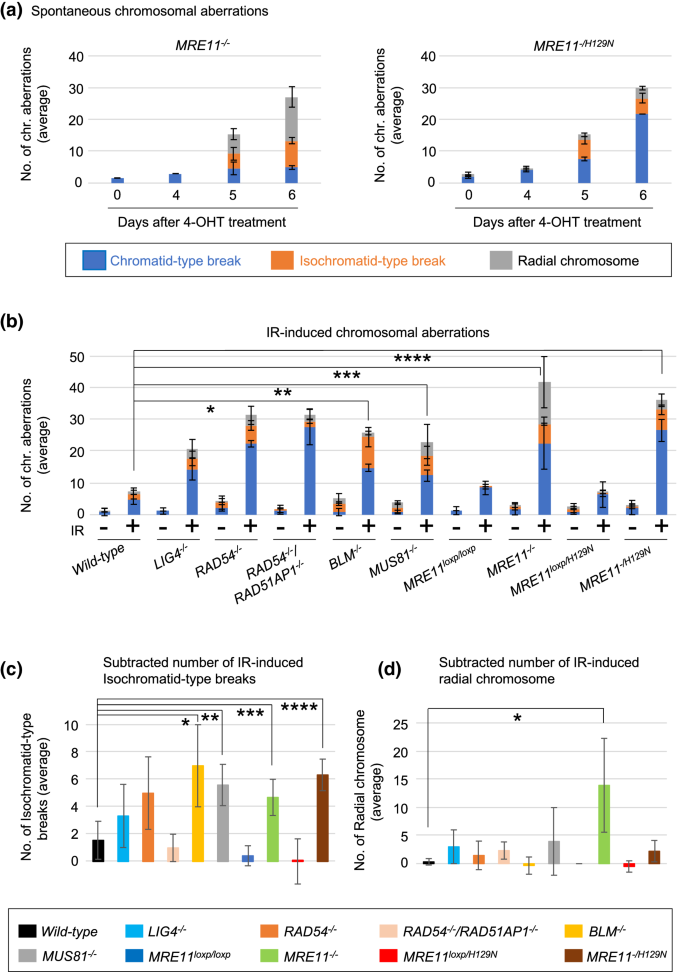
Loss of MRE11 increases the number of IR-induced isochromatid-type breaks and radial chromosomes. a The number of spontaneously-arising chromosomal aberrations in MRE11−/− and MRE11−/H129N TK6 cells. 4-OHT was added to the culture medium (day 0) and TK6 cells were harvested at the indicated time points following 3-h treatment of colcemid. The average number of aberrant chromosomes was calculated from three independent experiments. 100 metaphase cells were analyzed for each experiment. b The number of chromosomal aberrations induced by IR in the indicated genotypes. TK6 cells were exposed to 0.5 Gy irradiation and then treated with colcemid (0.1 μg/ml) for three hours. The average number of aberrant chromosomes was calculated from three independent experiments. Single, double, triple, and quadruple asterisks indicate p = 4.2 × 10–4, p = 1.5 × 10–2, p = 9.6 × 10–4, and p = 1.8 × 10–3, respectively, calculated by a two-tailed unpaired Student's t test. Error bars indicate the SD calculated from three independent experiments. 100 metaphase cells were analyzed for each experiment. c The numbers of IR-induced isochromatid-type breaks after subtracting these numbers by the background numbers of isochromatid-type breaks in non-irradiated TK6 cells. Single, double, triple, and quadruple asterisks indicate p = 4.6 × 10–2, p = 2.7 × 10–2, p = 4.7 × 10–2, and p = 1.0 × 10–2, respectively, calculated by a two-tailed unpaired Student's t test. d The numbers of IR-induced radial chromosomes after subtracting these numbers by the background numbers of radial chromosomes in non-irradiated TK6 cells. Single asterisk indicates p = 4.8 × 10–2, calculated by a two-tailed unpaired Student's t test
Full size imageInterestingly, the substantial fraction of the spontaneously-arising chromosome aberrations was radial chromosomes in MRE11−/− TK6 cells (Fig. 3a). Radial chromosomes result from a defective resolution of JMs followed by abnormal NHEJ events (Chan et al. 2018; West and Chan 2017; Kottemann and Smogorzewska 2013). Thus, increases in the numbers of both isochromatid-type breaks and radial chromosomes in MRE11−/− and MRE11−/H129N TK6 cells suggest the role of MRE11 in HR at post-resection. The higher number of radial chromosomes in MRE11−/− TK6 cells compared with MRE11-nuclease-deficient TK6 cells supports a non-catalytic, structural role for MRE11 in post-resection HR. Collectively, MRE11 may have catalytic and structural roles in post-resection HR.
MRE11 may maintain JMs for proper resolution
MRE11 plays multiple roles in DNA replication, and we wished to analyze HR-dependent repair of IR-induced DSBs in G2 phase but not in S phase (Kondratova et al. 2015; Bruhn et al. 2014; Lee and Dunphy 2013; Duursma et al. 2013; Shiotani et al. 2013; Schlacher et al. 2011; Doksani et al. 2009; Trenz et al. 2006). To examine the consequence of abnormal HR events in the absence of intact MRE11, we analyzed chromosome aberrations in the TK6 cells that had been γ-irradiated at the G2 phase. We pulse-labeled S-phase TK6 cells with BrdU immediately before γ-irradiation and confirmed that BrdU-labeled chromosomes were detectable in metaphase spreads at six hours (Fig. S3a) but not at three hours (Fig. S3b) after the addition of BrdU. Thus, we are able to evaluate the consequence of abnormal HR events occurring only in G2 phase by examining mitotic cells at three hours post-IR.
Analysis of chromosome aberrations in metaphase spreads allows for distinguishing defects in HR before and after the JM formation. Two sister chromatids are spatially separated in G2 phase as in situ hybridization detects a given locus as two distinct dots (Sonoda et al. 2001). γ-irradiation at the G2 phase generates DSBs at only one of two sister chromatids (Fig. S4). A defect in the formation of JMs keeps DSBs away from the intact sister chromatids, resulting in the formation of chromatid-type breaks (Fig. S4). On the other hand, a defect in HR after the JM formation, such as a defective dissolution/resolution of HJs, may keep both sisters entangled and interfere with chromosome condensation at the site of unresolved JMs, resulting in the formation of isochromatid-type breaks (Wechsler et al. 2011; Kikuchi et al. 2013). These ideas were in agreement with the following data. The loss of both RAD51AP1 and RAD54, which promote the formation of JMs (Mazin et al. 2010; Wiese et al. 2007; Sugawara et al. 2003; Petukhova et al. 1998), increased the fraction of chromatid-type breaks in TK6 cells (Fig. 3b). By contrast, the loss of either MUS81 or BLM, which promote the dissolution and resolution of dHJs, respectively (Wyatt and West 2014; Bizard and Hickson 2014), predominantly increased the fraction of isochromatid-type breaks in TK6 cells (Fig. 3b and c). Collectively, isochromatid-type breaks induced by γ-irradiation at the G2 phase are a specific biomarker for evaluating the resolution of JMs.
The MRE11−/− and MRE11−/H129N mutations in TK6 cells caused 4.8-fold and 7.7-fold increases in the total numbers of IR-induced chromosome aberrations, respectively (Fig. 3b). Importantly, these mutations also caused increases in the number of isochromatid-type breaks as did the loss of the MUS81 and BLM genes (Fig. 3c). The noticeable increase in the number of isochromatid-type breaks suggests that MRE11 plays a role in HR after the formation of JMs. Moreover, the over ten-times increase in the number of radial chromosomes in MRE11−/− TK6 cells, but not in MRE11−/H129N TK6 cells (Fig. 3b and d), suggests that MRE11 may have the structural role in the maintaining of JMs for their proper resolution.
Defective HR of MRE11−/H129N TK6 cells is suppressed by overexpression of GEN1, a HJ resolvase
To further investigate the role played by MRE11 in the processing of JMs, we examined whether the overexpression of the GEN1 HJ resolvase suppressed the defective HR of MRE11−/H129N TK6 cells. Using a lentivirus vector, we introduced a GEN1 transgene carrying the nuclear localization signal (GEN1NLS) (Chan and West 2014) into cells and analyzed the morphology of chromosomes in mitotic spreads. The expression level of GEN1NLS was increased approximately 8-times compared with the endogenous expression of GEN1 in wild-type (Fig. S5). The ectopic GEN1NLS expression suppressed the increased frequencies of IR-induced isochromatid-type breaks in MUS81−/− TK6 cells but not in wild-type TK6 cells, indicating that overexpressed GEN1 can efficiently resolve the JMs that accumulated in the absence of MUS81 (Fig. 4). The GEN1NLS expression suppressed the numbers of chromatid-type and isochromatid-type breaks by 43% and 74%, respectively, in MRE11−/H129N TK6 cells (Fig. 4). These results suggest that the nuclease activity of MRE11 may promote the resolution of JMs.
Fig. 4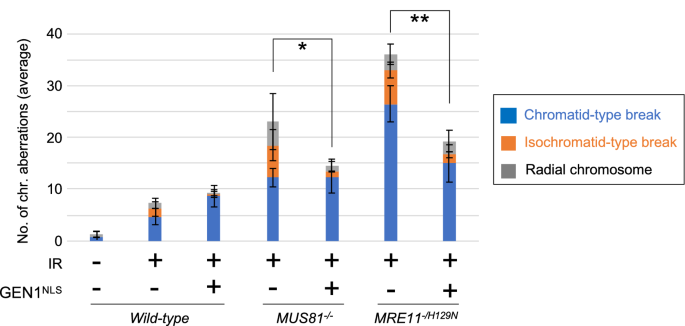
Suppression of HR defects in the MRE11-deficient TK6 cells by overexpression of GEN1, an HJ resolvase. The number of chromosomal aberrations induced by IR in the indicated genotypes. The analysis was done as in Fig. 3b. MRE11−/H129N TK6 cells were generated by 4-OHT as described in Fig. 1c. The average number of aberrant chromosomes was calculated from three independent experiments. Single and double asterisks indicate p = 2.7 × 10–2 and p = 6.1 × 10–3, respectively, determined by a two-tailed unpaired Student's t test. These p values were calculated by comparing the number of isochromatid-type breaks between wild-type and the indicated genotypes. Error bars indicate the SD calculated from three independent experiments. 100 metaphase cells were analyzed for each experiment
Full size imageDiscussion
In this study, we demonstrated that MRE11 plays a critical role in HR at steps after DSB resection. Both MRE11−/− and MRE11−/H129N TK6 cells exhibited a severe defect in HR despite normal DSB resection and thus provided a unique opportunity of investigating a potential role played by MRE11 in HR at steps other than DSB resection. The severe defect in HR in MRE11−/− and MRE11−/H129N TK6 cells was shown by a delay in the resolution of RAD51 foci induced by IR (Fig. 1c) and olaparib (Fig. 2). These data demonstrated the crucial role of MRE11 in HR through a mechanism other than DSB resection in TK6 cells. Like the loss of MUS81, the inactivation of MRE11-nuclease activity caused an increase in the number of isochromatid-type breaks following γ-irradiation at G2 phase (Fig. 3b and c), and this phenotype of MUS81−/− and MRE11−/H129N TK6 cells was reversed by ectopic expression of the GEN1 HJ resolvase (Fig. 4). We, therefore, suggest that MRE11 can significantly contribute to HR by processing JMs for proper resolution (Fig. S4).
An important question is whether MRE11 has a pivotal role in the post-resection step in mammalian cells other than TK6 cells. Genetic studies of S. cerevisiae have shown that the nuclease activity of MRE11 is essential for meiotic HR at DSB resection step (Garcia et al. 2011) (reviewed in Oh and Symington (2018), Paull (2018)). Mammalian MRE11 also plays an important role in HR, and it has been believed that this important role is to resect DSB ends (Buis et al. 2008; Shibata et al. 2014; Paull 2018). However, it remains unclear whether only a 50–70% decrease in DSB resection is solely responsible for a very severe defect in HR in MRE11-deficient TK6 cells (Buis et al. 2008; Zhou et al. 2014). The 50–70% decrease may not explain this very severe defect due to the following experimental evidence. The selective inactivation of nuclease activity in MRE11 reduces DSB resection by a few times and causes only a modest delay in HR-dependent repair of DSBs generated by the HO-restriction-enzyme in S. cerevisiae (Westmoreland and Resnick 2013; Moreau et al. 1999). Likewise, an approximately 70% decrease in DSB resection in CTIPlow/low TK6 cells had no detectable effect on the efficiency of HR (Fig. 1b and c) (Hoa et al. 2015a). These data suggest that in yeast and mammalian cells, DSB resection takes place at an excessive level, and a several-times decrease in DSB resection might not fully account for a very severe defect in HR seen in the MRE11-deficient mammalian cells. We propose that mammalian MRE11 has an additional role in HR at a post-resection step, which operates independent of the promotion of DSB resection by MRE11. Thus, MRE11 significantly contributes to HR at both DSB resection and post-resection in mammalian cells.
Analysis of IR-induced chromosome aberrations in metaphase spreads suggests the catalytic role for MRE11 in HR after the JM formation (Fig. 3b and c). The overexpression of GEN1 HJ resolvase reversed the defective HR of both MUS81−/− and MRE11−/H129N TK6 cells (Fig. 4). No biochemical studies showed the processing of JMs or HJs by MRE11. On the other hand, the processing of HJs by the MRE11-nuclease activity is supported by the previous genetic study, which analyzed the structure of stalled replication forks at a DSB in S. cerevisiae deficient in MRE11 (Doksani et al. 2009). Yeast MRE11 processes cruciform intermediates resulted as a consequence of fork reversal at DSBs. Considering the structural similarity between cruciform intermediates and HJs (Branzei and Szakal 2016; Giannattasio et al. 2014), this genetic study suggests the capability of MRE11 in processing HJs in vivo. In conclusion, the nuclease activity of MRE11 may be involved in the resolution of HJs, similar to GEN1 and MUS81.
In addition to the catalytic role, MRE11 has a structural role, as evidenced by the data that the number of radial chromosomes increased in MRE11−/− TK6 cells but not MRE11−/H129N TK6 cells (Fig. 3a, b, and d). Radial chromosomes result from complex chromosome rearrangements involving abnormal NHEJ (Kottemann and Smogorzewska 2013; Chan et al. 2018; West and Chan 2017). MRE11 complexes with RAD50, a member of the structural maintenance of chromosomes (SMC) family, which includes SMC subunits of cohesin and condensin protein complexes (Paull 2018; Kakui and Uhlmann 2018). Studies of S. cerevisiae have shown that MRE11-RAD50 complex facilitates DNA end-to-end tethering and juxtaposition of broken sister chromatid independently of the MRE11-nuclease activity (De Jager et al. 2001; Wiltzius et al. 2005; Hopfner et al. 2002; Seeber et al. 2016). JMs associate with RPA, and interactions between RPA and MRE11-RAD50 complex might also facilitate the tethering of broken sister chromatids with intact sisters. This tethering may play an important role in HR in mammalian cells, but not in yeast, considering that chromosome condensation pulls two sister chromatids apart to a much greater extent in mammalian cells than in budding yeast due to their several 100-fold larger genome size and a higher degree of chromosome compaction in mitosis (Kakui and Uhlmann 2018). Thus, mammalian cells may require an additional mechanism for counteracting the tensile force and stabilizing JMs by tethering two sister chromatids. One possible scenario is that the MRE11-RAD50 complex might tether sister chromatids and thereby maintain the proper structure of JMs. In the absence of MRE11-RAD50 complex, premature resolution of JMs followed by abnormal ligation of DNA ends by NHEJ might generate radial chromosomes (Fig. S4). We propose that metazoan cells have evolved a novel mechanism for the catalytic and structural roles played by the MRN complex in the maintenance of JMs for their proper resolution. To prove this idea, further studies analyzing mammalian cells other than TK6 cells are needed. Confirming the role of MRE11 in the maintenance of JMs in various malignant tumors may help understand how MRE11 suppresses tumorigenesis.
Materials and methods
Cell culture and reagents
Human TK6 B cells were incubated in RPMI1640 medium (Cat# 3026456, Nacalai Tesque, Japan) supplemented with horse serum (5%) (Gibco, US), penicillin (100 U/ml), streptomycin (100 μg/ml) (Nacalai, Japan), and sodium pyruvate (200 μg/ml) (ThermoFisher, US). 293 T cells were maintained in DMEM supplemented with fetal bovine serum (10%), penicillin (100 U/ml), streptomycin (100 μg/ml) (Nacalai, Japan), sodium pyruvate (200 μg/ml) and L-glutamine (Nacalai, Japan). TK6 and 293 T cells were maintained at 37 °C under a humidified atmosphere and 5% CO2.
Generation of TK6 mutant cells
The TK6 mutant cells used in this study are listed in Table S1. To generate gene-targeting constructs, we amplified left and right arms (~ 1 kb each) from genomic DNA. The amplified arms were assembled with the DT-ApA/MARKERR vector digested with ApaI and the AflII using GeneArt Seamless Cloning Enzyme Mix (ThermoFischer, US). Primer information about the left and right arms (~ 1 kb each) used for this study is described in Table S2. The DT-ApA/MARKERR was provided by the Laboratory for Animal Resources and Genetic Engineering, Center for Developmental Biology, RIKEN Kobe (https://www.clst.riken.jp/arg/cassette.html). The gRNAs were inserted into the BbsI site of pX330 vector (Cat# 42230, Addgene, US). The resulting pX330-gRNA and the targeting vectors containing two different antibiotic markers were transfected with pX330-gRNA into six million TK6 cells as previously described (Akagawa et al. 2020).
To generate RAD54−/−/RAD51AP1−/− TK6 mutant cells, we disrupted RAD51AP1 genes in RAD54−/− TK6 cells (Hoa et al. 2015b). PCR genotyping using the primers shown in Table S2 was performed as a primary screening. The gene-disruption events were confirmed by western blotting, reverse transcription-PCR (RT-PCR), or Southern blotting analysis. Disruption of MUS81 alleles was confirmed by genomic Southern blotting (Fig. S1d and S1e) using a 32P-labeled probe, which was amplified by the following primers, 5′-AGGAGACAGCAGTGCCAGGAGCAGCTT and 5′-GCCCTTCACCTGGGTCTCTAGGATTGGTCT. The genomic DNA was digested with DraI for Southern blotting.
Measurement of cellular sensitivity to DNA damaging agents
Cellular sensitivity of an asynchronous population of TK6 cells to IR and olaparib was measured by clonogenic cell-survival analysis. TK6 cells were grown in the respective medium described above, containing methylcellulose, for 10 days.
Generation of TK6 cells overexpressing GEN1
pMSCV-GEN1-NLS-IRES-GFP retroviral expression vector (a gift from Dr. Stephen C. West) was co-transfected into 293 T cells with a helper plasmid (pClampho) expressing the viral gag, pol and env proteins to produce viral supernatant. The viral supernatant was collected after 48 h and used to infect wild-type, MUS81−/−, and MRE11−/H129N TK6 cells. The efficiency of infection was assessed by quantifying the number of TK6 cells expressing GFP with flow cytometry (LSR Fortessa, BD Biosciences, US). The TK6 cells expressing GFP were enriched with a cell sorter (FACSAria III, BD Biosciences, US) and seeded into 96 well plates to isolate single colonies. The GEN1 overexpression was confirmed by quantitative RT-PCR (Fig. S5).
Chromosome analysis in mitotic chromosome spreads
TK6 cells were exposed to γ-rays (0.5 Gy, 137Cs) and subsequently treated with colcemid (0.1 μg/ml) (Invitrogen, Carlsbad, US) for three hours. TK6 cells were suspended in potassium chloride (75 mM) for 15 min, washed with Carnoy’s solution (a 3:1 mixture of methanol and acetic acid), dropped on slides and stained with a Giemsa solution (3%) for 25 min. For detection of BrdU-incorporated chromosomes (Fig. S3), the TK6 cells were incubated with BrdU (100 μM) for 10 min prior to IR exposure. After removing BrdU from the medium, the TK6 cells were splitted into two samples of equal volume and one of the two samples was induced with colcemid. Another sample was incubated with normal medium for 3 h and further incubated with colcemid. The experimental procedure is depicted in Fig. S3. The chromosomes dropped on the sides were incubated with HCl (2 M) containing TritonX-100 (0.5%) for 30 min to denature the duplex DNA. The denatured DNA molecules were incubated with α-BrdU antibody (1/100 dilution, BD, UK) for 30 min. The BrdU-incorporated mitotic chromosomes were visualized by α-mouse secondary antibody conjugated with Alexa Fluor 488.
Immuno-staining of RAD51 foci in TK6 cells
TK6 cells were exposed to IR (0.5 Gy) and incubated for the indicated times. TK6 cells were subsequently collected with cytospin and fixed with formaldehyde (4%) (Wako, Japan) in PBS, permeabilized with Tween-20 (0.1%) (23926-35, Nacalai Tesque) in PBS and blocked by BSA (5%) (01683-48, Nacalai Tesque) in PBS. We used α-RAD51 antibody (1/1000 dilution, #70-001, Bioacademia, Japan) and α-CyclinA (anti-CyclinA) antibody (1/500 dilution, BD, US). To distinguish cells in G1 phase from other phases. The slides were mounted in Fluoro-KEEPER containing 4′, 6-diamidino-2-phenylindole (DAPI) (12745-74, Nacalai Tesque, Japan). Table S3 includes the resources used in this study.
Focus counting and statistical analysis
The numbers of subnuclear foci in at least 100 cyclinA-positive S/G2-phase cells were counted per experiment. The foci were visualized under the confocal (SP8, Leica Microsystems, Germany) and the fluorescence microscope (BZ-9000, KEYENCE, Japan) microscopes. All data are represented as individual replicates and replicate number and error bars are explained in the figure legends. The statistical test (a two-tailed unpaired Student's t test) and resulting p values are indicated in the figure legends and have been generated using Excel and PRISM8 software (GraphPad, LLC).
References
Akagawa, R., Trinh, H. T., Saha, L. K., Tsuda, M., Hirota, K., Yamada, S., et al. (2020). UBC13-mediated ubiquitin signaling promotes removal of blocking adducts from DNA double-strand breaks. iScience. https://doi.org/10.1016/j.isci.2020.101027.
Bizard, A. H., & Hickson, I. D. (2014). The dissolution of double Holliday junctions. Cold Spring Harbor Perspectives in Biology,6, 7. https://doi.org/10.1101/cshperspect.a016477.
Branzei, D., & Szakal, B. (2016). DNA damage tolerance by recombination: Molecular pathways and DNA structures. DNA Repair,44, 68–75. https://doi.org/10.1016/j.dnarep.2016.05.008.
Bruhn, C., Zhou, Z. W., Ai, H., & Wang, Z. Q. (2014). The essential function of the MRN complex in the resolution of endogenous replication intermediates. Cell Reports,6(1), 182–195. https://doi.org/10.1016/j.celrep.2013.12.018.
Bryant, H. E., Schultz, N., Thomas, H. D., Parker, K. M., Flower, D., Lopez, E., et al. (2005). Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature,434(7035), 913–917. https://doi.org/10.1038/nature03443.
Buis, J., Wu, Y., Deng, Y., Leddon, J., Westfield, G., Eckersdorff, M., et al. (2008). Mre11 nuclease activity has essential roles in DNA repair and genomic stability distinct from ATM activation. Cell,135(1), 85–96. https://doi.org/10.1016/j.cell.2008.08.015.
Castor, D., Nair, N., Déclais, A. C., Lachaud, C., Toth, R., Macartney, T. J., et al. (2013). Cooperative control of Holliday junction resolution and DNA Repair by the SLX1 and MUS81-EME1 nucleases. Molecular Cell,52(2), 221–233. https://doi.org/10.1016/j.molcel.2013.08.036.
Chan, Y. W., & West, S. C. (2014). Spatial control of the GEN1 Holliday junction resolvase ensures genome stability. Nature Communications,5(1), 1–11. https://doi.org/10.1038/ncomms5844.
Chan, Y. W., Fugger, K., & West, S. C. (2018). Unresolved recombination intermediates lead to ultra-fine anaphase bridges, chromosome breaks and aberrations. Nature Cell Biology,20(1), 92–103. https://doi.org/10.1038/s41556-017-0011-1.
Chang, H. H. Y., Pannunzio, N. R., Adachi, N., & Lieber, M. R. (2017). Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nature Reviews Molecular Cell Biology,18(8), 495–506. https://doi.org/10.1038/nrm.2017.48.
De Jager, M., Van Noort, J., Van Gent, D. C., Dekker, C., Kanaar, R., & Wyman, C. (2001). Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Molecular Cell,8(5), 1129–1135. https://doi.org/10.1016/S1097-2765(01)00381-1.
Doksani, Y., Bermejo, R., Fiorani, S., Haber, J. E., & Foiani, M. (2009). Replicon dynamics, dormant origin firing, and terminal fork integrity after double-strand break formation. Cell,137(2), 247–258. https://doi.org/10.1016/j.cell.2009.02.016.
Duursma, A. M., Driscoll, R., Elias, J. E., & Cimprich, K. A. (2013). A role for the MRN complex in ATR activation via TOPBP1 recruitment. Molecular Cell,50(1), 116–122. https://doi.org/10.1016/j.molcel.2013.03.006.
Farmer, H., McCabe, H., Lord, C. J., Tutt, A. H. J., Johnson, D. A., Richardson, T. B., et al. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature,434(7035), 917–921. https://doi.org/10.1038/nature03445.
Fellows, M. D., & O’Donovan, M. R. (2010). Etoposide, cadmium chloride, benzo[a]pyrene, cyclophosphamide and colchicine tested in the in vitro mammalian cell micronucleus test (MNvit) in the presence and absence of cytokinesis block using L5178Y mouse lymphoma cells and 2-aminoanthracene tested in. Mutation Research/Genetic Toxicology and Environmental Mutagenesis,702(2), 163–170. https://doi.org/10.1016/J.MRGENTOX.2009.09.003.
Gaillard, P. H. L., Noguchi, E., Shanahan, P., & Russell, P. (2003). The endogenous Mus81-Eme1 complex resolves Holliday junctions by a nick and counternick mechanism. Molecular Cell,12(3), 747–759. https://doi.org/10.1016/S1097-2765(03)00342-3.
Garcia, V., Phelps, S. E. L., Gray, S., & Neale, M. J. (2011). Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature,479(7372), 241–244. https://doi.org/10.1038/nature10515.
Giannattasio, M., Zwicky, K., Follonier, C., Foiani, M., Lopes, M., & Branzei, D. (2014). Visualization of recombination-mediated damage bypass by template switching. Nature Structural and Molecular Biology,21(10), 884–892. https://doi.org/10.1038/nsmb.2888.
Haber, J. E. (2016). A life investigating pathways that repair broken chromosomes. Annual Review of Genetics,50(1), 1–28. https://doi.org/10.1146/annurev-genet-120215-035043.
Hoa, N. N., Akagawa, R., Yamasaki, T., Hirota, K., Sasa, K., Natsume, T., et al. (2015a). Relative contribution of four nucleases, CtIP, Dna2, Exo1 and Mre11, to the initial step of DNA double-strand break repair by homologous recombination in both the chicken DT40 and human TK6 cell lines. Genes to Cells,20(12), 1059–1076. https://doi.org/10.1111/gtc.12310.
Hoa, N. N., Kobayashi, J., Omura, M., Hirakawa, M., Yang, S.-H., Komatsu, K., et al. (2015b). BRCA1 and CtIP are both required to recruit Dna2 at double-strand breaks in homologous recombination (B. D. Price, Ed.). PLoS ONE,10(4), e0124495. https://doi.org/10.1371/journal.pone.0124495.
Hoa, N. N., Shimizu, T., Zhou, Z. W., Wang, Z.-Q., Deshpande, R. A., Paull, T. T., et al. (2016). Mre11 is essential for the removal of lethal topoisomerase 2 covalent cleavage complexes. Molecular Cell,64(3), 580–592. https://doi.org/10.1016/J.MOLCEL.2016.10.011.
Honma, M., Izumi, M., Sakuraba, M., Tadokoro, S., Sakamoto, H., Wang, W., et al. (2003). Deletion, rearrangement, and gene conversion; genetic consequences of chromosomal double-strand breaks in human cells. Environ Mol Mutagen, 42(4), 288–298. https://doi.org/10.1002/em.10201.
Hopfner, K. P., Craig, L., Moncalian, G., Zinkel, R. A., Usui, T., Owen, B. A. L., et al. (2002). The Rad50 zinc-hook is a structure joining Mre11 complexes in DNA recombination and repair. Nature,418(6897), 562–566. https://doi.org/10.1038/nature00922.
Ibrahim, M. A., Yasui, M., Saha, L. K., Sasanuma, H., Honma, M., & Takeda, S. (2020). Enhancing the sensitivity of the thymidine kinase assay by using DNA repair-deficient human TK6 cells. Environmental and Molecular Mutagenesis. https://doi.org/10.1002/em.22371.
Kakui, Y., & Uhlmann, F. (2018). SMC complexes orchestrate the mitotic chromatin interaction landscape. Current Genetics,64(2), 335–339. https://doi.org/10.1007/s00294-017-0755-y.
Keka, I. S., Mohiuddin, M. Y., Rahman, M. M., Sakuma, T., Honma, M., et al. (2015). Smarcal1 promotes double-strand-break repair by nonhomologous end-joining. Nucleic Acids Research,43(13), 6359–6372. https://doi.org/10.1093/nar/gkv621.
Kikuchi, K., Narita, T., Pham, V. T., Iijima, J., Hirota, K., Keka, I. S., et al. (2013). Structure-specific endonucleases Xpf and Mus81 play overlapping but essential roles in DNA repair by homologous recombination. Cancer Research,73(14), 4362–4371. https://doi.org/10.1158/0008-5472.CAN-12-3154.
Kondratova, A., Watanabe, T., Marotta, M., Cannon, M., Segall, A. M., Serre, D., et al. (2015). Replication fork integrity and intra-S phase checkpoint suppress gene amplification. Nucleic Acids Research,43(5), 2678–2690. https://doi.org/10.1093/nar/gkv084.
Kottemann, M. C., & Smogorzewska, A. (2013). Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature,493(7432), 356–363. https://doi.org/10.1038/nature11863.
Lee, J., & Dunphy, W. G. (2013). The Mre11-Rad50-Nbs1 (MRN) complex has a specific role in the activation of Chk1 in response to stalled replication forks. Molecular Biology of the Cell,24(9), 1343–1353. https://doi.org/10.1091/mbc.E13-01-0025.
Lorge, E. (2010). Comparison of different cytotoxicity measurements for the in vitro micronucleus assay using L5178Y and TK6 cells in support of OECD draft Test Guideline 487. Mutation Research - Genetic Toxicology and Environmental Mutagenesis,702(2), 199–207. https://doi.org/10.1016/j.mrgentox.2010.03.002.
Mao, Z., Bozzella, M., Seluanov, A., & Gorbunova, V. (2008). DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle,7(18), 2902–2906. https://doi.org/10.4161/cc.7.18.6679.
Mazin, A. V., Mazina, O. M., Bugreev, D. V., & Rossi, M. J. (2010). Rad54, the motor of homologous recombination. DNA Repair,9(3), 286–302. https://doi.org/10.1016/j.dnarep.2009.12.006.
Mehta, A., & Haber, J. E. (2014). Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harbor Perspectives in Biology,6(9), a016428. https://doi.org/10.1101/cshperspect.a016428.
Mimitou, E. P., & Symington, L. S. (2008). Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature,455(7214), 770–774. https://doi.org/10.1038/nature07312.
Moreau, S., Ferguson, J. R., & Symington, L. S. (1999). The nuclease activity of Mre11 is required for meiosis but not for mating type switching, end joining, or telomere maintenance. Molecular and Cellular Biology,19(1), 556–566. https://doi.org/10.1128/mcb.19.1.556.
Moynahan, M. E., & Jasin, M. (2010). Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nature Reviews Molecular Cell Biology,11(3), 196–207. https://doi.org/10.1038/nrm2851.
Murai, J., Huang, S. Y. N., Das, B. B., Renaud, A., Zhang, Y., Doroshow, J. H., et al. (2012). Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Research,72(21), 5588–5599. https://doi.org/10.1158/0008-5472.CAN-12-2753.
O’Driscoll, M., & Jeggo, P. A. (2006). The role of double-strand break repair—insights from human genetics. Nature Reviews Genetics,7(1), 45–54. https://doi.org/10.1038/nrg1746.
Oh, J., & Symington, L. S. (2018). Role of the Mre11 complex in preserving genome integrity. Genes,9, 12. https://doi.org/10.3390/genes9120589.
Paull, T. T. (2018). 20 Years of Mre11 biology: no end in sight. Molecular Cell,71(3), 419–427. https://doi.org/10.1016/j.molcel.2018.06.033.
Petukhova, G., Stratton, S., & Sung, P. (1998). Catalysis of homologous DNA pairing by yeast Rad51 and Rad54 proteins. Nature,393(6680), 91–94. https://doi.org/10.1038/30037.
Schlacher, K., Christ, N., Siaud, N., Egashira, A., Wu, H., & Jasin, M. (2011). Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell,145(4), 529–542. https://doi.org/10.1016/j.cell.2011.03.041.
Seeber, A., Hegnauer, A. M., Hustedt, N., Deshpande, I., Poli, J., Eglinger, J., et al. (2016). RPA mediates recruitment of MRX to forks and double-strand breaks to hold sister chromatids together. Molecular Cell,64(5), 951–966. https://doi.org/10.1016/j.molcel.2016.10.032.
Shibata, A., Moiani, D., Arvai, A. S., Perry, J., Harding, S. M., Genois, M. M., et al. (2014). DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Molecular Cell,53(1), 7–18. https://doi.org/10.1016/j.molcel.2013.11.003.
Shiotani, B., Nguyen, H. D., Håkansson, P., Maréchal, A., Tse, A., Tahara, H., et al. (2013). Two distinct modes of ATR activation orchestrated by Rad17 and Nbs1. Cell Reports,3(5), 1651–1662. https://doi.org/10.1016/j.celrep.2013.04.018.
Sonoda, E., Sasaki, M. S., Buerstedde, J. M., Bezzubova, O., Shinohara, A., Ogawa, H., et al. (1998). Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO Journal,17(2), 598–608. https://doi.org/10.1093/emboj/17.2.598.
Sonoda, E., Matsusaka, T., Morrison, C., Vagnarelli, P., Hoshi, O., Ushiki, T., et al. (2001). Scc1/Rad21/Mcd1 is required for sister chromatid cohesion and kinetochore function in vertebrate cells. Developmental cell,1(6), 759–770. https://doi.org/10.1016/s1534-5807(01)00088-0.
Stracker, T. H., & Petrini, J. H. J. (2011). The MRE11 complex: Starting from the ends. Nature Reviews Molecular Cell Biology. https://doi.org/10.1038/nrm3047.
Sugawara, N., Wang, X., & Haber, J. E. (2003). In vivo roles of Rad52, Rad54, and Rad55 proteins in Rad51-mediated recombination. Molecular Cell,12(1), 209–219. https://doi.org/10.1016/S1097-2765(03)00269-7.
Symington, L. S., & Gautier, J. (2011). Double-strand break end resection and repair pathway choice. Annual Review of Genetics,45(1), 247–271. https://doi.org/10.1146/annurev-genet-110410-132435.
Trenz, K., Smith, E., Smith, S., & Costanzo, V. (2006). ATM and ATR promote Mre11 dependent restart of collapsed replication forks and prevent accumulation of DNA breaks. The EMBO Journal,25(8), 1764–1774. https://doi.org/10.1038/sj.emboj.7601045.
Wechsler, T., Newman, S., & West, S. C. (2011). Aberrant chromosome morphology in human cells defective for Holliday junction resolution. Nature,471(7340), 642–646. https://doi.org/10.1038/nature09790.
West, S. C., & Chan, Y. W. (2017). Genome instability as a consequence of defects in the resolution of recombination intermediates. Cold Spring Harbor symposia on quantitative biology,82, 207–212. https://doi.org/10.1101/sqb.2017.82.034256.
Westmoreland, J. W., & Resnick, M. A. (2013). Coincident resection at both ends of random, γ–induced double-strand breaks requires MRX (MRN), Sae2 (Ctp1), and Mre11-Nuclease (M. Lichten, Ed.). PLoS Genetics,9(3), e1003420. https://doi.org/10.1371/journal.pgen.1003420.
Wiese, C., Dray, E., Groesser, T., San Filippo, J., Shi, I., Collins, D. W., et al. (2007). Promotion of homologous recombination and genomic stability by RAD51AP1 via RAD51 recombinase enhancement. Molecular Cell,28(3), 482–490. https://doi.org/10.1016/j.molcel.2007.08.027.
Wiltzius, J. J. W., Hohl, M., Fleming, J. C., & Petrini, J. H. J. (2005). The Rad50 hook domain is a critical determinant of Mre11 complex functions. Nature Structural and Molecular Biology,12(5), 403–407. https://doi.org/10.1038/nsmb928.
Wu, L., & Hickson, I. O. (2003). The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature,426(6968), 870–874. https://doi.org/10.1038/nature02253.
Wu, L., Bachrati, C. Z., Ou, J., Xu, C., Yin, J., Chang, M., et al. (2006). BLAP75/RMI1 promotes the BLM-dependent dissolution of homologous recombination intermediates. Proceedings of the National Academy of Sciences of the United States of America,103(11), 4068–4073. https://doi.org/10.1073/pnas.0508295103.
Wyatt, H. D. M., & West, S. C. (2014). Holliday junction resolvases. Cold Spring Harbor Perspectives in Biology,6(9), a023192. https://doi.org/10.1101/cshperspect.a023192.
Wyatt, H. D. M., Sarbajna, S., Matos, J., & West, S. C. (2013). Coordinated actions of SLX1-SLX4 and MUS81-EME1 for holliday junction resolution in human cells. Molecular Cell,52(2), 234–247. https://doi.org/10.1016/j.molcel.2013.08.035.
Yin, J., Sobeck, A., Xu, C., Meetei, A. R., Hoatlin, M., Li, L., et al. (2005). BLAP75, an essential component of Bloom’s syndrome protein complexes that maintain genome integrity. The EMBO Journal,24(7), 1465–1476. https://doi.org/10.1038/sj.emboj.7600622.
Zhou, Y., Caron, P., Legube, G., & Paull, T. T. (2014). Quantitation of DNA double-strand break resection intermediates in human cells. Nucleic Acids Research,42(3), e19. https://doi.org/10.1093/nar/gkt1309.
Zhu, Z., Chung, W. H., Shim, E. Y., Lee, S. E., & Ira, G. (2008). Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell,134(6), 981–994. https://doi.org/10.1016/j.cell.2008.08.037.
Acknowledgements
We thank the members of the Radiation Genetics lab in Kyoto for their helpful comments on the manuscript. We are also grateful to the staff of the Medical Research Support Center for technical assistance with flow cytometer and microscope (supported by Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS, AMED Grant JP19am0101092). This work was supported by the following grants; a Grant-in-Aid from the Ministry of Education, Science, Sport and Culture to S.T. (KAKENHI 25650006, 23221005, and 16H06306), and H.S. (KAKENHI 16H02953, 18H04900, and 19H04267), the Takeda research foundation, and Mitsubishi foundation (to H.S.) and JSPS Core-to-Core Program, A. Advanced Research Networks (to S.T.).
Author information
Affiliations
Department of Radiation Genetics, Graduate School of Medicine, Kyoto University, Yoshida Konoe, Sakyo-ku, Kyoto, 606-8501, Japan
Naoto Shimizu, Remi Akagawa, Shunichi Takeda & Hiroyuki Sasanuma
Corresponding authors
Correspondence to Shunichi Takeda or Hiroyuki Sasanuma.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Fig. S1 Kinetics of RAD51-focus formation after IR exposure. Kinetics of RAD51-focus formation in cyclinA-positive wild-type TK6 cells. after IR exposure. All foci counted from three independent experiments were plotted. Box plot represents the median (black bars) and 10-90th percentiles (box ranges). (b, c) Schematic diagram of the generation of CTIPlow/low TK6 cells (b) and western blotting analysis of CTIP expression in wild-type and CTIPlow/low TK6 cells (c). CTIP cDNA was inserted into the downstream of exon containing first ATG by TALEN. (d) Schematic diagram of MUS81 genomic locus, target location and guide RNA (gRNA) sequence in TK6 cells. Exon 1 was replaced by PUROR and BSRR markers. (e) Southern blotting analysis to confirm MUS81 deletion in TK6 cells. Lane 1 shows wild-type TK6 cells. Lane 2 and 3 show MUS81 -/- clones. MUS81 -/- #1 clone was used in this study. (f) Kinetics of RAD51-focus formation in cyclinA-positive cells for the indicated genotypes. All foci counted from three independent experiments were plotted. Box plot represents the median (bar in the box) and 10-90th percentiles (box ranges). Fig. S2 Generation of TK6 mutant cells and kinetics of olaparib-induced RAD51-focus formation. (a) Schematic diagram of RAD51AP1 genomic locus, target location and gRNA sequence in TK6 cells. Exon 1 was replaced by NEOR and PUROR markers. (b) Reverse transcription-polymerase chain reaction (RT-PCR) analysis of RAP51AP1 gene disruption in TK6 cells. Lane 1, 2, and 3 show wild-type, RAD51AP1-/-, and RAD54-/-/RAD51AP1-/- TK6 cells, respectively. β-actin is a loading control. (c, d) The sensitivity of RAD54-/-/RAD51AP1-/- TK6 cells to IR (c) and olaparib (d) was measured by clonogenic cell-survival analysis. The doses for IR and olaparib are displayed by the x-axis on a linear scale. The percentage of cell survival relative to that of untreated cells is shown by Y-axis on a logarithmic scale. Error bars show SD calculated from three independent experiments. (e) Distribution of RAD51 foci per cyclinA-positive cell following pulse exposure (one hour) to olaparib (1 μM) in the indicated genotypes. All foci counted from three independent experiments were plotted. Box plot represents the median (black bars) and 10-90th percentiles (box ranges). Asterisks indicate p < 1.0 × 10-4, calculated by a two-tailed unpaired Student's t test. Fig. S3 BrdU-labelled chromosome analysis. (a, b) Schematic representation showing BrdU-labelled chromosomal analysis. TK6 cells were incubated with BrdU for 0.17 h (10 minutes) and released into BrdU-free medium. TK6 cells then were exposed to 0.5 Gy irradiation and treated with 0.1 μg/ml colcemid for three hours. We harvested the TK6 cells at six hours (a) and three hours (b) After adding BrdU into the medium. The incorporated BrdU was visualized by immunofluorescence microscopy. Lower two panels in (a) and (b) indicate representative images containing both interphase and mitotic phase cells. Green dots indicate the incorporated BrdU. Fig. S4 Model for generation of isochromatid-type breaks and radial chromosomes following IR at the G2 phase. The irradiation generates DSBs in one of two sister chromatids. Defective initiation of HR leads to chromatid-type breaks in mitotic chromosome spreads. Defective formation of JMs such as HJs followed by failure of their resolution could generate isochromatid-type breaks, where two sister chromatids are broken at the same sites due to a defect in local chromosome condensation at HR intermediates. The defective resolution of JMs also leads to the formation of radial chromosomes by aberrant NHEJ. Fig. S5 Quantitative PCR analysis of GEN1 transcripts in TK6 cells. Reverse transcription quantitative polymerase chain reaction (RT-qPCR) analysis for wild-type, MUS81-/-, and MRE11-/H129N TK6 cells expressing GEN1NLS. Quantitative RT-PCR were performed from total RNA extracted from the indicated genotypes. Single, double, and triple asterisks indicate p = 1.0 × 10-4, p = 8.7 × 10-3, and p = 7.4 × 10-4, respectively, calculated by a two-tailed unpaired Student's t test. (PPTX 10840 kb)
Supplementary file2 (DOCX 61 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article
Cite this article
Shimizu, N., Akagawa, R., Takeda, S. et al. The MRE11 nuclease promotes homologous recombination not only in DNA double-strand break resection but also in post-resection in human TK6 cells. GENOME INSTAB. DIS. 1, 184–196 (2020). https://doi.org/10.1007/s42764-020-00015-w
Received 28 April 2020
Revised 28 May 2020
Accepted 04 June 2020
Published 30 June 2020
Issue Date July 2020
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
MRE11
Homologous recombination
Holliday junction
Isochromatid-type breaks
Resolution of Holliday junction
DNA double-strand break repair
PARP inhibitor
用户登录
还没有账号?
立即注册