






Autophagy and DNA damage repair
Review Article
Genome Instability & Disease 1,172–183(2020)
Abstract
DNA damage occurs frequently resulting from both exogenous and endogenous factors, which induce a series of downstream responses including autophagy and DNA damage repair. In the past few years, increasing evidence has indicated that the interplay between autophagy and DNA damage repair is essential for maintaining genome stability as well as cellular homeostasis, and have significant effects on cell fate. On one hand, autophagy is induced during the process of DNA damage repair, and can act as an upstream factor of DNA damage repair as well. On the other hand, autophagy plays a rather dual role in regulating DNA damage repair, as mild and repairable DNA damage repair can be restored with facilitation of autophagy, hyperactivation of autophagy can be cytotoxic and have negative impact on DNA damage repair. In this article, we review current understandings about the cross talk between autophagy and DNA damage repair, with particular attention to their significance to genome integrity and effects on cell fate.
Introduction
Macroautophagy (hereafter referred to as autophagy) is a process in which dysfunctional and aged proteins or organelles are delivered to lysosomes for degradation, recycling cell components and providing macromolecular precursors and energy for cellular metabolism (Levy et al. 2017). Autophagy is typically regarded as five steps: initiation, vesicle nucleation, vesicle elongation, vesicle fusion and cargo degradation (Levy et al. 2017). Take mammalian cells as example, initiation is induced by various conditions of stress, such as starvation, aggregation of protein and dysfunctional organelles, hypoxia, oxidative stress and others, as a result, the Unc-51-like kinase 1 (ULK1) complex [consisting of ULK1, ULK2, focal adhesion kinase family interacting protein of 200 kDa (FIP200), autophagy-related protein 13 (Atg13), Atg101] is activated, which in turn phosphorylate Class III Phosphatidylinositol 3-Kinase (PI3K) complex [consisting of vacuolar protein sorting 34 (VPS34), VPS15, Atg14, Beclin1, UV irradiation resistance-associated gene (UVRAG), activating molecule in Beclin1-regulated autophagy protein 1 (AMBRA1)]. The activated Class III PI3K complex produces phosphatidylinositol 3-phosphate (PI3P), recruiting downstream factors such as double FYVE-containing protein 1 (DFCP1) and WD-repeat proteins interacting with phosphoinositide (WIPIs) to conduct vesicle nucleation. Vesicle elongation is then performed by Atg5-Atg12-Atg16L conjugation system as well as Atg8-PE conjugation system. Finally, fusion of the autophagosome with the lysosome is completed, and acidic hydrolases in the vesicle degrade autophagic cargo, recycling nutrients to be used again by the cell (Mizushima et al. 2011; Levy et al. 2017; Dikic and Elazar 2018). Thus, autophagy plays a protective role in cell survival (Vessoni et al. 2013). Besides, taking a broader view on organism homeostasis, autophagy also contributes to dead-cell clearance during programmed cell death (Qu et al. 2007) as well as responding certain types of DNA damage (Crighton et al. 2006; Gao et al. 2011).
DNA damage occurs frequently in response to both exogenous and endogenous factors (Bader et al. 2020), for example, exposure to environmental carcinogens like ultraviolet A (UVA) and aromatic compound; medical side effects like genotoxic cancer therapeutics and some anti-inflammatory drugs; metabolic waste such as reactive oxygen species (ROS) (Roos et al. 2016). DNA damage may lead to genomic instabilities: chromosome alterations like aneuploidy and rearrangements, and base-pair abnormalities such as point mutations, insertions, deletions (Bader et al. 2020), other important lesions include double-strand breaks (DSB), single-strand breaks (SSB), pyrimidine dimers, interstrand cross links (ICLs), photoproducts (Roshani-Asl et al. 2020) and so on, which can ultimately result in cellular dysfunction, organism degenerative diseases or even develop into cancer (Surova and Zhivotovsky 2013). Nevertheless, the consequence of DNA damage relies largely on the magnitude of the damaging agents. DNA damage response (DDR) acts as an evaluation mechanism to classify the severity of DNA damages (Matt and Hofmann 2016). Mild DNA damages are those that can be repaired with or without cell-cycle arrest (Surova and Zhivotovsky 2013). Under regular growth conditions, occurrences of DNA lesions add up to 200,000 per cell per day (Atamna et al. 2000; Matt and Hofmann 2016), for example, genotoxic methylating agents such as N-nitroso compounds exist widely in the environment and can be found in multiple endogenous processes (Roos and Kaina 2013; Christmann and Kaina 2011). However, DNA damages caused by chronic low-level exposure to genotoxic agents are usually insufficient to trigger cell death (Roos and Kaina 2013). Damages of much severity can lead to cellular senescence, apoptosis and necrosis (Surova and Zhivotovsky 2013), which have also been used in the radiotherapy and chemotherapy targeting cancer cells (Huang and Zhou 2020). Ionizing radiation (IR) is usually used in cancer treatment to create breaks on DNA (Huang and Zhou 2020). It has been demonstrated that high-linear energy transfer (LET) irradiation can cause almost 500 DSBs/μm3 track volume, which create a heavy burden to repair and therefore result in cell death (Lorat et al. 2015).
The main regulators in DDR are three PI3-kinase-related protein kinases: ataxia-telangiectasia mutated (ATM), ATM and RAD3-related (ATR), and DNA-dependent protein kinase catalytic subunit (DNA-PKcs) (Menolfi and Zha 2020; Roos and Kaina 2013). These regulators together with downstream proteins are critically needed in the process of DDR. DNA damage repair is a step involved in DDR (Ciccia and Elledge 2010), following detection and signaling of the lesion (Vessoni et al. 2013).Basically, the repair process can be classified as five pathways: homologous recombination (HR), non-homologous end-joining (NHEJ), nucleotide excision repair (NER), base-excision repair (BER), and mismatch repair (MMR). Among these mechanisms, HR and NHEJ are used to repair DSBs and ICLs (Gomes et al. 2017; Nazio et al. 2018), while the other three excision repair pathways are responsible for impairments associated to single base-pair (Nazio et al. 2018). It should be noticed that lesions repaired by HR allows error-free product due to the use of a homologous template (Hewitt and Korolchuk 2017), and only short patches of DNA are synthesized during HR (Moldovan et al. 2010; Chapman et al. 2012). On the contrary, NHEJ connects the ends of DSB without a referential template, which can be a fallible pattern that frequently causing substitutions, insertions, and deletions at the break site, and thereby results in erroneous repair products (Chapman et al. 2012; Hewitt and Korolchuk 2017).
It has been demonstrated that autophagy, even being a cytoplasmic process, has direct impact on the proper activation of DNA repair (Nazio et al. 2018) and is vital for the genomic integrity of both nucleus and mitochondria (Vessoni et al. 2013). However, nowadays it is becoming widely believed that autophagic response also plays a detrimental role and accompanies the induction of cell death mechanisms under specific circumstances. In this article, we review the genome stabilizing effect of autophagy, with particular attention to its connection with DNA repair, as either of these two processes can be induced by the other. Autophagy’s cell-toxic role in DNA damage repair is also implicated in this chapter, albeit the findings so far are rather controversial. As our knowledge about the cross talk between DNA repair and autophagy expands, it may further contribute to clinical advantage.
Autophagy maintains genomic stability
Naturally, optimal autophagic responses can suppress the accumulation of genetic and genomic defects through a variety of mechanisms (Galluzzi et al. 2015) (Fig. 1).The term “nucleophagy” means autophagy degrading nuclear components so as to maintain nuclear integrity and function (Park et al. 2009). Considering nucleus is the place where eukaryotic cells store their genetic information, the accumulation of nuclear abnormalities can undermine cellular homeostasis. In fungus with more than on nucleus such as Aspergillus oryzae, whole nuclear degradation by autophagy can be observed (Shoji et al. 2010). In line with this, increasing findings support the concept of evolutionarily conserved nucleophagy in eukaryotes (Luo et al. 2016), for example, autophagosomes or autolysosomes enfolding partial nuclear materials like histone H1 is presented in cytoplasm (Vessoni et al. 2013). Meanwhile, inhibition of autophagy leads to nuclear disorders and reduced cell viability (Park et al. 2009), indicating its contribution in nuclear stability. Moreover, the co-localization of micronuclei and LC3, a wildly used marker for autophagosome, is observed in cells with intact autophagy while significantly decreases after abolishing autophagy by knocking down of Atg5 or Atg7 (Yang and Klionsky 2020; Rello-Varona et al. 2012). Notably, γH2AX+ DNA damage foci are also observed in GFP-LC3-positive envelopes, indicating that autophagy removes micronuclei therefore plays an important role in maintaining nuclear genome stability (Rello-Varona et al. 2012).
Fig. 1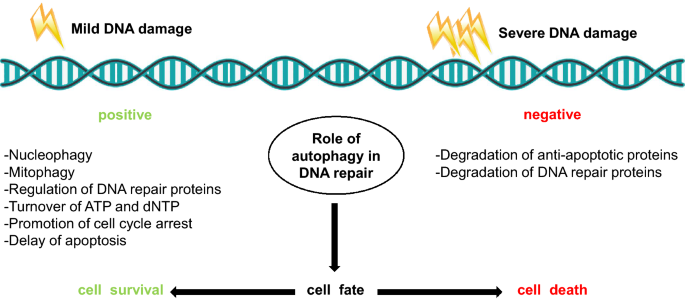
Dual role of autophagy in DNA repair. On one hand, mild DNA damage can trigger autophagy as a safeguard mechanism, and conduct a cytoprotective role through nucleophagy, mitophagy for clearance of excessive ROS, regulation of DNA repair proteins, turnover of ATP and dNTP, promotion of cell cycle-arrest, and delay of apoptosis. On the other hand, upon massive amount of DNA damage, hyperactivated autophagy can degrade anti-apoptotic proteins as well as DNA repair proteins, which consequently results in cell death
Full size imageROS, inevitable by-products in normal mitochondrial activity (Vessoni et al. 2013), participate in a variety of crucial metabolic pathways (Perillo et al. 2020). While contemporary studies have revealed its facilitation to normal physiology (Peoples et al. 2019), excessive accumulation of ROS can result in macromolecular damage (Richardson and Schadt 2014). For example, oxidative damage to proteins and lipids (Peoples et al. 2019), mtDNA mutations (Nissanka and Moraes 2018) and genome instability through affecting the spindle checkpoint pathway (D'Angiolella et al. 2007), which in reverse accelerate ROS production and ultimately develop a vicious circle. Autophagy acts as a safeguard mechanism that mitigates DNA damage by controlling ROS production through mitophagy (Li et al. 2015), which is a mitochondrial selective autophagy, removing dysfunctional mitochondria, thus prevent it from detrimental overload and downregulate ROS-mediated DNA damage events. Conditional deletion of Atg7 in mouse hematopoietic stem cell causes accumulation of dysfunctional mitochondria and ROS (Galluzzi et al. 2015), as well as increased proliferation and DNA damage (Mortensen et al. 2011), supporting the positive role of autophagy in protecting mitochondrial genome.
Consist with this, autophagy-incompetent cells are rather vulnerable in exposure of DNA damaging factors and are prone to tumorigenesis. Allelic loss of Beclinl, which is a positive regulator of autophagy, activates DDR both in vitro and in mammary tumors, facilitating breast cancer progression (Karantza-Wadsworth et al. 2007; Mathew et al. 2007). Likewise, depletion of Atg7 in keratinocytes exhibits increased DNA damage upon oxidative stress (Song et al. 2017). Another research using colorectal cancer cell finds that knockdown of Beclin1, UVRAG or Atg5 increases radiation-induced DSBs, and leads to cell death (Park et al. 2014). Autophagy deficiency by knocking down of other autophagy genes such as Bif1 and VPS34 is shown to result in poly-nucleation (Belaid et al. 2013; Mathew et al. 2007). Also, in lack of functional autophagy, factors jeopardizing genomic homeostasis are further promoted, such as ROS production, mitochondria disorder, protein aggregates like p62/SQSTM1, as well as DNA leision (Ariyoshi et al. 2016; Mathew et al. 2009). In the context of nuclear envelope defects, autophagy impairment aggravates artificial aneuploidy and cell-cycle perturbation, which consequently increase DNA damage and genomic instability (Ariyoshi et al. 2016; Hewitt and Korolchuk 2017). As a result, it can be speculated that compromised autophagy can drive gene amplification, aneuploidy as well as DNA damage processes that not only lead to genomic dysfunction but may also generate tumor (Belaid et al. 2013).
Thus, with the function of degrading nuclear components, removing ROS through mitophagy, and decomposing other genotoxic cellular materials, together with the impairment of autophagy causes genomic unsteadiness, again demonstrate the fact that autophagy is vital in sustaining genomic stability.
Interaction between autophagy and DNA repair
As mentioned above, autophagy plays a pivotal role in maintaining genome integrity. In line with this, increasing evidence has suggested a more compact interaction between autophagy and DNA damage repair (Eliopoulos et al. 2016). DDR signaling can activate autophagy (Roos et al. 2016). It has been proved that G-quadruplex ligand-mediated telomere damage can trigger autophagy as a safeguard mechanism against the cytotoxic effect of the agent (Folini et al. 2012). DNA damage signaling pathways activate autophagy by inhibition of mTORC1 or activation of ULK1 and Beclin1 complexes (Roos et al. 2016; Czarny et al. 2015). The proteins implicated in these pathways that have been elucidated so far are summarized as below (Fig. 2).
Fig. 2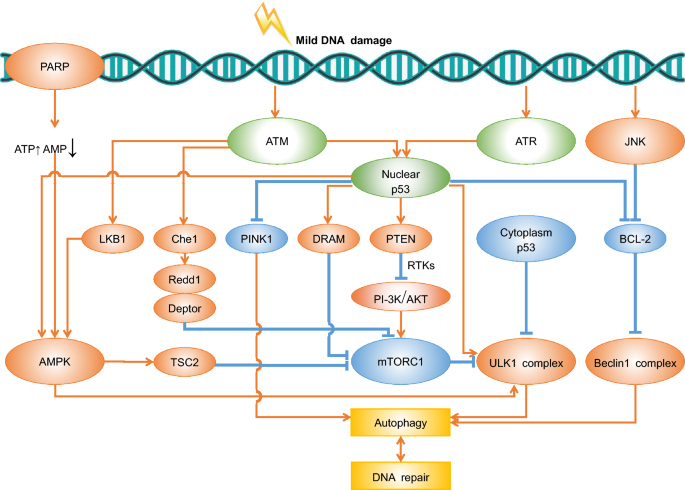
Overview of known pathways linking DDR and autophagy upon mild DNA damage. Detailed information about the pathways and proteins implicated are interpreted in the main text. Proteins inducing autophagy (red); Proteins inhibiting autophagy (blue); Proteins with dual effects to autophagy (green)
Full size imageWhile ATR is a major sensor functions in the replication stress response (Shubassi et al. 2012), ATM mainly senses DSBs and can be activated by other aberrant DNA structures (Roos et al. 2016; Menolfi and Zha 2020). ATM activates LKB1 and results in AMPK activation, which in turn phosphorylates the tumor suppressor TSC2, resulting in mTORC1 repression and therefore alleviate its inhibitory effect on autophagy (Alexander et al. 2010). mTORC1 inhibits the initiation of autophagy by suppressing ULK1 activation as well (Kim et al. 2011). In addition, AMPK can also directly activate autophagy by phosphorylating Ser 317 and Ser 777 of ULK1. Another study finds that ATM mediates the activation of Che-l, which then triggers Redd1 and Deptor expression, the two important mTOR inhibitors (Desantis et al. 2015; Nazio et al. 2018).
Poly (ADP-ribose) polymerase 1 (PARP-1) is a survival protein in DNA damage recovery, which binds with DNA and is involved in base-excision repair (Murcia et al. 1997). DNA damage activates PARP-1, leading to ATP depletion along with upregulated AMP levels that triggers AMPK, thus inducing autophagy (Rodriguez-Vargas et al. 2012; Eliopoulos et al. 2016). c-JUN N-terminal kinase (JNK) is a positive regulator of DNA damage (Zhang et al. 2018) and it can also promote autophagy via two distinct ways: JNK phosphorylates BCL-2, which inhibits autophagy through suppressing elevation of cytosolic calcium (Mizushima 2007), consequently disrupts the binding of BCL-2-Beclin1 complex and therefore induce autophagy (Wei et al. 2008). The other mechanism is by upregulation of damage-regulated autophagy modulator (DRAM), a downstream gene of p53 that encodes a lysosomal protein resulting in induction of autophagy (Crighton et al. 2006).
A most recent study reveals that checkpoint kinase 2 (CHK2)–FOXK (FOXK1 and FOXK2) axis is pivotal in DNA damage–mediated autophagy at transcriptional regulation level (Chen et al. 2020). FOXK proteins in the nucleus act as suppressors of Atgs. Upon Cisplatin-induced DNA damage, the transport of FOXK from nucleus to cytoplasm is mediated by CHK2, therefore alleviates the inhibition of FOXK on the transcription of Atgs in the nucleus, resulting in autophagy activation (Chen et al. 2020).
The proteins described above have either effect of inhibiting or promoting autophagy, interestingly, p53, which can be activated by both ATM and ATR (Blackford and Jackson 2017), regulates autophagy dually in DDR. On one hand, nuclear p53 induces autophagy through increasing the transcription of several pro-autophagic genes, for example, DRAM, sestrins-1, -2 (Prokhorova et al. 2020), Atg2b, Atg4a, Atg4c, Atg7, Atg10, ULK1, ULK2, UVRAG (Kenzelmann et al. 2013; Pitolli et al. 2019) and PTEN, which then inhibits the PI3K/Akt axis by receptor tyrosine kinases (RTKs) (Roos et al. 2016), resulting in repression of mTORC1 and thereby induces autophagy initiation. Activation of p53 inhibits mTORC1 activity by upregulating AMPK and subsequent activation of the TSC1/TSC2 complex (Feng et al. 2005). Moreover, expression of BCL-2 and certain growth factor receptors involved in autophagy inhibition is repressed upon nuclear p53 activation (Prokhorova et al. 2020; Zhang et al. 2010). On the other hand, notwithstanding the autophagy-activating role of nuclear p53, cytoplasmic p53 serves as a suppressor of autophagy as it binds to FIP200 and impedes its interaction with ULK1, Atg13 and Atg101 (Tasdemir et al. 2008; Maiuri et al. 2010; Prokhorova et al. 2020; Morselli et al. 2011). It seems that the alternative of p53 to either activate or inhibit autophagy is exclusively associated with its cellular localization (Pitolli et al. 2019). Nevertheless, a recent research shows that nuclear p53 can also negatively regulate autophagy through repressing transcriptional level of PTEN-induced kinase 1 (PINK1), a key protein involved in mitophagy (Goiran et al. 2018).
Given its bidirectional role towards autophagy, p53 is essential in determining cell fate. Upon mild DNA damage, Ser15-p53 is phosphorylated and triggers initiation of autophagy, nevertheless, pSer46-p53 elevates the expression of apoptotic genes after sensing stress with higher severity and consequently results in cell death (Prokhorova et al. 2020). Collectively therefore, albeit there is no explicit elaboration of the alternative effects of DNA damage towards autophagy, however, it can be speculated that different cellular fate can be evoked depending on the seriousness of DNA lesions and cell conditions. In some conditions with reparable lesions, DNA damage induces autophagy as a cell-protective mechanism, while may inhibit autophagy in other lethal context (Pitolli et al. 2019; Roshani-Asl et al. 2020; Eliopoulos et al. 2016).
Converse with DNA damage inducing autophagy, it has been proved that autophagy can also act as an upstream role, not just a consequence, of DDR signaling (Dan et al. 2015). Plenty of studies have suggested that deficient autophagy can lead to dysfunction in DDR. For example, depletion of Beclin1 and Atg5 elevate the sensitivity of mammalian cancer cell towards irradiation both in vitro and in vivo (Ko et al. 2014), indicating that autophagy could affect trafficking of damaged DNA (Dan et al. 2015), coherent with the finding that accumulation of damaged DNA is shown in autophagy-deficient cells (Dan et al. 2015). Inhibition of autophagy through knocking-out of FIP200 represses DNA repair and enhances DNA lesion due to an upregulation of p62 (Czarny et al. 2015; Hewitt and Korolchuk 2017). Also, after DNA damaging events, it is observed that the induction of autophagy precedes activation of p53 and ATM along with the increased expression of DNA repair genes (Dan et al. 2015), revealing that DDR can be regulated by autophagy as well.
Overall, robust evidences suggest that there is an apparent crosstalk between DNA damage repair and autophagy, raising the crucial question that whether autophagy impinges on positive consequence of DDR.
Dual role of autophagy in DNA repair
Autophagy functions positively in DNA repair by degrading proteins that have negative effects on the repair process. p62, a multifunctional protein which can be induced by various forms of cellular stress, is degraded by autophagy (Pankiv et al. 2010). Notably, p62 connects the autophagy pathway and the ubiquitin–proteasome system (UPS) in DNA damage repair (Liu et al. 2016; Hewitt et al. 2016). Filamin A (FLNA) is a protein crucial in DNA repair process that recruits RAD51, a key recombinase working at DSB site, and promotes HR (Hewitt et al. 2016; Gomes et al. 2017). In the context of irradiation, nuclear p62 promotes proteasomal degradation of FLNA, thus causing RAD51 decrease in the nucleus and restraining DNA repair (Hewitt et al. 2016). Consistently, p62 is also a major mediator in controlling HR and NHEJ (Gomes et al. 2017; Hewitt et al. 2016). As mentioned above, compared to HR, NHEJ is a rather error-prone manner, reliance on which results in spontaneous genomic instability even in the absence of exogenous DNA damage (Gillespie and Ryan 2016). p62 down-modulation by autophagy increases HR and reduces NHEJ, therefore leads to correct repaired products (Hewitt et al. 2016; Gomes et al. 2017). Likewise, p62-mediated inhibition of DNA repair is shown to be reversed in response to dietary restriction, which is a latent motivator for autophagy (Hewitt et al. 2016; Hewitt and Korolchuk 2017), further demonstrating that degradation of p62 through selective autophagy is pivotal in DNA damage repair. In line with this, upon autophagy inhibition, on one hand, p62 accumulation in the nucleus promotes ROS production and cell death (Wang et al. 2019; Czarny et al. 2015), as well as proteasome-mediated CHK1 and RAD51 protein degradation (Wang et al. 2019). This claim is further supported by findings that inhibition of autophagy resulted in decreased expression of both CHK1 and RAD51 (Gillespie and Ryan 2016; Dan et al. 2015; Mo et al. 2014). On the other hand, accumulated p62 induced by autophagy-deficiency binds with E3 ligase RNF168 and inhibits its activity (Feng and Klionsky 2017). As RNF168 is required for H2A ubiquitination in response to DSBs, a number of proteins such as RAP80, BRCA1, and RAD51 (Gomes et al. 2017), which are key factors for DNA repair cannot be recruited in autophagy-deficient cells. As a result, DNA damage repair is diminished while sensitizing cells towards oxidative stress (Feng and Klionsky 2017; Wang et al. 2016).
Another vital mechanism by which autophagy controls HR activity is the regulation of CHK1 levels. The ATR-CHKl pathway is a direct effector of DNA damage (Smith et al. 2010), upon which activated CHK1 phosphorylates RAD51 and promotes its recruitment to damaged foci and initiates recombination (Smith et al. 2010; Gillespie and Ryan 2016). It is widely believed that autophagy and UPS are the two main ways to degrade cellular products (Nazio et al. 2018), and previous studies have shown that CHK1 is degraded by UPS (Liu et al. 2015). As described above, autophagy does not control CHK1 level directly whereas conducts its regulation through inhibiting proteasome-mediated CHK1 degradation (Gomes et al. 2017). Compromised autophagy releases its inhibiting effect thereby depletes CHK1 and impairs HR (Gillespie and Ryan 2016). In contract to macroautophagy, chaperone-mediated autophagy (CMA), which is induced as well in the context of genotoxic insults, degrades activated CHK1 in a lysosome-dependent manner (Park et al. 2015). Declined CMA results in hazardous CHK1 aggregates and HR blockage (Park et al. 2015; Gomes et al. 2017). Thus, neither loss of macroautophagy nor CMA results in aberrant CHK1 levels and consequently leads to diminished HR as well as hyper-dependent on NHEJ (Gillespie and Ryan 2016), reducing cell survival and genomic stability.
Furthermore, activation of autophagy can promote DNA damage repair via degradation of KAP1, a signal transducer and activator of transcription 3 (STAT3) repressor, and up regulates STAT3-mediated transcription of BRCA1, removing DSBs through HR (Gomes et al. 2017; Fei et al. 2017). Another key protein involved in DNA repair is heterochromatin protein 1α (HP1α), which is well-known for its role in heterochromatin formation and regulation in gene transcription (Chen et al. 2015). Ubiquitinated by RAD6 at residue K154, HP1α is then degraded by autophagy and eventually leads to an open chromatin structure that facilitates efficient HR repair (Chen et al. 2015; Gomes et al. 2017). In addition, autophagy facilitates the degradation of Sae2, a protein participates in lesion repair, therefore eliminates cell-damaging extensive DSB resection (Vessoni et al. 2013; Robert et al. 2011; Surova and Zhivotovsky 2013).
Taken together, these findings suggest that autophagy influences the kinetics of DNA damage repair process by turnover of important proteins implicated in the repair process. Apart from proteins, energy-related factors are also necessary for DNA repair, leading to another crucial role of autophagy in regulation of ATP, NAD+ and dNTPs supplement (Dan et al. 2015; Vessoni et al. 2013). ATP is implicated in multiple energy-dependent processes and is indispensable for DNA repair (Vessoni et al. 2013), for example, ATP-dependent chromatin remodeling is essential in both NHEJ and HR (van Attikum and Gasser 2005), and ATP-dependent helicases are needed in NER (de Laat et al. 1999). A research using multiple glioma cell lines demonstrate that non-apoptotic cell death with relation to mitotic catastrophe and micronucleation significantly increased upon diminished autophagy-induced ATP production (Katayama et al. 2007). This result is further confirmed by the fact that DNA damage can be rescued by pyruvate, which elevates ATP levels and therefore protects cells (Katayama et al. 2007), suggesting an autophagy-associated ATP surge functions in DNA repair process. Specifically, the increase in AMP will activate AMPK which in turn stimulates autophagy to facilitate the restoration of ATP levels (Roos et al. 2016).
Imbalanced deoxyribonucleotide triphosphates (dNTPs) can lead to mutagenesis and jeopardize cell homeostasis in a pattern that is associated with the nature and severity of the imbalance (Kumar et al. 2011). Autophagy can fight against this DNA damaging event by reducing cellular levels of RRM2, a crucial subunit of ribonucleotide reductase (RNR) which is the rate limiting enzyme contributes in dNTPs production (Chen et al. 2016). Specifically, autophagy optimizes RNR activity through facilitating the moderate supply of deoxynucleotides required for DNA replication as well as DNA repair (Nordlund and Reichard 2006), thus ensures a balanced dNTP pool (Kumar et al. 2011; Vessoni et al. 2013). Notably, autophagy regulates dNTP pool levels in a negative feedback manner, as autophagy causes a reduction of dNTP pool levels, excessive amount of dNTP pools, in contrast, leads to a corresponding desensitization of cells toward the outset of autophagy (Chen et al. 2014).
Generally, functional cell cycle acts like a dynamo for mitosis whereas under the condition of DNA damage, improper chromosome segregation emerges constantly and may even end in tumorgenisis (Vessoni et al. 2013; Dotiwala et al. 2013). A study in Limbal stem cells (LSC) suggests that upon UVA exposure, PAX6, which is a master transcription factor governing cell cycle, translocates from nuclear to cytoplasm. Autophagy induced by elevated ROS can facilitate this export process and therefore promoting cell-cycle arrest (Laggner et al. 2017). In the context of autophagy deficiency, UVA induces massive amounts of ROS with retention of PAX6 in the nucleus, hampering cell-cycle response and resulting in cell proliferation (Laggner et al. 2017), which may contribute to early tumorigenesis.
In addition, it is observed in breast cancer cells that upon excessive DNA damage, apoptotic cell death can be delayed by autophagy (Abedin et al. 2007). This finding can be further demonstrated by the fact that with genotoxic temozolomide treatment, glioblastoma cells exhibits senescence stimulated by autophagy rather than apoptosis. Impairment of autophagy intensifies the harmful character of cytotoxic drugs and elevates drug-mediated apoptosis (Knizhnik et al. 2013; Prokhorova et al. 2020). These results suggest the underlying mechanism that autophagy stimulates cells to undergo senescence rather than apoptosis under DNA damage conditions, thus acting as a survival mechanism (Knizhnik et al. 2013).
Interestingly, serving as a cytoprotective role in normal cells, autophagy in malignant tumor cells can help its metabolism upon nutrient deprivation as well as massive DNA damage caused by cancer therapy such as ionizing radiation (IR) and DNA methylating agents (Goldstein and Kastan 2015). Multiple researches have shown that non-cell-autonomous autophagy in both tumor microenvironment and remote tissues is induced, thereby promoting cancer cell survival as well as radio-chemoresistance (Katheder et al. 2017). A number of inhibitors function in different steps of autophagy have been used, such as ULK inhibitors, PI3K inhibitors, Atg inhibitors, and lysosome inhibitors (Pérez-Hernández et al. 2019). Among which, chloroquine (CQ) and hydroxychloroquine (HCQ) are clinical available drugs that have put into use (Levy et al. 2017). The first clinical trial using autophagy inhibitors in treatment of glioblastoma multiforme has demonstrated that CQ together with radiation and temozolomide significantly prolongs the survival time of patients and precludes resistant clones during radiotherapy and chemotherapy (Briceño et al. 2003). A phase I clinical trial using HCQ in conjunction with doxorubicin has emerged superior outcome as the overall response rate (ORR) in modeled patients with non-hodgkin lymphoma (NHL) is up to 93.3% (Barnard et al. 2014). Another human clinical trial reveals that combination of HCQ and temsirolimus in patients with advanced solid tumors and melanoma exhibit significant antitumor effects (Rangwala et al. 2014). Trials using HCQ in relapsed/refractory myeloma (Vogl et al. 2014) and newly diagnosed glioblastoma (Rosenfeld et al. 2014) achieve positive outcomes as well. Therefore, adding autophagy inhibitor into several cancer treatment again indicates the pro-survival role of autophagy in cells (Pérez-Hernández et al. 2019).
All of the aforementioned findings can lead to a conclusion that in the context of DNA damage, the induction of autophagy is promoted, paralleling to its contribution to DNA damage repair process not only by direct nucleophagy and mitophagy but also through recycling key proteins, ATP, NAD+ and dNTPs and regulating cell-cycle arrest, as well as retarding apoptotic cell death. However, despite its predominant role as a survival pathway (Mathew et al. 2007), the cell fate following autophagy induction extensively relies on cellular condition, nature and magnitude of stress. Small amount of DNA damage usually can be fixed through DNA repair with the positive support of autophagy, whereas more severe and irreparable DNA impairment cannot be rescued by autophagy, which, in contrast, may even induce autophagic cell death (Surova and Zhivotovsky 2013).
With its main function in turnover of cellular material, autophagy may degrade anti-apoptotic proteins as well as proteins involved in DNA repair. dBruce is a classic inhibitor of apoptosis (IAP) in Drosophila, which is responsible for DNA fragmentation in nurse cells and can be decomposed by autophagy, revealing a novel mechanism that the counteracting effect of autophagy towards IAP can lead to cell apoptosis and therefore, harmful for cells (Nezis et al. 2010). Catalase is known as a major enzymatic ROS scavenger, the degradation of which by selective autophagy can cause ROS accumulation and oxidatively generated damage (Li et al. 2006).
It is mentioned above that autophagy can clear Sae2, a recombination protein playing a key role in DSB site and is regulated by histone acetyltransferases (HATs) and deacetylases (HDACs). However, autophagy induced by HDAC inhibitor valproic acid (VPA), which impairs checkpoint activation, triggers Sae2 degradation that significantly causes DSB repair defects (Robert et al. 2011). Further studies have proved that inactivation of autophagy, specifically the cytoplasm to vacuole (CVT) pathway, rescues Sae2 degradation in VPA (Shubassi et al. 2012). Consistently, nuclease Exol, which also functions positively in the DSB site is degraded by autophagy in an acetylation-dependent manner (Li et al. 2017; Shubassi et al. 2012). Collectively, these findings suggest that autophagy negatively influences DSB repair in a hyperacetylated state (Shubassi et al. 2012). Besides DSB repair, autophagy also interferes with BER repair by degrading OGG1, an enzyme participates in BER, under nutrient-deprived condition in cardiomyocytes (Siggens et al. 2012), which may contribute to persistence of DNA damage (Vessoni et al. 2013).
Thus, it is plausible that massive exposure to ROS and other DNA damaging events may upregulate autophagy, and with its enhanced function of degradation, autophagy can affect the dynamic of DNA repair process and even accelerate cell death (Vessoni et al. 2013; Kuo et al. 2011). Notably, autophagy inhibitors in conjunction with radio-chemotherapy are efficient treatment in multiple malignant tumors, activators of autophagy have also been described as feasible therapeutic agents suppressing tumorigenesis due to its negative role in cancer cells (Pérez-Hernández et al. 2019). mTOR inhibitor rapamycin is used to stimulate autophagy, which delays growth of murine S180 sarcoma (Shi et al. 2019) and inhibits cell proliferation in human neuroblastoma (Lin et al. 2018). HDAC inhibitor MHY2256 possesses anticancer activity in MCF-7 human breast cancer cells and induces apoptosis and autophagic cell death in hormone-related endometrial cancer (De et al. 2018). BH3 mimetics ( −)-gossypol (AT-101) can trigger autophagy and inhibits growth of malignant mesothelioma (Benvenuto et al. 2018). Therefore, with stimulated autophagy, some newly developed tumors may undergo autophagic cell death before the outburst of tumorigenesis (Pérez-Hernández et al. 2019). Serving as a disadvantage for tumor cell itself, inducing autophagic cell death in primal cancers is a potential treatment in connection with tumor type and progress. Altogether, despite autophagy may confer an advantage for cells under stress, hyperactivation of autophagy can be rather cytotoxic and indeed have negative impact on DDR if it is not switched off.
However, as the pro-death and pro-survival mechanism in one cell can be activated simultaneously by DDR, the clear threshold discriminating mild and severe DNA damage is context-dependent (Roos et al. 2016; Wirawan et al. 2010; Surova and Zhivotovsky 2013; Vessoni et al. 2013), thus making the boundary between cytoprotective and cytotoxic autophagy opaque. A study in bone-marrow-derived pro-B-cell line Ba/F3 shows that the switch from cytoprotective autophagy to cell apoptosis is characterized by caspase cleavage of Beclin1 and PI3KC3 in condition of persistent DNA damage and long-lasting DDR by interleukin-3 (IL-3) depletion after 16 h (Wirawan et al. 2010; Roos et al. 2016). Another research in Caenorhabditis Elegans shows that upon treatment with DNA damaging agents such as N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) and methyl viologen (MV) for 1 h, DSBs accumulates and autophagy-related proteins like VPS34 significantly upregulates (SenGupta et al. 2013; Moriwaki et al. 2015), and ultimately autophagic cell death is induced due to “futile repair” and hyperactivated MMR (Moriwaki et al. 2015). Altogether, albeit the exact biochemical mechanism of how DDR distinguish whether the DNA lesion is repairable is still unclear, it can be speculated that autophagy confers an advantage for normal cells with low and non-persistent DNA damage, while when the lesion is beyond repair or sustains too long, hyperactivation of autophagy can be rather cytotoxic and can initiate cell death program if it is not switched off. (Dan et al. 2015).
Conclusion
To draw a conclusion, it is clear that there is an apparent cross talk between autophagy and DNA repair, while the underlying mechanisms remain largely unknown, it can be speculated that this interaction rely on the severity of DNA damage to a great extent. With the important role of maintaining genome integrity and cellular homeostasis, better understanding of DNA damage repair and autophagy process as well as their counteraction would contribute to a more intrinsic learning about cellular fate in response to genotoxic insults. Future studies need to be done to probe into the mechanisms concerning this interplay as well as the boundary of autophagy’s cell-protective and cell-toxic influence after DNA damage, as some controversial results are obtained so far. Moreover, with a more thorough study of the interaction between autophagy and DNA repair, promising therapeutic potentials will be generated that make contribution to treatment of cancer and other diseases.
References
Abedin, M. J., Wang, D., McDonnell, M. A., Lehmann, U., & Kelekar, A. (2007). Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death and Differenciation,14(3), 500–510. https://doi.org/10.1038/sj.cdd.4402039.
Alexander, A., Kim, J., & Walker, C. L. (2010). ATM engages the TSC2/mTORC1 signaling node to regulate autophagy. Autophagy,6(5), 672–673. https://doi.org/10.4161/auto.6.5.12509.
Ariyoshi, K., Miura, T., Kasai, K., Fujishima, Y., Oshimura, M., & Yoshida, M. (2016). Induction of genomic instability and activation of autophagy in artificial human aneuploid cells. Fundamental and Molecular Mechanisms of Mutagenesis,790(1), 19–30. https://doi.org/10.1016/j.mrfmmm.2016.06.001.
Atamna, H., Cheung, I., & Ames, B. N. (2000). A method for detecting abasic sites in living cells: Age-dependent changes in base excision repair. Proceedings of the National Academy of Sciences,97(2), 686–691. https://doi.org/10.1073/pnas.97.2.686.
Bader, A., Hawley, B., Wilczynska, A., & Bushell, M. (2020). The roles of RNA in DNA double-strand break repair. British Journal of Cancer,122(1), 1–11. https://doi.org/10.1038/s41416-019-0624-1.
Barnard, R., Wittenburg, L., Amaravadi, R., Gustafson, D., Thorburn, A., & Thamm, D. (2014). Phase I clinical trial and pharmacodynamic evaluation of combination hydroxychloroquine and doxorubicin treatment in pet dogs treated for spontaneously occurring lymphoma. Autophagy,10(8), 1415–1425. https://doi.org/10.4161/auto.29165.
Belaid, A., Cerezo, M., Chargui, A., Corcelle-Termeau, E., Pedeutour, F., Giuliano, S., et al. (2013). Autophagy plays a critical role in the degradation of active RHOA, the control of cell cytokinesis, and genomic stability. Cancer Research,73(14), 4311–4322. https://doi.org/10.1158/0008-5472.Can-12-4142.
Benvenuto, M., Mattera, R., Sticca, J., Rossi, P., Cipriani, C., Giganti, M., et al. (2018). Effect of the BH3 mimetic polyphenol (–)-gossypol (AT-101) on the in vitro and in vivo growth of malignant mesothelioma. Frontiers in Pharmacology,9, 1269. https://doi.org/10.3389/fphar.2018.01269.
Blackford, A. N., & Jackson, S. P. (2017). ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Molecular Cell,66(6), 801–817. https://doi.org/10.1016/j.molcel.2017.05.015.
Briceño, E., Reyes, S., & Sotelo, J. (2003). Therapy of glioblastoma multiforme improved by the antimutagenic chloroquine. Neurosurgical Focus,14(2), 1–6. https://doi.org/10.3171/foc.2003.14.2.4.
Chapman, J. R., Taylor, M. R., & Boulton, S. J. (2012). Playing the end game: DNA double-strand break repair pathway choice. Molecular Cell,47(4), 497–510. https://doi.org/10.1016/j.molcel.2012.07.029.
Chen, S., Wang, C., Sun, L., Wang, D. L., Chen, L., Huang, Z., et al. (2015). RAD6 promotes homologous recombination repair by activating the autophagy-mediated degradation of heterochromatin protein HP1. Molecular and Cellular Biology,35(2), 406–416. https://doi.org/10.1128/MCB.01044-14.
Chen, W., Zhang, L., Zhang, K., Zhou, B., Kuo, M. L., Hu, S., et al. (2014). Reciprocal regulation of autophagy and dNTP pools in human cancer cells. Autophagy,10(7), 1272–1284. https://doi.org/10.4161/auto.28954.
Chen, Y., Wu, J., Liang, G., Geng, G., Zhao, F., Yin, P., et al. (2020). CHK2-FOXK axis promotes transcriptional control of autophagy programs. Science advances,6(1), eaax5819. https://doi.org/10.1126/sciadv.aax5819.
Chen, Y. R., Tsou, B., Hul, S., Ma, H., Liul, X., Yen, Y., et al. (2016). Autophagy induction causes a synthetic lethal sensitization to ribonucleotide reductase inhibition in breast cancer cells. Oncotarget,7(2), 1984–1999. https://doi.org/10.18632/oncotarget.6539.
Christmann, M., & Kaina, B. (2011). O 6-methylguanine-DNA methyltransferase (MGMT): Impact on cancer risk in response to tobacco smoke. Mutation Research,736, 64–74. https://doi.org/10.1016/j.mrfmmm.2011.06.004.
Ciccia, A., & Elledge, S. J. (2010). The DNA damage response: making it safe to play with knives. Molecular Cell,40(2), 179–204. https://doi.org/10.1016/j.molcel.2010.09.019.
Crighton, D., Wilkinson, S., O'Prey, J., Syed, N., Smith, P., Harrison, P. R., et al. (2006). DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell,126(1), 121–134. https://doi.org/10.1016/j.cell.2006.05.034.
Czarny, P., Pawlowska, E., Bialkowska-Warzecha, J., Kaarniranta, K., & Blasiak, J. (2015). Autophagy in DNA damage response. International Journal of Molecular Sciences,16(2), 2641–2662. https://doi.org/10.3390/ijms16022641.
D'Angiolella, V., Santarpia, C., & Grieco, D. (2007). Oxidative stress overrides the spindle checkpoint. Cell Cycle,6(5), 576–579. https://doi.org/10.4161/cc.6.5.3934.
Dan, Z., Tang, B., Xie, X., Xiao, Y.-F., Yang, S.-M., & Zhang, J.-W. (2015). The interplay between DNA repair and autophagy in cancer therapy. Cancer Biology and Therapy,16(7), 1005–1013. https://doi.org/10.1080/15384047.2015.1046022.
de Laat, W. L., Jaspers, N. G., & Hoeijmakers, J. H. (1999). Molecular mechanism of nucleotide excision repair. Genes and Development,13(7), 768–785. https://doi.org/10.1101/gad.13.7.768.
De, U., Son, J., Sachan, R., Park, Y., Kang, D., Yoon, K., et al. (2018). A new synthetic histone deacetylase inhibitor, MHY2256, induces apoptosis and autophagy cell death in endometrial cancer cells via p53 acetylation. International Journal of Molecular Sciences,19, 2743. https://doi.org/10.20944/preprints201808.0146.v1.
Desantis, A., Bruno, T., Catena, V., De Nicola, F., Goeman, F., Iezzi, S., et al. (2015). Che-1-induced inhibition of mTOR pathway enables stress-induced autophagy. The European Molecular Biology Organization Journal,34(9), 1214–1230. https://doi.org/10.15252/embj.201489920.
Dikic, I., & Elazar, Z. (2018). Mechanism and medical implications of mammalian autophagy. Nature Reviews Molecular Cell Biology,19(6), 349–364. https://doi.org/10.1038/s41580-018-0003-4.
Dotiwala, F., Eapen, V. V., Harrison, J. C., Arbel-Eden, A., Ranade, V., Yoshida, S., et al. (2013). DNA damage checkpoint triggers autophagy to regulate the initiation of anaphase. Proceedings of the National Academy of Sciences of the United States of America,110(1), 41–49. https://doi.org/10.1073/pnas.1218065109.
Eliopoulos, A. G., Havaki, S., & Gorgoulis, V. G. (2016). DNA damage response and autophagy: a meaningful partnership. Frontiers in Genetics,7(204), 1–13. https://doi.org/10.3389/fgene.2016.00204.
Fei, X., Xin, L., & Lili, Y. (2017). Autophagy promotes the repair of radiation-induced DNA damage in bone marrow hematopoietic cells via enhanced STAT3 signaling. Radiation Research,187(3), 382–396. https://doi.org/10.1667/RR14640.1.
Feng, Y., & Klionsky, D. J. (2017). Autophagy regulates DNA repair through SQSTM1/p62. Autophagy,13(6), 995–996. https://doi.org/10.1080/15548627.2017.1317427.
Feng, Z., Zhang, H., Levine, A. J., & Jin, S. (2005). The coordinate regulation of the p53 and mTOR pathways in cells. Proceedings of the National Academy of Sciences of the United States of America,102(23), 8204–8209.
Folini, M., Orlotti, N. I., Cimino-Reale, G., Borghini, E., Daidone, M. G., Palumbo, M., et al. (2012). Autophagy acts as a safeguard mechanism against G-quadruplex ligand-mediated telomere damage. Molecular Cancer Therapeutics,8(8), 1185–1196.
Galluzzi, L., Pietrocola, F., Bravo-San Pedro, J. M., Amaravadi, R. K., Baehrecke, E. H., Cecconi, F., et al. (2015). Autophagy in malignant transformation and cancer progression. The European Molecular Biology Organization Journal,34(7), 856–880. https://doi.org/10.15252/embj.201490784.
Gao, W., Shen, Z., Shang, L., & Wang, X. (2011). Upregulation of human autophagy-initiation kinase ULK1 by tumor suppressor p53 contributes to DNA-damage-induced cell death. Cell Death and Differenciation,18(10), 1598–1607. https://doi.org/10.1038/cdd.2011.33.
Gillespie, D. A., & Ryan, K. M. (2016). Autophagy is critically required for DNA repair by homologous recombination. Molecular and Cellular Oncology,3(1), e1030538. https://doi.org/10.1080/23723556.2015.1030538.
Goiran, T., Duplan, E., Rouland, L., El Manaa, W., Lauritzen, I., Dunys, J., et al. (2018). Nuclear p53-mediated repression of autophagy involves PINK1 transcriptional down-regulation. Cell Death and Differenciation,25(5), 873–884. https://doi.org/10.1038/s41418-017-0016-0.
Goldstein, M., & Kastan, M. B. (2015). The DNA damage response: implications for tumor responses to radiation and chemotherapy. Annual Review of Medicine,66, 129–143. https://doi.org/10.1146/annurev-med-081313-121208.
Gomes, L. R., Menck, C. F. M., & Leandro, G. S. (2017). Autophagy roles in the modulation of DNA repair pathways. International Journal of Molecular Sciences,18(11), 1–21. https://doi.org/10.3390/ijms18112351.
Hewitt, G., Carroll, B., Sarallah, R., Correia-Melo, C., Ogrodnik, M., Nelson, G., et al. (2016). SQSTM1/p62 mediates crosstalk between autophagy and the UPS in DNA repair. Autophagy,12(10), 1917–1930. https://doi.org/10.1080/15548627.2016.1210368.
Hewitt, G., & Korolchuk, V. I. (2017). Repair, reuse, recycle: the expanding role of autophagy in genome maintenance. Trends in Cell Biology,27(5), 340–351. https://doi.org/10.1016/j.tcb.2016.11.011.
Huang, R. X., & Zhou, P. K. (2020). DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduction and Targeted Therapy,5(1), 60. https://doi.org/10.1038/s41392-020-0150-x.
Karantza-Wadsworth, V., Patel, S., Kravchuk, O., Chen, G., Mathew, R., Jin, S., et al. (2007). Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes and Development,21(13), 1621–1635. https://doi.org/10.1101/gad.1565707.
Katayama, M., Kawaguchi, T., Berger, M. S., & Pieper, R. O. (2007). DNA damaging agent-induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells. Cell Death and Differenciation,14(3), 548–558. https://doi.org/10.1038/sj.cdd.4402030.
Katheder, N. S., Khezri, R., O’Farrell, F., Schultz, S. W., Jain, A., Rahman, M. M., et al. (2017). Microenvironmental autophagy promotes tumour growth. Nature,541(7637), 417–420. https://doi.org/10.1038/nature20815.
Kenzelmann, B. D., Spano, M. S., Bieging, K. T., Jiang, D., Dusek, R. L., Brady, C. A., et al. (2013). Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes and Development,27(9), 1016–1031. https://doi.org/10.1101/gad.212282.112.
Kim, J., Kundu, M., Viollet, B., & Guan, K. L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature Cell Biology,13(2), 132–141. https://doi.org/10.1038/ncb2152.
Knizhnik, A. V., Roos, W. P., Nikolova, T., Quiros, S., Tomaszowski, K. H., Christmann, M., et al. (2013). Survival and death strategies in glioma cells: autophagy, senescence and apoptosis triggered by a single type of temozolomide-induced DNA damage. PLoS ONE,8(1), e55665. https://doi.org/10.1371/journal.pone.0055665.
Ko, A., Kanehisa, A., Martins, I., Senovilla, L., Chargari, C., Dugue, D., et al. (2014). Autophagy inhibition radiosensitizes in vitro, yet reduces radioresponses in vivo due to deficient immunogenic signalling. Cell Death and Differenciation,21(1), 92–99. https://doi.org/10.1038/cdd.2013.124.
Kumar, D., Abdulovic, A. L., Viberg, J., Nilsson, A. K., Kunkel, T. A., & Chabes, A. (2011). Mechanisms of mutagenesis in vivo due to imbalanced dNTP pools. Nucleic Acids Research,39(4), 1360–1371. https://doi.org/10.1093/nar/gkq829.
Kuo, T. C., Chen, C. T., Baron, D., Onder, T. T., Loewer, S., Almeida, S., et al. (2011). Midbody accumulation through evasion of autophagy contributes to cellular reprogramming and tumorigenicity. Nature Cell Biology,13(10), 1214–1223. https://doi.org/10.1038/ncb2332.
Laggner, M., Pollreisz, A., Schmidinger, G., Schmidt-Erfurth, U., & Chen, Y. T. (2017). Autophagy mediates cell cycle response by regulating nucleocytoplasmic transport of PAX6 in limbal stem cells under ultraviolet-A stress. PLoS ONE,12(7), e0180868. https://doi.org/10.1371/journal.pone.0180868.
Levy, J. M. M., Towers, C. G., & Thorburn, A. (2017). Targeting autophagy in cancer. Nature Reviews Cancer,17(9), 528–542. https://doi.org/10.1038/nrc.2017.53.
Li, F., Zheng, L. D., Chen, X., Zhao, X., Briggs, S. D., & Du, H. N. (2017). Gcn5-mediated Rph1 acetylation regulates its autophagic degradation under DNA damage stress. Nucleic Acids Research,45(9), 5183–5197. https://doi.org/10.1093/nar/gkx129.
Li, L., Tan, J., Miao, Y., Lei, P., & Zhang, Q. (2015). ROS and autophagy: interactions and molecular regulatory mechanisms. Cellular and Molecular Neurobiology,35(5), 615–621. https://doi.org/10.1007/s10571-015-0166-x.
Li, Y., Fengyi, W., Sudeshna, D., & Sarah, W. (2006). Autophagic programmed cell death by selective catalase degradation. Proceedings of the National Academy of Sciences of the United States of America,103, 49–52.
Lin, X., Han, L., Weng, J., Wang, K., & Chen, T. (2018). Rapamycin inhibits proliferation and induces autophagy in human neuroblastoma cells. Bioscience Reports. https://doi.org/10.1042/bsr20181822.
Liu, E. Y., Xu, N., O'Prey, J., Lao, L. Y., Joshi, S., Long, J. S., et al. (2015). Loss of autophagy causes a synthetic lethal deficiency in DNA repair. Proceedings of the National Academy of Sciences of the United States of America,112(3), 773–778. https://doi.org/10.1073/pnas.1409563112.
Liu, W. J., Ye, L., Huang, W. F., Guo, L. J., Xu, Z. G., Wu, H. L., et al. (2016). p62 links the autophagy pathway and the ubiquitin-proteasome system upon ubiquitinated protein degradation. Cell and Molecular Biology Letters,21(29), 1–14. https://doi.org/10.1186/s11658-016-0031-z.
Lorat, Y., Brunner, C., Schanz, S., Jakob, B., & Taucher-Scholz, G. (2015). Nanoscale analysis of clustered DNA damage after high-LET irradiation by quantitative electron microscopy—The heavy burden to repair. DNA Repair,28, 93–106. https://doi.org/10.1016/j.dnarep.2015.01.007.
Luo, M., Zhao, X., Song, Y., Cheng, H., & Zhou, R. (2016). Nuclear autophagy: an evolutionarily conserved mechanism of nuclear degradation in the cytoplasm. Autophagy,12(11), 1973–1983. https://doi.org/10.1080/15548627.2016.1217381.
Maiuri, M. C., Galluzzi, L., Morselli, E., Kepp, O., Malik, S. A., & Kroemer, G. (2010). Autophagy regulation by p53. Current Opinion in Cell Biology,22(2), 181–185. https://doi.org/10.1016/j.ceb.2009.12.001.
Mathew, R., Karp, C. M., Beaudoin, B., Vuong, N., Chen, G., Chen, H. Y., et al. (2009). Autophagy suppresses tumorigenesis through elimination of p62. Cell,137(6), 1062–1075. https://doi.org/10.1016/j.cell.2009.03.048.
Mathew, R., Kongara, S., Beaudoin, B., Karp, C. M., Bray, K., Degenhardt, K., et al. (2007). Autophagy suppresses tumor progression by limiting chromosomal instability. Genes and Development,21(11), 1367–1381. https://doi.org/10.1101/gad.1545107.
Matt, S., & Hofmann, T. G. (2016). The DNA damage-induced cell death response: a roadmap to kill cancer cells. Cellular and Molecular Life Sciences,73(15), 2829–2850. https://doi.org/10.1007/s00018-016-2130-4.
Menolfi, D., & Zha, S. (2020). ATM, DNA-PKcs and ATR: shaping development through the regulation of the DNA damage responses. Genome Instability and Disease,1(2), 47–68. https://doi.org/10.1007/s42764-019-00003-9.
Mizushima, N. (2007). Autophagy: process and function. Genes and Development,21(22), 2861–2873. https://doi.org/10.1101/gad.1599207.
Mizushima, N., Yoshimori, T., & Ohsumi, Y. (2011). The role of Atg proteins in autophagosome formation. Annual Review of Cell and Developmental Biology,27(1), 107–132. https://doi.org/10.1146/annurev-cellbio-092910-154005.
Mo, N., Lu, Y. K., Xie, W. M., Liu, Y., Zhou, W. X., Wang, H. X., et al. (2014). Inhibition of autophagy enhances the radiosensitivity of nasopharyngeal carcinoma by reducing Rad51 expression. Oncology Reports,32(5), 1905–1912. https://doi.org/10.3892/or.2014.3427.
Moldovan, G. L., Madhavan, M. V., Mirchandani, K. D., McCaffrey, R. M., Vinciguerra, P., & D'Andrea, A. D. (2010). DNA polymerase POLN participates in cross-link repair and homologous recombination. Molecular and Cellular Biology,30(4), 1088–1096. https://doi.org/10.1128/MCB.01124-09.
Moriwaki, T., Kato, Y., Nakamura, C., Ishikawa, S., & Zhang-Akiyama, Q. M. (2015). A novel DNA damage response mediated by DNA mismatch repair in Caenorhabditis elegans: induction of programmed autophagic cell death in non-dividing cells. Genes Cancer,6(7–8), 341–355. https://doi.org/10.18632/genesandcancer.70.
Morselli, E., Shen, S., Ruckenstuhl, C., Bauer, M. A., Marino, G., Galluzzi, L., et al. (2011). p53 inhibits autophagy by interacting with the human ortholog of yeast Atg17, RB1CC1/FIP200. Cell Cycle,10(16), 2763–2769. https://doi.org/10.4161/cc.10.16.16868.
Mortensen, M., Soilleux, E. J., Djordjevic, G., Tripp, R., Lutteropp, M., Sadighi-Akha, E., et al. (2011). The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. Journal of Experimental Medicine,208(3), 455–467. https://doi.org/10.1084/jem.20101145.
Murcia, J. M. D., Niedergang, C., & Trucco, C. (1997). Requirement of poly (ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proceedings of the National Academy of Sciences of the United States of America,94(14), 7303–7307.
Nazio, F., Maiani, E., & Cecconi, F. (2018). The cross talk among autophagy, ubiquitination, and DNA repair: an overview. Ubiquitination Governing DNA Repair—Implications in Health and Disease, Chapter,6, 103–122. https://doi.org/10.5772/intechopen.71404.
Nezis, I. P., Shravage, B. V., Sagona, A. P., Lamark, T., Bjorkoy, G., Johansen, T., et al. (2010). Autophagic degradation of dBruce controls DNA fragmentation in nurse cells during late Drosophila melanogaster oogenesis. Journal of Cell Biology,190(4), 523–531. https://doi.org/10.1083/jcb.201002035.
Nissanka, N., & Moraes, C. T. (2018). Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Letters,592(5), 728–742. https://doi.org/10.1002/1873-3468.12956.
Nordlund, P., & Reichard, P. (2006). Ribonucleotide reductases. Annual Review of Biochemistry,75(1), 681–706. https://doi.org/10.1146/annurev.biochem.75.103004.142443.
Pankiv, S., Lamark, T., Bruun, J. A., Overvatn, A., Bjorkoy, G., & Johansen, T. (2010). Nucleocytoplasmic shuttling of p62/SQSTM1 and its role in recruitment of nuclear polyubiquitinated proteins to promyelocytic leukemia bodies. Journal of Biological Chemistry,285(8), 5941–5953. https://doi.org/10.1074/jbc.M109.039925.
Park, C., Suh, Y., & Cuervo, A. M. (2015). Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nature Communications,16(6), 6823. https://doi.org/10.1038/ncomms7823.
Park, J., Tougeron, D., Huang, S., Okamoto, K., & Sinicrope, F. (2014). Beclin 1 and UVRAG confer protection from radiation-induced DNA damage and maintain centrosome stability in colorectal cancer cells. PLoS ONE,9(6), e100819. https://doi.org/10.1371/journal.pone.0100819.
Park, Y. E., Hayashi, Y. K., Bonne, G., Arimura, T., Noguchi, S., Nonaka, I., et al. (2009). Autophagic degradation of nuclear components in mammalian cells. Autophagy,5(6), 795–804. https://doi.org/10.4161/auto.8901.
Peoples, J. N., Saraf, A., Ghazal, N., Pham, T. T., & Kwong, J. Q. (2019). Mitochondrial dysfunction and oxidative stress in heart disease. Experimental & Molecular Medicine,51(12), 1–13. https://doi.org/10.1038/s12276-019-0355-7.
Pérez-Hernández, M., Arias, A., Martínez-García, D., Pérez-Tomás, R., Quesada, R., & Soto-Cerrato, V. (2019). Targeting autophagy for cancer treatment and tumor chemosensitization. Cancers (Basel),11(10), 1599. https://doi.org/10.3390/cancers11101599.
Perillo, B., Di Donato, M., Pezone, A., Di Zazzo, E., Giovannelli, P., Galasso, G., et al. (2020). ROS in cancer therapy: the bright side of the moon. Experimental & Molecular Medicine,52(2), 192–203. https://doi.org/10.1038/s12276-020-0384-2.
Pitolli, C., Wang, Y., Candi, E., Shi, Y., Melino, G., & Amelio, I. (2019). p53-mediated tumor suppression: DNA-damage response and alternative mechanisms. Cancers,11(12), 1–14. https://doi.org/10.3390/cancers11121983.
Prokhorova, E. A., Egorshina, A. Y., Zhivotovsky, B., & Kopeina, G. S. (2020). The DNA-damage response and nuclear events as regulators of nonapoptotic forms of cell death. Oncogene,39(1), 1–16. https://doi.org/10.1038/s41388-019-0980-6.
Qu, X., Zou, Z., Sun, Q., Luby-Phelps, K., Cheng, P., Hogan, R. N., et al. (2007). Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell,128(5), 931–946. https://doi.org/10.1016/j.cell.2006.12.044.
Rangwala, R., Chang, Y., Hu, J., Algazy, K., Evans, T., Fecher, L., et al. (2014). Combined MTOR and autophagy inhibition. Autophagy,10(8), 1391–1402. https://doi.org/10.4161/auto.29119.
Rello-Varona, S., Lissa, D., Shen, S., Niso-Santano, M., Senovilla, L., Marino, G., et al. (2012). Autophagic removal of micronuclei. Cell Cycle,11(1), 170–176. https://doi.org/10.4161/cc.11.1.18564.
Richardson, A. G., & Schadt, E. E. (2014). The role of macromolecular damage in aging and age-related disease. Journals of Gerontology Series A-Biological Sciences and Medical Sciences,69(Suppl 1), 28–32. https://doi.org/10.1093/gerona/glu056.
Robert, T., Vanoli, F., Chiolo, I., Shubassi, G., Bernstein, K. A., Rothstein, R., et al. (2011). HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature,471(7336), 74–79. https://doi.org/10.1038/nature09803.
Rodriguez-Vargas, J. M., Ruiz-Magana, M. J., Ruiz-Ruiz, C., Majuelos-Melguizo, J., Peralta-Leal, A., Rodriguez, M. I., et al. (2012). ROS-induced DNA damage and PARP-1 are required for optimal induction of starvation-induced autophagy. Cell Research,22(7), 1181–1198. https://doi.org/10.1038/cr.2012.70.
Roos, W. P., & Kaina, B. (2013). DNA damage-induced cell death: from specific DNA lesions to the DNA damage response and apoptosis. Cancer Letters,332(2), 237–248. https://doi.org/10.1016/j.canlet.2012.01.007.
Roos, W. P., Thomas, A. D., & Kaina, B. (2016). DNA damage and the balance between survival and death in cancer biology. Nature Reviews Cancer,16(1), 20–33. https://doi.org/10.1038/nrc.2015.2.
Rosenfeld, M., Ye, X., Supko, J., Desideri, S., Grossman, S., Brem, S., et al. (2014). A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy,10(8), 1359–1368. https://doi.org/10.4161/auto.28984.
Roshani-Asl, E., Mansori, B., Mohammadi, A., Najafi, S., Danesh-Pouya, F., & Rasmi, Y. (2020). Interaction between DNA damage response and autophagy in colorectal cancer. Gene,730(1), 144323. https://doi.org/10.1016/j.gene.2019.144323.
SenGupta, T., Torgersen, M. L., Kassahun, H., Vellai, T., Simonsen, A., & Nilsen, H. (2013). Base excision repair AP endonucleases and mismatch repair act together to induce checkpoint-mediated autophagy. Nature Communications,4(1), 2674. https://doi.org/10.1038/ncomms3674.
Shi, H., Zhang, L., Zhang, C., Hao, Y., & Zhao, X. (2019). Rapamycin may inhibit murine S180 sarcoma growth by regulating the pathways associated with autophagy and cancer stem cells. J Cancer Res Ther,15(2), 398–403. https://doi.org/10.4103/jcrt.JCRT_639_18.
Shoji, J. Y., Kikuma, T., Arioka, M., & Kitamoto, K. (2010). Macroautophagy-mediated degradation of whole nuclei in the filamentous fungus Aspergillus oryzae. PLoS ONE,5(12), e15650. https://doi.org/10.1371/journal.pone.0015650.
Shubassi, G., Robert, T., Vanoli, F., Minucci, S., & Foiani, M. (2012). Acetylation: a novel link between double-strand break repair and autophagy. Cancer Research,72(6), 1332–1335. https://doi.org/10.1158/0008-5472.CAN-11-3172.
Siggens, L., Figg, N., Bennett, M., & Foo, R. (2012). Nutrient deprivation regulates DNA damage repair in cardiomyocytes via loss of the base-excision repair enzyme OGG1. Faseb Journal Official Publication of the Federation of American Societies for Experimental Biology,26(5), 2117–2124. https://doi.org/10.1096/fj.11-197525.
Smith, J., Tho, L. M., & Xu, N. (2010). The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Advances in Cancer Research,108(C), 73–112. https://doi.org/10.1016/B978-0-12-380888-2.00003-0.
Song, X., Narzt, M. S., Nagelreiter, I. M., Hohensinner, P., Terlecki-Zaniewicz, L., Tschachler, E., et al. (2017). Autophagy deficient keratinocytes display increased DNA damage, senescence and aberrant lipid composition after oxidative stress in vitro and in vivo. Redox Biology,11, 219–230. https://doi.org/10.1016/j.redox.2016.12.015.
Surova, O., & Zhivotovsky, B. (2013). Various modes of cell death induced by DNA damage. Oncogene,32(33), 3789–3797. https://doi.org/10.1038/onc.2012.556.
Tasdemir, E., Maiuri, M. C., Galluzzi, L., Vitale, I., Djavaheri-Mergny, M., D'Amelio, M., et al. (2008). Regulation of autophagy by cytoplasmic p53. Nature Cell Biology,10(6), 676–687. https://doi.org/10.1038/ncb1730.
van Attikum, H., & Gasser, S. M. (2005). ATP-dependent chromatin remodeling and DNA double-strand break repair. Cell Cycle,4(8), 1011–1014. https://doi.org/10.4161/cc.4.8.1887.
Vessoni, A. T., Filippi-Chiela, E. C., Menck, C. F., & Lenz, G. (2013). Autophagy and genomic integrity. Cell Death and Differentiation,20(11), 1444–1454. https://doi.org/10.1038/cdd.2013.103.
Vogl, D., Stadtmauer, E., Tan, K.-S., Heitjan, D., Davis, L., Pontiggia, L., et al. (2014). Combined autophagy and proteasome inhibition. Autophagy,10(8), 1380–1390. https://doi.org/10.4161/auto.29264.
Wang, L., Howell, M. E. A., Sparks-Wallace, A., Hawkins, C., Nicksic, C. A., Kohne, C., et al. (2019). p62-Mediated selective autophagy endows virus-transformed cells with insusceptibility to DNA damage under oxidative stress. PLoS Pathogens,15(4), e1007541. https://doi.org/10.1371/journal.ppat.1007541.
Wang, Y., Zhang, N., Zhang, L., Li, R., Fu, W., Ma, K., et al. (2016). Autophagy regulates chromatin ubiquitination in DNA damage response through elimination of SQSTM1/p62. Molecular Cell,63(1), 34–48. https://doi.org/10.1016/j.molcel.2016.05.027.
Wei, Y., Sinha, S., & Levine, B. (2008). Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy,4(7), 949–951. https://doi.org/10.4161/auto.6788.
Wirawan, E. V. W., Walle, L., Kersse, K., Cornelis, S., Claerhout, S., Vanoverberghe, I., et al. (2010). Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death and Disease,1(1), e18. https://doi.org/10.1038/cddis.2009.16.
Yang, Y., & Klionsky, D. J. (2020). Autophagy and disease: unanswered questions. Cell Death and Differentiation,27(3), 858–871. https://doi.org/10.1038/s41418-019-0480-9.
Zhang, X., Qin, Z., & Wang, J. (2010). The role of p53 in cell metabolism. Acta Pharmacologica Sinica,31(9), 1208–1212. https://doi.org/10.1038/aps.2010.151.
Zhang, Z., Ren, Z., Chen, S., Guo, X., Liu, F., Guo, L., et al. (2018). ROS generation and JNK activation contribute to 4-methoxy-TEMPO-induced cytotoxicity, autophagy, and DNA damage in HepG2 cells. Archives of Toxicology,92(2), 717–728. https://doi.org/10.1007/s00204-017-2084-9.
Author information
Affiliations
Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education), Beijing Key Laboratory of Protein Posttranslational Modifications and Cell Function, Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Peking University Health Science Center, Beijing, 100191, China
Congting Guo & Ying Zhao
Corresponding author
Correspondence to Ying Zhao.
Rights and permissions
About this article
Cite this article
Guo, C., Zhao, Y. Autophagy and DNA damage repair. GENOME INSTAB. DIS. 1, 172–183 (2020). https://doi.org/10.1007/s42764-020-00016-9
Received 07 May 2020
Revised 01 June 2020
Accepted 17 June 2020
Published 30 June 2020
Issue Date July 2020
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
Autophagy
DNA damage repair
Genome stability
Cell fate
用户登录
还没有账号?
立即注册