






ATM-deficient lung, prostate and pancreatic cancer cells are acutely sensitive to the combination of olaparib and the ATR inhibitor AZD6738
Short Communication Open Access
Nicholas R. Jette, Suraj Radhamani, Ruiqiong Ye, Yaping Yu, Greydon Arthur, Siddhartha Goutam, Tarek A. Bismar, Mehul Kumar, Pinaki Bose, Steven Yip, Michael Kolinsky & Susan P. Lees-Miller
Genome Instability & Disease 1,197–205(2020)
Abstract
The Ataxia Telangiectasia Mutated (ATM) protein kinase is mutated in several human cancers, presenting potential opportunities for targeted cancer therapy. We previously reported that the poly-ADP-ribose polymerase (PARP) inhibitor olaparib induces transient G2 arrest but not cell death in ATM-deficient lung cancer cells, while the combination of olaparib with the ATM- and Rad3-related (ATR) inhibitor VE-821 induced cell death. Here, we show that combination of olaparib plus the clinically relevant ATR inhibitor AZD6738 also induces cell death in ATM-deficient lung, prostate and pancreatic cancer cells with little effect on their ATM-proficient counterparts. Together, our data suggest that lung, prostate and pancreatic patients whose tumours exhibit loss or inactivation of ATM may benefit from combination of a PARP inhibitor plus an ATR inhibitor.
Introduction
A goal of precision oncology is to achieve better tumour control by targeting tumours based on their specific genetic or epigenetic characteristics. Perhaps one of the best examples of such an approach is the development of poly-ADP-ribose polymerase (PARP) inhibitors to target tumours with defects in DNA damage response (DDR) genes (Pommier et al. 2016). PARP inhibitors were first shown to target breast cancer cells with depletion of the breast and ovarian cancer susceptibility proteins BRCA1 and BRCA2 (Bryant et al. 2005; Farmer et al. 2005) and are now FDA approved for use in BRCA-deficient ovarian cancer with clinical trials showing potential in BRCA-deficient breast, prostate and pancreatic cancers (Lord and Ashworth 2017; Coleman et al. 2017; Golan et al. 2019; Mateo et al. 2019; Mirza et al. 2016; Moore et al. 2018; Robson et al. 2017; Mateo et al. 2015). BRCA1 and 2 play important roles in the detection and repair of DNA double-strand breaks (DSBs) through the homologous recombination (HR) repair pathway (Boulton 2006), leading to the idea that cells with deficiency in DSB repair pathways may be sensitive to PARP inhibitors. Indeed, siRNA depletion of other proteins involved in the cellular response to DSBs, including Ataxia Telangiectasia Mutated (ATM), also sensitized cancer cells to PARP inhibition (McCabe et al. 2006; Bryant and Helleday 2006).
ATM is an apex signalling kinase that plays a central role in the cellular response to DNA damage (Shiloh 2014). Loss of both copies of ATM leads to ataxia telangiectasia (A-T), a devastating childhood condition characterized by neurological degeneration, cancer predisposition and immune deficiencies (Shiloh and Lederman 2016; Rothblum-Oviatt et al. 2016). In addition, ATM is mutated in a variety of human cancers (reviewed in (Choi et al. 2016; Jette et al. 2020)), leading us to hypothesize that PARP inhibitors could have therapeutic potential in cancers with loss or mutation of ATM. Indeed, up to 40% of mantle cell lymphoma (MCL) have loss of ATM (Greiner et al. 2006) and we showed that ATM-deficient MCL cells are sensitive to the PARP inhibitor olaparib in both cell line and animal models (Williamson et al. 2010). Similarly, ATM-deficient chronic lymphocytic leukaemia (CLL) cells are sensitive to PARP inhibitors (Weston et al. 2010). PARP inhibitor sensitivity in ATM-deficient MCL cells was enhanced when p53 was also deleted or inactivated and p53-deficient MCL cell lines were sensitive to PARP inhibitors in the presence of an ATM kinase inhibitor, KU55933 (Williamson et al. 2012). ATM has also been reported to be mutated in ~ 12.5% of gastric cancers (Kim et al. 2013a, b) and we found that p53-deficient gastric cancer cell lines with shRNA-mediated depletion of ATM, or in which ATM was inhibited using KU55933, were also sensitive to olaparib (Kubota et al. 2014). Similarly, ATM is mutated in approximately 16% of colorectal cancers and we showed that ATM-deficient colorectal cancer cell lines are sensitive to olaparib and this sensitivity was enhanced when TP53 was also deleted (Wang et al. 2017). In these studies, olaparib sensitivity was inferred from either trypan blue exclusion assays (Williamson et al. 2010, 2012) or clonogenic survival assays (Kubota et al. 2014; Wang et al. 2017), however, the mechanism of olaparib-induced sensitivity in these cell lines was unclear.
Recently, we deleted ATM from p53-proficient A549 cells using CRISPR/Cas9 and showed that olaparib reduced cell proliferation and caused reversible G2 arrest but not cell death in ATM-deficient A549 cells (Jette et al. 2019). We hypothesized that addition of an ATR inhibitor would abolish the G2 checkpoint and lead to cell death. Indeed, we found that addition of the ATR inhibitor VE-821 led to apoptosis in olaparib-treated ATM-deficient A549 cells, while no cell death was detected in isogenic, olaparib and VE821-treated ATM-proficient cells (Jette et al. 2019). These studies suggested that combination of a PARP inhibitor and an ATR inhibitor could be an effective treatment for ATM-deficient tumour cells, however, it was important to determine whether the same effect was observed in ATM-deficient cell lines from other cancer types.
Prostate cancer is the most commonly diagnosed cancer in men in Canada (Canadian Cancer Statistics 2019), accounting for almost 10% of all cancer deaths amongst males. Next-generation sequencing has indicated that up to 10% of prostate cancers harbour mutations in at least one DDR gene, with BRCA2 and ATM being the most commonly mutated (Mateo et al. 2017). Moreover, the results of the recent TOPARP-B clinical trial support a role for olaparib in metastatic castration-resistant prostate cancer with DDR gene aberrations (Mateo et al. 2019).
Pancreatic cancer is one of the most lethal forms of cancer with a 10-year survival of less than 10% and accounts for approximately 6% of all cancer deaths in Canadians (Canadian Cancer Statistics 2019). ATM is mutated in some hereditary forms of pancreatic cancer (Grant et al. 2014; Roberts et al. 2012) as well as ~ 6% of sporadic pancreatic adenocarcinoma, with 4% of overall mutations resulting in deletion of the ATM protein (Kim et al. 2014). In patients with metastatic pancreatic cancer and germline BRCA1/2 mutations, olaparib maintenance treatment following first-line platinum-based chemotherapy resulted in a significant improvement in progression-free survival, in contrast to placebo (Golan et al. 2019).
The encouraging evidence of clinical efficacy of therapy targeting the heterogeneous group of DDR alterations in these tumour groups (Moore et al. 2018; Mirza et al. 2016; Robson et al. 2017; Golan et al. 2019; Mateo et al. 2015, 2019) prompted us to ask whether prostate and pancreatic cancer cell lines with ATM deficiency would be sensitive to PARP inhibitor either alone or in combination with an ATR inhibitor. We depleted ATM from prostate and pancreatic cancer cell lines and show that, as in lung cancer cells, olaparib alone reduces cell proliferation but that addition of the ATR inhibitor, AZD6738 (Foote et al. 2015, 2018) is required to induce apoptosis in ATM-deficient prostate and pancreatic cancer cells. Importantly, no cell death was observed in isogenic ATM-proficient cells treated with olaparib or AZD6738 alone or in combination.
Materials and methods
Cell lines
A549 lung adenocarcinoma, Panc 10.05 pancreatic cancer and PC-3 prostate cancer cell lines were obtained from ATCC and checked regularly for mycoplasma. Cells were maintained in a humidified incubator in the presence of 5% CO2 at 37 °C in either Dulbecco’s Modified Eagle Medium (DMEM) (ThermoFisher Scientific, MA, USA) plus 10% (w/v) Hyclone Fetalclone III Serum (A549 cells), F-12K nutrient mixture (1 ×) Kaighn’s modification (ThermoFisher Scientific, MA, USA) with 10% (w/v) fetal bovine serum (FBS) (PC-3), or RPMI 1649 (1 ×) (ThermoFisher Scientific, MA, USA) in the presence of 15% (w/v) Hyclone Fetalclone III Serum (Panc 10.05 cells). All media was supplemented with 50 μg/mL penicillin–streptomycin (Gibco, ThermoFisher Scientific). BT and L3 cells were cultured as described previously (Wang et al. 2017).
Immunoblots
Cells were harvested by NETN lysis as described previously (Jette et al. 2019). The antibody to ATM was from Upstate (#05-513), the antibody to Ku was from Abcam (#33242), and antibodies to DNA-PKcs were generated in house as described previously (Douglas et al. 2010).
Generation of ATM-deficient cell lines
A549 cells with CRISPR/Cas9 deletion of ATM or DNA-PKcs have been described previously (Jette et al. 2019). Stable knock down of ATM in pancreatic cancer Panc 10.05 cells was achieved using shRNA as described previously (Elkon et al. 2005; Williamson et al. 2010). For CRISPR/Cas9 depletion of ATM in PC-3 cells, three short guide RNAs targeting exons 9, 16 and 23 of human ATM were designed, synthesized and ligated into pSpCas9 (BB)-2A-GFP (pX458) vector, a gift from Dr. Feng Zhang (Addgene plasmid #48138). Constructs were transfected into PC-3 cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s recommended procedures and GFP-containing cells were sorted by flow cytometry. Genomic DNA was isolated from wild-type cells and PC-3 cells transfected with sgRNA constructs targeting ATM exons 9, 16 or 23. The DNA fragment around the target site was amplified by PCR and PCR products were cleaved by Surveyor Nuclease S as per the manufacturer’s recommended conditions. Surveyor Nuclease S assay confirmed the presence of CRISPR activity in exon 9, 16 and 23 sgRNA constructs. TIDE analysis showed that over 90% of cells transfected with the sgRNA construct targeting exon 16 contained insertion/deletion mutations. No cells were recovered from cells transfected with the sgRNA construct targeting exon 9 (out of over 800 colonies screened) or targeting exon 23 (out of over 800 colonies screened). Only one clone, PC-3 E16 was recovered from the sgRNA construct targeting exon 16 (out of 2400 colonies screened). Clone PC-3-E16 was expanded and DNA sequencing revealed that one allele of ATM had a one nucleotide (Adenine) deletion in exon 16 which created a premature stop codon, while the other allele had a 12-nucleotide deletion in exon 16 predicted to cause a one amino acid mutation and a four amino acid deletion. This clone was used for all subsequent experiments.
Inhibitors
Olaparib and AZD6738 were purchased from Selleck Chemicals, dissolved in DMSO and stored as stock solutions at 10 mM at − 20 °C and − 80 °C, respectively.
Trypan blue viability assays
Trypan blue assays were carried out using trypan blue solution (BioRad) and a TC-20 automated cell counter (BioRad) according to the manufacturers’ instructions. Briefly, 50,000–100,000 cells were seeded for each experiment. Cells were allowed to attach for 16 h, then treated with either 1 μM olaparib, 0.3 μM AZD6738, a combination of olaparib (1 µM) and AZD6738 (0.3 µM) or an equivalent volume of DMSO. Cells were harvested after 48, 96, 120 and 144 h. The mean with SEM of three independent experiments is shown. Statistical significance was determined by one-way ANOVA. p values of less than 0.05 were considered statistically significant and are indicated by an asterisk.
Annexin assays
Cells were seeded at 50,000–100,000 cells per 6 cm dish, treated as above and then analysed by annexin staining and flow cytometry using an Annexin Apoptosis assay (ThermoFisher Scientific Cat#V13241). Cells were harvested after 48, 96, 120 and 144 h. The mean with SEM of three independent experiments is shown and statistical significance was determined by one-way ANOVA. p values of less than 0.05 were considered statistically significant and are indicated by an asterisk.
Results
ATM-deficient A549 lung cancer cells are sensitive to the combination of olaparib and AZD6738
ATM or the related protein kinase DNA-PKcs (DNA-dependent protein kinase catalytic subunit) were deleted from A549 lung cancer cells as described previously (Jette et al. 2019). Western blot confirmed that no ATM protein was detected in the A549–CRISPR–ATM cells compared to A549 control cells (Fig. 1a). As reported previously, CRISPR deletion of DNA-PKcs also resulted in a significant reduction in ATM expression, however, this did not translate into sensitivity to olaparib (Jette et al. 2019) (Fig. 1a, b). We next treated the ATM-deficient, DNA-PKcs-deficient and control A549 cells with olaparib (1 µM), AZD6738 (0.3 µM) or a combination of both inhibitors and the number of viable cells was determined using the trypan blue exclusion assay. The number of viable control A549 cells was unaffected by incubation with either inhibitor alone or by a combination of both inhibitors (Fig. 1b, c), although as reported previously, A549–CRISPR–DNA-PKcs cells grew more slowly than control cells (Jette et al. 2019; Ruis et al. 2008) (Fig. 1b). Incubation with olaparib or AZD6738 alone decreased the number of viable ATM-deficient cells by about 50% at the 114-h time point, whereas incubation with both compounds decreased the number of viable cells by about 90% at the 114-h time point (Fig. 1b, c). However, as shown previously for VE-821 (Jette et al. 2019), neither olaparib nor AZD6738 induced apoptosis as single agents, suggesting that the decrease in the number of viable cells is due to a decrease in cell proliferation rather than an increase in cell death. In contrast, the combination of olaparib plus AZD6738 induced apoptosis in the ATM-deficient cells as determined by annexin staining, while leaving control ATM-proficient and DNA-PKcs-deficient cells unaffected (Fig. 1d). Thus, as with the ATR inhibitor VE-821 (Jette et al. 2019), the combination of olaparib plus an ATR inhibitor (AZD6738) induces apoptosis in ATM-deficient A549 cells, while ATM-competent cells are relatively unaffected by either agent alone or in combination.
Fig. 1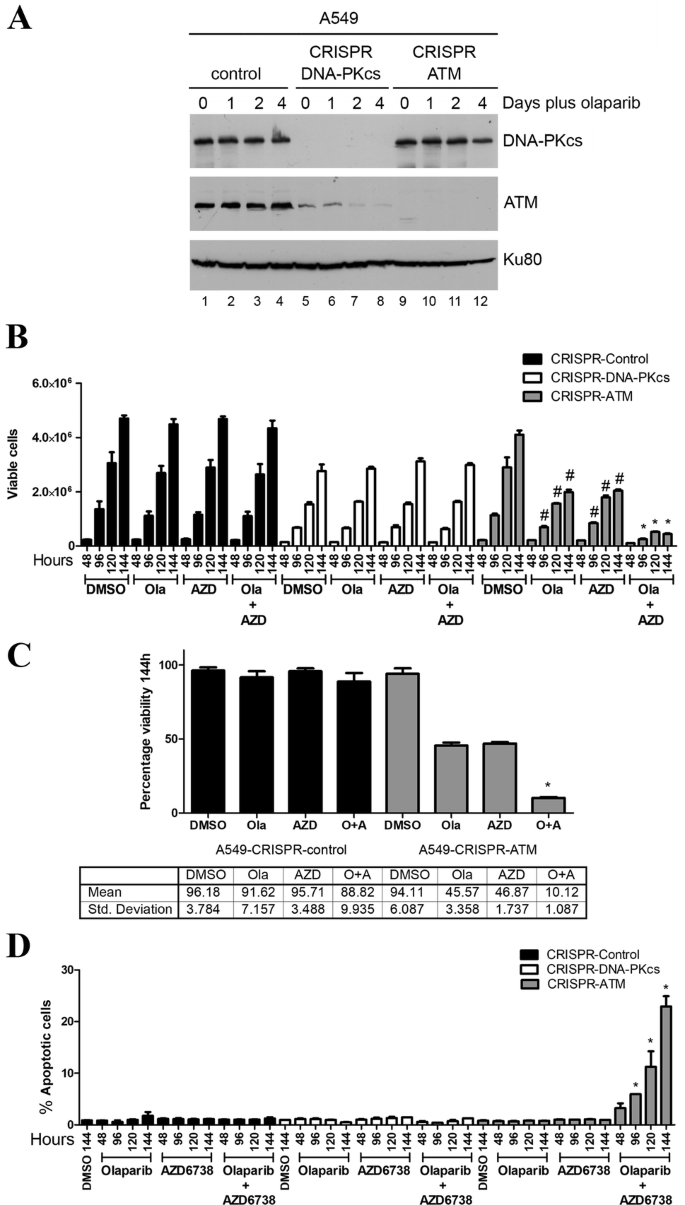
ATM-deficient A549 lung cancer cells undergo cell death when incubated with olaparib plus the ATR inhibitor AZD6738 but not either agent alone. a A549–CRISPR–control, A549–CRISPR–DNA-PKcs and A549–CRISPR–ATM cells were incubated with olaparib (1 µM) for 0, 1, 2 or 4 days then harvested by NETN lysis and 50 µg total protein was run on SDS PAGE and immunoblotted for DNA-PKcs, ATM or Ku80 (loading control) as indicated on the right. b–d A549–CRISPR–control (black bars), A549–CRISPR–DNA-PKcs (white bars) and A549–CRISPR–ATM cells (grey bars) were incubated with olaparib (1 µM), AZD6738 (0.3 µM) or both olaparib (Ola) and AZD6738 (Azd) for 48–144 h and assayed for either cell viability using the trypan blue exclusion assay (b, c) or apoptosis using annexin staining (d). Control samples were incubated with an equal volume of DMSO. c Quantitation of a at the 144-h time point for ATM-proficient (left) and ATM-deficient (right) lung cancer cell lines. O represents olaparib and A represents AZD6738. Results show the mean with SEM of three separate experiments. Statistical significance was determined by one-way ANOVA. *p < 0.05 when compared to other treatment groups of that cell line and the DMSO control. In b, #p < 0.05 when compared to DMSO control of that cell line only
Full size imageATM-deficient PC-3 prostate cancer cells are sensitive to the combination of olaparib and AZD6738
We next depleted ATM from PC-3 cells using CRISPR/Cas9 (Fig. 2a). PC-3 cells are derived from bone metastasis of a prostate cancer patient and have homozygous TP53 mutation. No mutations are reported for ATM, BRCA1 or 2 or PTEN in the Catalogue of Somatic Mutations in Cancer (COSMIC) database (Bamford et al. 2004; Forbes et al. 2011), however, PC-3 cells do not express PTEN protein (Fraser et al. 2012), possibly due to promoter methylation. Despite multiple attempts, we were unable to obtain complete deletion of ATM using CRISPR/Cas9, but we isolated one clone, E16-1, in which one allele of ATM had a one nucleotide (adenine) deletion in exon 16 which created a premature stop codon, while the other allele had a 12-nucleotide deletion in exon 16 predicted to cause a one amino acid mutation and a four amino acid deletion. Western blot indicated that this clone expressed very low levels of ATM protein, approximately 10% of that in control PC-3 cells (Fig. 2a). ATM-deficient and control PC-3 cells were treated with olaparib (1 µM), AZD6738 (0.3 µM) or a combination of both inhibitors and assayed for the number of viable cells and cell death as above. The decrease in viable cells at the 144-h time point in the ATM-deficient cell lines was 28% and 39% for olaparib and AZD6738 alone, respectively, and 83% for the combination of olaparib plus AZD6738 (Fig. 2b, c). Importantly, cell death, as determined by the annexin staining, was only observed in ATM-deficient PC-3 cells treated with both olaparib and AZD6738 (Fig. 2d), so, as in ATM-deficient lung cancer cells, olaparib alone reduced proliferation without inducing cell death while the combination of olaparib plus AZD6738 induced cell death.
Fig. 2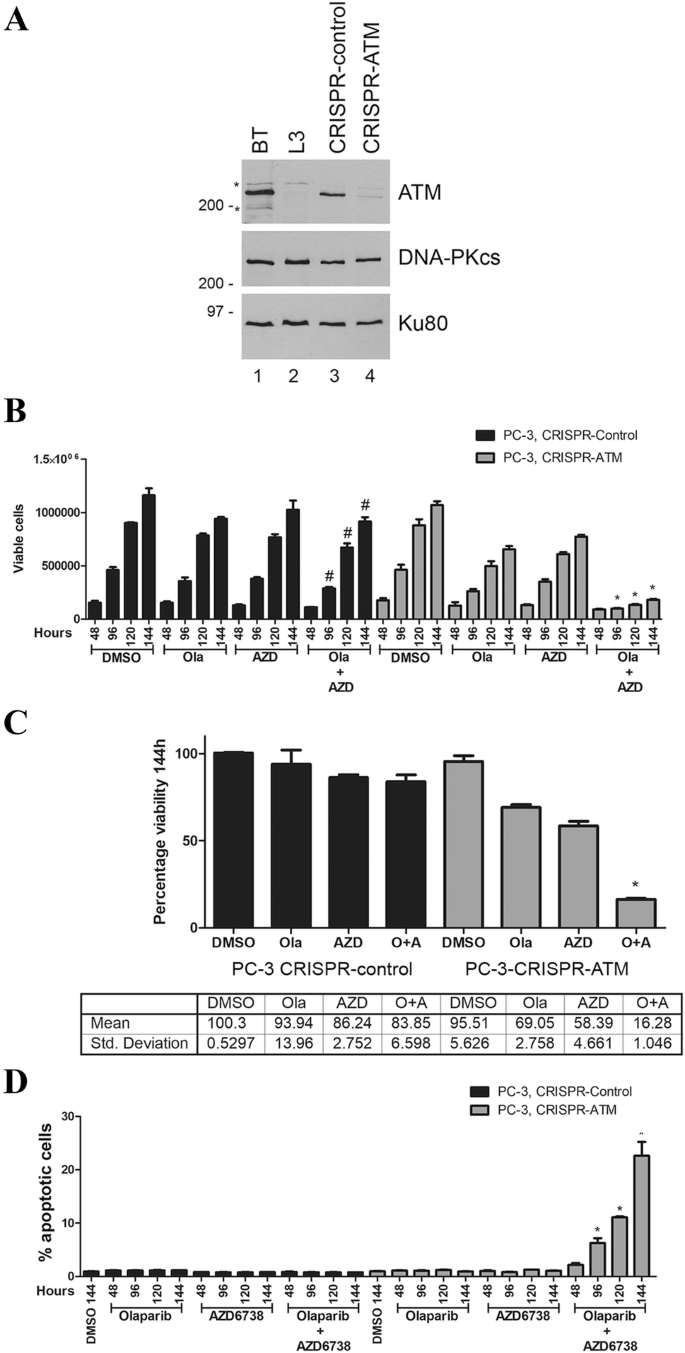
ATM-deficient PC-3 prostate cancer cells undergo cell death when incubated with olaparib plus the ATR inhibitor AZD6738 but not either agent alone. a ATM was depleted from PC-3 prostate cancer cells using CRISPR/Cas9 as described above. Extracts were generated by NETN lysis and 50 µg total protein run on SDS PAGE and immunoblotted for ATM, DNA-PKcs or Ku80 as shown (a). BT and L3 are lymphoblastoid cell lines from a control (BT) and an A-T patient (L3) as described previously (Williamson et al. 2010). Molecular weight markers are indicated in kDa on the right-hand side. Asterisk represents non-specific cross-reacting bands. The effects of olaparib and AZD6738 on cell viability and apoptosis are shown in b, c (viability) and d (apoptosis). *p < 0.05 when compared to other treatment groups of that cell line and the DMSO control. #p < 0.05 when compared to DMSO control of that cell line only. c Quantitation of a at the 144-h time point for ATM-proficient (left) and ATM-deficient (right) prostate cancer cell lines
Full size imageATM-deficient Panc-10.05 pancreatic cancer cells are sensitive to the combination of olaparib and AZD6738
To test whether ATM-deficient pancreatic cancer cells are also sensitive to olaparib and AZD6738, we depleted ATM from Panc 10.05 cells using shRNA, as described previously (Williamson et al. 2010, 2012; Wang et al. 2017). Effective depletion of ATM protein was achieved as shown by western blot (Fig. 3a). Panc 10.05 cells are heterozygous for c.3077+103A>G mutation in ATM and homozygous for I255N in TP53. ATM-deficient and shGFP Panc 10.05 (control) cells were treated with olaparib (1 µM), AZD6738 (0.3 µM) or a combination of both inhibitors as above. As in lung and prostate cancer cells, olaparib and AZD6738 alone had no significant effect on viability in ATM-proficient pancreatic cancer cells, whereas the combination of olaparib plus AZD6738 reduced the number of viable cells by almost 70% compared to a 6% or 17% decrease in viability for either treatment alone (Fig. 3b, c). Moreover, as in lung and prostate cancer cells, cell death was observed in ATM-deficient cells treated with the combination of olaparib plus AZD6738 but not either agent alone. Similarly, no cell death was observed in ATM-proficient pancreatic cancer cells treated with olaparib and AZD6738 alone or in combination (Fig. 3d).
Fig. 3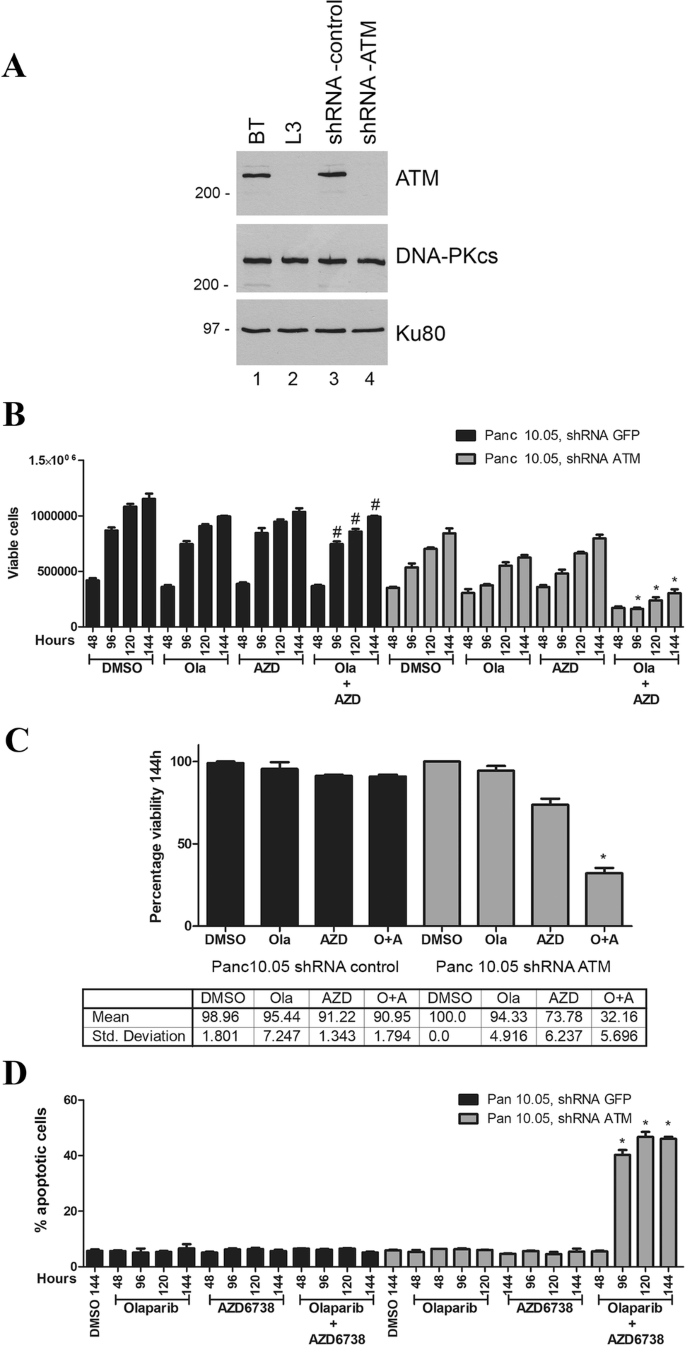
ATM-deficient Panc 10.05 pancreatic cancer cells undergo cell death when incubated with olaparib plus the ATR inhibitor AZD6738 but not either agent alone. a ATM was depleted from Panc 10.05 pancreatic cancer cells using shRNA as described previously (Williamson et al. 2010). Extracts were generated by NETN lysis and 50 µg total protein run on SDS PAGE and immunoblotted for ATM, DNA-PKcs or Ku80 as shown. BT and L3 are ATM positive and negative controls as in Fig. 2. The effect of olaparib and AZD6738 on viability and apoptosis is shown in b, c (viability) and d (apoptosis). Quantitation of viability at the 114-h time point is shown in c. Statistical analysis and asterisk are as in Figs. 1 and 2
Full size imageDiscussion
PARP inhibitors are proving to be highly effective in targeting cancers with mutations in the BRCA1 and BRCA2 genes. Since BRCA1 and BRCA2 proteins play key roles in the detection and repair of DSBs, these findings open up the possibility that cancers with mutations in other DNA repair genes may also respond to PARP inhibitor therapy. One such candidate is ATM, a DNA damage activated protein kinase that plays an important role in regulation of cell cycle checkpoints, cell survival after DNA damage and repair of complex DNA DSBs (Blackford and Jackson 2017). Moreover, the ATM gene is mutated in a number of cancers including lymphoma, lung, prostate and pancreatic cancers (Gao et al. 2013; Cerami et al. 2012), offering opportunities for precision therapy.
We previously showed that the PARP inhibitor olaparib reduces cell proliferation and induces reversible G2 arrest in ATM-deficient A549 lung cancer cells (Jette et al. 2019). We hypothesized that disruption of the G2 checkpoint using an ATR inhibitor would overcome the olaparib-induced G2/M arrest and induce cell death. Indeed, we found that the combination of the ATR inhibitor VE-821 with olaparib induced apoptosis in ATM-depleted A549 lung cancer cells with little effect on viability of ATM-proficient A549 cells (Jette et al. 2019). Here, we extend these studies to show that the ATR inhibitor AZD6738, which is in clinical trials for a number of solid tumours (Foote et al. 2015, 2018), also induces cell death in olaparib-treated ATM-deficient lung, prostate and pancreatic cancer cells with little effect on ATM-proficient control cells. This suggests that the combination of a PARP inhibitor with an ATR inhibitor may be useful in the treatment of a variety of cancer types with ATM deficiency. We speculate that combination of an ATR inhibitor with olaparib could also have benefit in cases of PARP inhibitor resistance and/or in patients with mutations in other DDR gene subsets that make tumours less responsive to PARP inhibition, however, this remains to be determined.
As discussed, PARP inhibitors are FDA approved for use in ovarian cancer and are in clinical trials for a number of other cancers, including breast, prostate and pancreatic cancer. In an initial clinical trial of 60 patients with metastatic prostate cancer, five were shown to have somatic or germline mutations in ATM and four of the five responded favourably to olaparib (Mateo et al. 2017), suggesting that ATM is a promising target in prostate cancer. Subsequently, olaparib treatment was shown to result in a statistically significant increase in radiographic progression-free survival (rPFS) compared to physician’s choice therapy in patients with metastatic castration-resistant prostate cancer and DDR gene alterations, for example in BRCA1/2, ATM and PALB2 (Hussain et al. 2019). However, exploratory analyses suggest that patients with ATM gene defects may not experience as impressive rates of progression-free survival in response to olaparib treatment alone as patients with other DDR gene alterations. This early evidence supports the need to examine combination therapy to enhance the clinical efficacy in this unique DDR gene alteration. Our observation that olaparib alone does not cause cell death in ATM-deficient prostate cancer cell lines may explain why men with mutation in ATM exhibited inferior outcome to olaparib treatment compared to those with BRCA1 mutation (Marshall et al. 2019), supporting our hypothesis that combination of PARP inhibitor with an ATR inhibitor may be more appropriate for tumours with ATM deficiency.
Critical to adoption of PARP inhibitors in the clinic will be the accurate identification of patients who may benefit from the selected treatment. Data from TCGA Pan Cancer Atlas studies, available on c-Bioportal (Cerami et al. 2012; Gao et al. 2013) (accessed March 15, 2020) reveal that ATM is mutated in approximately 6% of all cancers (677 of 10,967 samples), with 766 identified mutations (522 missense, 230 truncating, 8 in-frame and 6 other) that are scattered throughout the coding sequence, with very few occurring in multiple tumour samples. An exception is R337C/H that occurs in 22 out of 677 (3%) of the samples analysed (discussed in Jette et al. 2020). Moreover, the functional consequences of the vast majority of ATM mutations are unknown, thus screening patients by DNA sequence alone may not be informative. However, analysis of ATM mutations in Ataxia Telangiectasia, a cancer predisposition syndrome caused by loss or inactivation of both copies of the ATM gene (Rothblum-Oviatt et al. 2016) has shown that many missense mutations result in protein truncation (Gilad et al. 1998). Indeed, immunohistochemistry of ATM protein expression in lung adenocarcinoma patient samples revealed that over 40% had low protein expression, despite only 10% showing gene mutation (Villaruz et al. 2016). In addition, promoter methylation has been shown to lead to ATM gene silencing in some cancer cells (Hirakawa et al. 2019; Kim et al. 2002; Jette et al. 2020). Therefore, it is possible that more patient samples may lack ATM expression or function than indicated by exome sequencing alone and that methods for assaying ATM protein expression and/or activity will be required for patient selection.
Our results also highlight how the type of assay used to measure sensitivity of cultured cells to a compound can influence interpretation of results. Our results using clonogenic survival assays indicated that ATM-deficient cells are highly sensitive to olaparib (less than 1% survival at 1 µM olaparib) (Jette et al. 2019), however, subsequent results based on a better understanding of the mechanism of action of olaparib indicated that these cells were only arrested in G2 and did not show evidence of cell death by either sub-G1 DNA or annexin staining. Only after including the ATR inhibitor VE-821 did we observe evidence of cell death (Jette et al. 2019). These data illustrate how inferring cell viability from a clonogenic survival/colony formation assay (Franken et al. 2006) can be misleading, as the assay measures the ability of a single cell to proliferate to form a colony, usually of 50 or more cells, not viability/cell death, and, as shown by our work, a cell that is arrested in G2 is still viable; it is just unable to form colonies. Therefore, the type of viability assay used for determining sensitivity to olaparib and possibly other PARP inhibitors needs to be carefully chosen.
Availability of data and material
All original data is archived at the Charbonneau Cancer Centre, Cumming School of Medicine, University of Calgary.
References
Bamford, S., Dawson, E., Forbes, S., Clements, J., Pettett, R., Dogan, A., et al. (2004). The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. British Journal of Cancer,91(2), 355–358. https://doi.org/10.1038/sj.bjc.6601894.
Blackford, A. N., & Jackson, S. P. (2017). ATM, ATR, and DNA-PK: The trinity at the heart of the DNA damage response. Molecular Cell,66(6), 801–817. https://doi.org/10.1016/j.molcel.2017.05.015.
Boulton, S. J. (2006). Cellular functions of the BRCA tumour-suppressor proteins. Biochemical Society Transactions,34(Pt 5), 633–645. https://doi.org/10.1042/bst0340633.
Bryant, H. E., & Helleday, T. (2006). Inhibition of poly (ADP-ribose) polymerase activates ATM which is required for subsequent homologous recombination repair. Nucleic Acids Research,34(6), 1685–1691.
Bryant, H. E., Schultz, N., Thomas, H. D., Parker, K. M., Flower, D., Lopez, E., et al. (2005). Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature,434(7035), 913–917. https://doi.org/10.1038/nature03443.
Canadian Cancer Statistics (2019). http://www.cancer.ca/statistics.
Cerami, E., Gao, J., Dogrusoz, U., Gross, B. E., Sumer, S. O., Aksoy, B. A., et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discovery,2(5), 401–404. https://doi.org/10.1158/2159-8290.cd-12-0095.
Choi, M., Kipps, T., & Kurzrock, R. (2016). ATM mutations in cancer: Therapeutic implications. Molecular Cancer Therapeutics,15(8), 1781–1791. https://doi.org/10.1158/1535-7163.mct-15-0945.
Coleman, R. L., Oza, A. M., Lorusso, D., Aghajanian, C., Oaknin, A., Dean, A., et al. (2017). Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet,390(10106), 1949–1961. https://doi.org/10.1016/s0140-6736(17)32440-6.
Douglas, P., Zhong, J., Ye, R., Moorhead, G. B., Xu, X., & Lees-Miller, S. P. (2010). Protein phosphatase 6 interacts with the DNA-dependent protein kinase catalytic subunit and dephosphorylates gamma-H2AX. Molecular and Cellular Biology,30(6), 1368–1381. https://doi.org/10.1128/mcb.00741-09.
Elkon, R., Rashi-Elkeles, S., Lerenthal, Y., Linhart, C., Tenne, T., Amariglio, N., et al. (2005). Dissection of a DNA-damage-induced transcriptional network using a combination of microarrays, RNA interference and computational promoter analysis. Genome Biology,6(5), R43.
Farmer, H., McCabe, N., Lord, C. J., Tutt, A. N., Johnson, D. A., Richardson, T. B., et al. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature,434(7035), 917–921. https://doi.org/10.1038/nature03445.
Foote, K. M., Lau, A., & Nissink, J. W. (2015). Drugging ATR: progress in the development of specific inhibitors for the treatment of cancer. Future Medicinal Chemistry,7(7), 873–891. https://doi.org/10.4155/fmc.15.33.
Foote, K. M., Nissink, J. W. M., McGuire, T., Turner, P., Guichard, S., Yates, J. W. T., et al. (2018). Discovery and characterization of AZD6738, a potent inhibitor of Ataxia Telangiectasia mutated and Rad3 related (ATR) kinase with application as an anticancer agent. Journal of Medicinal Chemistry,61(22), 9889–9907. https://doi.org/10.1021/acs.jmedchem.8b01187.
Forbes, S. A., Bindal, N., Bamford, S., Cole, C., Kok, C. Y., Beare, D., et al. (2011). COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Research,39(Database issue), D945–D950. https://doi.org/10.1093/nar/gkq929.
Franken, N. A., Rodermond, H. M., Stap, J., Haveman, J., & van Bree, C. (2006). Clonogenic assay of cells in vitro. Nature Protocols,1(5), 2315–2319. https://doi.org/10.1038/nprot.2006.339.
Fraser, M., Zhao, H., Luoto, K. R., Lundin, C., Coackley, C., Chan, N., et al. (2012). PTEN deletion in prostate cancer cells does not associate with loss of RAD51 function: Implications for radiotherapy and chemotherapy. Clinical Cancer Research,18(4), 1015–1027. https://doi.org/10.1158/1078-0432.Ccr-11-2189.
Gao, J., Aksoy, B. A., Dogrusoz, U., Dresdner, G., Gross, B., Sumer, S. O., et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science Signaling. https://doi.org/10.1126/scisignal.2004088.
Gilad, S., Chessa, L., Khosravi, R., Russell, P., Galanty, Y., Piane, M., et al. (1998). Genotype-phenotype relationships in ataxia-telangiectasia and variants. American Journal of Human Genetics,62(3), 551–561.
Golan, T., Hammel, P., Reni, M., Van Cutsem, E., Macarulla, T., Hall, M. J., et al. (2019). Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. New England Journal of Medicine,381(4), 317–327. https://doi.org/10.1056/NEJMoa1903387.
Grant, R. C., Selander, I., Connor, A. A., Selvarajah, S., Borgida, A., Briollais, L., et al. (2014). Prevalence of germline mutations in cancer predisposition genes in patients with pancreatic cancer. Gastroenterology. https://doi.org/10.1053/j.gastro.2014.11.042.
Greiner, T. C., Dasgupta, C., Ho, V. V., Weisenburger, D. D., Smith, L. M., Lynch, J. C., et al. (2006). Mutation and genomic deletion status of ataxia telangiectasia mutated (ATM) and p53 confer specific gene expression profiles in mantle cell lymphoma. Proceedings of the National Academy of Sciences of the United States of America,103(7), 2352–2357. https://doi.org/10.1073/pnas.0510441103.
Hirakawa, T., Nasu, K., Aoyagi, Y., Takebayashi, K., Zhu, R., & Narahara, H. (2019). ATM expression is attenuated by promoter hypermethylation in human ovarian endometriotic stromal cells. Molecular Human Reproduction,25(6), 295–304. https://doi.org/10.1093/molehr/gaz016.
Hussain, M., Mateo, J., Fizazi, K., Saad, F., Shore, N., Sandhu, S., et al. (2019). LBA12_PR-PROfound: Phase III study of olaparib versus enzalutamide or abiraterone for metastatic castration-resistant prostate cancer (mCRPC) with homologous recombination repair (HRR) gene alterations. Paper presented at the ESMO Congress 2019, Barcelona.
Jette, N., Kumar, M., Radhamani, S., Arthur, G., Goutam, S., Yip, S., et al. (2020). ATM-deficient cancers provide new opportunities for precision oncology [Review]. Cancers (Basel). https://doi.org/10.3390/cancers12030687.
Jette, N. R., Radhamani, S., Arthur, G., Ye, R., Goutam, S., Bolyos, A., et al. (2019). Combined poly-ADP ribose polymerase and ataxia-telangiectasia mutated/Rad3-related inhibition targets ataxia-telangiectasia mutated-deficient lung cancer cells. British Journal of Cancer,121(7), 600–610. https://doi.org/10.1038/s41416-019-0565-8.
Kim, H. S., Choi, S. I., Min, H. L., Kim, M. A., & Kim, W. H. (2013a). Mutation at intronic repeats of the ataxia-telangiectasia mutated (ATM) gene and ATM protein loss in primary gastric cancer with microsatellite instability. PLoS One,8(12), e82769. https://doi.org/10.1371/journal.pone.0082769.
Kim, H. S., Kim, M. A., Hodgson, D., Harbron, C., Wellings, R., O’Connor, M. J., et al. (2013b). Concordance of ATM (ataxia telangiectasia mutated) immunohistochemistry between biopsy or metastatic tumor samples and primary tumors in gastric cancer patients. Pathobiology,80(3), 127–137. https://doi.org/10.1159/000346034.
Kim, H., Saka, B., Knight, S., Borges, M., Childs, E., Klein, A., et al. (2014). Having pancreatic cancer with tumoral loss of ATM and normal TP53 protein expression is associated with a poorer prognosis. Clinical Cancer Research,20(7), 1865–1872. https://doi.org/10.1158/1078-0432.ccr-13-1239.
Kim, W. J., Vo, Q. N., Shrivastav, M., Lataxes, T. A., & Brown, K. D. (2002). Aberrant methylation of the ATM promoter correlates with increased radiosensitivity in a human colorectal tumor cell line. Oncogene,21(24), 3864–3871.
Kubota, E., Williamson, C. T., Ye, R., Elegbede, A., Peterson, L., Lees-Miller, S. P., et al. (2014). Low ATM protein expression and depletion of p53 correlates with olaparib sensitivity in gastric cancer cell lines. Cell Cycle,13(13), 2129–2137. https://doi.org/10.4161/cc.29212.
Lord, C. J., & Ashworth, A. (2017). PARP inhibitors: Synthetic lethality in the clinic. Science,355(6330), 1152–1158. https://doi.org/10.1126/science.aam7344.
Marshall, C. H., Sokolova, A. O., McNatty, A. L., Cheng, H. H., Eisenberger, M. A., Bryce, A. H., et al. (2019). Differential response to olaparib treatment among men with metastatic castration-resistant prostate cancer harboring BRCA1 or BRCA2 versus ATM mutations. European Urology,76(4), 452–458. https://doi.org/10.1016/j.eururo.2019.02.002.
Mateo, J., Boysen, G., Barbieri, C. E., Bryant, H. E., Castro, E., Nelson, P. S., et al. (2017). DNA repair in prostate cancer: Biology and clinical implications. European Urology,71(3), 417–425. https://doi.org/10.1016/j.eururo.2016.08.037.
Mateo, J., Carreira, S., Sandhu, S., Miranda, S., Mossop, H., Perez-Lopez, R., et al. (2015). DNA-repair defects and olaparib in metastatic prostate cancer. New England Journal of Medicine,373(18), 1697–1708. https://doi.org/10.1056/NEJMoa1506859.
Mateo, J., Porta, N., Bianchini, D., McGovern, U., Elliott, T., Jones, R., et al. (2019). Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): a multicentre, open-label, randomised, phase 2 trial. Lancet Oncology. https://doi.org/10.1016/s1470-2045(19)30684-9.
McCabe, N., Turner, N. C., Lord, C. J., Kluzek, K., Bialkowska, A., Swift, S., et al. (2006). Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Research,66(16), 8109–8115. https://doi.org/10.1158/0008-5472.can-06-0140.
Mirza, M. R., Monk, B. J., Herrstedt, J., Oza, A. M., Mahner, S., Redondo, A., et al. (2016). Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. New England Journal of Medicine,375(22), 2154–2164. https://doi.org/10.1056/NEJMoa1611310.
Moore, K., Colombo, N., Scambia, G., Kim, B. G., Oaknin, A., Friedlander, M., et al. (2018). Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. New England Journal of Medicine,379(26), 2495–2505. https://doi.org/10.1056/NEJMoa1810858.
Pommier, Y., O’Connor, M. J., & de Bono, J. (2016). Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Science Translational Medicine,8(362), 362ps317. https://doi.org/10.1126/scitranslmed.aaf9246.
Roberts, N. J., Jiao, Y., Yu, J., Kopelovich, L., Petersen, G. M., Bondy, M. L., et al. (2012). ATM mutations in patients with hereditary pancreatic cancer. Cancer Discovery,2(1), 41–46. https://doi.org/10.1158/2159-8290.cd-11-0194.
Robson, M., Im, S. A., Senkus, E., Xu, B., Domchek, S. M., Masuda, N., et al. (2017). Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. New England Journal of Medicine,377(6), 523–533. https://doi.org/10.1056/NEJMoa1706450.
Rothblum-Oviatt, C., Wright, J., Lefton-Greif, M. A., McGrath-Morrow, S. A., Crawford, T. O., & Lederman, H. M. (2016). Ataxia telangiectasia: A review. Orphanet Journal of Rare Diseases,11(1), 159. https://doi.org/10.1186/s13023-016-0543-7.
Ruis, B. L., Fattah, K. R., & Hendrickson, E. A. (2008). The catalytic subunit of DNA-dependent protein kinase regulates proliferation, telomere length, and genomic stability in human somatic cells. Molecular and Cellular Biology,28(20), 6182–6195.
Shiloh, Y. (2014). ATM: Expanding roles as a chief guardian of genome stability. Experimental Cell Research. https://doi.org/10.1016/j.yexcr.2014.09.002.
Shiloh, Y., & Lederman, H. M. (2016). Ataxia-telangiectasia (A–T): An emerging dimension of premature ageing. Ageing Research Reviews. https://doi.org/10.1016/j.arr.2016.05.002.
Villaruz, L. C., Jones, H., Dacic, S., Abberbock, S., Kurland, B. F., Stabile, L. P., et al. (2016). ATM protein is deficient in over 40% of lung adenocarcinomas. Oncotarget. https://doi.org/10.18632/oncotarget.9757.
Wang, C., Jette, N., Moussienko, D., Bebb, D. G., & Lees-Miller, S. P. (2017). ATM-deficient colorectal cancer cells are sensitive to the PARP inhibitor olaparib. Translational Oncology,10(2), 190–196. https://doi.org/10.1016/j.tranon.2017.01.007.
Weston, V. J., Oldreive, C. E., Skowronska, A., Oscier, D. G., Pratt, G., Dyer, M. J., et al. (2010). The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood,116(22), 4578–4587. https://doi.org/10.1182/blood-2010-01-265769.
Williamson, C. T., Kubota, E., Hamill, J. D., Klimowicz, A., Ye, R., Muzik, H., et al. (2012). Enhanced cytotoxicity of PARP inhibition in mantle cell lymphoma harbouring mutations in both ATM and p53. EMBO Molecular Medicine,4(6), 515–527. https://doi.org/10.1002/emmm.201200229.
Williamson, C. T., Muzik, H., Turhan, A. G., Zamo, A., O’Connor, M. J., Bebb, D. G., et al. (2010). ATM deficiency sensitizes mantle cell lymphoma cells to poly(ADP-ribose) polymerase-1 inhibitors. Molecular Cancer Therapeutics,9(2), 347–357. https://doi.org/10.1158/1535-7163.mct-09-0872.
Acknowledgements
We thank the Flow Cytometry Facility and the Center for Genome Engineering at the Cumming School of Medicine, University of Calgary for expert assistance.
Funding
This work was funded by grants from the Cancer Research Society and the Pancreatic Cancer Society of Canada (#20229), the Engineered Air Chair in Cancer Research (# 21202) and the Alberta Cancer Foundation (# 27042). N. R. Jette was a recipient of a Rejeanne Taylor Graduate scholarship in translational cancer research as well as Achievers in Medical Science Leaders in Medicine scholarship.
Author information
Nicholas R. Jette and Suraj Radhamani contributed equally.
Affiliations
Department of Biochemistry and Molecular Biology, University of Calgary, 3330 Hospital Drive NW, Calgary, AB, T2N 1N4, Canada
Nicholas R. Jette, Suraj Radhamani, Ruiqiong Ye, Yaping Yu, Greydon Arthur, Siddhartha Goutam, Tarek A. Bismar, Mehul Kumar, Pinaki Bose & Susan P. Lees-Miller
Robson DNA Science Centre, University of Calgary, 3330 Hospital Drive NW, Calgary, AB, T2N 1N4, Canada
Nicholas R. Jette, Suraj Radhamani, Ruiqiong Ye, Yaping Yu, Greydon Arthur, Siddhartha Goutam & Susan P. Lees-Miller
Department of Oncology, University of Calgary, 3330 Hospital Drive NW, Calgary, AB, T2N 1N4, Canada
Tarek A. Bismar, Pinaki Bose, Steven Yip & Susan P. Lees-Miller
Department of Pathology, University of Calgary, 3330 Hospital Drive NW, Calgary, AB, T2N 1N4, Canada
Tarek A. Bismar
Charbonneau Cancer Institute, Cumming School of Medicine, University of Calgary, 3330 Hospital Drive NW, Calgary, AB, T2N 1N4, Canada
Nicholas R. Jette, Suraj Radhamani, Ruiqiong Ye, Yaping Yu, Greydon Arthur, Siddhartha Goutam, Tarek A. Bismar, Mehul Kumar, Pinaki Bose, Steven Yip & Susan P. Lees-Miller
Tom Baker Cancer Centre, 1331 29 St NW, Calgary, AB, T2N 4N2, Canada
Tarek A. Bismar & Steven Yip
Cross Cancer Institute, 11560 University Avenue NW, Edmonton, AB, T6G 1Z2, Canada
Michael Kolinsky
Contributions
Experiments and data analysis were carried out by NRJ, SR, RY, YY, GA, MKu, SG and SPLM. The study was conceived by MK and SPLM with intellectual contributions from PB, SY and TAB. All authors contributed to writing and editing of the manuscript and have approved the final submission.
Corresponding author
Correspondence to Susan P. Lees-Miller.
Ethics declarations
Conflict of interest
SY has received honoraria and/or consulting fees from Janssen, Pfizer, Roche, BMS, Merck, AstraZeneca, Bayer, and Novartis. MKo has accepted honoraria and/or consulting fees from Janssen, Ipsen, Astellas, BMS, Merck, AstraZeneca, Bayer, and travel support from Novartis and TAB has accepted an honorarium from AstraZeneca. NRJ, SR, RY, YY, GA, SG, MKu, PB and SPLM declare no conflict of interest.
Consent for publication
All authors have approved the final submission and consented to publication.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article
Cite this article
Jette, N.R., Radhamani, S., Ye, R. et al. ATM-deficient lung, prostate and pancreatic cancer cells are acutely sensitive to the combination of olaparib and the ATR inhibitor AZD6738. GENOME INSTAB. DIS. 1, 197–205 (2020). https://doi.org/10.1007/s42764-020-00011-0
Received 18 March 2020
Revised 27 March 2020
Accepted 28 March 2020
Published 08 April 2020
Issue Date July 2020
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
ATM
ATR inhibitor
Pancreatic adenocarcinoma
Prostate cancer
Lung adenocarcinoma
Olaparib
VE-821
AZD6738
用户登录
还没有账号?
立即注册