






Deacetylation of a deacetylase drives the DNA damage response
Commentary
Raissa Ng & Michael S. Y. Huen
Genome Instability & Disease 1,151–154(2020)
Abstract
The lifespan regulator protein SIRT6 is an NAD + -dependent deacetylase that plays an early role in cellular responses to DNA damage. In a recent study from Liu, Xu and team (2020), the authors describe a novel lysine acetylation/deacetylation switch on SIRT6 that modulates its ability to polymerize and sense DNA breaks. They also identified SIRT1 as the major deacetylase that regulates this lysine modification, unveiling a novel SIRT1-SIRT6 axis that drives the DNA damage response.
SIRT6, hailed as a “longevity protein”, exerts protective effects against aging and its associated pathologies (Mostoslavsky et al. 2006; Kanfi et al. 2012) through its pleiotropic roles in the DNA damage response (DDR) and in genome maintenance (Mostoslavsky et al. 2006; Mao et al. 2011; Toiber et al. 2013). SIRT6 is best characterized as a histone deacetylase that targets acetylated lysines on histone H3, namely H3K9ac (Michishita et al. 2008) and H3K56ac (Michishita et al. 2009; Yang et al. 2009), to orchestrate telomeric chromatin compaction, regulation of transcription, and responses to DNA damage. Indeed, SIRT6 can deacetylate H3K9ac (McCord et al. 2009) and H3K56ac (Toiber et al. 2013) on damaged chromatin, thereby recruiting the chromatin remodelers SNF2H (Toiber et al. 2013) and CHD4 (Hou et al. 2020) to promote chromatin relaxation (see Fig. 1). This in turn drives a full-blown DDR to protect genome integrity (McCord et al. 2009; Toiber et al. 2013; Hou et al. 2020; Atsumi et al. 2015).
Fig. 1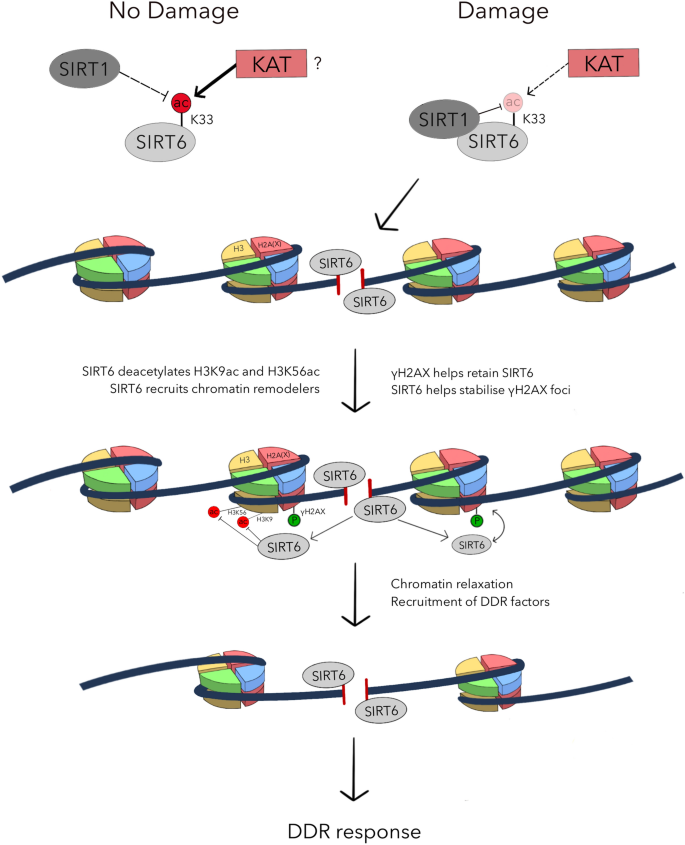
Lysine 33 (K33) of SIRT6 is deacetylated by SIRT1 and acetylated by unknown acetyltransferase(s) under normal physiological conditions. When DNA is damaged, binding between SIRT1 and SIRT6 is enhanced. As a result, SIRT6 is preferentially deacetylated by SIRT1. Hypoacetylation of SIRT6 at K33 facilitates its polymerization and DSB binding. γH2AX (red) promotes SIRT6 retention in the proximity of the damaged chromatin, facilitating efficient SIRT6-dependent deacetylation of H3K9ac and H3K56ac on histone H3 (yellow). Concurrently, SIRT6 recruits chromatin remodelers to the DSB site and promotes chromatin relaxation. The open chromatin conformation allows downstream DDR factors to associate with the damaged chromatin and initiate DSB signaling and repair
SIRT6 binds DNA in vitro, and has been implicated as a candidate DSB sensor alongside the likes of PARP1, MRN, and Ku70/80 (Onn et al. 2020). Inspired by observations where SIRT6 polymerizes in solution (Pan et al. 2011), and that in its multimeric configuration directly recognizes DSBs (Onn et al. 2020), Meng et al. sought to pinpoint the residues responsible for SIRT6 association with DSBs, reasoning that positively-charged residues (R32, K33, R39) on its N terminus might display affinity for the negatively-charged phosphate groups on the DNA backbone (Meng et al. 2020).
Post-translational modifiers play established roles as molecular switches to modulate protein activities and functions. Hence, it is likely that post-translational modifications on SIRT6 may underlie its swift mobilization and subsequent binding to DNA breaks. Accordingly, Meng et al. proposed that adding acetyl groups on the putative DNA binding motif on the SIRT6 N terminal peptide might neutralize its positive charge, thereby suppressing its ability to recognize DSBs. To explore this idea, the researchers first conducted LC–MS/MS experimentations, and showed that SIRT6 can be acetylated at K33 in vivo. To test the significance of SIRT6 acetylation, they generated both hypoacetylated (K33R) and hyperacetylated (K33Q) SIRT6 mutants, and uncovered that only hypoacetylated SIRT6 is efficiently recruited to DNA breaks to mediate deacetylation of its histone targets. By contrast, the hyperacetylated SIRT6 mutant was unable to polymerize (as determined by coimmunoprecipitation and bimolecular fluorescence complementation (BiFC) assays) and displayed an attenuated ability to accumulate at DSBs.
In search of possible factors that may facilitate SIRT6 docking at DNA breaks, Meng et al. turned to γH2AX, which serves as a common platform to recruit many DDR factors. Intriguingly, the researchers unprecedentedly showed that hypoacetylated SIRT6 specifically bound to phosphorylated H2AX (γH2AX) in vitro, leading them to postulate that such a phospho-specific interaction could be attributed to a novel mode of protein–protein interaction that relies on the electrostatic force between the negatively charged phosphate groups and the positively charged lysine residues (Wang et al. 2016; Errington and Doig 2005). As depletion of H2AX and ATM/ATR inhibition hampered SIRT6 concentration at laser-induced DNA damage sites, they concluded that DSB association of SIRT6 is dependent on γH2AX, and that the initial recognition of γH2AX may depend on its acetylation status.
To isolate genetic factors that mediate the dynamic SIRT6 acetylation status, the researchers assayed SIRT6 acetylation against a panel of HDAC inhibitors, and subsequently identified SIRT1 as a candidate SIRT6 deacetylase. Congruent with this idea, the authors demonstrated a novel interaction between SIRT1 and SIRT6, and that SIRT1 targets and deacetylates K33ac on the SIRT6 polypeptide. Consistently, SIRT6 recruitment to sites of DNA damage is compromised in SIRT1-depleted cells, placing SIRT1 deacetylation activity upstream of SIRT6 recruitment to DNA breaks.
To link SIRT6 as an SIRT1 substrate in the DNA damage setting, Meng et al. found that DNA damage enhanced the SIRT1–SIRT6 interaction, and that SIRT6 becomes deacetylated upon DNA damage. Based on these findings, the authors propose that SIRT1 deacetylates SIRT6 at K33 in response to DNA damage, which in turn unleashes SIRT6-mediated histone deacetylation at the damaged chromatin for effective DNA repair.
While this study identified SIRT1 as the major SIRT6 deacetylase that fuels the DDR, the acetyltransferase that mediates SIRT6 acetylation at K33 during unperturbed cell proliferation remains to be identified. The researchers postulated that ectopically-expressed SIRT6 might not be efficiently acetylated, implying that this acetyltransferase may be limiting in cells, although it is required to suppress any unscheduled DNA-binding activity of SIRT6. Thus, it will be important to define how SIRT1 and this hitherto unidentified acetyltransferase competes for SIRT6 to maintain cellular homeostasis. Under DNA damage conditions, it is possible that the balance between acetyltransferase and deacetylase is tipped to favor SIRT6 K33 deacetylation, making it a damage-specific modification.
The model suggested by Meng et al. also places SIRT1 upstream of SIRT6 in its ability to sense DNA damage, as SIRT6 deacetylation by SIRT1 may be a prerequisite for its recruitment to DSBs. Given its DNA binding property, the model also predicts that SIRT6 does not rely on SIRT1 as a scaffold at the damaged chromatin. So where does SIRT1 deacetylate SIRT6? While it remains formally plausible that SIRT1 deacetylates SIRT6 prior to their accumulation at DSBs, further experimentations will be needed to fully dissect the tempo-spatial control of SIRT6 deacetylation.
In summary, although the mechanisms that regulate SIRT6 acetylation status in both physiological and DNA damage conditions remain enigmatic, given the emerging evidence that implicates SIRT6 as a bona fide DSB sensor, the longevity protein likely serves as a stress sensing hub to affect its pleiotropic roles in DNA repair and genome stability maintenance. The ever-expanding SIRT1 repertoire also reminds us of the endless possibilities and complexities of post-translational modifications as regulatory bases that drive the many forms of the DDR.
References
Atsumi, Y., Minakawa, Y., Ono, M., Dobashi, S., Shinohe, K., Shinohara, A., et al. (2015). ATM and SIRT6/SNF2H mediate transient H2AX stabilization when DSBs form by blocking HUWE1 to allow efficient gammaH2AX foci formation. Cell Reports,13(12), 2728–2740. https://doi.org/10.1016/j.celrep.2015.11.054.
Errington, N., & Doig, A. J. (2005). A phosphoserine-lysine salt bridge within an alpha-helical peptide, the strongest alpha-helix side-chain interaction measured to date. Biochemistry,44(20), 7553–7558. https://doi.org/10.1021/bi050297j.
Hou, T., Cao, Z., Zhang, J., Tang, M., Tian, Y., Li, Y., et al. (2020). SIRT6 coordinates with CHD4 to promote chromatin relaxation and DNA repair. Nucleic Acids Research,48(6), 2982–3000. https://doi.org/10.1093/nar/gkaa006.
Kanfi, Y., Naiman, S., Amir, G., Peshti, V., Zinman, G., Nahum, L., et al. (2012). The sirtuin SIRT6 regulates lifespan in male mice. Nature,483(7388), 218–221. https://doi.org/10.1038/nature10815.
Mao, Z., Hine, C., Tian, X., Van Meter, M., Au, M., Vaidya, A., et al. (2011). SIRT6 promotes DNA repair under stress by activating PARP1. Science,332(6036), 1443–1446. https://doi.org/10.1126/science.1202723.
McCord, R. A., Michishita, E., Hong, T., Berber, E., Boxer, L. D., Kusumoto, R., et al. (2009). SIRT6 stabilizes DNA-dependent protein kinase at chromatin for DNA double-strand break repair. Aging (Albany NY),1(1), 109–121. https://doi.org/10.18632/aging.100011.
Meng, F., Qian, M., Peng, B., Peng, L., Wang, X., Zheng, K., et al. (2020). Synergy between SIRT1 and SIRT6 helps recognize DNA breaks and potentiates the DNA damage response and repair in humans and mice. Elife. https://doi.org/10.7554/eLife.55828.
Michishita, E., McCord, R. A., Berber, E., Kioi, M., Padilla-Nash, H., Damian, M., et al. (2008). SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature,452(7186), 492–496. https://doi.org/10.1038/nature06736.
Michishita, E., McCord, R. A., Boxer, L. D., Barber, M. F., Hong, T., Gozani, O., et al. (2009). Cell cycle-dependent deacetylation of telomeric histone H3 lysine K56 by human SIRT6. Cell Cycle,8(16), 2664–2666. https://doi.org/10.4161/cc.8.16.9367.
Mostoslavsky, R., Chua, K. F., Lombard, D. B., Pang, W. W., Fischer, M. R., Gellon, L., et al. (2006). Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell,124(2), 315–329. https://doi.org/10.1016/j.cell.2005.11.044.
Onn, L., Portillo, M., Ilic, S., Cleitman, G., Stein, D., Kaluski, S., et al. (2020). SIRT6 is a DNA double-strand break sensor. Elife. https://doi.org/10.7554/eLife.51636.
Pan, P. W., Feldman, J. L., Devries, M. K., Dong, A., Edwards, A. M., & Denu, J. M. (2011). Structure and biochemical functions of SIRT6. Journal of Biological Chemistry,286(16), 14575–14587. https://doi.org/10.1074/jbc.M111.218990.
Toiber, D., Erdel, F., Bouazoune, K., Silberman, D. M., Zhong, L., Mulligan, P., et al. (2013). SIRT6 recruits SNF2H to DNA break sites, preventing genomic instability through chromatin remodeling. Molecular Cell,51(4), 454–468. https://doi.org/10.1016/j.molcel.2013.06.018.
Wang, D., Kon, N., Lasso, G., Jiang, L., Leng, W., Zhu, W. G., et al. (2016). Acetylation-regulated interaction between p53 and SET reveals a widespread regulatory mode. Nature,538(7623), 118–122. https://doi.org/10.1038/nature19759.
Yang, B., Zwaans, B. M., Eckersdorff, M., & Lombard, D. B. (2009). The sirtuin SIRT6 deacetylates H3 K56Ac in vivo to promote genomic stability. Cell Cycle,8(16), 2662–2663. https://doi.org/10.4161/cc.8.16.9329.
Acknowledgements
The authors thank Ms. Tina Wang for help with illustrations, and all members of the Huen Laboratory for discussion and input. The work is in part sponsored by fund from Research Grants Council to Michael Huen (Project no. C7007-17GF and 17104618). Raissa Ng is supported by the Hong Kong PhD Fellowship.
Author information
Affiliations
School of Biomedical Sciences, LKS Faculty of Medicine, The University of Hong Kong, 21 Sassoon Road, Pokfulam, Hong Kong
Raissa Ng & Michael S. Y. Huen
State Key Laboratory of Brain and Cognitive Sciences, The University of Hong Kong, 5 Sassoon Road, Pokfulam, Hong Kong
Michael S. Y. Huen
Corresponding author
Correspondence to Michael S. Y. Huen.
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article
Cite this article
Ng, R., Huen, M.S.Y. Deacetylation of a deacetylase drives the DNA damage response. GENOME INSTAB. DIS. 1, 151–154 (2020). https://doi.org/10.1007/s42764-020-00018-7
Published 13 July 2020
Issue Date July 2020
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
SIRT1
SIRT6
DNA damage response (DDR)
Deacetylation
Post-translational modification
Double strand break (DSB) sensor
用户登录
还没有账号?
立即注册