






The roles of TRAF3 mutation in the oncogenic progression and drug response of multiple myeloma
Review Article
Genome Instability & Disease 1,278–285(2020)
Abstract
Multiple myeloma (MM) is a malignant plasma cell proliferating in the bone marrow. Oncogenesis of MM is a multi-stage cytogenetic event. Among these, aberrant activation of the non-canonical nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling pathway is critical for the oncogenic progression of MM. Tumor necrosis factor receptor‐associated factor 3 (TRAF3) mutation is among the most common tumor suppressor mutations in MM that allows constitutive activation of the non-canonical NF-кB pathway, thereby enhancing de novo survival of MM cells. Although there are some promising developments on current therapeutic regimens for MM patients that target such NF-кB signature, drug resistance perhaps remains a major concern. So far, TRAF3 mutation has been reported to modulate proteasome inhibitor response of MM. Mechanistically, concomitant to TRAF3 mutation-associated NIK stability that leads to the pathway activation, it has been reported that bortezomib treatment also causes a drastic increase in the level of NIK that causes pathway cross talk activating the canonical pathway, thereby triggering an acquired proteasome inhibitor resistance (PIR) pathway. Concomitantly targeting such NIK-driven acquired PIR or else targeting NIK than TRAF3 mutation and associated phenotype is likely to be the better option and thus remains to be elucidated. Hence, this review explains the roles of TRAF3-mutation-associated NF-кB pathway activation in the oncogenic progression and drug response of MM.
Introduction
Multiple myeloma is a malignant plasma cell proliferation in the bone marrow. It is the second most prevalent hematological malignancy and accounts for about 1% of all cancers, 2% of all cancer deaths and 20% of deaths caused by hematological malignancies (Stuhmer et al. 2005; Yuregir et al. 2009). The incidence of the disease is rising in Asia, following the pattern of the aging population. Risk groups are those over 65 years of age with about 5 years of median survival time.
The oncogenesis of MM is a multi-stage process preceded by monoclonal gammopathy of undetermined clinical significance (MGUS) which progresses to smoldering multiple myeloma (SMM), followed by an overt MM with the increasing rate of transformation per year and trend of tumor burden across these stages (Anderson and Carrasco 2011; Fonseca et al. 2009; Kyle et al. 2007). The myeloma cells damage and weaken the bone and eventually cause anemia, lytic bone lesions that may cause bone fractures or pain, renal failure and hypercalcemia (Raab et al. 2016). The consequence of expansion and survival within the bone marrow represents the major clinical manifestations of MM and chemotherapy resistance as the final sequel.
The primary cytogenetic events in the tumorigenesis of MM include chromosomal instability that leads to IgH translocation, deletion of chromosome 13 and dysregulation of cyclin D (Korde et al. 2011; Kumar et al. 2012), while the secondary cytogenetic events include activating mutations of RAS, loss of chromosome 17p and deletion of TP53, MYC dysregulation and amplification of chromosome 1q21. Among these, mutations that cause aberrant activation of the NF-κB pathway are the most common (Annunziata et al. 2007; Keats et al. 2007; Lohr et al. 2014).
Although there are also some promising developments on current therapeutic regimens for MM patients targeting NF-кB signature, drug resistance perhaps remains a major concern. For instance, approximately 20% of MM patients who received the first-in-class drug (bortezomib) do not respond (San Miguel et al. 2008). Many patients may be naturally resistant to it or perhaps develop resistance during the course of treatment. As a result, the disease is still regarded incurable. Hence, this review was made to give an overview of MM and to elucidate the roles of TRAF3-associated NF-кB pathway mutation in the oncogenic progression as well as drug response of MM.
Multiple myeloma: overview
Oncogenesis of multiple myeloma
The tumorigenesis of myeloma is often preceded by a pre-malignant stage called monoclonal gammopathy of undetermined clinical significance (MGUS). MGUS that progresses to an intermediate stage is represented by smoldering multiple myeloma (SMM). SMM eventually advances to an overt MM (Saltarella et al. 2015). The tumor burden at this stage is more than in MGUS and has a higher risk of progression to symptomatic myeloma and these patients require therapy (Kyle et al. 2007). The two primary cytogenetic events that occur as a result of genomic/chromosomal instability during the evolution of MGUS are hyperdiploid (HRD) and non-hyperdiploid (NHRD) karyotypes. The most common early cytogenetic events is IgH translocation: t(4;14), t(14;16), t(6;14), t(11;14), and t(14;20) (Anderson and Carrasco 2011; Fonseca et al. 2009). MM with t(14;16) belongs to the high-risk group, t(4;14) to intermediate-risk group and t(11;14) to standard-risk group group. IgH translocation occurs in 10% of HRD, while it amounts to 70% in NHRD (Kumar et al. 2012) and dysregulates the expression of some juxtaposed proto-oncogene fibroblast growth factor receptor 3 (FGFR3) and epigenetic regulator multiple myeloma SET domain (MMSET). Deletion of chromosome 13 and dysregulation of cyclin D are among unifying early oncogenic events that often persist across the stages of MM progression (Korde et al. 2011). The prevalence of IgH translocations increases with disease stages: about 50% in MGUS, 55–70% for intramedullary MM, 85% in extramedullary MM and > 90% in multiple myeloma cell lines (MMCLs) (Bergsagel and Chesi 2013).
The other secondary cytogenetic events associated with the progression of MM include activating mutations of RAS, loss of chromosome 17p and deletion of TP53, MYC dysregulation and amplification of chromosome 1q21. Among these, mutations that cause aberrant activation of the NF-κB pathway are the most common (Annunziata et al. 2007; Keats et al. 2007; Lohr et al. 2014).
Treatment of multiple myeloma
The current therapeutics modality for MM is a combination therapy of mainly proteasome inhibitors and immunomodulatory drugs. Recent advances in chemotherapeutic development for MM include approval of bortezomib, thalidomide and lenalidomide, as active pharmacological agents in MM. A study by Jakubowiak et al. found that the combination of bortezomib, lenalidomide, dexamethasone, and pegylated liposomal doxorubicin was generally well tolerated and highly active in newly diagnosed MM patients (Jakubowiak et al. 2011). Targeted at the deregulated NF-κB signaling pathway, the development of proteasome inhibitor, bortezomib and immunomodulatory drugs (such as lenalidomide, thalidomide, and pomalidomide) has been a promising induction therapy for MM patients (Wong et al. 2020). Transplant-eligible patient also undergoes collateral therapy, while maintenance therapy also is given based on the age of the patient and disease progression (Saad et al. 2014). Though these therapeutic regimens improved patient survival, most of the patients eventually relapse underscoring the need for finding alternative treatment options. In this regard, further understanding of the pathobiology of MM in the context of the bone marrow microenvironment is crucial.
The role of the NF-кB pathway mutation in the oncogenesis of MM
The NF-κB consists of a family of transcription factors that normally play important roles in multiple biologic processes, including innate and adaptive immunity, regulation of cell growth, proliferation, survival, death and development processes (Cildir et al. 2016; Hayden and Ghosh 2008; Vallabhapurapu and Karin 2009). There are two NF-κB pathways, namely the canonical and non-canonical pathways (Fig. 1). The canonical NF-κB signaling pathway is the most studied classical pathway and has a rapid, transient response to numerous stimuli. This pathway is activated upon the binding of soluble and membrane-bound extracellular ligands and cytokines to the tumor necrosis factor (TNF) family receptors, such as Fas, tumor necrosis factor ligand superfamily member (TRAIL), and TUMOR necrosis factor receptor 1 (TNFR1), interleukin-1 receptor (IL-1R) family (Medzhitov 2008), pattern recognition receptors (PRRs) such as Toll-like receptor 4 (TLR4) (Hacker et al. 2006; Oganesyan et al. 2006) and cytoplasmic RIG-I-like receptor (RLR) family (Takeuchi and Akira 2010), thereby triggering the activation of transforming growth factor-beta-activated kinase 1 (TAK1) that phosphorylates the inhibitor of nuclear factor kappa-B kinase subunit beta (IKKβ), which subsequently phosphorylates and degrades IкBα (the inhibitors of NF-κB). The proteasomal degradation of IкBα allows the release of RelA and p50 from the complexes into the cytoplasm. This functionally active transcription factors can then localize to the nucleus to activate transcription of the target gene. The inactivation of the inhibitor of kappaB alpha (IкBα) complex is thus a rate-limiting step in the canonical pathway activation (Fig. 1).
Fig. 1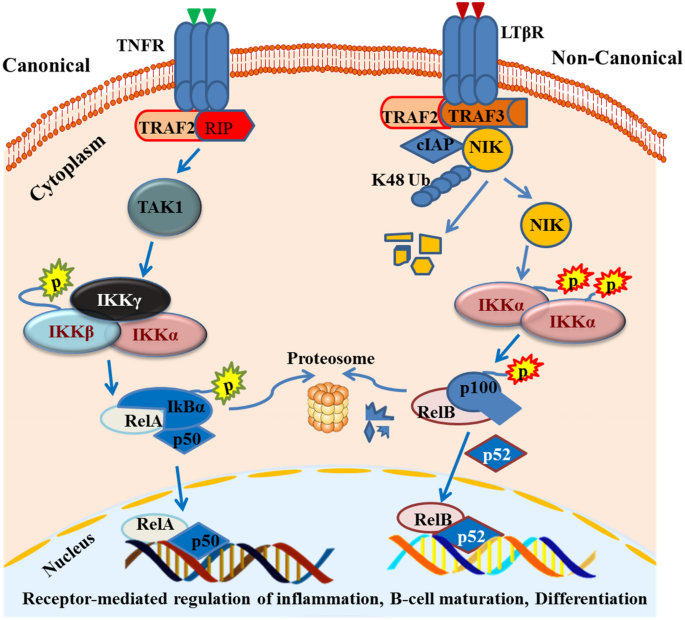
The canonical and non-canonical NF-кB pathway. The canonical NF-кB pathway (left side) uses TNFR receptors and recruits TRAF2/RIP adaptor proteins that activate TAK1. This kinase phosphorylates IKKβ and, subsequently, IкBα phosphorylation allows the release of RelA/p65 and p50. The non-canonical pathway (right side) used LTBR and other receptors discussed above. It binds to the TRAF2/TRAF3/BIRC3 complex that recruits NIK. NIK phosphorylates IKKα and IKKα phosphorylates the p100/RelB complex, thereby allowing the p100 to p52 processing to activate the pathway
Full size imageThe non-canonical (alternative) NF-κB signaling pathway uses a subset of receptors, including lymphotoxin-β receptor (LTβR), CD40, fibroblast growth factor-inducible 14 (Fn14) and B cell-activating factor receptor (BAFF-R). At resting state, regulatory adaptor molecules such as a cellular inhibitor of apoptosis protein 1 (cIAP1), cIAP2, TRAF3 and TRAF2 and the kinase NIK are assembled (Matsushima et al. 2001; Shinkura et al. 1999; Yin et al. 2001; Zarnegar et al. 2008) and NF-κB2 p100/RelB complexes are inactive in the cytoplasm. When receptor-mediated signaling is activated, NIK phosphorylates the IKKα complex that subsequently phosphorylates NF-κB2 p100. Phosphorylation of NF-κB2 p100 leads to its ubiquitination and proteasomal processing of p100 to p52. This creates transcriptionally active p52/RelB dimers that translocate to the nucleus and induce target gene expression. In contrast to the canonical pathway, the non-canonical NF-κB pathway is slow and persistently responds to specific sub-sets of signals (Sun 2012). It is negatively regulated by TRAF3 and tightly controlled by the ubiquitination and proteasomal degradation of the NIK (Liao et al. 2004). In the physiological state, this pathway has been reported to control B cell development, survival and secondary lymphoid organogenesis (Cildir et al. 2013; Sun 2011). Lethal phenotypes have been seen in TRAF3-deficient mice due to deranged activation of the non-canonical NF-кB pathway and rescued by the combined deletion of the NF-ΚB2 gene (He et al. 2006).
NF-κB-activating mutations are one of the secondary cytogenetic events that drive MGUS and SMM progression to the clinical phases of MM (Chng et al. 2007a, b). Indeed, previous reports found that 20% of untreated MM patients and 45% of MMCLs have one or more genetic aberrations in the components of the NF-κB pathway that leads to constitutive activation of canonical and/or non-canonical NF-κB pathways (Annunziata et al. 2007; Chapman et al. 2011; Keats et al. 2007). These studies found amplification or rearrangement of genes including NIK, LTBR, TACI, NF-ΚB1, NF-ΚB2, and CD40, as well as deletion or loss-of-function mutations in genes including CYLD, BIRC2/BIRC3, TRAF2, and TRAF3, each of which results in the activation of NF-κB-associated gene expression.
TRAF3 mutation and associated phenotype in MM
TRAF3, a cytoplasmic adaptor protein, has previously been described as a novel tumor suppressor in B lymphocytes (Lalani et al. 2015). As a negative regulator of NF-κB2 activation, it destabilizes NIK and activates IKKα-dependant NF-κB 2 p100 processing to p52/RelB. Deregulated accumulation of NIK has been associated with lymphoid malignancies indicating that tight control of NIK fate is important(Sun 2011).
The recent whole-genome sequencing studies identified TRAF3 as one of the most frequently mutated tumor suppressors in MM (Annunziata et al. 2007). TRAF3 is inactivated in 17% of human MMCLs and 12.3% of MM patients (Keats et al. 2007). In fact, 21 out of 83 cell lines (25%) derived from a variety of hematological malignancies (lymphoblastic leukemia, mantle cell lymphoma and Hodgkin lymphoma) also lacked detectable TRAF3 by immunohistochemistry (Zapata et al. 2000), indicating the importance of TRAF3 in the pathogenesis of hematological malignancies at large.
TRAF3 mutation is the most common tumor suppressor mutation in MM that allows non-canonical NF-кB pathway activation (Matsushima et al. 2001; Shinkura et al. 1999; Yin et al. 2001; Zarnegar et al. 2008). As summarized in Table 1, MMCLs with TRAF3 mutation and those with no NF-кB mutation have different patterns of cytogenetic profile.
| MMCLs | NF-κB mutation | Genetic rearrangement | Fusion genes | |
|---|---|---|---|---|
| MM.1S | TRAF3 | 9aa deletion | t(14;16)(q32;q23) | IGH/MAF |
| RPMI8226 | TRAF3 | Biallelic deletion | t(14;16)(q32;q23) | IGH/MAF |
| t(16;22)(q23;q11) | c-maf and Igλ | |||
| U266 | TRAF3 | Frame-shift | t(14;16)(q32;q23) | IGF/MAF |
| JJN3 | NIK | Rearrangement | t(14;16)(q32;q23) | IGF/MAF |
| KMS11 | TRAF3 | MATH deletion | t(4;14)(p16;q32) | FGFR3 and MMSET |
| XG7 | no | no | t(4;14)(p16;q32) | FGFR3 and MMSET |
| KMS28BM | no | no | t(4;14)(p16;q32) | FGFR3 and MMSET |
| NCI-H929 | no | no | t(4; 14)(p16;q32) | FGFR3 and MMSET |
Table 1 Cytogenetic profile of MMCLs (as reported in (Demchenko et al. 2010) and (Shin et al. 2020))
Full size tableBased on the cytogenetic features, similar to translocation in TRAF3/NIK mutant MMCLs (Demchenko et al. 2010), patients with t(14;16) translocations are a high-risk group with poor prognosis. On the contrary, TRAF3 wild-type MMCLs have t(4;14) translocations and patients with this cytogenetic feature belong to the intermediate-risk group. In fact, the TRAF3 gene is also located on chromosome 14q32, where the most frequent IgH rearrangement t(14;16)(q32;q23) (Lim et al. 2013) affecting this region happens and might cause ofTRAF3 partial or biallelic deletion. Such alterations may underlie the high occurrence of TRAF3 mutation. In a recent study, albeit its small sample size, TRAF3 mutation showed a strong impact on diffuse large B-cell lymphoma survival (Zhang et al. 2015). The cBioPortal, which also compiled TRAF3 mutation data across all types of cancers to date, indicates the loss of the C-terminal region called the meprin and TRAF homology (MATH) domain where it interacts with the N-terminal domain of NIK to elicit ubiquitin-dependent degradation of NIK (Liao et al. 2004). It is also worthy to note that, due to the rearrangement at N-terminal of NIK where TRAF3 interacts, JJN3 cell line happens to have the phenocopy of other cell lines with TRAF3 mutation.
Drug resistance in MM
Despite current developments in the treatment regimen for MM patients, drug response perhaps remains a major concern. For example, bortezomib is the first-in-class drug that continued to be used in MM; approximately, 20% of MM patients do not respond to bortezomib (San Miguel et al. 2008). Patients may be intrinsically resistant to it or perhaps may develop resistance during the course of treatment. For the DR in MM, the role of cytogenetic and subsequent epigenetic alterations and the pro-survival advantage conferred by the bone marrow milieu as well as the new concept of MM cancer stem cell (Abdi et al. 2013), the mutation in β5-subunit of the proteasome (PSMB5) (Franke et al. 2012; Oerlemans et al. 2008), aberrant activation of oncogenic signaling pathways associated with cell survival, anti-apoptosis and stress response have been indicated (Lu and Wang 2013).
The 26S proteasome consists of a 20S proteolytic core and two 19S regulatory complexes that bind the proteins tagged with ubiquitin and direct them to the 20S core. The 20S proteolytic core further consists of different α and β subunits of which β5-subunit of the proteasome (PSMB5) contains the bortezomib-binding pocket (Pandey et al. 2015). The proteasome inhibitor reversibly inhibits the chymotrypsin-like activity at the PSMB5 and has marked efficacy against multiple myeloma, several non-Hodgkin’s lymphoma subtypes and other hematological malignancy. But mutation of this PSMB5 subunit has been associated with proteasome inhibitor resistance in cancers of malignancy including multiple myeloma (Acosta-Alvear et al. 2015; Franke et al. 2012; Lu and Wang 2013). Recently post-proteasome inhibitor exposition report showed that MM patients harbor acquired regulatory DNA mutations as well as epimutations that affect different proteasomal subunits and, by different modes of action, compromise proteasome activity to escape PI therapy (Haertle et al. 2019). Nevertheless, the actual mechanisms of acquired bortezomib resistance in MM patients are not yet defined.
As the pathogenesis of MM is complex and multiple cellular events are involved, the subsequent drugs used for therapy were designed to target multiple events by combined drug modality. This therapeutic intervention also further complicates the current study of the drug response of MM. Contrary to the widely held paradigm for the presence of constitutive NF-κB activation associated with a TRAF3 inactivation, the TRAF3 gene expression was predicted to have an inverse correlation with clinical response to bortezomib, a proteasome inhibitor drug commonly used in MM (Chng et al. 2007a, b). Although the physiological pattern shows that receptor-mediated TRAF3 degradation allows NIK stabilization, and thus NF-κB2 activation, it remains unclear whether receptor-mediated signaling for TRAF3 degradation or inactivating mutation is necessary and sufficient for NF-κB2 activation. While to target NIK, small molecule from Amgen (ETC-2066) was tested on human myeloma cell lines, so far there has been no clinically approved NIK inhibitor (NI) (Demchenko et al. 2014).
TRAF3 mutation modulates proteasome inhibitor response of MM
The non-redundant biological functions of TRAF3 from other TRAFs are essential for survival in mammals. In a mouse model, homozygous deletion of TRAF3 causes postnatal mortality (Xu et al. 1996) and conditional KO causes enhanced B-cell survival and development of B lymphoma (Xie et al. 2007). Taking the fact that TRAF3 mutation-driven persistent activation of non-canonical NF-κB pathway triggers the potencial hallmarks for the oncogenic progression of MM (Basseres and Baldwin 2006), investigating wither such pathway addicted survival advantage at a basal level can give a window for myeloma cell to acquire PIR was very important. In this regard, the recent comparative study conducted on TRAF3-mutant and wild-type MMCLs (Table 1) found that MM cells with TRAF3 mutations are less sensitive to proteasome inhibitors and LCP1 was discovered to be highly activated exclusively in TRAF3 mutant MM cells and, more importantly, LCP1 activation is dependent on NIK activity. Large datasets in MM patients also demonstrated the correlation of LCP1 expression with poor prognostic markers (Shin et al. 2020).
On top of this, it is known that one of the functions of NF-κB is to promote cell survival through activation of target genes, whose product inhibits the components of the apoptotic machinery in normal and cancerous cells (Luo et al. 2005). Such anti-apoptotic genes’ effect is complemented with other genes that collaterally reinforce and promote cell survival in pathway-mutated MM setting. Prominently, the basal level derangement and abnormal expression of such genes could act as a good buffering system for resilience against the apoptosis/killing effect of the drugs (Luo et al. 2005).
The unusual PIR pathway activation indicated in Fig. 2 is adopted from what has been previously reported (O’Connor et al. 2005; Shumway and Miyamoto 2004), in which the canonical pathway is activated (through non-canonical pathway cross-talk) independent of proteasomal degradation of IkBα. The other two independent studies also reported that bortezomib induces canonical NF-κB activation in non-canonical NF-кB pathway mutant MM cells (Hideshima et al. 2009; Markovina et al. 2008). Furthermore, constitutive PIR NF-κB activity observed in RPMI8226 cells was found to be augmented by the presence of patient-derived bone marrow stromal cells, resulting in increased NF-κB-dependent transcription and resistance to bortezomib-induced apoptosis.
Fig. 2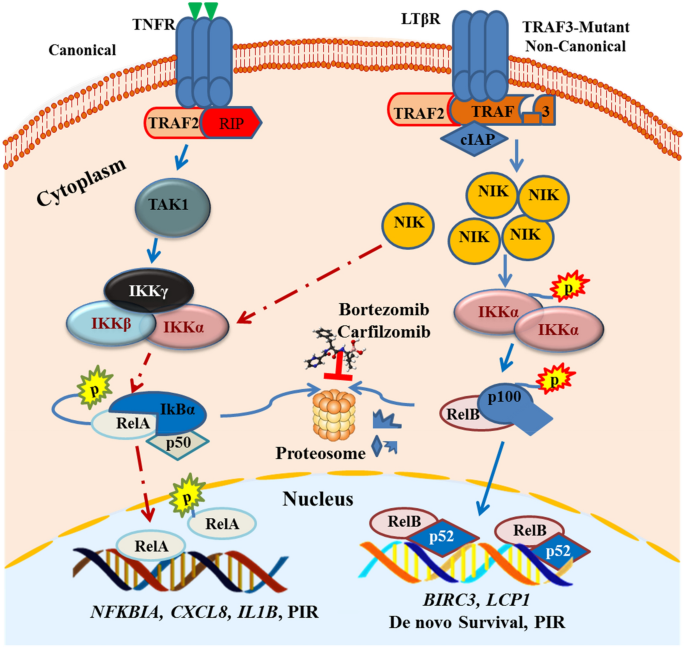
Predicted model of PIR pathway utilized by TRAF3 mutant MM cells. TRAF3 mutation allows an increase in the level of NIK that phosphorylates the IKKα homodimer and subsequently allows p100 processing to p52. Such constitutive activation of the non-canonical pathway gives a strong de novo survival signal to MM. On top of that when TRAF3 mutant cells are treated with bortezomib (which inhibits the proteasomal processing of p100), it increases the phosphorylation of p65 and IKBα degradation independent of the proteasome system. This unusual canonical pathway activation may act as a mechanism of PIR in TRAF3-mutated MM. The expression of the downstream molecular signature of TRAF3 mutation such as BIRC3 and lymphocyte cytosolic protein 1 (LCP1) can be used as a prognostic biomarker for MM
Full size imageAlthough such bortezomib-resistant NF-κB activity in multiple myeloma cells has been defined as “proteasome inhibitor-resistant” (PIR) pathway (Markovina et al. 2008), the exact molecular mechanism by which it happens and how TRAF3 mutation-associated NIK stability triggers PIR is largely unknown. But it has been reported that bortezomib treatment triggers a drastic increase in the level of NIK, followed by phosphorylation of p65 as reported in rhabdomyosarcoma (Rapino et al. 2015) and MM cell lines (Annunziata et al. 2007). Besides the universal event that can happen due to the general proteasome inhibitory effect, this stabilized NIK is still an enzymatically active kinase and thus MM cells can rather use it to maintain the survival signal under drug stress. The more recent study used TRAF3-mutant and WT cells listed in Table 1 to compare their drug response and found that MM cells with TRAF3 mutations were less sensitive to proteasome inhibitors (Shin et al. 2020). By doing genome-wide CHIP-Sequencing and RNA-sequencing, LCP1 was identified as a novel TRAF3-mutation exclusive biomarker with potential prognostic value, and LCP1 hyperactivation in TRAF3 mutant cells promotes survival under bortezomib treatment in an NIK‑dependent manner (Shin et al. 2020).
On top of this, the cross talk between the two NF-кB pathways and other stress-responsive pathways such as JUNK pathway has been reported to be mediated by NIK (Karin and Delhase 1998). The p53 pathway feedback loop also co-ordinately allows mutant cells to apparently bypass bortezomib-induced apoptosis. For example, constitutive activation of the NF-кB pathway in mutant cells allows the expression of genes encoding antioxidant and anti-apoptotic proteins (Papa et al. 2004) which enhance de novo survival of TRAF3 mutant cells. Nevertheless, so far there has been no clinically approved inhibitor of NIK; the experimental trial of crude NIK inhibitor (Demchenko et al. 2014) gives a clue that NIK inhibitor may be better for the therapy of TRAF3-mutated subpopulation of MM.
Perspectives
Taking the central role of the non-canonical NF-κB pathway in the pathogenesis of MM and the prominent frequency of TRAF3 mutation, a study on the downstream molecular signatures might be more important than our speculations. In this regard, LCP1 was recently identified as a novel NIK-driven TRAF3 dysregulation biomarker with potential prognostic value in multiple myeloma, and targeting LCP1 can provide a valuable therapeutic intervention in TRAF3-mutated MM (Shin et al. 2020). Hence, further study of the investigation of the effect of the TRAF3 mutation on the de novo survival or the emergence of drug resistance in MM, as well as to identify potential therapeutic targets for a TRAF3-mutated subpopulation of MM patients, is by far important. Since TRAF3 mutation mediates basal derangement of the non-canonical NF-кB pathway and promotes de novo survival of MM cells, whether TRAF3 mutation-associated NIK stability triggers NIK-driven acquired PIR or else whether targeting NIK than TRAF3 mutation and associated phenotype is better than to use proteasome inhibitor remains to be elucidated.
Conclusion
Multiple myeloma develops as a result of multistep tumorigenic events. The early cytogenetic events in MMGUS and SMM are often followed by the oncogenic mutation in the NF-кB pathway that drives the progression to MM. TRAF3 mutation is one of the most important prognostic factors in MM. TRAF3 mutation-mediated basal derangement of non-canonical NF-кB targets trigger de novo survival as well as the acquisition of PIR. Nevertheless, disease groups of MM patients with TRAF3 mutation need clinically relevant biomarkers. Hence, a detailed study is needed on the effect of a TRAF3 mutation in the clinical outcomes of MM, specifically to unravel the resistance mechanisms of commonly used proteasome inhibitors. On top of this, identifying alternative therapeutic targets to be used for future therapy of TRAF3 mutation-harboring subpopulation of MM patients is also very important.
References
Abdi, J., Chen, G., & Chang, H. (2013). Drug resistance in multiple myeloma: Latest findings and new concepts on molecular mechanisms. Oncotarget, 4, 2186–2207.
Acosta-Alvear, D., Cho, M. Y., Wild, T., Buchholz, T. J., Lerner, A. G., Simakova, O., et al. (2015). Paradoxical resistance of multiple myeloma to proteasome inhibitors by decreased levels of 19S proteasomal subunits. Elife, 4, e08153.
Anderson, K. C., & Carrasco, R. D. (2011). Pathogenesis of myeloma. Annual Review of Pathology, 6, 249–274.
Annunziata, C. M., Davis, R. E., Demchenko, Y., Bellamy, W., Gabrea, A., Zhan, F., et al. (2007). Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell, 12, 115–130.
Basseres, D. S., & Baldwin, A. S. (2006). Nuclear factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progression. Oncogene, 25, 6817–6830.
Bergsagel, P. L., & Chesi, M. (2013). V. Molecular classification and risk stratification of myeloma. Hematological Oncology, 31(Suppl 1), 38–41.
Chapman, M. A., Lawrence, M. S., Keats, J. J., Cibulskis, K., Sougnez, C., Schinzel, A. C., et al. (2011). Initial genome sequencing and analysis of multiple myeloma. Nature, 471, 467–472.
Chng, W. J., Glebov, O., Bergsagel, P. L., & Kuehl, W. M. (2007a). Genetic events in the pathogenesis of multiple myeloma. Best Practice & Research Clinical Haematology, 20, 571–596.
Chng, W. J., Kumar, S., Vanwier, S., Ahmann, G., Price-Troska, T., Henderson, K., et al. (2007b). Molecular dissection of hyperdiploid multiple myeloma by gene expression profiling. Cancer Research, 67, 2982–2989.
Cildir, G., Akincilar, S. C., & Tergaonkar, V. (2013). Chronic adipose tissue inflammation: all immune cells on the stage. Trends in Molecular Medicine, 19, 487–500.
Cildir, G., Low, K. C., & Tergaonkar, V. (2016). Noncanonical NF-kappaB signaling in health and disease. Trends in Molecular Medicine, 22, 414–429.
Demchenko, Y. N., Brents, L. A., Li, Z., Bergsagel, L. P., McGee, L. R., & Kuehl, M. W. (2014). Novel inhibitors are cytotoxic for myeloma cells with NFkB inducing kinase-dependent activation of NFkB. Oncotarget, 5, 4554–4566.
Demchenko, Y. N., Glebov, O. K., Zingone, A., Keats, J. J., Bergsagel, P. L., & Kuehl, W. M. (2010). Classical and/or alternative NF-kappaB pathway activation in multiple myeloma. Blood, 115, 3541–3552.
Fonseca, R., Bergsagel, P. L., Drach, J., Shaughnessy, J., Gutierrez, N., Stewart, A. K., et al. (2009). International Myeloma Working Group molecular classification of multiple myeloma: Spotlight review. Leukemia, 23, 2210–2221.
Franke, N. E., Niewerth, D., Assaraf, Y. G., van Meerloo, J., Vojtekova, K., van Zantwijk, C. H., et al. (2012). Impaired bortezomib binding to mutant beta5 subunit of the proteasome is the underlying basis for bortezomib resistance in leukemia cells. Leukemia, 26, 757–768.
Hacker, H., Redecke, V., Blagoev, B., Kratchmarova, I., Hsu, L. C., Wang, G. G., et al. (2006). Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature, 439, 204–207.
Haertle, L., Barrio, S. C., Simicek, M., Munawar, U., Sánchez, R., Bittrich, M., et al. (2019). Mechanisms of proteasome inhibitor resistance selected by clonal evolution in multiple myeloma. Blood, 134, 4349–4349.
Hayden, M. S., & Ghosh, S. (2008). Shared principles in NF-kappaB signaling. Cell, 132, 344–362.
He, J. Q., Zarnegar, B., Oganesyan, G., Saha, S. K., Yamazaki, S., Doyle, S. E., et al. (2006). Rescue of TRAF3-null mice by p100 NF-kappa B deficiency. The Journal of Experimental Medicine, 203, 2413–2418.
Hideshima, T., Ikeda, H., Chauhan, D., Okawa, Y., Raje, N., Podar, K., et al. (2009). Bortezomib induces canonical nuclear factor-kappaB activation in multiple myeloma cells. Blood, 114, 1046–1052.
Jakubowiak, A. J., Griffith, K. A., Reece, D. E., Hofmeister, C. C., Lonial, S., Zimmerman, T. M., et al. (2011). Lenalidomide, bortezomib, pegylated liposomal doxorubicin, and dexamethasone in newly diagnosed multiple myeloma: a phase 1/2 Multiple Myeloma Research Consortium trial. Blood, 118, 535–543.
Karin, M., & Delhase, M. (1998). JNK or IKK, AP-1 or NF-kappaB, which are the targets for MEK kinase 1 action? Proceedings of the National Academy of Sciences of the United States of America, 95, 9067–9069.
Keats, J. J., Fonseca, R., Chesi, M., Schop, R., Baker, A., Chng, W. J., et al. (2007). Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell, 12, 131–144.
Korde, N., Kristinsson, S. Y., & Landgren, O. (2011). Monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM): Novel biological insights and development of early treatment strategies. Blood, 117, 5573–5581.
Kumar, S., Fonseca, R., Ketterling, R. P., Dispenzieri, A., Lacy, M. Q., Gertz, M. A., et al. (2012). Trisomies in multiple myeloma: Impact on survival in patients with high-risk cytogenetics. Blood, 119, 2100–2105.
Kyle, R. A., Remstein, E. D., Therneau, T. M., Dispenzieri, A., Kurtin, P. J., Hodnefield, J. M., et al. (2007). Clinical course and prognosis of smoldering (asymptomatic) multiple myeloma. The New England Journal of Medicine, 356, 2582–2590.
Lalani, A. I., Luo, C., Han, Y., & Xie, P. (2015). TRAF3: a novel tumor suppressor gene in macrophages. Macrophage, 2, e1009.
Liao, G., Zhang, M., Harhaj, E. W., & Sun, S. C. (2004). Regulation of the NF-kappaB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. The Journal of Biological Chemistry, 279, 26243–26250.
Lim, A. S., Lim, T. H., See, K. H., Ng, Y. J., Tan, Y. M., Choo, N. S., et al. (2013). Cytogenetic and molecular aberrations of multiple myeloma patients: A single-center study in Singapore. Chinese Medical Journal, 126, 1872–1877.
Lohr, J. G., Stojanov, P., Carter, S. L., Cruz-Gordillo, P., Lawrence, M. S., Auclair, D., et al. (2014). Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell, 25, 91–101.
Lu, S., & Wang, J. (2013). The resistance mechanisms of proteasome inhibitor bortezomib. Biomarker Research, 1, 13.
Luo, J. L., Kamata, H., & Karin, M. (2005). IKK/NF-kappaB signaling: balancing life and death—a new approach to cancer therapy. The Journal of Clinical Investigation, 115, 2625–2632.
Markovina, S., Callander, N. S., O'Connor, S. L., Kim, J., Werndli, J. E., Raschko, M., et al. (2008). Bortezomib-resistant nuclear factor-kappaB activity in multiple myeloma cells. Molecular cancer research: MCR, 6, 1356–1364.
Matsushima, A., Kaisho, T., Rennert, P. D., Nakano, H., Kurosawa, K., Uchida, D., et al. (2001). Essential role of nuclear factor (NF)-kappaB-inducing kinase and inhibitor of kappaB (IkappaB) kinase alpha in NF-kappaB activation through lymphotoxin beta receptor, but not through tumor necrosis factor receptor I. The Journal of Experimental Medicine, 193, 631–636.
Medzhitov, R. (2008). Origin and physiological roles of inflammation. Nature, 454, 428–435.
O'Connor, S., Markovina, S., & Miyamoto, S. (2005). Evidence for a phosphorylation-independent role for Ser 32 and 36 in proteasome inhibitor-resistant (PIR) IkappaBalpha degradation in B cells. Experimental Cell Research, 307, 15–25.
Oerlemans, R., Franke, N. E., Assaraf, Y. G., Cloos, J., van Zantwijk, I., Berkers, C. R., et al. (2008). Molecular basis of bortezomib resistance: proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood, 112, 2489–2499.
Oganesyan, G., Saha, S. K., Guo, B., He, J. Q., Shahangian, A., Zarnegar, B., et al. (2006). Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature, 439, 208–211.
Pandey, M. K., Amin, S. G., Zangari, M., & Talamo, G. (2015). Drug resistance in multiple myeloma: how to cross the border. Annals of Hematology & Oncology, 2, 1025.
Papa, S., Zazzeroni, F., Pham, C. G., Bubici, C., & Franzoso, G. (2004). Linking JNK signaling to NF-kappaB: A key to survival. Journal of Cell Science, 117, 5197–5208.
Raab, M. S., Cavo, M., Delforge, M., Driessen, C., Fink, L., Flinois, A., et al. (2016). Multiple myeloma: Practice patterns across Europe. British Journal of Haematology, 175, 66–76.
Rapino, F., Abhari, B. A., Jung, M., & Fulda, S. (2015). NIK is required for NF-kappaB-mediated induction of BAG3 upon inhibition of constitutive protein degradation pathways. Cell Death & Disease, 6, e1692.
Saad, A., Mahindra, A., Zhang, M. J., Zhong, X., Costa, L. J., Dispenzieri, A., et al. (2014). Hematopoietic cell transplant comorbidity index is predictive of survival after autologous hematopoietic cell transplantation in multiple myeloma. Biology of Blood and Marrow Transplantation: Journal of the American Society for Blood and Marrow Transplantation, 20(402–408), e401.
Saltarella, I., Lamanuzzi, A., Reale, A., Vacca, A., & Ria, R. (2015). Identify multiple myeloma stem cells: Utopia? World J Stem Cells, 7, 84–95.
San Miguel, J. F., Schlag, R., Khuageva, N. K., Dimopoulos, M. A., Shpilberg, O., Kropff, M., et al. (2008). Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. The New England Journal of Medicine, 359, 906–917.
Shin, E. M., Neja, S. A., Fidan, K., Chua, J. Y. H., Chung, T.-H., Bertin, N., et al. (2020). Lymphocyte cytosolic protein 1 (LCP1) is a novel TRAF3 dysregulation biomarker with potential prognostic value in multiple myeloma. Genome Instability & Disease,. https://doi.org/10.1007/s42764-020-00014-x.
Shinkura, R., Kitada, K., Matsuda, F., Tashiro, K., Ikuta, K., Suzuki, M., et al. (1999). Alymphoplasia is caused by a point mutation in the mouse gene encoding Nf-kappa b-inducing kinase. Nature Genetics, 22, 74–77.
Shumway, S. D., & Miyamoto, S. (2004). A mechanistic insight into a proteasome-independent constitutive inhibitor kappaBalpha (IkappaBalpha) degradation and nuclear factor kappaB (NF-kappaB) activation pathway in WEHI-231 B-cells. The Biochemical Journal, 380, 173–180.
Stuhmer, T., Chatterjee, M., Hildebrandt, M., Herrmann, P., Gollasch, H., Gerecke, C., et al. (2005). Nongenotoxic activation of the p53 pathway as a therapeutic strategy for multiple myeloma. Blood, 106, 3609–3617.
Sun, S. C. (2011). Non-canonical NF-kappaB signaling pathway. Cell Research, 21, 71–85.
Sun, S. C. (2012). The noncanonical NF-kappaB pathway. Immunological Reviews, 246, 125–140.
Takeuchi, O., & Akira, S. (2010). Pattern recognition receptors and inflammation. Cell, 140, 805–820.
Vallabhapurapu, S., & Karin, M. (2009). Regulation and function of NF-kappaB transcription factors in the immune system. Annual Review of Immunology, 27, 693–733.
Wong, A. H., Shin, E. M., Tergaonkar, V., & Chng, W. J. (2020). Targeting NF-kappaB signaling for multiple myeloma. Cancers (Basel), 12, 2202.
Xie, P., Stunz, L. L., Larison, K. D., Yang, B., & Bishop, G. A. (2007). Tumor necrosis factor receptor-associated factor 3 is a critical regulator of B cell homeostasis in secondary lymphoid organs. Immunity, 27, 253–267.
Xu, Y., Cheng, G., & Baltimore, D. (1996). Targeted disruption of TRAF3 leads to postnatal lethality and defective T-dependent immune responses. Immunity, 5, 407–415.
Yin, L., Wu, L., Wesche, H., Arthur, C. D., White, J. M., Goeddel, D. V., et al. (2001). Defective lymphotoxin-beta receptor-induced NF-kappaB transcriptional activity in NIK-deficient mice. Science, 291, 2162–2165.
Yuregir, O. O., Sahin, F. I., Yilmaz, Z., Kizilkilic, E., Karakus, S., & Ozdogu, H. (2009). Fluorescent in situ hybridization studies in multiple myeloma. Hematology, 14, 90–94.
Zapata, J. M., Krajewska, M., Krajewski, S., Kitada, S., Welsh, K., Monks, A., et al. (2000). TNFR-associated factor family protein expression in normal tissues and lymphoid malignancies. Journal of Immunology, 165, 5084–5096.
Zarnegar, B. J., Wang, Y., Mahoney, D. J., Dempsey, P. W., Cheung, H. H., He, J., et al. (2008). Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nature Immunology, 9, 1371–1378.
Zhang, B., Calado, D. P., Wang, Z., Frohler, S., Kochert, K., Qian, Y., et al. (2015). An oncogenic role for alternative NF-kappaB signaling in DLBCL revealed upon deregulated BCL6 expression. Cell Report, 11, 715–726.
Acknowledgements
The author acknowledges Hawassa University for providing Internet facility and reading materials used to prepare this manuscript.
Funding
Nil.
Author information
Affiliations
Faculty of Veterinary Medicine, Hawassa University, P.O.Box 05, Awassa, Ethiopia
Sultan Abda Neja
Contributions
This manuscript was written entirely by the author.
Corresponding author
Correspondence to Sultan Abda Neja.
Ethics declarations
Conflict of interest
The author declares that he has no competing interests.
Rights and permissions
About this article
Cite this article
Neja, S.A. The roles of TRAF3 mutation in the oncogenic progression and drug response of multiple myeloma. GENOME INSTAB. DIS. 1, 278–285 (2020). https://doi.org/10.1007/s42764-020-00022-x
Received 06 June 2020
Revised 18 August 2020
Accepted 21 August 2020
Published 30 August 2020
Issue Date September 2020
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
Multiple myeloma
NIK
NF-κb
TRAF3
Resistance
Bortezomib
用户登录
还没有账号?
立即注册