






A new insight into base excision repair (BER) in targeted cancer therapy
Review Article
Genome Instability & Disease (2020)
Abstract
Dysregulation of redox homeostasis and the resulting generation of excessive reactive oxygen species (ROS) and oxidative DNA damage play an obligatory role in the progression of human cancer as well as therapeutic sensitivity to a variety of genome-targeting cancer drugs. In a recent study, Bao et al. (Free Radic Biol Med, 2020), present a novel mechanism that governs the sensitivity of certain cancer cells to DNA damaging therapies. The authors have identified a novel, ROS-associated, lysine acetylation modification in Uracil-DNA N-glycosylase 2 (UNG2)—a key understudied component of the base excision repair pathway, which primes UNG2 for subsequent ubiquitination by UHRF1 ligase, leading to coupling the UNG2 degradation to cancer cell death in a ROS-sensitive manner. Translational innovation of these mechanistic findings resides by the ability of epigenetic therapeutic inhibitors to elevate the levels of UNG2-Lys78 acetylation, leading to sensitizing ROS-resistant cancer cells to DNA damaging therapeutic agents. I close this commentary by briefly summarizing some outstanding questions and postulations in the field in the context of the novel findings presented by Bao et al.
Introduction
All living beings have the instinct to resist their demise by triggering the activation of survival mechanisms to defend against any arising death signals and overtime, develop coping mechanisms to both tolerate and endure repeat survival threats. This underlining principle of living beings, i.e. an attempt to rapidly build up and engage survival mechanisms to counteract threat signals, is also true at the cellular, biochemical and molecular levels for both the healthy and cancer cells. The ratio of the cell death versus survival signals and their successful and timely execution also creates a threshold tipping point for an impending outcome, cell death versus cell survival. In this context, modulation of such a threshold in cancer cells in favor of cell death—largely by impairing the ability of cancer cells to repair damaged DNA for maintaining the genomic integrity of target cells—constitutes an underlying goal for developing effective anti-cancer therapies. Here I will attempt to highlight Bao’s article, “UNG2 deacetylation confers cancer cell resistance to hydrogen peroxide-induced cytotoxicity” (Bao et al. 2020) which offers new mechanistic connections between the inner working of the molecular network which governs the functionality of reactive oxygen species (ROS) at the interface of damaging the integrity of DNA and ROS-associated acquired therapeutic resistance.
Dysregulation of redox homeostasis and resulting generation of excessive reactive oxygen species (ROS) and oxidative DNA damage play an obligatory role in the progression of human cancer as well as therapeutic sensitivity to DNA damaging therapeutic agents. Although ROS-centered mechanisms have been actively targeted in the past in the context of cancer prevention and treatment (Ursini et al. 2016; Schumacker 2006), the findings by Bao et al. add a new perspective to the field and presents a valuable cancer treatment strategy to overcome elevated ROS-associated therapeutic resistance in certain human tumors. To fully appreciate the significance of these findings and the obvious contribution to translational cancer medicine, I will first highlight the underlying big picture leading to Bao et al. observations by briefly discussing the historical perspective, advances made, continuing limitations, and how studies by Bao might serve as a guidepost for re-sensitizing certain subsets of ROS-loaded tumors to targeted therapies, in addition to increasing the effectiveness of DNA-damaging cancer therapeutics.
The DNA damage and repair pathways—historical perspective
Two of the most commonly used cancer treatments, radiation and chemotherapy, function by causing double-strand DNA breaks (DSBs), modulating the cellular redox homeostasis, elevating the levels of reactive oxygen species (ROS) which, in-turn, also trigger DNA damage—all contributing to cancer cell death. In addition, cancer cells also respond to DNA-damaging cancer drugs by stimulating the activities of several components of the DNA repair machinery at multiple regulatory levels—many of which are overexpressed, mutated, dysregulated, and/or modulated by intrinsic factors in cancer cells. In general, these DNA damage repair pathways help cancer cells to maintain their genomic integrity and survive the signals from DNA-damaging therapies. In addition to impacting the levels of accumulated DNA damage, dysregulation of the DNA repair pathways plays an inherent role in the development of therapeutic resistance to ROS and most, if not all, currently used DNA-targeting anticancer drugs over a period to posttreatment. There are several underlying causative mechanisms of the acquired therapeutic resistance such as hyperactivation of the DNA-repair machinery, activation of the cell survival pathways, compromised cell death pathways, altered drug metabolism, etc. Hence, cancer therapeutic agents designed to target the inner working of the DNA repair pathways are also useful for sensitizing relatively therapy-resistant cancer cells to a given treatment and/or reverse the therapeutic resistance. In brief, the core of an effective DNA-damaging cancer therapy resides in the nature of the cumulative DNA-damage by the drug, how efficiently damaged DNA is repaired by endogenous repair pathways despite high ROS in certain cancer-types, genomic status of molecules which guard the genomic integrity, etc.
Inklings of an association between ionizing radiation and cancer were initially discussed in the last century when radiation was thoughts to be a causative agent for certain cancers such as leukemia. For example, a strong association was noted between the increased incidence of leukemia among radiologists in the early 1900s, summarized in a later review (March 1944); experimental demonstration of radiation induced leukemia in mice by Krebs et al. (1930) and by Kirschbaum and Kaplan (1944); and epidemiological evidence of leukemia in victims of atomic bombs as summarized by Folley et al. (1952). In addition to establishing a cancer causative nature of radiation, Henry Kaplan along with Edward Ginzton also coinvented the first linear accelerator in 1954 in the Western world to use ionizing radiation to treat cancer patients (Cinzton et al.), including, Hodgkin lymphoma, which was considered a fetal, incurable cancer in 1957.
Similarly, the concept of treating cancer patients with chemotherapy could be traced back to the 1940s with its root in the early findings of the inhibitory effects of sulfur-based mustard upon the size of lymph nodes in the victims of World Wars I and II (Krumbhaar and Krumbhaar 1919). While exploring the therapeutic effects of nitrogen-based mustard in 1946, Alfred Gilman and Louis Goodman demonstrated the effectiveness of bis and tris b-chloroethyl amines derivatives of mustard in inhibiting the growth of grafted lymphomas in mice and in non-Hodgkin’s lymphoma patients in 1946 (Gilman and Philips 1946; Goodman et al. 1946). In the ensuing years, it was established that nitrogen-based mustard compounds function by creating DSBs and that such damaged DNA lesions hinder the orderly progression of the replication fork during the “S” phase of the cell cycle, leading to cellular toxicity if the damaged DNA can not be repaired in a timely manner. Oncologists use several DNA damaging agents (i.e. doxorubicin, gemcitabine, cisplatin, etoposide, etc.) which utilize various mechanisms to create DNA damage to treat a whole range of human cancers. Because DNA-damaging anti-cancer drugs are designed to promote the levels of accumulated DNA-damage, such agents also target healthy cells and cause non-specific toxicity. The issue of drug-associated toxicity and development of therapeutic resistance are two principle limitations of the current DNA-damaging agents (and perhaps, of most anti-cancer drugs). To overcome these exemplified limitations, the field is moving towards the notion of targeted therapy aimed at preferentially impairing the activity of a specific molecule/pathway of the DNA repair machinery by unearthing new aspects of DNA repair pathways, introducing new regulatory players, or assigning new functions to established molecules—as is the case in Bao et al. 2020.
Like the mid-twentieth century foundational work on DNA damage research, the history of DNA repair research tracks back to the 1940s, during the early days of research on the cellular effects of radiation on biological organisms (Friedberg 1997, 2008). This was the time when the significance of atomic energy was just beginning to be realized, with the observation that living system have an ability to tolerate and recover from the toxic cellular effects of radiation-induced cellular effects (Friedberg 2008; Hollaender and Curtis 1935). Like many early observations in late 1900s, the process of excising a damaged base was initially discovered in bacteria when scientists were attempting to understand the details underlying the removal of uracil base or pyrimidine dimers. The work led to the discovery of DNA glycosylase enzymes by Lindahl (1974) and the work from Lawrence Grossman’s team in 1980 established that the process of Base Excision Repair (BER)—the focus of Bao et al. 2020—is distinct from the nucleotide excision repair (Haseltine et al.1980), and involves excision of damaged DNA bases by a glycosylase enzyme.
Reactive oxygen species (ROS)—modulators of DNA damage sensitivity
One of the outstanding questions in DNA repair biology is the regulatory switch(s) underlying the paradox between the ability of ROS to promote oxidative DNA damage versus therapeutic resistance. This is important as modifying the functionality of ROS-processing pathways will provide a novel therapeutic strategy to overcome ROS-associated therapeutic resistance and promotes cytotoxic effects of cancer drugs. Among various types of natural DNA damage, DNA oxidization is the most common form of DNA damage in cancer cells, with an average occurrence of up to 10,000 events per day in a single cell (Meas et al. 2017). Notably, the levels of ROS are commonly elevated in cancer cells due to a variety of dysregulated biological pathways, including, high metabolic needs of cancer cells, upregulation of inflammatory cascades, reduced oxygenation leading to tumor hypoxia, inflammation, epigenetic modifications, downregulation of antioxidants affecting redox homeostasis, etc. (Schumacker 2006; Perillo et al. 2020). In addition, many cancer therapies increase the levels of cellular ROS to kill cancer cells (Perillo et al. 2020).
An efficient timely repair of damaged DNA due to intrinsic factors such as oxidative damage by free radicals and/or by extrinsic factors such as radiotherapy and chemotherapy in cancer cells is fundamental for genomic integrity. As components of DNA repair machinery are widely dysregulated in cancer cells, modulated in response to DNA-damaging anticancer agents and contribute to the acquired therapeutic resistance, targeting such molecules by targeted therapy has emerged as a preferred strategy to enhance the effectiveness of the DNA-damaging agents by overcoming therapeutic resistance (Kasai et al. 1991). Due to differing nature of the damage in double-strand DNA (dsRNA) and single-strand DNA (ssDNA), human cells have evolved distinct mechanisms to repair the damaged ssDNA and dsDNA. One such repair mechanism for ssDNA repair is BER, wherein DNA bases damaged by oxidation (or alkylation, or deamination) are excised by a glycosylase enzyme and a correct base is inserted by the sequential actions of endonucleases and DNA polymerase, and consequently, preventing mutagenesis and resisting cytotoxicity caused by ROS (David et al. 2007). Though radiation therapy and chemotherapy induce cytotoxic effects by introducing damage to DNA bases (Kelley and Fishel 2008 ), the effectiveness of such drugs is compromised in certain physiological settings such as elevated levels of ROS due to an active BER process, leading to therapeutic resistance.
Uracil-DNA N-glycosylase2 (UNG2)—a new tie that binds ROS to DNA repair
A timely repair of oxidative DNA damage is important for the survival of cancer cells in a ROS-abundant environment induced either by intrinsic biological activities or ROS-generating therapies, and therefore, such repair mechanisms contribute to ROS-resistant phenotypes. In this context, increasing efforts are being made to develop inhibitory agents to target the process of BER of oxidized DNA (Visnes et al. 2018). Unlike traditional chemotherapy and radiotherapy, which generally induce DSBs in cancer cells, these newly discovered BER inhibitors inhibit the clearance of accumulated oxidized bases from cancer cells, resulting in a toxic effect from these aberrant bases and thereby fulfilling their anti-neoplastic therapeutic purpose. Now, Bao et al. have provides further insights into BER by highlighting the critical role played by uracil DNA N-glycosylase 2 (UNG2) in the excision of genomic oxidized base lesions, leading to the survival of cancer cells in the presence of high ROS levels (Bao et al. 2020). This suggests that UNG2 might be a promising therapeutic target for cancer treatment by modifying the outcome of high ROS levels in favor of cytotoxic effects of ROS-generating therapies.
UNG is an evolutionarily conserved glycosylase from prokaryotes such as E. coli and mycoplasma to higher vertebrates, including mice and humans, suggesting that it may be essential in most living organisms. In eukaryotes, two UNG isoforms have been characterized: UNG1 resides in mitochondria and UNG2 in the nucleus. UNG1 is found proximal to the mitochondrial respiratory chain, where ROS are actively generated; therefore, it has received more attention than UNG2 in earlier studies that have explored the mechanisms via which UNG1 help cancer cells to resist ROS (Liu et al. 2016). Consequently, the role of UNG2 in therapeutic cellular resistance to ROS and its therapeutic potential, remain elusive until the present study. In this context, Bao et al. have now revealed that UNG2 and UNG1 play distinct roles in allowing cancer cells to resist ROS-associated cytotoxic effects (Bao et al. 2020). The team also established that UNG2 is the major isoform involved in repairing oxidized bases in genomic DNA and thereby, conferring therapeutic resistance against the cytotoxic effects of elevated levels of ROS in cancer cells (Bao et al. 2020). These findings also offer a valuable perspective on the individual roles of UNG2 and UNG1 in the repair of oxidized base damage and, importantly, highlights the therapeutic potential of the nuclear isoform of UNG2 in cancer prevention and treatment.
Acetylation enables UNG2 ubiquitination—a window for cancer therapy
Key components of the BER pathway are frequently modulated by post-translational modifications (PTM). For example, UNG2 glycosylase phosphorylation at Ser23, Thr60, and Ser64 increases its catalytic activity and regulates ubiquitination-associated UNG2 turnover in the late S-G2 phase of the cell cycle (Hagen et al. 2008); GSK3 phosphorylation of UNG2 at Thr60 also regulates its half-life (Baehr et al. 2016), and UNG2 phosphorylation at Thr6 or Tyr8 interrupts its binding to replication protein A and proliferating cell nuclear antigen in vitro (Weiser et al. 2017). In addition, although UNG2 protein has long been known to undergo ubiquitination with a limited understanding of the underlying mechanism (Ahn et al. 2010), while UNG2 relationship with its ubiquitination in the context of ROS biology was not investigated before. In this context, Bao et al. now report for the first time that UNG2 acetylation at Lys78 can lead to increased affinity for UHRF1, a RING domain E3 ligase that further catalyzes UNG2 ubiquitination, presumably, on Lys other than Lys78 residues, prior to its degradation (Bao et al. 2020). Interestingly, degradation of UNG2 was accompanied by a decreased BER activity and increased cell death in a ROS-sensitive manner. This finding underlines the exquisite mechanism by which UNG2 ubiquitination and consequently, BER activity and DNA damage are modulated by the status its acetylation on Lys78. Based on these new details of the inner working of UNG2 and UHRF1 in a ROS-rich physiological setting, the research team decided to modulate the status UNG2 acetylation using a histone deacetylase (HDAC) inhibitor and unexpectedly discovered an accelerated UNG2 degradation and ROS sensitivity, implying that HDAC inhibitors could be potentially be useful to covert a ROS-resistant tumor to ROS-sensitive tumor. The team went on to connect these newly discovered mechanistic events to the ROS biology by demonstrating a sensitizing effect of modifying UNG2 acetylation to an ROS-generating agent, piperlongumine, and hence, opening a translational strategy to combine an HDAC inhibitor with piperlongumine for the treatment of certain cancer-types.
Rethinking the mechanisms underlying synergistic epigenetic therapy
Work conducted over the past two decades has showed that a combined treatment of an HDAC inhibitor and decitabine (5′-aza-2′-deoxycytidine, 5-aza-CdR), a DNA methyltransferase (DNMT) inhibitor, induces a highly synergistic anti-neoplastic effect in various cancer-types (Zhu et al. 2001; Quintascardama et al. 2011), marking a breakthrough in the field of chemotherapy for cancer treatment. However, the mechanism of the synergistic effect induced by these two reagents remains unclear. Cancer cells are characterized by hypermethylation in the promoter of tumor suppressor genes, which suppresses their transcription. In addition, MeCP2, a protein that binds to methylated DNA, recruits HDAC1 to hypermethylated regions of DNA, which mediates histone deacetylation to further suppress gene transcription (Nan et al. 1998). Consistent with this finding, Cameron et al. discovered that the HDAC inhibitor trichostatin A (TSA) or 5-aza-CdR alone could only mildly alter gene expression levels, whereas a co-treatment with TSA and 5-aza-CdR leads to a robust increase in gene expression (Cameron et al. 1999). In addition, Zhu et al. discovered that this synergistic treatment resulted in remarkable increases in cancer cell death, by inducing p53-independent apoptosis from an albeit cell death by either agents alone (Zhu et al. 2001). As a result, epigenetic modulation of gene expression is thought to be one of the mechanisms underlying this exemplified synergistic therapy. However, this view somewhat contradicts the fact that the mutual silencing of DNMT1 and DNMT3 using RNAi, which mimics the inhibition of DNMT by 5-aza-CdR, failed to increase the cancer-killing effects of an HDAC inhibitor (Chai et al. 2008). In addition, although apoptosis is markedly enhanced by synergistic treatment involving an HDAC inhibitor and 5-aza-CdR, the transcription of apoptosis-related genes was seldom activated by this combined treatment. These findings suggest that a mechanism based on gene transcriptional activation resulting from the epigenetic chromatin remodeling might not be adequate to explain the synergistic anti-neoplastic effect of 5-aza-CdR and an HDAC inhibitor—both with a broad range of genome-wide targets, and we may have to think about the issue of target specificity and resulting functionality.
Interestingly, in addition to act as a DNMT inhibitor, 5-aza-CdRis also a nucleotide analog, and therefore, represents one of the earliest classes of chemicals used to treat cancer. Generally, nucleotide analogs inhibit cancer cell proliferation by their incorporation into DNA; this stalls DNA replication and leads to replication stress and DNA damage. Therefore, as a cytidine analog, 5-aza-CdR can be incorporated into DNA and induce DNA damage in cancer cells (Jones and Taylor 1980; Zhu et al. 2004; Wang et al. 2008), suggesting that 5-aza-CdR could inhibit cancer cells in a DNMT-independent manner. Importantly, a combined treatment comprising an HDAC inhibitor and 5-aza-CdR can result in enhanced levels of 5-aza-CdR in DNA and hence, increased DNA damage in cancer cells (Chai et al. 2008). This raises the possibility of regulating the level of 5-aza-CdR incorporated into DNA by HDAC inhibitors. Interestingly, the current study by Bao et al. found that an HDAC inhibitor induces UNG2 degradation, resulting in a decreased ability to excise aberrant bases from DNA. These findings might also offer an reasonable explanation why increased levels of 5-aza-CdR in DNA in cells treated with HDAC inhibitors are generally associated with a decrease in UNG2 activity. In addition, although the major substrate of UNG2 is pyrimidine uracil, for which it has the highest affinity, various pyrimidine derivatives, such as alloxan, 5-hydroxyuracil, and 5-fluorouracil, can be recognized and excised by UNG2 (Krokan and Bjørås 2013). Therefore, it is conceivable that the pyrimidine compartment of 5-aza-CdR nucleotides might also undergo UNG2-dependent excision and subsequently be repaired by the BER pathway. If these hypotheses are proven to be correct, then our understanding of the functions of UNG2 and BER could be extended to develop a synergistic strategy that combines an HDAC inhibitor with many other nucleotide analogs.
Outstanding questions and postulations
One of the key findings in the current study is to bring out the functional significance and translational novelty of UNG2-Lys78 acetylation which may create an epitope for binding to UHRF1 E3 ligase, as suggested by Bao et al. (Fig. 1a). In addition to the above translational novel possibility of combining the BER and HDAC inhibitors to overcome ROS-associated therapeutic resistance, observations made by Bao et al. have raised a few mechanistic questions for the field. It is anticipated that addressing some of these outstanding questions will not only enrich our understanding of the role played by the ROS-UNG2 axis as a determinant of ROS-sensitivity in the context of DNA damage but may help the scientific community to further refine the current therapeutic options to overcome ROS-associated resistance.
Fig. 1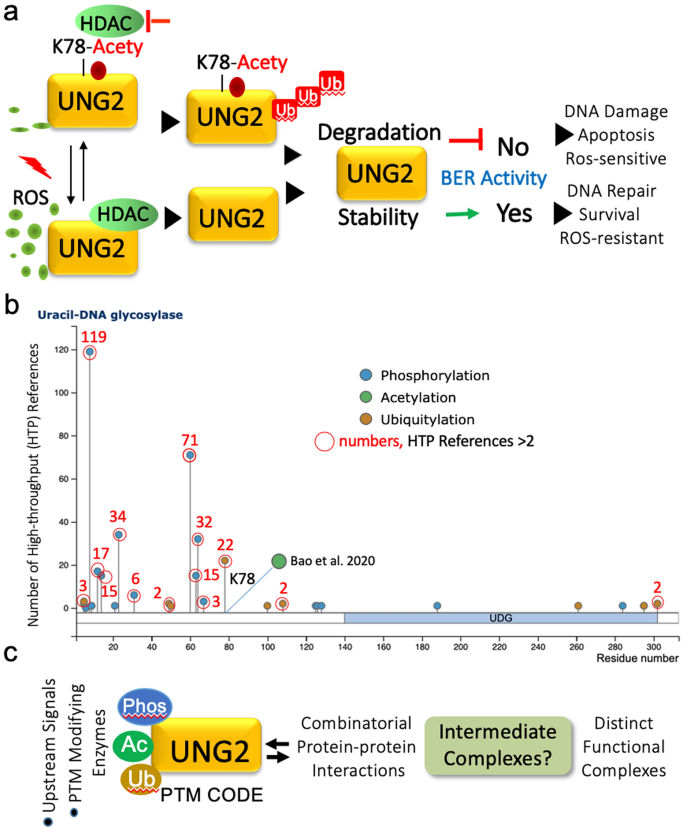
Coordinated regulation of post-translational modifications of UNG2 in the BER pathway. a Summary of the new findings by Bao et al. and its outlook. b Status of three most prevent post-translational modifications in UNG2. C, Trans-regulation of signaling-dependent PTMs and resulting possibilities
Full size imageLooking at the advances made since the recognition of ROS as a major intrinsic factor in cancer research and treatment, it is anticipated that the focus on UNG2 will continue to influence the field of DNA damage research in the coming decade. Here I will try to briefly summarize some outstanding questions in the field and our postulations in the coming decade.
As results using deletion mutants of UNG2 and UHRF1 studies suggest that multiple domains of UHRF1 interact with the UNG2 residues within the first 100 amino acids. As Lys78 has been found to be ubiquitinated in over 22 high-throughput proteomic studies (Fig. 1a) and the fact that now found to be acetylated (Bao et al. 2020), it will be important to have a better understanding of UNG2 target Lys residues of UHRF1. There are multiple lysine residues in UNG2 which have been shown to be ubiquitinated, i.e. Lys-3, -49, -50, -100, -108, -261, -295 and -302 (Fig. 1b). This raises a conceptual issue about the nature of the molecular switch or switches underlying the cellular decision whether a given lysine will be acetylated versus ubiquitinated beyond the reasons of flanking sequences and derived motifs. Based on the lessons from nuclear receptor coregulators (Kumar and O’Malley 2008), one could envision several possibilities (Fig. 1c). These include, (a) existence of two more pools of UNG2-Lys78-Ub and Lys78-Acty-containing protein–protein complexes; (b) relative activities of enzymes involved in conferring a specific PTM; (c) nature and kinetics of Lys78-Ub versus Lys78-Acety modifications and corresponding intermediate and stable protein-complexes—considering that although intermediate regulatory complexes are challenging to detect but could serve an important router for the formation of easily detectable protein complexes; (d) the switch resides in the nature and dynamic of combinatorial cross-talks among PTMs (Hunter 2007; Nguyen et al. 2013).
Some of the above postulations could be tentatively evaluated by homology modeling and computational tools as the crystal structure of a full-length UNG2 protein or with an interacting partner remains to be solved. The author further hypothesize that one of the potential switch may reside in the trans-regulation of PTMs, nature of priming PTM that cause sufficient intramolecular conformational changes to bring certain amino acid residues in proximity for a given modification(s) via creating an appropriate biophysical surface variables and accessibility to enzymes or cofactors. As signaling-dependent protein phosphorylation is one of most versatile and efficient cellular router and sensor to transduce dynamic changes in the extracellular milieu (Kumar et al. 2017; Li et al. 2012), I postulate that the nature of phosphorylation of UNG2 may be a switch, at-least in-part, to decide whether Lys78 will undergo ubiquitination or acetylation and/or prime to formation of two different pools of UNG2-containing protein–protein complexes. In the context, its noteworthy to mention that UNG2 has been found to be phosphorylated on Tyr8, Ser12, Ser14, Ser23, Thr31, Ser60, Ser63, Ser64 and Ser67 in numerous proteomic studies (Fig. 1b). Apart from Ser23, Ser60 and Ser64, functions of other phosphorylation sites in UNG2 remain orphan. Because of these phosphorylation sites are in the immediate proximity of Lys78, the nature of combinatorial phosphorylation in physiologically relevant settings may create distinct three-dimension Lys78 flanking epitope for a given modification. This thinking also lend support from the following examples of phosphorylation-dependent modulation of regulatory functions: phosphorylation of estrogen receptor α on Ser305 inhibit its acetylation on Lys303 and modulate hormone responsiveness (Cui et al. 2004); phosphorylation of CtBP1 chromatin modifier on Ser158 hinders NAD + binding within the putative regulatory loop interfacing Ser158 (Barnes et al. 2003); phosphorylation of phosphoglucomutase on Thr466 stimulates its catalytic activity (Gururaj et al. 2004); phosphorylation of p41Arc—a subunit of the Arp2/3 complex—supports its interaction with other subunits of the complex and required for overall functional activity of the complex (Vadlamudi et al. 2004), etc. Any resulting outcome of these experimentally testable postulations will not only add to the structure function relationship of UNG2 but to the broad concept of signaling-dependent trans-regulation of PTMs as a central modifier of functional outcome of regulatory proteins in a context-dependent manner.
In summary, components of the BER pathway are increasingly perceived as promising surrogate biomarkers, therapeutic cancer targets and an avenue to gain a deeper mechanistic understanding of the processes involved in Radiation Biology. A comprehensive understanding of the regulatory mechanisms of key BER components might unravel new therapeutic strategies that will contribute to a cure for cancer.
References
Ahn, J., Vu, T., Novince, Z., Guerrero-Santoro, J., Rapic-Otrin, V., & Gronenborn, A. M. (2010). HIV-1 Vpr loads uracil DNA glycosylase-2 onto DCAF1, a substrate recognition subunit of a cullin 4A-ring E3 ubiquitin ligase for proteasome-dependent degradation. Journal of Biological Chemistry, 285, 37333–37341.
Baehr, C. A., Huntoon, C. J., Hoang, S.-M., Jerde, C. R., & Karnitz, L. M. (2016). Glycogen synthase kinase 3 (GSK-3)-mediated phosphorylation of uracil N-glycosylase 2 (UNG2) facilitates the repair of floxuridine-induced DNA lesions and promotes cell survival. Journal of Biological Chemistry, 291, 26875–26885.
Bao, Y., Tong, L., Song, B., Liu, G., Zhu, Q., Lu, X., et al. (2020). UNG2 deacetylation confers cancer cell resistance to hydrogen peroxide-induced cytotoxicity. Free Radical Biology and Medicine. https://doi.org/10.1016/j.freeradbiomed.2020.06.010.
Barnes, C. J., Vadlamudi, R. K., Mishra, S. K., Jacobson, R. H., Li, F., & Kumar, R. (2003). Functional inactivation of a transcriptional corepressor by a signaling kinase. Natural Structural Biology, 10, 662–668.
Cameron, E. E., Bachman, K. E., Myohanen, S., Herman, J. G., & Baylin, S. B. (1999). Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nature Genetics, 21(1), 103–107. https://doi.org/10.1038/5047.
Chai, G., Li, L., Zhou, W., Wu, L., Zhao, Y., Wang, D., et al. (2008). HDAC inhibitors act with 5-aza-2′-deoxycytidine to inhibit cell proliferation by suppressing removal of incorporated abases in lung cancer cells. PLoS ONE. https://doi.org/10.1371/journal.pone.0002445.
Cinzton, E. L., Mallory, K. B., & Kaplan, H. S. (1957). The Stanford medical linear accelerator. I. Design and development. Stanford Medical Bulletin, 15, 123–140.
Cui, Y., Zhang, M., Pestell, R., Curran, E. M., Welshons, W. V., & Fuqua, S. A. (2004). Phosphorylation of estrogen receptor alpha blocks its acetylation and regulates estrogen sensitivity. Canadian Research, 64, 9199–9208.
David, S. S., O’Shea, V. L., & Kundu, S. (2007). Base-excision repair of oxidative DNA damage. Nature, 447, 941–950.
Folley, J. H., Borges, W., & Yamasaki, T. (1952). Incidence of leukemia in survivors of the atom bomb in Hiroshima and Nagasaki, Japan. American Journal of Medicine, 13, 311–321.
Friedberg, E. C. (1997). Correcting the blueprint of life. An historical accounting of the discovery DNA repairing mechanisms. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Friedberg, E. C. (2008). A brief history of the DNA repair field. Cell Research, 18, 3–7.
Friedberg, E. C., & King, J. J. (1969). Endonucleolytic cleavage of UV-irradiated DNA controlled by the V + gene in phage T4. Biochemical Biophysics Research Communications, 37, 646–651.
Gilman, A. (1963). The initial clinical trial of nitrogen mustard. American Journal of Surgery, 105, 574–578.
Gilman, A., & Philips, F. S. (1946). The biological actions and therapeutic applications of the B-chloroethyl amines and sulfides. Science, 103, 409–436.
Goodman, L. S., Wintrobe, M. M., Dameshek, W., Goodman, M. J., Gilman, A., & McLennan, M. T. (1946). Nitrogen mustard therapy; use of methyl-bis (beta-chloroethyl) amine hydrochloride and tris (beta-chloroethyl) amine hydrochloride for Hodgkin’s disease, lymphosarcoma, leukemia and certain allied and miscellaneous disorders. Journal of the American Medical Association, 132, 126–132.
Gururaj, A., Barnes, G. J., Vadlamudi, R. K., & Kumar, R. (2004). Phosphoglucomutase regulation by P21-activated kinases. Oncogene, 23, 8118–8127.
Hagen, L., Kavli, B., Sousa, M. M. L., Torseth, K., Liabakk, N. B., Sundheim, O., et al. (2008). Cell cycle-specificUNG2 phosphorylations regulate protein turnover, activity and association with RPA. The EMBO Journal, 27(1), 51–61. https://doi.org/10.1038/sj.emboj.7601958.
Haseltine, W. A., Gordon, L. K., Lindan, C. P., Grafstrom, N., Grossman, l, & Grossman, L. (1980). Cleavage of pyrimidine dimers in specific DNA sequences by a pyrimidine dimer DNA glycosylase of M. luteus. Nature, 285, 634–641.
Hollaender, A., & Curtis, J. T. (1935). Effect of sublethal doses of monochromatic ultraviolet radiation on bacteria in liquid suspension. Proceedings of the Society for Experimental Biology and Medicine, 33, 61–62.
Hunter, T. (2007). The age of crosstalk:phosphorylation, ubiquitination, and beyond. Molecular Cell, 28, 730–738.
Jones, P. A., & Taylor, S. M. (1980). Cellular differentiation, cytidine analogs and DNA methylation. Cell, 20(1), 85–93. https://doi.org/10.1016/0092-8674(80)90237-8.
Kasai, H., et al. (1991). Hydroxyguanine, a DNA adduct formed by oxygen radicals: Its implication on oxygen radical-involved mutagenesis/carcinogenesis. Journal of Toxicology Science, 1, 95–105.
Kelley, M. R., & Fishel, M. L. (2008). DNA repair proteins as molecular targets for cancer therapeutics. Anti-Cancer Agents in Medicinal Chemistry, 8, 417–425.
Kirschbaum, A., & Kaplan, H. S. (1944). Induction of leukemia in mice. Science, 100, 360–361.
Krebs, C., Rask-Nielsen, H. C., & Wagner, A. (1930). The origin of lymphosarcomatosis and relation to other forms of leucosis in white mice. Acta Radiologica, 10, 1–53.
Krokan, H. E., & Bjørås, M. (2013). Base excision repair. Cold Spring Harbor perspectives in biology, 5(4), a012583. https://doi.org/10.1101/cshperspect.a012583.
Krumbhaar, E. B., & Krumbhaar, H. D. (1919). The blood and bone marrow in yellow cross gas (mustard gas) poisoning: Changes produced in the bone marrow of fatal cases. Journal of Medical Research, 40, 497–508.
Kumar, R., Deivendran, S., Santhosh Kumar, T. R., & Pillai, M. R. (2017). Signaling coupled epigenomic regulation of gene expression. Oncogene, 36, 5917–5926.
Kumar, R., & O’Malley, B. W. (2008). NR coregulators and human diseases (p. 602). Singapore: World Scientific Publishers.
Li, D.-Q., Nair, S. S., Ohshiro, K., Kumar, A., Nair, V. S., Pakala, S. B., et al. (2012). MORC2 signaling facilitates phosphorylation-dependent, ATPase-coupled chromatin remodeling during the DNA damage response. Cell Report, 2, 1657–1669.
Lindahl, T. (1974). An N-glycosidase from Escherichia coli that releases free uracil from DNA containing deaminated cytosine residues. Proceedings of the National academy of Sciences of the United States of America, 71, 3649–3653.
Liu, Z., Hu, Y., Gong, Y., Zhang, W., Liu, C., Wang, Q., et al. (2016). Hydrogen peroxide mediated mitochondrial UNG1-PRDX3 interaction and UNG1 degradation. Free Radical Biology and Medicine, 99, 54–62. https://doi.org/10.1016/j.freeradbiomed.2016.07.030.
March, H. C. (1944). Leukemia in radiologists. Radiology, 43, 275–278.
Meas, R., Wyrick, J. J., Smerdon, & Michael, J. (2017). Nucleosomes regulate base excision repair in chromatin. Mutation Research—Reviews in Mutation Research, 780, 29–36. https://doi.org/10.1016/j.mrrev.2017.10.002.
Nan, X., Ng, H., Johnson, C. A., Laherty, C. D., Turner, B. M., Eisenman, R. N., et al. (1998). Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature, 393(6683), 386–389. https://doi.org/10.1038/30764.
Nguyen, L. K., Kolch, W., & Kholodenko, B. N. (2013). When ubiquitination meets phosphorylation: A systems biology perspective of EGFR/MAPK signaling. Cell Communication and Signaling, 11, 52.
Perillo, B., Di Donato, M., Pezone, A., Di Zazzo, E., Giovannelli, P., Galasso, G., et al. (2020). ROS in cancer therapy: The bright side of the moon. Experimental and Molecular Medicine, 52(2), 192–203. https://doi.org/10.1038/s12276-020-0384-2.
Pilie, P. G., Tang, C., Mills, G. B., & Yap, T. A. (2019). State-of-the-art strategies for targeting the DNA damage response in cancer. Nature Reviews Clinical Oncology, 16(2), 81–104. https://doi.org/10.1038/s41571-018-0114-z.
Quintascardama, A., Santos, F., & Garciamanero, G. (2011). Histone deacetylase inhibitors for the treatment of myelodysplastic syndrome and acute myeloid leukemia. Leukemia, 25(2), 226–235. https://doi.org/10.1038/leu.2010.276.
Schumacker, P. T. (2006). Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell, 10(3), 175–176. https://doi.org/10.1016/j.ccr.2006.08.015.
Šoštarić, N., O’Reilly, F. J., Giansanti, P., Gavin, A. J. R., & Noort, V. V. (2018). Effects of acetylation and phosphorylation on subunit interactions in three large eukaryotic complexes. Molecular and Cellular Proteomics, 17, 2387–2401.
Ursini, F., Maiorino, M., & Forman, H. J. (2016). Redox homeostasis: The golden mean of healthy living. Redox Biology, 8, 205–215.
Vadlamudi, R. K., Li, F., Barnes, C. J., Bagheri-Yarmand, R., & Kumar, R. (2004). p41 subunit of human Arp2/3 complex is a p21-activated kinase 1 interacting substrate. EMBO Reports, 5, 154–160.
Visnes, T., Cazareskorner, A., Hao, W., Wallner, O., Masuyer, G., Loseva, O., et al. (2018). Small-molecule inhibitor of OGG1 suppresses proinflammatory gene expression and inflammation. Science, 362(6416), 834–839. https://doi.org/10.1126/science.aar8048.
Wang, H., Zhao, Y., Li, L., McNutt, M. A., Wu, L., Lu, S., et al. (2008). An ATM- and Rad3-related (ATR) signaling pathway and a phosphorylation-acetylation cascade are involved in activation of p53/p21Waf1/Cip1 in response to 5-aza-2′-deoxycytidine treatment. Journal of Biological Chemistry, 283(5), 2564–2574. https://doi.org/10.1074/jbc.M702454200.
Weiser, B. P., Stivers, J. T., & Cole, P. A. (2017). Investigation of N-terminal phospho-regulation of uracil DNA glycosylase using protein semisynthesis. Biophysical Journal, 113, 393–401.
Zhu, W.-G., Hileman, T., Ke, Y., Wang, P., Lu, S., Duan, W., et al. (2004). 5-Aza-2′-deoxycytidine activates the p53/p21Waf1/Cip1 pathway to inhibit cell proliferation. Journal of Biological Chemistry, 279(15), 15161–15166. https://doi.org/10.1074/jbc.M311703200.
Zhu, W.-G., Lakshmanan, R. R., Beal, M. D., & Otterson, G. A. (2001). DNA methyltransferase inhibition enhances apoptosis induced by histone deacetylase inhibitors. Cancer Research, 61(4), 1327–1333.
Acknowledgements
The author apologizes to many of his colleagues whose deserving work could not be discussed in the present article here due to a paucity of space.
Author information
Affiliations
Cancer Research Program, Rajiv Gandhi Centre for Biotechnology, Trivandrum, Kerala, India
Rakesh Kumar
Division of Hematology and Oncology, Department of Medicine, Rutgers New Jersey Medical School, Newark, USA
Rakesh Kumar
Department of Human and Molecular Genetics, Virginia Commonwealth University Medical Center, Richmond, USA
Rakesh Kumar
Corresponding author
Correspondence to Rakesh Kumar.
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article
Cite this article
Kumar, R. A new insight into base excision repair (BER) in targeted cancer therapy. GENOME INSTAB. DIS. (2020). https://doi.org/10.1007/s42764-020-00024-9
Received19 August 2020
Revised05 September 2020
Accepted15 September 2020
Published06 October 2020
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
DNA damaging therapies
BER
UNG2
Therapeutic resistance
ROS biology
Epigenetic combination therapy
用户登录
还没有账号?
立即注册