






Targeting DNA repair pathways: mechanisms and potential applications in cancer therapy
Review Article
Genome Instability & Disease (2020)
Abstract
DNA damage can be caused by both endogenous and environmental factors, such as replication errors, reactive oxygen species (ROS) and chemotherapeutic drugs. Accurate and effective repair of DNA damage can preserve the integrity of the genome. However, if left unrepaired or repaired inappropriately, DNA damage could lead to the development of a variety of diseases, including cancers. DNA damage response (DDR) pathways are regarded as attractive tumor therapeutic targets. In recent years, representative drugs like poly(ADP-ribose) (PAR) polymerase inhibitors (PARPis) target DNA damage repair pathways and thus have generated profound breakthroughs in the treatment of tumors. Therefore, it is necessary to understand the mechanisms behind these drugs for treating tumors. In this review, we will discuss the mechanisms of representative tumor therapeutic drugs in the field of DNA damage repair and our current understanding of the related inhibitors that are expected to be used in future cancer therapies. We believe that these underlying mechanisms will provide significant support for directing drug use and future drug development.
Introduction
DNA damage can be caused either by exogenous factors such as ionizing radiation, chemical toxicity drugs, and ultraviolet radiation, or by endogenous factors including DNA replication stress, reactive oxygen species (ROS), and cellular metabolites like formaldehyde (Barzilai and Yamamoto 2004; Hoeijmakers 2009; Burgos-Barragan et al. 2017). Tens of thousands of DNA single-strand breaks (SSBs) are produced in mammalian cells every day, and these SSBs need to be repaired by DNA repair machinery (Hossain et al. 2018). One of the important proteins involved in SSB repair is poly(ADP-ribose) (PAR) polymerase 1 (PARP1), which also participates in the repair of DNA double-strand breaks (DSBs) and stalled replication forks. Delayed or incorrect repair of DNA damage can lead to diseases such as immunodeficiency, aging, and cancers.
Different DNA repair pathways and their cancer relevance
According to the different forms of DNA damage, the repair mechanisms can be divided into base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), DNA double-strand break repair, and Fanconi anaemia (FA) repair pathways (Nalepa and Clapp 2018; David et al. 2007; Marteijn et al. 2014; Jiricny 2006; Scully et al. 2019). BER is the primary mechanism for removing small base changes in DNA structures, such as those caused by oxidation, alkylation and deamination (David et al. 2007). UV irradiation mainly produces the DNA photoproduct cyclobutane pyrimidine dimer (CPD) and pyrimidine (6–4) photoproducts (6–4 PPs), which are repaired by the NER pathway (Vermeulen 2011). MMR mainly corrects mismatched base pairs in the DNA double helix and repairs the insertion or deletion of nucleotides fewer than 4 nt due to replication slip (Fishel 2015; Gupta and Heinen 2019; Li 2008). DNA nonhomologous end joining (NHEJ) and homologous recombination (HR) are two main types of double-strand break repair. NHEJ is an error-prone repair that occurs throughout the cell cycle; while, since HR-mediated repair requires sister chromatids, it mainly occurs in the S/G2 phase and is largely error free (Her and Bunting 2018). Current HR-mediated DSB repair models show that a DSB is first recognized and bound by the MRE11-RAD50-NBS1 (MRN) complex, which initiates the cleavage of the 5′-terminated DNA strand (Paull 2018; Oh and Symington 2018). The FA pathway coordinates DNA replication and might regulate mitotic checkpoints through homologous recombination to ensure error-free chromosome separation during cell division and maximizes the efficiency of DNA damage repair (Nalepa and Clapp 2018). The DNA damage response in vertebrates is controlled by three related kinases: ataxia telangiectasia mutated (ATM), ATM and RAD3-related (ATR) protein kinase, and DNA-dependent protein kinase catalytic subunit (DNA-PKcs) (Blackford and Jackson 2017). ATM and ATR often work together to signal DNA damage and regulate downstream processes. The DNA damage response is coordinated by multiple signal transduction processes, the key to which is the ATM-Chk2 and ATR-Chk1 pathways (Smith et al. 2010). DNA-PKcs is crucial to NHEJ, and NHEJ is thought to proceed through three key steps: recognition of the DNA double-strand breaks by a Ku heterodimer (KU70/KU80) and DNA-PKcs, DNA processing to remove unattached ends or other forms of terminal damage by Artemis and polynucleotide kinase/phosphatase (PNKP), and finally ligation of the DNA ends, which is carried out by XRCC4, DNA ligase IV and XLF (Dobbs et al. 2010; Wang et al. 2014).
DNA damage repair is closely related to tumorigenesis. In recent years, PARP inhibitors (PARPis) have emerged as an effective drug for the treatment of tumors. PARPis constitute a class of drugs that have been found to target BRCA1/2-deficient tumor cells and cause synthetic lethality. Here, we will review the specific mechanisms critical to the antitumor effect of PARPis. In addition to PARPis, we will summarize and discuss other DDR inhibitors, such as inhibitors of topoisomerase, ATM, ATR, DNA-PKcs and MRE11 to inspire some insightful ideas for the development of other promising DDR inhibitors that may have potential use in antitumor therapy.
PARPs and PARP inhibitors
There are 17 members of the poly(ADP-ribose) (PAR) polymerase (PARP) family, among which PARP1 and PARP2 are the most clearly studied. Lack of PARP1 or PARP2 led to cell sensitivity to genotoxic substances, but mice remained viable (Menissier de Murcia et al. 2003). Interestingly, the absence of both PARP1 and PARP2 led to early embryonic death of mice, at day 8, suggesting that the two proteins were functionally complementary. PARP1 binds damaged DNA at single-strand breaks (SSBs), DNA double-strand breaks (DSBs) or stalled DNA replication forks, which leads to a series of allosteric changes in the structure of PARP1, thereby activating its catalytic function (Lord and Ashworth 2017). The primary structure of PARP1 includes an N-terminal tri-zinc finger domain, an internal BRCA C-terminal (BRCT) domain, a Trp-Gly-Arg (WGR) domain, and a C-terminal catalytic helical domain (HD) closely related to the ART domain (Azarm and Smith 2020) (Fig. 1a). The HD regulates catalytic activity by blocking NAD + binding. After DNA damage, Zn finger and WGR structural domains surrounded DNA in wounded tissues, leading to HD allosteric instability and NAD + entry into the catalytic active site. The PARP2 structure is highly similar to the PARP1 structure, with both having the WGR, HD and ART domains (Fig. 1a).
Fig. 1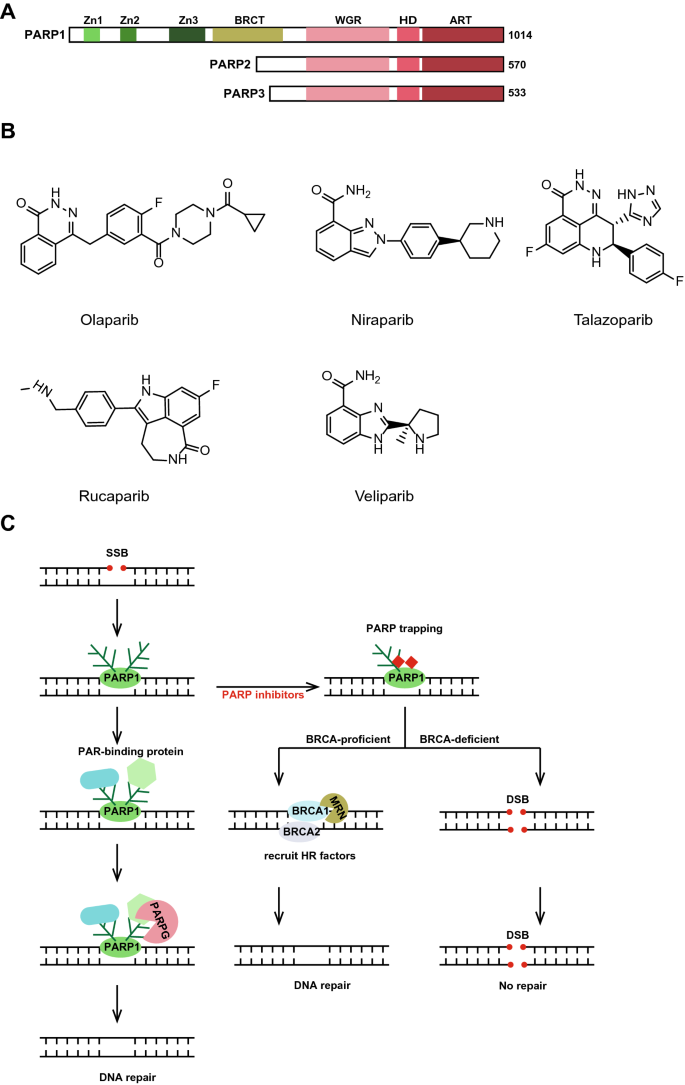
PARP inhibitors and related mechanisms. a Schematic representation of the human PARP1, PARP2 and PARP3 molecular architecture. b The chemical structure of PARP inhibitors olaparib, niraparib, talazoparib, rucaparib and veliparib. c The role of PARP1 in DNA damage repair, especially in DNA single-strand breaks (SSBs) repair, and the mechanism of PARP inhibition. When DNA single-strand breaks occur, PARP1 is rapidly recruited to the SSB site where it undergoes its own PAR modification. Then, a PAR-binding protein is recruited and repairs the damaged DNA. When the repair is completed, PARG will mediate the dissociation of PARP1 from the SSB site by facilitating the removal of the PAR chain. PARP inhibitors can trap PARP1 on DNA, preventing auto-PARylation and PARP1 release from DNA damage sites and interfering with the catalytic activity of PARP1. When the function of PARP1 is inhibited, SSBs that undergo replication are converted to DSBs, which require the involvement of key proteins such as BRCA1 and BRCA2, and when BRCA1 or BRCA2 is loss, the damage cannot be repaired
Full size imagePARP1 is known to be recruited to DNA damage sites quickly, and several studies have sought to determine the upstream signal of PARP1. Research groups have found that PARP1 also plays a critical role in the assembly of ribosomes (Boamah et al. 2012; Kim et al. 2019). In one study, PARP1 is activated by snoRNAs rather than damaged DNA and activated PARP1 ADP-ribosylated RNA helicase DDX21, further controlling rDNA transcription and ribosome biosynthesis (Kim et al. 2019). For BRCA1/2-expressing wild-type cells, PARPis can be used to block enhanced ribosomal biogenesis and cancer cell growth. Zimmermann et al. used the CRISPR screening method to identify genomic ribonucleotides as a source of PARP-trapping lesions (Zimmermann et al. 2018).
PARP1 positively regulates the activation of macrophages, microglia, granulocytes, lymphocytes, and TNF-α-challenged endothelial cells (Aldinucci et al. 2007). Recently, PARPis were shown to play roles in cancer-related immunity. Shen et al. found that PARPi-mediated activation of the cGAS-STING pathway caused by unrepaired single-strand DNA contributes to their therapeutic effects independent of BRCA1/2 mutations (Shen et al. 2019). Correspondingly, a PARPi has been reported to upregulate PD-L1 expression by inactivating GSK3β (Jiao et al. 2017). In addition, PARPis showed an immunomodulatory potential to enhance the efficacy of ICIs (immune checkpoint inhibitors) in non-small cell lung cancer (NSCLC) patients with ERCC1 deficiency (Chabanon et al. 2019). In contrast, for immunotherapy, several studies have found that immunotherapy is more effective in patients with defective DNA damage repair than in patients whose repair mechanisms are intact. For example, the clinical benefits of immune checkpoint blocking PD-1 inhibitor pembrolizumab can be predicted with DNA mismatch repair status, and due to increased numbers of point mutations and the resulting accumulation of neoantigens, patients with mismatch repair defects respond better (Le et al. 2015, 2017). The combination of DDR inhibitors and immunotherapy has the potential to expand and deepen the response of cancer patients, and the first clinical trial investigating the combination of PARPis and immunotherapy is now underway (O'Connor 2015; Lee and Konstantinopoulos 2019). PARP1 is also involved in chromatin remodelling and transcription (Fujimoto et al. 2017; Huang et al. 2017; Zhao et al. 2015; Yu et al. 2018a, b; Wang et al. 2018b; Ding et al. 2019). PARP1 can function as a structural component of chromatin and regulate chromatin structure through intrinsic enzyme activity (Kim et al. 2004). PARP1 maintains histone 3 lysine 4 trimethylation (H3K4me3) levels by PARylating histone demethylase KDM5B and inhibiting its recruitment, which promotes the loading of RNA polymerase II (RNA pol II) onto the promoters of positively regulated target genes (Krishnakumar and Kraus 2010; Ke et al. 2019). PARP1 is enriched at many RNA polymerase II-transcribed promoters (Krishnakumar et al. 2008). PARylation also can modify certain RNA-binding proteins (RBPs) to regulate RNA processing, including splicing, polyadenylation, and mRNA turnover (Ke et al. 2017; Ji and Tulin 2013).
Other PARP family members also play important roles in DNA repair pathways and may have great potential in future cancer therapies. PARP3, the structure of which is very similar to that of PARP2 (Fig. 1a), catalyzes the covalent binding of a single ADP-ribose derived from the oxidative form of NAD + to target proteins, which then induce ligation and repair of DNA breaks through the BER pathway (Rodriguez-Vargas et al. 2019). Consequently, loss of PARP3 delays repair efficiency and enhances the cytotoxicity of the DNA damage caused by ionizing radiation or chemotherapy drugs. PARP10 interacts with PCNA via its PIP-box and is required for DNA repair (Nicolae et al. 2014). Mono-ADP-ribosyltransferase PARP14 is involved not only in homologous recombination necessary for repairing DNA damage but also in regulating macrophage activation together with PARP9 (Nicolae et al. 2015; Iwata et al. 2016).
In 2014, olaparib (Lynparza, AstraZeneca) (Fig. 1b) was approved by the US Food and Drug Administration (FDA) for the treatment of BRCA1 or BRCA2 mutant ovarian cancers. Currently, PARPi application is being extended to treating HR-deficient tumor cells or those cells with a defect in BRCAness-related genes. PARPis have been successfully applied to patients in the clinic by inducing synthetic lethality. Synthetic lethality is an extreme example of a genetic interaction in which two independent viable mutations combine to produce a lethal phenotype (Hengel et al. 2017; Lord and Ashworth 2017; Murai et al. 2012). As described above, PARP1 is critical to the repair of SSBs. When the function of PARP is inhibited, replication coverts unrepaired SSBs to one-end breaks, whose repair requires the involvement of the key proteins BRCA1/2, and loss of BRCA1 or BRCA2 leads to cell death. Mechanistically, the main function of PARPis is to trap PARP1 onto DNA, preventing auto-PARylation and PARP1 release from the DNA damage site (Lord and Ashworth 2017) (Fig. 1c).
To date, the FDA has approved four PARPis: olaparib (Lynparza, AstraZeneca), rucaparib (Rubraca, Clovis), niraparib (Zejula, Tesaro), and talazoparib (Talzenna, Pfizer). Veliparib (ABT-888, AbbVie) is a PARPi that has failed in previous clinical trials. The chemical structures of these PARPis are shown in Fig. 1b. All PARPis used clinically contain the benzamide portions (similar to nicotinamide) that compete with NAD + to bind PARP, therefore, their ability to restrain the catalytic activity of PARP is similar. However, due to structural differences, the selectivity for different PARPs differs among them. PARPis in clinical applications vary greatly in their ability to trap PARP1 with following effectiveness levels: talazoparib >> niraparib > olaparib ≈ rucaparib >> veliparib. Notably, this does not mean that talazoparib has the best effect, and its adverse reactions need to be considered. It was approved for treating germline BRCA-mutated, Her2-negative locally advanced or metastatic breast cancer. Rarely, a bone marrow problem called myelodysplastic syndrome has occurred in patients treated with talazoparib (https://www.drugs.com). Olaparib has been granted for the treatment of stage IV ovarian cancer with BRCA mutations, stage IV triple-negative breast cancer (TNBC) with BRCA mutations, stage IV castration-resistant prostate cancer as well as maintenance therapy for stage IV pancreatic cancer with BRCA mutations. Niraparib, an oral PARP1 and PARP2 inhibitor, was recently approved by the FDA as a maintenance treatment for adult patients with advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer who exhibit a complete or partial response to first-line platinum-based chemotherapy (https://www.asco.org/). Niraparib and rucaparib received approvals from FDA for prostate cancer treatment too. It is interesting that testing for BRCAs or other markers is not required prior to the use of niraparib.
PARPis have been shown to be used in combination with chemotherapy drugs or ionizing radiation therapy. The combination of PARPis and other anticancer drugs, such as PI3K inhibitors, CDK inhibitors, Wee1 inhibitors, DNA topoisomerase I inhibitors, DNA methyltransferase inhibitors (DNMTis) and bromodomain containing 4 (BRD4) inhibitors, is increasingly used in tumor therapy, where it has been shown to enhance the cytotoxic effects of PARPis (Jiao et al. 2017; Ibrahim et al. 2012; Juvekar et al. 2012; Liu et al. 2019; Fang et al. 2019; Zander et al. 2010; Abbotts et al. 2019; Sun et al. 2018). Among the compounds used in combination with PARPis, CDK inhibitors are very popular. In particular, the CDK4/6 inhibitor palbociclib was among the top ten tumor drugs sold globally in 2019. CDK1 phosphorylates BRCA1, which is essential for the formation of BRCA1 foci. Therefore, CDK1 inhibitors combined with PARPis can broaden the application range of PARPis (Johnson et al. 2011). The inhibition of CDK12 was also found to be an effective strategy to inhibit tumor growth and promote synthetic lethal interactions in conjunction with PARP inhibition (Lui et al. 2018). This effect is mainly due to changes in the expression of critical HR genes, such as ATM and ATR, which are profoundly decreased by CDK12 loss (Dubbury et al. 2018). Similarly, DNA methyltransferase inhibitors can achieve the effect of a combination of two inhibitors by reducing the expression of key HR factors (Abbotts et al. 2019). It is interesting that PARP1 and DNMT1 interact with each other (Reale et al. 2005). Therefore, we may be able to screen drugs that downregulate the expression of BRCAness-related genes and, importantly, combine these drugs with PARPis to expand the use of the PARPis. DNA topoisomerase I inhibitors combined with olaparib are effective, because they increase the number of SSBs. In the absence of functional PARPs, TOP1 inhibitors significantly increased the number of SSBs, thereby increasing the synthetic lethality effect in BRCA1/2-deficient cells (Zander et al. 2010).
In terms of tumor treatment, PARPis have achieved good results in treating breast cancer, ovarian cancer, prostate cancer and pancreatic cancer. In addition, PARPis are being used in small-molecule therapy for small cell lung cancer (SCLC), and SLFN11 is a predictive biomarker for the sensitivity of SCLC to single drug therapy with PARPis (Lok et al. 2017; Zoppoli et al. 2012).
Drug resistance of PARP inhibitors and their future development
Despite the clinical success of PARPis, drug resistance remains a long-term clinical problem. There are many primary factors with PARPis resistance: (1) secondary recovery mutations in the BRCA1 or BRCA2; (2) decreased poly(ADP-ribose) glycohydrolase (PARG); (3) loss of PARP1; (4) HR recovery in BRCA mutant tumor cells caused by the loss of NHEJ key factors and (5) increased expression of P-glycoprotein and decrease in PTIP and EZH2 (Cleary et al. 2020).
Secondary recovery mutations in the BRCA gene pose a threat to the treatment with PARPis. To prevent this recovery, it is crucial to find other combinations of drugs or to find the mutation site through genome sequencing.
Many research groups have carried out CRISPR screening or omics analysis on PARPis and many proteins or pathways related to the drug resistance to PARPis (Gogola et al. 2018). Poly(ADP-ribose) glycohydrolase (PARG) has recently been found to be a major cause of PARPi resistance. Excessive PARylation can induce PARP1 dissociation from DNA, and it is noteworthy that the removal of the PAR chain is also rapidly regulated by the glycation enzyme PARG (Azarm and Smith 2020). Therefore, PARG plays a critical role in controlling the life-and-death balance driven by PAR modification (Fig. 1c). It is hoped that PARGi will offer hope to patients who are resistant to PARPis. COH34, an inhibitor of PARG, can efficiently kill PARPi-resistant cancer cells (Chen and Yu 2019).
According to the mechanism of PARPis, the absence of PARP1 expression also leads to drug resistance to PARPis. However, few studies on PARP1 deficiency show the cause of drug resistance of PARPis. More broader studies have shown that the loss of 53BP1 or the shieldin complex (REV7, SHLD1, SHLD2, SHLD3) leads to HR recovery in BRCA mutant tumor cells (Gupta et al. 2018; Guo et al. 2018; Ghezraoui et al. 2018). The shieldin complex functions as a downstream factor of 53BP1-RIF1 by restraining DNA end resection facilitated by BRCA1. The conformation change of REV7 affects its interactions with other shieldin members. TRIP13 may be used as a therapeutic target for PARPi-resistant tumors by fine-tuning the conformation of the REV7 protein (Clairmont et al. 2020).
There are still many problems with PARPis that need to be solved. How cancer cells escape the lethal effects of PARPis in addition to using HR mechanisms is poorly understood (Gogola et al. 2018). Moreover, only 5–10% of cancers have BRCA1 or BRCA2 mutations that make them sensitive to PARPis, but the role of PARPis in BRCA-proficient cancers is poorly understood. In addition, PARPis are only effective against tumors in the ovary, breast, prostate, and pancreas. To expand the use of PARPis, the most important strategy is to combine them with other therapeutic approaches, such as immunotherapy. The mechanism of action of the conjugate still needs to be further explored. Additionally, the discovery of new HR factors in tumors may broaden the therapeutic potential of PARPis. In future clinical trials, choosing the most potent PARPis is critical; in this respect, we may need to measure the extent of PARP trapping, levels of drug toxicity and breadth of the treatment spectrum of the tumors.
Topoisomerases and TOP poisons
Topoisomerases play a crucial role in the metabolism of DNA within cells, releasing the topological stress that cells generate during replication, transcription, and DNA repair (Alonso et al. 2016). There are two main types of DNA topoisomerases, type I (TOP1, TOP1mt, TOP3α and TOP3β) and type II (TOP2α, TOP2β and SPO11) (McKinnon 2016). The type I topoisomerases break one strand of double-stranded DNA, allowing the other strand to rotate at the fracture around the break or downstream double-stranded DNA and then resealing the broken strand. Type II topoisomerases undergo a series of large conformational changes when catalyzed by a “chain channel” mechanism. When a type II topoisomerase splits two DNA strands, they form a phosphotyrosine bond at the two active sites (a tyrosine and a pair of 5′-phosphates), ensuring that DNA integrity is restored (Delgado et al. 2018).
Similar to the mechanism of PARPis, the function of topoisomerase (TOP) inhibitors is to trap the TOP protein onto the DNA and form stable topoisomerase-drug-DNA ternary complexes (Fig. 2a, b). Clinically successful topoisomerase-targeting anticancer drugs acting through poisoning of topoisomerase result in replication fork arrest and the formation of DNA double-strand breaks.
Fig. 2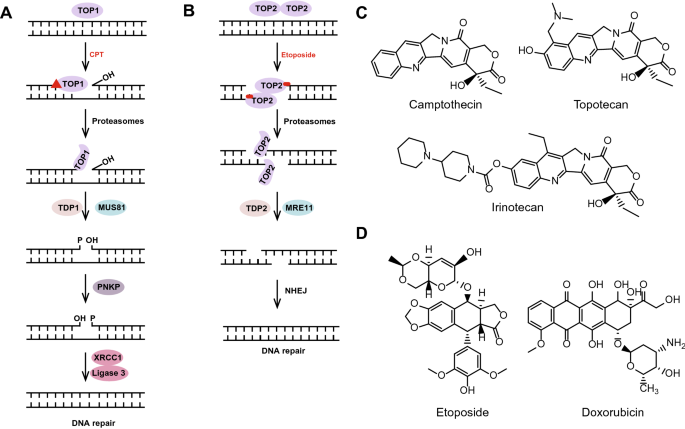
Topoisomerases and mechanisms of TOP inhibitors. a, b Topoisomerases introduce transient single or double strand breaks into DNA and rejoin the broken DNA ends to resolve DNA supercoiling during transcription and replication. In this process, both CPT and etoposide can inhibit the rejoining step and trap the TOP protein onto the DNA to form stable topoisomerase-drug-DNA ternary complexes. a Function of TOP1 in DNA damage repair and the mechanism of the TOP1 inhibitor camptothecin. There are three ways involved in TOP1cc removal: tyrosyl-DNA phosphodiesterase 1 (TDP1), an endonuclease such as MUS81, and the ubiquitin–proteasome pathway. Before DNA re-ligation by the XRCC1-DNA ligase 3 complex, TDP1 and PNKP are processed successively at the DNA 3′ end. b Function of TOP2 in DNA damage repair and mechanism of TOP2 inhibitor etoposide. TOP2 causes DNA double-strand breaks over time. Under the action of etoposide, TOP2 will be trapped to form a stable TOP2 cleavage complex (TOP2cc) at the 5′ end of the DNA sequence. Aborted TOP2ccs needed to be removed by one of three pathways: (1) proteasomal degradation of TOP2; (2) hydrolytic removal of TOP2 peptide from the DNA break site by TDP2; and (3) an endonuclease, such as MRE11. Degradation of TOP2cc by the proteasome pathway is completed before Top2cc is processed. c Chemical structure of TOP1 inhibitors, including camptothecin, topotecan and irinotecan. d Chemical structure of the TOP2 inhibitors etoposide and doxorubicin
Full size imageThe representative TOP1 inhibitor is camptothecin (CPT) (Fig. 2c), which is isolated from camptotheca plant. It functions by stabilizing the topoisomerase I cleavage complex (TOP1cc), where TOP1 covalently connects to the 3′-end of the broken DNA, which also leads to the subsequent activation of DDR (Huang et al. 2010). Without special treatment, such as CPT, after DNA relaxation, TOP1cc is rapidly reversed, and TOP1 is released. 3′ TOP1 adducts of stalled TOP1ccs need to be removed. (1) The proteasome pathway degrades 3′ TOP1 adducts at stalled TOP1ccs and contributes to the recovery from transcription block by stalled TOP1ccs (Cristini et al. 2019, 2016); (2) before DNA re-ligation by the DNA ligase III-XRCC1 complex, tyrosyl-DNA phosphodiesterase 1 (TDP1) resolves the tyrosyl-DNA phosphodiester bond to generate a 3′-phosphate, and PNKP further processes it to 3′-OH (Cristini et al. 2019); and (3) endonucleases such as MUS81 have also been shown to be involved in the removal of TOP1cc (Regairaz et al. 2011; Sacho and Maizels 2011) (Fig. 2a). More recently, high-mobility group box 2 (HMGB2) was reported to stabilize TOP1cc and further enhances recognition of cytoplasmic chromatin fragments (CCFs) by cyclic cGMP-AMP synthase (cGAS) (Zhao et al. 2020). Replication is associated with CPT-activated DDR, including RPA phosphorylation, NF-κB activation, and RAD51-mediated double-strand repair pathways, as well as p53 protein stabilization (Huang et al. 2010). Derivatives of CPT, such as topotecan and irinotecan (Fig. 2c), are currently used in the systemic treatment of colon, ovarian and non-small cell lung cancers. Although TOP1 inhibitors have a promising therapeutic effect on tumors, their use is limited because of their instability and severe side effects, as well as the resistance of P-glycoproteins. To overcome these limitations, non-camptothecin Top1 inhibitors have been developed and studied. For example, NB-506, nine-isoquinoline, and dibenzodiazepine (ARC-111) are in clinical development (Delgado et al. 2018).
The most studied TOP2 inhibitor is etoposide (Fig. 2d), which can rapidly stabilize and trap the short-lived and reversible TOP2 cleavage complex (TOP2cc), leading to the repair of stalled TOP2-linked DSBs. Aborted TOP2ccs need to be removed by these three cellular DNA repair pathways: (1) proteasomal degradation of TOP2; (2) hydrolytic removal of the TOP2 peptide from the DNA break by tyrosyl-DNA phosphodiesterase 2 (TDP2) (Ledesma et al. 2009); and (3) an endonuclease, such as MRE11, which can also remove covalently linked TOP2 from DSBs (Sasanuma et al. 2018; Hoa et al. 2016).
Degradation of TOP2 adducts through the proteasome pathway is the step before Top2cc processing, which precedes TDP2 removal of the remaining 5′ tyrosine DNA adducts (Lee et al. 2018). Finally, the DNA ends processed by TDP2 are repaired through the NHEJ pathway (Gomez-Herreros et al. 2013) (Fig. 2b). Many studies have shown that some new molecules can contribute to TOP2cc clearance by participating in these three pathways. Schellenberg et al. showed that the SUMO ligase ZATT promotes the resolution of TOP2cc both by enhancing the recruitment of TDP2 to SUMOylated TOP2 and by enhancing the hydrolase activity of TDP2 (Schellenberg et al. 2017). Sasanuma et al. demonstrated that loss of BRCA1 caused significant increases in pathological TOP2cc levels following exposure to etoposide and a decrease in etoposide-induced MRE11 foci in the G1 phase. These results suggested that BRCA1 promotes the MRE11-mediated removal TOP2cc in the G1 phase (Sasanuma et al. 2018; Akagawa et al. 2020). The endonuclease activity of MRE11 requires CtIP phosphorylation, but it has been reported that the phosphorylation of CtIP mainly occurs in the S/G2 phase (Anand et al. 2016; Ahn et al. 2015; Sartori et al. 2007). Therefore, although several studies have shown that CtIP can also be phosphorylated in the G1 phase, to elucidate the mechanism by which BRCA1 and MRE11 promote the clearance of TOP2cc in the G1 phase (Barton et al. 2014; Biehs et al. 2017; Quennet et al. 2011), further study is needed. In addition, the occurrence of abortive TOP2 catalysis and resulting stable TOP2cc formation largely depend on the level of transcription. In the region of low or no transcriptional activity, TOP2cc tends to be in a reversible state. In contrast, TOP2cc is more easily converted to an irreversible state in a region of high transcriptional activity, and the proteasome pathway plays an important role in this process (Canela et al. 2019). Notably, replication also plays an important role in this process. However, replication- and transcription-initiated processing (RIP and TIP) pathways contribute to the differential activation of various DNA damage signalling pathways and downstream cellular responses (Fan et al. 2008). END-seq was used to comprehensively capture TOP2ccs and protein-free DSBs and to provide genome-wide expression of all sites of TOP2 action (Canela et al. 2017). The development of these sequencing technologies will further elucidate the unknown mechanisms of TOP2cc action. Anthracyclines, such as doxorubicin (Fig. 2d), are widely used in the treatment of tumors, because they disrupt tumor growth by binding to topoisomerase II (TOP2) and blocking its function. Mechanistically, anthracyclines prevent TOP2 catalysis at the religation step, leading to stable DSB formation and cell death (Sawyer 2013). Although evidence suggests that reactive oxygen species may be important triggers of cardiotoxicity induced by anthracyclines, other causes of cardiotoxicity are also worth considering. Moreover, the development of anthracyclines that induce only low cardiotoxicity levels is urgently needed.
In addition to being directly related to TOP1 and TOP2, as described above, some antitumor compounds with unclear mechanisms are related to the formation of TOP1cc or TOP2cc. Carnosol is a natural compound that has anticancer activity; however, the mechanism of its anticancer effects remains unclear. More recently, carnosol was found to induce the formation of both TOP1cc and TOP2cc (Feng et al. 2020). 4-Nitroquinoline-1-oxide (4NQO), a compound that has been shown to be hypersensitive to BLM-deficient cells, induces higher levels of Top1cc formation in BLM-deficient cells than it does in BLM-complemented cells (Miao et al. 2006).
Targeting ATM, ATR and DNA-PKcs in cancer treatment
In addition to chemotherapy and surgery, radiotherapy is one of the main treatments currently used. Approximately 50% of all treatments administered to cancer patients involve radiation, either alone or, more commonly, in a combination with surgery and chemotherapy for a variety of cancers (Allen et al. 2017; Delaney et al. 2005). Radiation therapy causes DNA damage, including DNA double-strand breaks. Thus, it is important to understand the relationship of certain histological or oncogenic mutations or tumor-related hypoxia and radiotherapy dose delivery (Schaue and McBride 2015). As explained above, in addition to some exogenous factors such as ionizing radiation, chemical toxic drugs and ultraviolet radiation, which can cause DNA damage, endogenous factors such as DNA replication stress, the misaligned transcription and replication, the production of reactive oxygen species, and cellular metabolites such as formaldehyde can also cause DNA damage.
DNA damage response signals are driven by ATM, ATR (ataxia telangiectasia and RAD3-related proteins), and DNA-PKcs (DNA-dependent protein kinase catalytic subunit)-mediated protein phosphorylation. They are serine/threonine kinases of the phosphatidylinositol-3-kinase-like kinase family (PIKK), and they are very similar in structural domain composition. The kinase domain of PIKKs is located near its carboxy terminus and flanked by the conserved FAT (FRAP-ATM-TRRAP) domain and the FAT carboxy terminus (FATC) (Perry and Kleckner 2003) (Fig. 3a). Ataxia telangiectasia (A-T), a human syndrome caused by an inherited biallelic mutation of ATM, is characterized by radiosensitivity, neurodegeneration, immunodeficiency and predisposition to tumors, including thymic lymphoma. ATM, ATR, and DNA-PKcs must be tightly regulated to prevent aberrant activation that can lead to DNA repair defects and cell cycle arrest (Blackford and Jackson 2017).
Fig. 3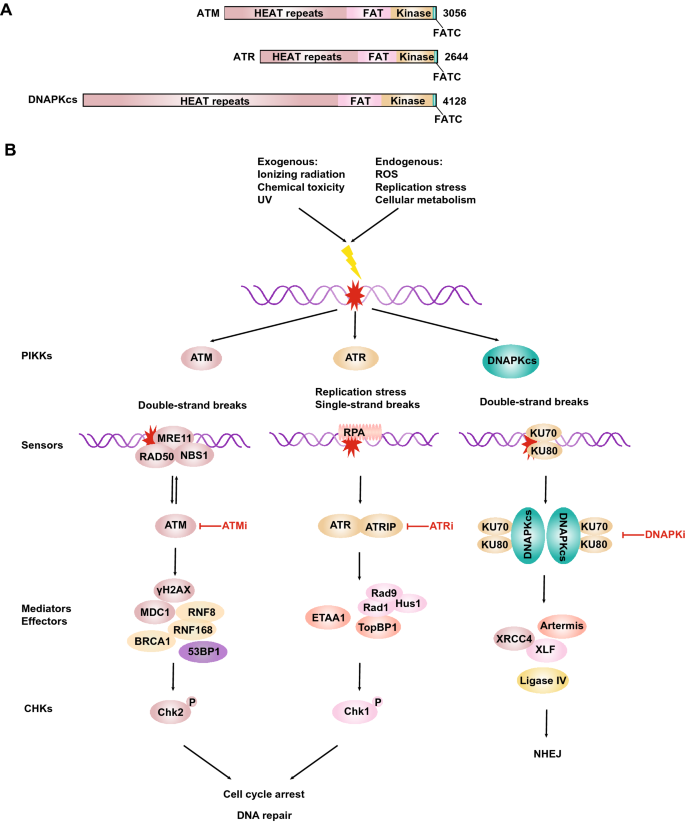
ATM, ATR and DNA-PKcs in cancer treatment. a Schematic representation of human ATM, ATR and DNA-PKcs molecular architecture. b DNA damage signals are driven by ATM, ATR and DNA-PKcs kinase (see text for details)
Full size imageCofactors are also needed for the activation of these proteins and their recruitment to DNA damage sites. DNA-PKcs is activated and recruited by Ku70/80-bound DSB ends. ATR is recruited to RPA-coated ssDNA by ATRIP (Zou and Elledge 2003). ATM is activated and recruited to DSBs by the MRE11-RAD50-NBS1 (MRN) complex (Lee and Paull 2005; Paull 2015). In turn, the MRN complex is also the direct substrate of ATM (Fig. 3b). UFMylation of MRE11 is also required for optimal DSB-induced ATM activation (Wang et al. 2019). Notably, ATM can be activated by MRN-independent mechanisms, for example, in response to oxidative stress or viral infection (Guo et al. 2010; Shah and O'Shea 2015). Tumorigenic events early in the progression of major human cancer types activate ATR/ATM-regulated checkpoints by releasing the control on DNA replication and causing DNA damage, thereby activating an induced barrier to tumor progression and genetic instability (Bartkova et al. 2005; Gorgoulis et al. 2005).
ATM and related cancer therapy
ATM is the master regulator of the DDR pathway, with many substrates (there are a large number of substrates common to ATR), which orchestrates a complex network of mediator and effector proteins to protect human genome integrity against exogenous and endogenous damage (Matsuoka et al. 2007). In undamaged cells, ATM is a dimer, and upon radiation exposure, ATM transforms into a monomer and self-phosphorylates. Human ATM is phosphorylated at S1981, which is often regarded as a marker of DNA damage repair signal activation (Bakkenist and Kastan 2003). When DNA damage occurs, ATM phosphorylates H2AX that is adjacent to the DSB sites, and γH2AX recruits MDC1 to transmit the phosphorylation signal. RNF8 binds to ATM-target motifs on MDC1 and is recruited to DSB sites (Kolas et al. 2007; Stewart et al. 2003; Lou et al. 2003). Lethal(3) malignant brain tumor-like protein 2 (L3MBTL2) is ubiquitylated by RNF8 and facilitates the recruitment of RNF168 (Nowsheen et al. 2018). RNF168 then triggers the ubiquitination of histone H2A/H2AX, which is crucial for the recruitment of 53BP1 and BRCA1 to DSB sites (Mattiroli et al. 2012) (Fig. 3b). ATM transmits signals to a large number of sensor enzymes, including checkpoint kinase 2 (Chk2) and p53. Thus, Chk2-T68 phosphorylation is also routinely detected to evaluate the status of ATM activation. ATM phosphorylates the tumor suppressor gene p53 at multiple sites, and this phosphorylation mediates biological processes such as cell cycle arrest in G1, ageing, or apoptosis (Shiloh and Ziv 2013; Banin et al. 1998).
As a key protein in the DDR pathway, ATM mainly leads to radiation resistance of tumors by promoting DNA damage repair, cell cycle arrest and chromatin remodelling. Therefore, targeting the DDR signalling pathway can reduce tumor radiotherapy resistance, and many small-molecule inhibitors targeting ATM have been shown to enhance radiotherapy sensitivity. Small-molecule inhibitors, such as AZD0156 and AZD1390, are currently available for use against kinase activity of ATM and ATM-mediated signalling, which are under investigation in phase I clinical trials for solid tumors. They have a strong selective inhibition effect on ATM kinase and can effectively penetrate the blood–brain barrier (Jin and Oh 2019). Additionally, growing evidence suggests that hyperactive DDR signalling is associated with various neurodegenerative diseases. Lu et al. showed that a reduction in ATM signalling ameliorates pathologies in models of Huntington’s disease (Lu et al. 2014).
ATR and ATR inhibitors
ATR stabilizes stalled replication and activates the cell cycle checkpoint at the S/G2 phase (Karnitz and Zou 2015). The recognition of RPA-ssDNA by ATR-ATRIP and the loading of 9–1–1 at the junction of ssDNA and dsDNA are both necessary conditions for TopBP1 to stimulate ATR at the DNA damage site (Marechal and Zou 2013) (Fig. 3b). There are at least three Mec1 (ATR ortholog)-activated proteins in budding yeast, namely, Dpb11 (TopBP1 ortholog), Ddc1, and Dna2 (Mordes et al. 2008; Kumar and Burgers 2013; Navadgi-Patil and Burgers 2009). In mammals, in addition to TopBP1 activating ATR, the ETAA1 protein has also been found to activate ATR (Haahr et al. 2016; Bass et al. 2016; Lee et al. 2016; Zou 2017; Feng et al. 2016). In contrast to TopBP1, ETAA1 can interact not only with ATR at the ATR activation domain but also directly with RPA1 and RPA2. In addition, ETAA1 is essential for promoting RPA phosphorylation but is dispensable for activating Chk1 (Haahr et al. 2016; Bass et al. 2016). Even though ETAA1 can be quickly recruited to the DSB site, the absence of ETAA1 does not increase cell sensitivity to IR or PARPis. Therefore, the function of ETAA1 in the processing of DSBs needs to be further studied. Whether the activation of ATR induced by ETAA1 is the same as that in the case of replication fork stalling needs to be confirmed. During mitosis, ATR locates at the centromere through Aurora A-regulated binding to centromere protein F (CENP-F), enabling ATR to be involved in replication protein A (RPA)-coated centromeric R-loops (Kabeche et al. 2018). The R-loop-ATR pathway promotes the faithful segregation of chromosomes (Kabeche et al. 2018). ATR is a particularly attractive target for anticancer therapy, because tumor cells may be heavily dependent on ATR for survival under high replication pressure. Correspondingly, replication stress is a hallmark of cancer, which not only represents an important aspect of cancer aetiology but also has the potential to direct cancer treatment development. Anticancer treatments do not necessarily require the inhibition of cell cycle progression. Instead, cancer cells generally grow out of control, prompting them to accelerate DNA replication could trigger a stress state that may lead to cancer cell death (Dobbelstein and Sorensen 2015). By measuring the quantitative positioning behaviour of hundreds of proteins on replication forks, Dungrawala et al. combined iPOND with SILAC mass spectrometry to understand how replication checkpoint functions in the maintenance of genomic stability (Dungrawala et al. 2015). In recent years, we have made significant progress in understanding the replication stress response of cancer cells. However, most of the work has been done in cell or in vitro. In the future, this work will benefit from complementary studies of related in vivo models, such as patient-derived explant (PDE) models or based on samples from relevant tumor patients.
ATR inhibitors are currently in clinical trials to be tested as cancer therapeutics. For example, VX-970 (berzosertib or M6620) is a promising anticancer drug that can be used either as a monotherapy or in combination with chemotherapeutic or radiotherapeutic strategies (Gorecki et al. 2020). Hypoxic cells are often resistant to chemotherapy and radiotherapy but very sensitive to ATR inhibitors (Bristow and Hill 2008; Hammond et al. 2004; Prevo et al. 2012). This finding indicates a good solution for the treatment of hypoxic solid tumors. AZD6738, an inhibitor of ATR, showed selective cytotoxicity in ATM- or p53-deficient chronic lymphocytic leukaemia (CLL) cells (Kwok et al. 2015). The synthetic lethality caused by ATR inhibition and ATM defects in gastric cancer cells also suggests a new strategy for the treatment of gastric cancer (Min et al. 2017).
DNA-PKcs and related cancer therapy
DNA-PKcs, a colossal ∼ 460-kDa polypeptide, is crucial for DSB repair through a pathway involving nonhomologous end joining (NHEJ). NHEJ is initiated by Ku and DNA-PKcs binding to DSBs, which promotes DNA-terminal connectivity through subsequent action by a DNA-PK holoenzyme. Other NHEJ core factors, including Artemis, XRCC4, XLF, and DNA ligase IV, are subsequently recruited to the end of the tight junction (Graham et al. 2016) (Fig. 3b). As a key protein kinase in the DDR pathway, DNA-PKcs catalytic activity also plays a role in V(D)J recombination (Blunt et al. 1995). A number of novel DNA-PK inhibitors have recently entered clinical development. For example, MSC2490484A (Merck KGaA; NCT02316197) is being evaluated in a phase I trial combined with radiotherapy for the treatment of patients with locally advanced tumors, and a phase I trial of VX-984 (Vertex pharmaceuticals) combined with liposomal doxorubicin has been initiated (NCT02644278) (Brown et al. 2017).
There are still some problems that need to be solved. First, the results of clinical trials are needed to confirm the efficacy of these drugs. Second, because of the structural complexity of these three important kinases, high-resolution crystal structures need to be resolved. Third, ATR inhibitors do not affect DNA synthesis or γH2AX foci formation in mitotic cells (Kabeche et al. 2018), since they have a broad spectrum of effects, which may make it more toxic. Fourth, chromatin structure and nuclear architecture critically influence the dynamics and extent of DNA damage and repair, and thus, the DNA damage response of heterochromatin and euchromatin is substantially different (Clouaire and Legube 2019; Janssen et al. 2019). Hence, DNA damage in different chromosomal structures may have different response effects and mechanisms related to these kinases, and thus need to be treated differently. New and emerging technologies, such as single-cell sequencing, CRISPR library screening, super-resolution microscopy and cryo-electron microscopy, will undoubtedly help address these remaining issues.
The MRN complex and cancer treatment
The MRN (MRE11/RAD50/NBS1) complex has been recognized as a DNA damage sensor that can be rapidly recruited to DNA double-strand break sites. Deletion or mutation of any member of the MRN complex will lead to genomic instability: the deletion and mutation of MRE11 is manifested in patients with ATLD (ataxia-telangiectasia-like disorder). Mutations in NBS1 in patients with Nijmegen breakage syndrome (NBS) are associated with tumor susceptibility. DSBs can be chemically heterogeneous and include covalent protein adducts (e.g., Top-DNA adducts). These "dirty" DNA ends require nucleolytic processing by the MRN complex and its cofactor CtIP for subsequent repair or DNA damage signalling (Hoa et al. 2016; Wright et al. 2018; Kashammer et al. 2019). MRE11 has both endonuclease activity and 3′–5′ exonuclease activity. In the HR repair pathway, based on the endonuclease activity, MRE11 first cuts DNA strand to generate a nick, and then uses its 3′–5′ exonuclease activity to chew back to the DNA end. The resultant DNA structures could serve as entry sites for DNA2/BLM or EXO1 to facilitate long-range DNA end resection (Garcia et al. 2011). This process, called DNA end resection, is the key difference between the NHEJ and HR repair pathways. Next, RAD51 mediates the replacement of replication protein A (RPA) to form RAD51 nuclear protein filaments, which catalyze a homology search and strand invasion into the sister chromatid (Fig. 4a). RAD51 foci can be detected to assess the progress of the HR repair pathway. The BRCA2-DSS1 complex is critical to RAD51 filament nucleation. However, in yeast, there are no BRCA2 homologous proteins, and RAD52 replaces the BRCA2 function. In vertebrates, RAD52-knockout mice exhibit a nearly normal phenotype, and RAD52-deficient embryonic stem cells are not hypersensitive to agents that induce DSBs (Rijkers et al. 1998; Sotiriou et al. 2016; Hanamshet et al. 2016). Research findings have shown that loss of RAD52 function is synthetically lethal in combination with BRCA2 deficiency (Feng et al. 2011).
Fig. 4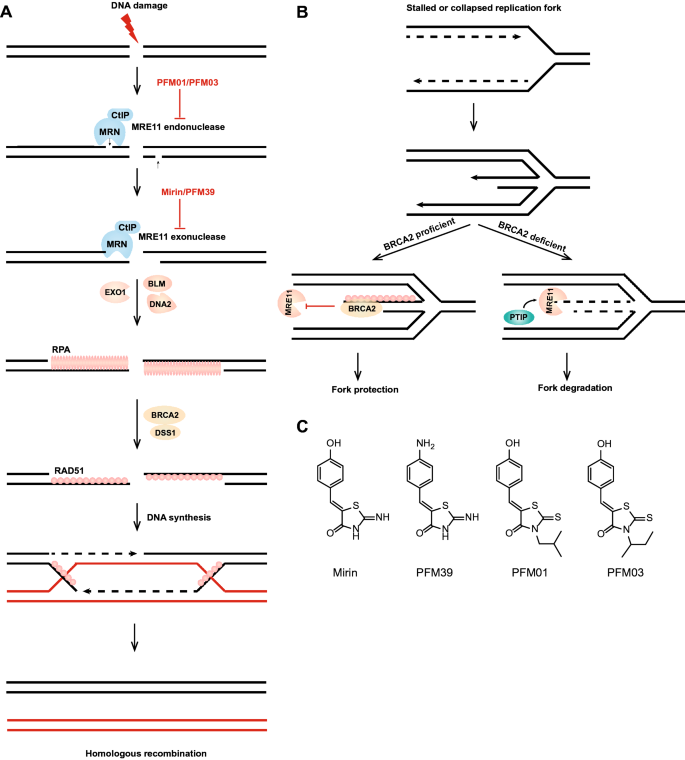
The MRN complex and cancer treatment. a Homologous recombination repair pathway and the role of MRE11 inhibitors; MRE11 has both endonuclease activity and 3′–5′ exonuclease activity. In the HR repair pathway, the MRN complex and CtIP first cut DNA to generate a short overhang to prepare for DNA2/BLM- or EXO1-mediated long-range DNA end resection. The exposed 3′ ssDNA binds to RPA, and under the action of BRCA2 and DSS1, RPA is replaced by RAD51, and then, new DNA is synthesized through chain invasion and other steps. b The role of MRE11 in disturbed replication forks. In the absence of BRCA2 protection for stabilizing RAD51 filaments, PTIP recruits the MRE11 nuclease to the stalled or collapsed replication forks to degrade nascent DNA strands and promote fork degradation-based restart. c Structure of the MRE11 exonuclease inhibitors mirin and PFM39 and the MRE11 endonuclease inhibitors PFM01 and PFM03
Full size imageBiological functions of MRE11
MRE11 can remove a variety of nucleic acid and protein blocks at DNA ends, such as covalent Spo11-DNA during meiosis, TOP1cc and TOP2cc (Moreau et al. 1999; Hoa et al. 2016; Huang et al. 2010). The study of the crystal structure of MRE11 greatly promotes the functional study of MRE11. These crystal structures revealed an interesting phenomenon: when the SMC-like RAD50 core domains are bound to ATP, the catalytic domain of MRE11 is completely blocked (Paull 2018). ATP is essential for the function of MRE11-RAD50 (MR) complex in repairing DSBs. It can regulate the structure of the MR complex and the DNA-binding activity (Lammens et al. 2011). The MRE11 dimer is flexible and stabilized by its cofactor NBS1 (Schiller et al. 2012). NBS1 promotes MR exonuclease activity at protein-blocked DNA ends while blocking the resection of clean DNA ends (Deshpande et al. 2016). In addition to promoting the exonuclease activity of MR, NBS1 functions as a sensor of CtIP phosphorylation and can also promote the endonuclease activity of MR (Anand et al. 2019). Phosphorylation of CtIP promotes MR endonuclease activity, a process of a widely accepted model (Anand et al. 2016; Wang et al. 2017; Cannavo et al. 2018; Sartori et al. 2007). In contrast, interesting studies have shown that MRE11 can regulate CtIP directly: MRE11 controls CtIP protein levels and phosphorylation status by directly interacting with cyclin-dependent kinase 2 (CDK2) (Buis et al. 2012). While the critical role of MRE11 at the initial step of homologous recombination is established, a recent study published in Genome Stability and Diseases indicates a critical role of MRE11 in homologous recombination after the formation of Holliday junctions, homologous recombination intermediates (Genome Instability and Disease, https://doi.org/10.1007/s42764-020-00015-w).
In addition to playing an important role in homologous recombination repair, MRE11 is also involved in alt-NHEJ after removal of 5′ blocking adducts, the degradation of replication forks and R-loop formation. The MRN complex and DNA ligase IIIα/XRCC1 act together in alt-NHEJ, which contributes to genome instability in cancer cells (Della-Maria et al. 2011). The identity of alt-NHEJ remains controversial, and alt-NHEJ may reflect the MRE11-mediated removal of blocking adducts from DSB ends (Hoa et al. 2016; Akagawa et al. 2020). In the absence of BRCA2 protection for stabilizing RAD51 filaments, the MLL3/4 complex protein PTIP recruits the MRE11 nuclease to stalled or collapsed replication forks, which degrades nascent DNA strands and promotes fork degradation-based restart (Chaudhuri et al. 2016; Schlacher et al. 2011) (Fig. 4b). However, it is not clear whether the function of MRE11 in the replication fork depends on the entire MRN complex. As a co-factor of MRN, CtIP is also involved in the degradation of replication forks. However, in contrast to MRE11, CtIP protects disturbed replication forks from over-excision by DNA2 (Przetocka et al. 2018). Recently, the MRN complex was found to be involved in the formation of R-loops but not dependent on the nuclease activity of MRE11, showing that the MRN complex suppresses R-loop formation and is required for FANCM and BLM recruitment to R-loop sites (Chang et al. 2019). Similarly, CtIP, BRCA1 and BRCA2 prevent R-loop accumulation in eukaryotic cells (Makharashvili et al. 2018; Chiang et al. 2019; Bhatia et al. 2014).
MRE11 is also involved in immunoregulation. Recently, the MRE11 and cGAS-STING pathways were found to be interrelated. The ssDNA generated by MRE11 cleavage may dissociate and move to the cytoplasm through the nuclear pore and activate the cGAS-STING pathway (Coquel et al. 2018; Mariggio et al. 2017; Unterholzner 2013). The cGAS-STING signalling pathway was initially discovered to be an important part of the natural immune system. Its function is to detect the presence of cytoplasmic dsDNA, including dsDNA from a DNA virus source, dsDNA produced by RNA virus reverse transcription and dsDNA released by its own nucleus and mitochondria, and to trigger the expression of inflammatory genes (Lau et al. 2015; Wang et al. 2018a; Ma et al. 2015). The differences in the ability of MRE11 and cGAS to recognize DNA have not yet been demonstrated, and the role of MRE11 in immunotherapy needs to be further studied. The biological functions involved in MRE11 are summarized in Table 1.
| Functions or related pathways | Representative participating molecules | References |
|---|---|---|
| Homologous recombination repair | RAD50, NBS1, CtIP, ATM, BRCA1, BLM, DNA2, BRCA2, RAD51, PARP1, Top-DNA adducts, cIAP2, UBQLN4, C1QBP, DYNLL1 | Anand et al. (2016), Garcia et al. (2011), Lee and Paull (2004), Sasanuma et al. (2018), Hoa et al. (2016), Haince et al. (2008), Zhao et al. (2017), Nicholson et al. (2017), Jachimowicz et al. (2019), Bai et al. (2019) and He et al. (2018) |
| Alt-NHEJ (alternative-non-homologous end joining) | RAD50, NBS1, DNA ligase IIIα, XRCC1, PARP1, Polymerase θ | Della-Maria et al. (2011) and Ceccaldi et al. (2015) |
| Replication fork stability | ATR, TopBP1, SAMHD1, PTIP, BRCA2, CtIP, DNA2, CHD4, PARP1 | Trenz et al. (2006), Duursma et al. (2013), Coquel et al. (2018), Chaudhuri et al. (2016), Przetocka et al. (2018), Ying et al. (2012) and Thangavel et al. (2015) |
| Removal of 5′ blocking adducts from DSB ends | RAD50, NBS1, BRCA1, RAP80, UBC13 | Neale et al. (2005), Garcia et al. (2011), Hoa et al. (2016) and Akagawa et al. (2020) |
| Mitochondrial DNA damage repair | RAD50, NBS1, mitochondria DNA, FXR1, RAD51 | Qi et al. (2020), Li et al. (2019), Dmitrieva et al. (2011) and Dahal et al. (2018) |
| R-loop regulation | RAD50, NBS1, RNase H, CtIP, FANCM, BLM, BRCA1 | Chang et al. (2019), Makharashvili et al. (2018) and Chiang et al. (2019) |
| Immunoregulation | ssDNA, dsDNA, cGAS, STING, Caspase1, SAMHD1 | Coquel et al. (2018) and Li et al. (2019) |
| Meiosis | RAD50, NBS1, Spo11 | Moreau et al. (1999) |
The fine-tuned regulation of MRE11 results in precise enzyme activity and protein levels as regulated by other co-factors. Previously, a series of molecules were reported to promote the exonuclease activity of MRE11, for example, NBS1, CtIP, and SAMHD1 (Coquel et al. 2018; Anand et al. 2019, 2016). The overexpression of the E3 ligase cellular inhibitor of apoptosis protein 2 (cIAP2) resulted in the downregulation of MRE11 protein levels in response to HDAC inhibition (Nicholson et al. 2017). Loss of proteasomal shuttle factor UBQLN4 leads to MRE11 accumulation in chromatin, thereby promoting HR repair activity (Jachimowicz et al. 2019). However, which molecules negatively regulate the exonuclease activity of MRE11? Recently, our group found that C1QBP stabilizes MRE11-RAD50 while inhibiting MRE11 nuclease activity by preventing its binding to DNA or chromatin (Bai et al. 2019). We speculate that the design of a small-molecule inhibitor to disrupt the interaction between MRE11 and C1QBP allows MRE11 to cut DNA without restriction, which may be helpful for diseases with limited MRE11 function. Correspondingly, inhibitors that enhance their interactions can be screened for inhibition of MRE11 enzyme activity. In BRCA1-deficient cells, He et al. demonstrated that LC-8 (DYNLL1) functions by inhibiting the exonuclease activity of MRE11 (He et al. 2018). The precise recruitment of MRE11 revealed the molecular nature of Cas activity in single-base resolution cells. Wienert et al. followed the recruitment of MRE11 to DNA breaks induced by CRISPR-Cas9, which enabled them to offer unbiased descriptions of off-target editing in cellular and animal models (Wienert et al. 2019).
MRE11 inhibitors
The first MRE11 inhibitor, mirin (6-(4-hydroxyphenyl)-2-thiophen-2,3-dihydro-4(1H)-pyrimidone) (Fig. 4c), emerged from compounds screened by forward chemical genetics and has been widely used in functional studies of MRE11, because it can inhibit the exonuclease activity of MRE11 and inhibit the activation of MRN-dependent ATM (Garner et al. 2009; Dupre et al. 2008). Selective inhibitors of endo- or exonuclease activity were developed using key libraries of mirin derivatives and by understanding the structure of the MRE11-mirin active site. Specific inhibitors of MRE11 exonuclease or endonuclease activity have been developed based on the structure of mirin. PFM01 or PFM03 (Fig. 4c) can specifically inhibit the endonuclease activity of MRE11, while PFM39 (Fig. 4c) can specifically inhibit the exonuclease activity of MRE11 (Shibata et al. 2014). In summary, the discovery of these inhibitors will play an important role in the functional study of MRE11 and contribute to the further study of DSB repair pathways (Fig. 4a).
Many key questions about the MRN complex need to be addressed. First, it has been reported that MRE11 is also located in mitochondria and is involved in the repair of mitochondrial DNA, and its binding partner FXR1 might contribute to cellular defence against mitochondrial ROS (Qi et al. 2020; Dmitrieva et al. 2011; Li et al. 2019). The inhibition of MRE11 resulted in the leakage of mitochondrial DNA into the cytosol, triggering inflammasome assembly and caspase-1-dependent pyroptosis (Li et al. 2019). HR proteins such as the MRN complex and RAD51 preserve the maintenance of mitochondrial genome stability (Dahal et al. 2018). However, it is not clear how MRE11 functions in mitochondrial DNA damage repair. What are the differences between mitochondrial DNA damage repair and genomic DNA damage repair? Second, we examined the association of MRE11 with neurodegenerative diseases and viral infections. For inhibitors of MRE11, in addition to having promising applications in the treatment of tumors, they may be useful for experimentation in neurodegenerative diseases. Moreover, to study the function of MRE11 on DSBs, it is necessary to distinguish its endonuclease and exonuclease activities. Furthermore, some showed that variants of MRN complex genes were not associated with increased risks of breast cancer (Couch et al. 2017), but are recently found to be associated with clonal expansion of hematopoietic cells in aged people (Terao et al. 2020). Thus, the mechanistic correlation between the MRN complex and breast cancer remains to be further elucidated. Finally, a longstanding question remains unanswered: how exactly does MR recognize and bind DNA ends? Obtaining an answer may require further efforts to complete the models of the MRN complex structure in mammals, possibly requiring a joint effort by researchers in structural biology and biochemistry.
Perspectives
Efficient DDR pathways can help tumor cells overcome endogenous and exogenous damage. When one of these pathways is defective, a specific pathway to repair will be triggered in the tumor cell, and at that point, a specific pathway inhibitor can be used to effectively kill the tumor cells. Therefore, DDR pathways may be ideal targets for tumor therapy. Tumor cells have more efficient or more specific DNA damage repair pathways and undergo more replication stress; therefore, we can identify tumor therapeutic targets and mechanisms of drug resistance. It is wise to look for hallmarks of cancer and then use the differences between tumor cells and normal cells to select a therapeutic regimen. In addition, some special structures related to DNA, such as G-quadruplexes and R-loops, are gradually attracting increasing research attention as possible targets of antitumor drugs (Zimmer et al. 2016). Moreover, in addition to targeting tumors with DDR inhibitors, it will also be important to maximize the therapeutic window by identifying the correct dose and schedule to treat the patient, those require an understanding of the mechanism of action, target engagement, and downstream pharmacodynamic biomarkers of the drug (O'Connor 2015). Clinical successes, such as PARPis, used with targeted DDR agents is likely possible. Recently, many inhibitors have been found to inhibit RAD51 and RAD52. Chandramouly et al. identified 6-hydroxy-DL-dopa, which interferes with RAD52 foci formation in BRCA1-deficient cells (Chandramouly et al. 2015). In addition, D-103 and D-G23 inhibit the ssDNA annealing activity of RAD52 (Huang et al. 2016), and inhibitors like DIDS and B02 affect the function of RAD51 (Huang et al. 2011; Ishida et al. 2009). Other inhibitors related to the DDR pathway may also offer hope for the treatment of tumors, such as the Ku70 inhibitor and DNA2 inhibitor (Liu et al. 2016; Li et al. 2018; Yu et al. 2018a, b).
The treatment of cancer has always been an important issue in the scientific community. With the development of science and technology, the understanding of tumors is growing, leading to advances from previous surgical treatment and chemotherapy to the targeted therapy and immunotherapy used today. Recently, antibodies against PD-1/PD-L1 and CTLA-4 have arrived on the stage of tumor treatment, bringing hope for the treatment of tumors and driving the direction of tumor treatment to immunotherapy.
In brief, advancement from learning basic mechanisms to conducting clinical research is the correct path to success, and understanding the mechanisms is critical to screening the most effective PARPis, which will also lead to more opportunities for drug development through synthetic death induction. It will be exciting to study each mechanism when we understand how the synergy of these drugs benefits each patient.
Code availability
Not applicable.
References
Abbotts, R., Topper, M. J., Biondi, C., Fontaine, D., Goswami, R., Stojanovic, L., et al. (2019). DNA methyltransferase inhibitors induce a BRCAness phenotype that sensitizes NSCLC to PARP inhibitor and ionizing radiation. Proceedings of the National Academy of Sciences, 116(45), 22609–22618. https://doi.org/10.1073/pnas.1903765116
Ahn, J. W., Kim, S., Na, W., Baek, S. J., Kim, J. H., Min, K., et al. (2015). SERBP1 affects homologous recombination-mediated DNA repair by regulation of CtIP translation during S phase. Nucleic Acids Research, 43(13), 6321–6333. https://doi.org/10.1093/nar/gkv592
Akagawa, R., Trinh, H. T., Saha, L. K., Tsuda, M., Hirota, K., Yamada, S., et al. (2020). UBC13-mediated ubiquitin signaling promotes removal of blocking adducts from DNA double-strand breaks. iScience, 23(4), 101027. https://doi.org/10.1016/j.isci.2020.101027
Aldinucci, A., Gerlini, G., Fossati, S., Cipriani, G., Ballerini, C., Biagioli, T., et al. (2007). A key role for poly(ADP-ribose) polymerase-1 activity during human dendritic cell maturation. The Journal of Immunology, 179(1), 305–312. https://doi.org/10.4049/jimmunol.179.1.305
Allen, C., Her, S., & Jaffray, D. A. (2017). Radiotherapy for cancer: present and future. Advanced Drug Delivery Reviews, 109, 1–2. https://doi.org/10.1016/j.addr.2017.01.004
Alonso, C., Fuertes, M., Gonzalez, M., Rubiales, G., Tesauro, C., Knudsen, B. R., et al. (2016). Synthesis and biological evaluation of indeno[1,5]naphthyridines as topoisomerase I (TopI) inhibitors with antiproliferative activity. European Journal of Medicinal Chemistry, 115, 179–190. https://doi.org/10.1016/j.ejmech.2016.03.031
Anand, R., Jasrotia, A., Bundschuh, D., Howard, S. M., Ranjha, L., Stucki, M., et al. (2019). NBS1 promotes the endonuclease activity of the MRE11-RAD50 complex by sensing CtIP phosphorylation. EMBO Journal. https://doi.org/10.15252/embj.2018101005
Anand, R., Ranjha, L., Cannavo, E., & Cejka, P. (2016). Phosphorylated CtIP functions as a co-factor of the MRE11-RAD50-NBS1 endonuclease in DNA end resection. Molecular Cell, 64(5), 940–950. https://doi.org/10.1016/j.molcel.2016.10.017
Azarm, K., & Smith, S. (2020). Nuclear PARPs and genome integrity. Genes and Development, 34(5–6), 285–301. https://doi.org/10.1101/gad.334730.119
Bai, Y., Wang, W., Li, S., Zhan, J., Li, H., Zhao, M., et al. (2019). C1QBP promotes homologous recombination by stabilizing MRE11 and controlling the assembly and activation of MRE11/RAD50/NBS1 complex. Molecular Cell, 75(6), 1299–1314. https://doi.org/10.1016/j.molcel.2019.06.023
Bakkenist, C. J., & Kastan, M. B. (2003). DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature, 421(6922), 499–506. https://doi.org/10.1038/nature01368
Banin, S., Moyal, L., Shieh, S., Taya, Y., Anderson, C. W., Chessa, L., et al. (1998). Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science, 281(5383), 1674–1677. https://doi.org/10.1126/science.281.5383.1674
Bartkova, J., Horejsi, Z., Koed, K., Kramer, A., Tort, F., Zieger, K., et al. (2005). DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature, 434(7035), 864–870. https://doi.org/10.1038/nature03482
Barton, O., Naumann, S. C., Diemer-Biehs, R., Kunzel, J., Steinlage, M., Conrad, S., et al. (2014). Polo-like kinase 3 regulates CtIP during DNA double-strand break repair in G1. Journal of Cell Biology, 206(7), 877–894. https://doi.org/10.1083/jcb.201401146
Barzilai, A., & Yamamoto, K. (2004). DNA damage responses to oxidative stress. DNA Repair, 3(8–9), 1109–1115. https://doi.org/10.1016/j.dnarep.2004.03.002
Bass, T. E., Luzwick, J. W., Kavanaugh, G., Carroll, C., Dungrawala, H., Glick, G. G., et al. (2016). ETAA1 acts at stalled replication forks to maintain genome integrity. Nature Cell Biology, 18(11), 1185–1195. https://doi.org/10.1038/ncb3415
Bhatia, V., Barroso, S. I., Garcia-Rubio, M. L., Tumini, E., Herrera-Moyano, E., & Aguilera, A. (2014). BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature, 511(7509), 362–365. https://doi.org/10.1038/nature13374
Biehs, R., Steinlage, M., Barton, O., Juhasz, S., Kunzel, J., Spies, J., et al. (2017). DNA double-strand break resection occurs during non-homologous end joining in G1 but is distinct from resection during homologous recombination. Molecular Cell, 65(4), 671–684. https://doi.org/10.1016/j.molcel.2016.12.016
Blackford, A. N., & Jackson, S. P. (2017). ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Molecular Cell, 66(6), 801–817. https://doi.org/10.1016/j.molcel.2017.05.015
Blunt, T., Finnie, N. J., Taccioli, G. E., Smith, G. C., Demengeot, J., Gottlieb, T. M., et al. (1995). Defective DNA-dependent protein kinase activity is linked to V(D)J recombination and DNA repair defects associated with the murine scid mutation. Cell, 80(5), 813–823. https://doi.org/10.1016/0092-8674(95)90360-7
Boamah, E. K., Kotova, E., Garabedian, M., Jarnik, M., & Tulin, A. V. (2012). Poly(ADP-Ribose) polymerase 1 (PARP-1) regulates ribosomal biogenesis in Drosophila nucleoli. PLoS Genetics, 8(1), e1002442. https://doi.org/10.1371/journal.pgen.1002442
Bristow, R. G., & Hill, R. P. (2008). Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nature Review Cancer, 8(3), 180–192. https://doi.org/10.1038/nrc2344
Brown, J. S., O’Carrigan, B., Jackson, S. P., & Yap, T. A. (2017). Targeting DNA repair in cancer: beyond PARP inhibitors. Cancer Discovery, 7(1), 20–37. https://doi.org/10.1158/2159-8290.CD-16-0860
Buis, J., Stoneham, T., Spehalski, E., & Ferguson, D. O. (2012). Mre11 regulates CtIP-dependent double-strand break repair by interaction with CDK2. Nature Structural and Molecular Biology, 19(2), 246–252. https://doi.org/10.1038/nsmb.2212
Burgos-Barragan, G., Wit, N., Meiser, J., Dingler, F. A., Pietzke, M., Mulderrig, L., et al. (2017). Mammals divert endogenous genotoxic formaldehyde into one-carbon metabolism. Nature, 548(7669), 549–554. https://doi.org/10.1038/nature23481
Canela, A., Maman, Y., Huang, S. N., Wutz, G., Tang, W., Zagnoli-Vieira, G., et al. (2019). Topoisomerase II-induced chromosome breakage and translocation is determined by chromosome architecture and transcriptional activity. Molecular Cell, 75(2), 252–266. https://doi.org/10.1016/j.molcel.2019.04.030
Canela, A., Maman, Y., Jung, S., Wong, N., Callen, E., Day, A., et al. (2017). Genome organization drives chromosome fragility. Cell, 170(3), 507–521. https://doi.org/10.1016/j.cell.2017.06.034
Cannavo, E., Johnson, D., Andres, S. N., Kissling, V. M., Reinert, J. K., Garcia, V., et al. (2018). Regulatory control of DNA end resection by Sae2 phosphorylation. Nature Communication, 9(1), 4016. https://doi.org/10.1038/s41467-018-06417-5
Ceccaldi, R., Liu, J. C., Amunugama, R., Hajdu, I., Primack, B., Petalcorin, M. I., et al. (2015). Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature, 518(7538), 258–262. https://doi.org/10.1038/nature14184
Chabanon, R. M., Muirhead, G., Krastev, D. B., Adam, J., Morel, D., Garrido, M., et al. (2019). PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer. The Journal of Clinical Investigation, 129(3), 1211–1228. https://doi.org/10.1172/JCI123319
Chandramouly, G., McDevitt, S., Sullivan, K., Kent, T., Luz, A., Glickman, J. F., et al. (2015). Small-molecule disruption of RAD52 rings as a mechanism for precision medicine in BRCA-deficient cancers. Chemistry and Biology, 22(11), 1491–1504. https://doi.org/10.1016/j.chembiol.2015.10.003
Chang, E. Y., Tsai, S., Aristizabal, M. J., Wells, J. P., Coulombe, Y., Busatto, F. F., et al. (2019). MRE11-RAD50-NBS1 promotes Fanconi Anemia R-loop suppression at transcription-replication conflicts. Nature Communication, 10(1), 4265. https://doi.org/10.1038/s41467-019-12271-w
Chen, S. H., & Yu, X. (2019). Targeting dePARylation selectively suppresses DNA repair-defective and PARP inhibitor-resistant malignancies. Science Advances, 5(4), eaav4340. https://doi.org/10.1126/sciadv.aav4340
Chiang, H. C., Zhang, X., Li, J., Zhao, X., Chen, J., Wang, H. T., et al. (2019). BRCA1-associated R-loop affects transcription and differentiation in breast luminal epithelial cells. Nucleic Acids Research, 47(10), 5086–5099. https://doi.org/10.1093/nar/gkz262
Clairmont, C. S., Sarangi, P., Ponnienselvan, K., Galli, L. D., Csete, I., Moreau, L., et al. (2020). TRIP13 regulates DNA repair pathway choice through REV7 conformational change. Nature Cell Biology, 22(1), 87–96. https://doi.org/10.1038/s41556-019-0442-y
Cleary, J. M., Aguirre, A. J., Shapiro, G. I., & D’Andrea, A. D. (2020). Biomarker-guided development of DNA repair inhibitors. Molecular Cell, 78(6), 1070–1085. https://doi.org/10.1016/j.molcel.2020.04.035
Clouaire, T., & Legube, G. (2019). A Snapshot on the cis chromatin response to DNA double-strand breaks. Trends in Genetics, 35(5), 330–345. https://doi.org/10.1016/j.tig.2019.02.003
Coquel, F., Silva, M. J., Techer, H., Zadorozhny, K., Sharma, S., Nieminuszczy, J., et al. (2018). SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature, 557(7703), 57–61. https://doi.org/10.1038/s41586-018-0050-1
Couch, F. J., Shimelis, H., Hu, C., Hart, S. N., Polley, E. C., Na, J., et al. (2017). Associations between cancer predisposition testing panel genes and breast cancer. JAMA Oncology, 3(9), 1190–1196. https://doi.org/10.1001/jamaoncol.2017.0424
Cristini, A., Park, J. H., Capranico, G., Legube, G., Favre, G., & Sordet, O. (2016). DNA-PK triggers histone ubiquitination and signaling in response to DNA double-strand breaks produced during the repair of transcription-blocking topoisomerase I lesions. Nucleic Acids Research, 44(3), 1161–1178. https://doi.org/10.1093/nar/gkv1196
Cristini, A., Ricci, G., Britton, S., Salimbeni, S., Huang, S. N., Marinello, J., et al. (2019). Dual processing of R-Loops and topoisomerase I induces transcription-dependent DNA double-strand breaks. Cell Reports, 28(12), 3167–3181. https://doi.org/10.1016/j.celrep.2019.08.041
Dahal, S., Dubey, S., & Raghavan, S. C. (2018). Homologous recombination-mediated repair of DNA double-strand breaks operates in mammalian mitochondria. Cellular and Molecular Life Sciences, 75(9), 1641–1655. https://doi.org/10.1007/s00018-017-2702-y
David, S. S., O’Shea, V. L., & Kundu, S. (2007). Base-excision repair of oxidative DNA damage. Nature, 447(7147), 941–950. https://doi.org/10.1038/nature05978
Delaney, G., Jacob, S., Featherstone, C., & Barton, M. (2005). The role of radiotherapy in cancer treatment: estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer, 104(6), 1129–1137. https://doi.org/10.1002/cncr.21324
Delgado, J. L., Hsieh, C. M., Chan, N. L., & Hiasa, H. (2018). Topoisomerases as anticancer targets. The Biochemical Journal, 475(2), 373–398. https://doi.org/10.1042/BCJ20160583
Della-Maria, J., Zhou, Y., Tsai, M. S., Kuhnlein, J., Carney, J. P., Paull, T. T., et al. (2011). Human Mre11/human Rad50/Nbs1 and DNA ligase IIIalpha/XRCC1 protein complexes act together in an alternative nonhomologous end joining pathway. Journal of Biological Chemistry, 286(39), 33845–33853. https://doi.org/10.1074/jbc.M111.274159
Deshpande, R. A., Lee, J. H., Arora, S., & Paull, T. T. (2016). Nbs1 converts the human Mre11/Rad50 nuclease complex into an endo/exonuclease machine specific for protein-DNA adducts. Molecular Cell, 64(3), 593–606. https://doi.org/10.1016/j.molcel.2016.10.010
Ding, L., Chen, X., Xu, X., Qian, Y., Liang, G., Yao, F., et al. (2019). PARP1 suppresses the transcription of PD-L1 by poly(ADP-Ribosyl)ating STAT3. Cancer Immunology Research, 7(1), 136–149. https://doi.org/10.1158/2326-6066.CIR-18-0071
Dmitrieva, N. I., Malide, D., & Burg, M. B. (2011). Mre11 is expressed in mammalian mitochondria where it binds to mitochondrial DNA. American Journal of Physiology Regulatory, Integrative and Comparative Physiology, 301(3), R632-640. https://doi.org/10.1152/ajpregu.00853.2010
Dobbelstein, M., & Sorensen, C. S. (2015). Exploiting replicative stress to treat cancer. Nature Reviews Drug Discovery, 14(6), 405–423. https://doi.org/10.1038/nrd4553
Dobbs, T. A., Tainer, J. A., & Lees-Miller, S. P. (2010). A structural model for regulation of NHEJ by DNA-PKcs autophosphorylation. DNA Repair, 9(12), 1307–1314. https://doi.org/10.1016/j.dnarep.2010.09.019
Dubbury, S. J., Boutz, P. L., & Sharp, P. A. (2018). CDK12 regulates DNA repair genes by suppressing intronic polyadenylation. Nature, 564(7734), 141–145. https://doi.org/10.1038/s41586-018-0758-y
Dungrawala, H., Rose, K. L., Bhat, K. P., Mohni, K. N., Glick, G. G., Couch, F. B., et al. (2015). The replication checkpoint prevents two types of fork collapse without regulating replisome stability. Molecular Cell, 59(6), 998–1010. https://doi.org/10.1016/j.molcel.2015.07.030
Dupre, A., Boyer-Chatenet, L., Sattler, R. M., Modi, A. P., Lee, J. H., Nicolette, M. L., et al. (2008). A forward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nature Chemical Biology, 4(2), 119–125. https://doi.org/10.1038/nchembio.63
Duursma, A. M., Driscoll, R., Elias, J. E., & Cimprich, K. A. (2013). A role for the MRN complex in ATR activation via TOPBP1 recruitment. Molecular Cell, 50(1), 116–122. https://doi.org/10.1016/j.molcel.2013.03.006
Fan, J. R., Peng, A. L., Chen, H. C., Lo, S. C., Huang, T. H., & Li, T. K. (2008). Cellular processing pathways contribute to the activation of etoposide-induced DNA damage responses. DNA Repair, 7(3), 452–463. https://doi.org/10.1016/j.dnarep.2007.12.002
Fang, Y., McGrail, D. J., Sun, C., Labrie, M., Chen, X., Zhang, D., et al. (2019). Sequential therapy with PARP and WEE1 inhibitors minimizes toxicity while maintaining efficacy. Cancer Cell, 35(6), 851–867. https://doi.org/10.1016/j.ccell.2019.05.001
Feng, S., Zhao, Y., Xu, Y., Ning, S., Huo, W., Hou, M., et al. (2016). Ewing tumor-associated antigen 1 interacts with replication protein A to promote restart of stalled replication forks. Journal of Biological Chemistry, 291(42), 21956–21962. https://doi.org/10.1074/jbc.C116.747758
Feng, X., Wu, X., Wu, Y., Zhao, Z., Xiang, C., Bai, X., et al. (2020). Critical roles of tyrosyl-DNA phosphodiesterases in cell tolerance to carnosol-induced DNA damage. Cell Biology International. https://doi.org/10.1002/cbin.11357
Feng, Z., Scott, S. P., Bussen, W., Sharma, G. G., Guo, G., Pandita, T. K., et al. (2011). Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proceedings of National Academy of Sciences, 108(2), 686–691. https://doi.org/10.1073/pnas.1010959107
Fishel, R. (2015). Mismatch repair. Journal of Biological Chemistry, 290(44), 26395–26403. https://doi.org/10.1074/jbc.R115.660142
Fujimoto, M., Takii, R., Takaki, E., Katiyar, A., Nakato, R., Shirahige, K., et al. (2017). The HSF1-PARP13-PARP1 complex facilitates DNA repair and promotes mammary tumorigenesis. Nature Communication, 8(1), 1638. https://doi.org/10.1038/s41467-017-01807-7
Garcia, V., Phelps, S. E., Gray, S., & Neale, M. J. (2011). Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature, 479(7372), 241–244. https://doi.org/10.1038/nature10515
Garner, K. M., Pletnev, A. A., & Eastman, A. (2009). Corrected structure of mirin, a small-molecule inhibitor of the Mre11-Rad50-Nbs1 complex. Nature Chemical Biology, 5(3), 129–130. https://doi.org/10.1038/nchembio0309-129
Ghezraoui, H., Oliveira, C., Becker, J. R., Bilham, K., Moralli, D., Anzilotti, C., et al. (2018). 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature, 560(7716), 122–127. https://doi.org/10.1038/s41586-018-0362-1
Gogola, E., Duarte, A. A., de Ruiter, J. R., Wiegant, W. W., Schmid, J. A., de Bruijn, R., et al. (2018). Selective loss of PARG restores PARylation and counteracts PARP inhibitor-mediated synthetic lethality. Cancer Cell, 33(6), 1078–1093. https://doi.org/10.1016/j.ccell.2018.05.008
Gomez-Herreros, F., Romero-Granados, R., Zeng, Z., Alvarez-Quilon, A., Quintero, C., Ju, L., et al. (2013). TDP2-dependent non-homologous end-joining protects against topoisomerase II-induced DNA breaks and genome instability in cells and in vivo. PLoS Genetics, 9(3), e1003226. https://doi.org/10.1371/journal.pgen.1003226
Gorecki, L., Andrs, M., Rezacova, M., & Korabecny, J. (2020). Discovery of ATR kinase inhibitor berzosertib (VX-970, M6620): clinical candidate for cancer therapy. Pharmacology and Therapeutics. https://doi.org/10.1016/j.pharmthera.2020.107518
Gorgoulis, V. G., Vassiliou, L. V., Karakaidos, P., Zacharatos, P., Kotsinas, A., Liloglou, T., et al. (2005). Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature, 434(7035), 907–913. https://doi.org/10.1038/nature03485
Graham, T. G., Walter, J. C., & Loparo, J. J. (2016). Two-stage synapsis of DNA ends during non-homologous end joining. Molecular Cell, 61(6), 850–858. https://doi.org/10.1016/j.molcel.2016.02.010
Guo, X., Bai, Y., Zhao, M., Zhou, M., Shen, Q., Yun, C. H., et al. (2018). Acetylation of 53BP1 dictates the DNA double strand break repair pathway. Nucleic Acids Research, 46(2), 689–703. https://doi.org/10.1093/nar/gkx1208
Guo, Z., Kozlov, S., Lavin, M. F., Person, M. D., & Paull, T. T. (2010). ATM activation by oxidative stress. Science, 330(6003), 517–521. https://doi.org/10.1126/science.1192912
Gupta, D., & Heinen, C. D. (2019). The mismatch repair-dependent DNA damage response: mechanisms and implications. DNA Repair, 78, 60–69. https://doi.org/10.1016/j.dnarep.2019.03.009
Gupta, R., Somyajit, K., Narita, T., Maskey, E., Stanlie, A., Kremer, M., et al. (2018). DNA repair network analysis reveals Shieldin as a key regulator of NHEJ and PARP inhibitor sensitivity. Cell, 173(4), 972–988. https://doi.org/10.1016/j.cell.2018.03.050
Haahr, P., Hoffmann, S., Tollenaere, M. A., Ho, T., Toledo, L. I., Mann, M., et al. (2016). Activation of the ATR kinase by the RPA-binding protein ETAA1. Nature Cell Biology, 18(11), 1196–1207. https://doi.org/10.1038/ncb3422
Haince, J. F., McDonald, D., Rodrigue, A., Dery, U., Masson, J. Y., Hendzel, M. J., et al. (2008). PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. Journal of Biological Chemistry, 283(2), 1197–1208. https://doi.org/10.1074/jbc.M706734200
Hammond, E. M., Dorie, M. J., & Giaccia, A. J. (2004). Inhibition of ATR leads to increased sensitivity to hypoxia/reoxygenation. Cancer Research, 64(18), 6556–6562. https://doi.org/10.1158/0008-5472.CAN-04-1520
Hanamshet, K., Mazina, O. M., & Mazin, A. V. (2016). Reappearance from obscurity: mammalian Rad52 in homologous recombination. Genes (Basel). https://doi.org/10.3390/genes7090063
He, Y. J., Meghani, K., Caron, M. C., Yang, C., Ronato, D. A., Bian, J., et al. (2018). DYNLL1 binds to MRE11 to limit DNA end resection in BRCA1-deficient cells. Nature, 563(7732), 522–526. https://doi.org/10.1038/s41586-018-0670-5
Hengel, S. R., Spies, M. A., & Spies, M. (2017). Small-molecule inhibitors targeting DNA repair and dna repair deficiency in research and cancer therapy. Cell Chemical Biology, 24(9), 1101–1119. https://doi.org/10.1016/j.chembiol.2017.08.027
Her, J., & Bunting, S. F. (2018). How cells ensure correct repair of DNA double-strand breaks. Journal of Biological Chemistry, 293(27), 10502–10511. https://doi.org/10.1074/jbc.TM118.000371
Hoa, N. N., Shimizu, T., Zhou, Z. W., Wang, Z. Q., Deshpande, R. A., Paull, T. T., et al. (2016). Mre11 is essential for the removal of lethal topoisomerase 2 covalent cleavage complexes. Molecular Cell, 64(3), 580–592. https://doi.org/10.1016/j.molcel.2016.10.011
Hoeijmakers, J. H. (2009). DNA damage, aging, and cancer. New England Journal of Medicine, 361(15), 1475–1485. https://doi.org/10.1056/NEJMra0804615
Hossain, M. A., Lin, Y., & Yan, S. (2018). Single-strand break end resection in genome integrity: mechanism and regulation by APE2. International Journal of Molecular Sciences. https://doi.org/10.3390/ijms19082389
Huang, F., Goyal, N., Sullivan, K., Hanamshet, K., Patel, M., Mazina, O. M., et al. (2016). Targeting BRCA1- and BRCA2-deficient cells with RAD52 small molecule inhibitors. Nucleic Acids Research, 44(9), 4189–4199. https://doi.org/10.1093/nar/gkw087
Huang, F., Motlekar, N. A., Burgwin, C. M., Napper, A. D., Diamond, S. L., & Mazin, A. V. (2011). Identification of specific inhibitors of human RAD51 recombinase using high-throughput screening. ACS Chemical Biology, 6(6), 628–635. https://doi.org/10.1021/cb100428c
Huang, K., Du, M., Tan, X., Yang, L., Li, X., Jiang, Y., et al. (2017). PARP1-mediated PPARalpha poly(ADP-ribosyl)ation suppresses fatty acid oxidation in non-alcoholic fatty liver disease. Journal of Hepatology, 66(5), 962–977. https://doi.org/10.1016/j.jhep.2016.11.020
Huang, T. H., Chen, H. C., Chou, S. M., Yang, Y. C., Fan, J. R., & Li, T. K. (2010). Cellular processing determinants for the activation of damage signals in response to topoisomerase I-linked DNA breakage. Cell Research, 20(9), 1060–1075. https://doi.org/10.1038/cr.2010.95
Ibrahim, Y. H., Garcia-Garcia, C., Serra, V., He, L., Torres-Lockhart, K., Prat, A., et al. (2012). PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discovery, 2(11), 1036–1047. https://doi.org/10.1158/2159-8290.CD-11-0348
Ishida, T., Takizawa, Y., Kainuma, T., Inoue, J., Mikawa, T., Shibata, T., et al. (2009). DIDS, a chemical compound that inhibits RAD51-mediated homologous pairing and strand exchange. Nucleic Acids Research, 37(10), 3367–3376. https://doi.org/10.1093/nar/gkp200
Iwata, H., Goettsch, C., Sharma, A., Ricchiuto, P., Goh, W. W., Halu, A., et al. (2016). PARP9 and PARP14 cross-regulate macrophage activation via STAT1 ADP-ribosylation. Nature Communication, 7, 12849. https://doi.org/10.1038/ncomms12849
Jachimowicz, R. D., Beleggia, F., Isensee, J., Velpula, B. B., Goergens, J., Bustos, M. A., et al. (2019). UBQLN4 represses homologous recombination and is overexpressed in aggressive tumors. Cell, 176(3), 505–519. https://doi.org/10.1016/j.cell.2018.11.024
Janssen, A., Colmenares, S. U., Lee, T., & Karpen, G. H. (2019). Timely double-strand break repair and pathway choice in pericentromeric heterochromatin depend on the histone demethylase dKDM4A. Genes and Development, 33(1–2), 103–115. https://doi.org/10.1101/gad.317537.118
Ji, Y., & Tulin, A. V. (2013). Post-transcriptional regulation by poly(ADP-ribosyl)ation of the RNA-binding proteins. International Journal of Molecular Sciences, 14(8), 16168–16183. https://doi.org/10.3390/ijms140816168
Jiao, S., Xia, W., Yamaguchi, H., Wei, Y., Chen, M. K., Hsu, J. M., et al. (2017). PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clinical Cancer Research, 23(14), 3711–3720. https://doi.org/10.1158/1078-0432.CCR-16-3215
Jin, M. H., & Oh, D. Y. (2019). ATM in DNA repair in cancer. Pharmacology and Therapeutics, 203, 107391. https://doi.org/10.1016/j.pharmthera.2019.07.002
Jiricny, J. (2006). The multifaceted mismatch-repair system. Nature Reviews Molecular Cell Biology, 7(5), 335–346. https://doi.org/10.1038/nrm1907
Johnson, N., Li, Y. C., Walton, Z. E., Cheng, K. A., Li, D., Rodig, S. J., et al. (2011). Compromised CDK1 activity sensitizes BRCA-proficient cancers to PARP inhibition. Nature Medicine, 17(7), 875–882. https://doi.org/10.1038/nm.2377
Juvekar, A., Burga, L. N., Hu, H., Lunsford, E. P., Ibrahim, Y. H., Balmana, J., et al. (2012). Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discovery, 2(11), 1048–1063. https://doi.org/10.1158/2159-8290.CD-11-0336
Kabeche, L., Nguyen, H. D., Buisson, R., & Zou, L. (2018). A mitosis-specific and R loop-driven ATR pathway promotes faithful chromosome segregation. Science, 359(6371), 108–114. https://doi.org/10.1126/science.aan6490
Karnitz, L. M., & Zou, L. (2015). Molecular pathways: targeting ATR in cancer therapy. Clinical Cancer Research, 21(21), 4780–4785. https://doi.org/10.1158/1078-0432.CCR-15-0479
Kashammer, L., Saathoff, J. H., Lammens, K., Gut, F., Bartho, J., Alt, A., et al. (2019). Mechanism of DNA end sensing and processing by the Mre11-Rad50 complex. Molecular Cell, 76(3), 382–394. https://doi.org/10.1016/j.molcel.2019.07.035
Ke, Y., Han, Y., Guo, X., Wen, J., Wang, K., Jiang, X., et al. (2017). PARP1 promotes gene expression at the post-transcriptiona level by modulating the RNA-binding protein HuR. Nature Communication, 8, 14632. https://doi.org/10.1038/ncomms14632
Ke, Y., Zhang, J., Lv, X., Zeng, X., & Ba, X. (2019). Novel insights into PARPs in gene expression: regulation of RNA metabolism. Cellular and Molecular Life Sciences, 76(17), 3283–3299. https://doi.org/10.1007/s00018-019-03120-6
Kim, D. S., Camacho, C. V., Nagari, A., Malladi, V. S., Challa, S., & Kraus, W. L. (2019). Activation of PARP-1 by snoRNAs controls ribosome biogenesis and cell growth via the RNA helicase DDX21. Molecular Cell, 75(6), 1270–1285. https://doi.org/10.1016/j.molcel.2019.06.020
Kim, M. Y., Mauro, S., Gevry, N., Lis, J. T., & Kraus, W. L. (2004). NAD+-dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP-1. Cell, 119(6), 803–814. https://doi.org/10.1016/j.cell.2004.11.002
Kolas, N. K., Chapman, J. R., Nakada, S., Ylanko, J., Chahwan, R., Sweeney, F. D., et al. (2007). Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science, 318(5856), 1637–1640. https://doi.org/10.1126/science.1150034
Krishnakumar, R., Gamble, M. J., Frizzell, K. M., Berrocal, J. G., Kininis, M., & Kraus, W. L. (2008). Reciprocal binding of PARP-1 and histone H1 at promoters specifies transcriptional outcomes. Science, 319(5864), 819–821. https://doi.org/10.1126/science.1149250
Krishnakumar, R., & Kraus, W. L. (2010). PARP-1 regulates chromatin structure and transcription through a KDM5B-dependent pathway. Molecular Cell, 39(5), 736–749. https://doi.org/10.1016/j.molcel.2010.08.014
Kumar, S., & Burgers, P. M. (2013). Lagging strand maturation factor Dna2 is a component of the replication checkpoint initiation machinery. Genes and Development, 27(3), 313–321. https://doi.org/10.1101/gad.204750.112
Kwok, M., Davies, N., Agathanggelou, A., Smith, E., Petermann, E., Yates, E., et al. (2015). Synthetic lethality in chronic lymphocytic leukaemia with DNA damage response defects by targeting the ATR pathway. Lancet, 385(Suppl 1), S58. https://doi.org/10.1016/S0140-6736(15)60373-7
Lammens, K., Bemeleit, D. J., Mockel, C., Clausing, E., Schele, A., Hartung, S., et al. (2011). The Mre11:Rad50 structure shows an ATP-dependent molecular clamp in DNA double-strand break repair. Cell, 145(1), 54–66. https://doi.org/10.1016/j.cell.2011.02.038
Lau, L., Gray, E. E., Brunette, R. L., & Stetson, D. B. (2015). DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science, 350(6260), 568–571. https://doi.org/10.1126/science.aab3291
Le, D. T., Durham, J. N., Smith, K. N., Wang, H., Bartlett, B. R., Aulakh, L. K., et al. (2017). Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science, 357(6349), 409–413. https://doi.org/10.1126/science.aan6733
Le, D. T., Uram, J. N., Wang, H., Bartlett, B. R., Kemberling, H., Eyring, A. D., et al. (2015). PD-1 blockade in tumors with mismatch-repair deficiency. New England Journal of Medicine, 372(26), 2509–2520. https://doi.org/10.1056/NEJMoa1500596
Ledesma, F. C., El Khamisy, S. F., Zuma, M. C., Osborn, K., & Caldecott, K. W. (2009). A human 5′-tyrosyl DNA phosphodiesterase that repairs topoisomerase-mediated DNA damage. Nature, 461(7264), 674–678. https://doi.org/10.1038/nature08444
Lee, E. K., & Konstantinopoulos, P. A. (2019). Combined PARP and immune checkpoint inhibition in ovarian cancer. Trends Cancer, 5(9), 524–528. https://doi.org/10.1016/j.trecan.2019.06.004
Lee, J. H., & Paull, T. T. (2004). Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science, 304(5667), 93–96. https://doi.org/10.1126/science.1091496
Lee, J. H., & Paull, T. T. (2005). ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science, 308(5721), 551–554. https://doi.org/10.1126/science.1108297
Lee, K. C., Swan, R. L., Sondka, Z., Padget, K., Cowell, I. G., & Austin, C. A. (2018). Effect of TDP2 on the level of TOP2-DNA complexes and SUMOylated TOP2-DNA complexes. International Journal of Molecular Sciences. https://doi.org/10.3390/ijms19072056
Lee, Y. C., Zhou, Q., Chen, J., & Yuan, J. (2016). RPA-binding protein ETAA1 is an ATR activator involved in DNA replication stress response. Current Biology, 26(24), 3257–3268. https://doi.org/10.1016/j.cub.2016.10.030
Li, G. M. (2008). Mechanisms and functions of DNA mismatch repair. Cell Research, 18(1), 85–98. https://doi.org/10.1038/cr.2007.115
Li, Y., Shen, Y., Jin, K., Wen, Z., Cao, W., Wu, B., et al. (2019). The DNA repair nuclease MRE11A functions as a mitochondrial protector and prevents T cell pyroptosis and tissue inflammation. Cell Metabolism, 30(3), 477–492. https://doi.org/10.1016/j.cmet.2019.06.016
Li, Z., Liu, B., Jin, W., Wu, X., Zhou, M., Liu, V. Z., et al. (2018). hDNA2 nuclease/helicase promotes centromeric DNA replication and genome stability. EMBO Journal. https://doi.org/10.15252/embj.201796729
Liu, B., Li, L., Yang, G., Geng, C., Luo, Y., Wu, W., et al. (2019). PARP inhibition suppresses GR-MYCN-CDK5-RB1-E2F1 signaling and neuroendocrine differentiation in castration-resistant prostate cancer. Clinical Cancer Research, 25(22), 6839–6851. https://doi.org/10.1158/1078-0432.CCR-19-0317
Liu, W., Zhou, M., Li, Z., Li, H., Polaczek, P., Dai, H., et al. (2016). A selective small molecule DNA2 inhibitor for sensitization of human cancer cells to chemotherapy. EBioMedicine, 6, 73–86. https://doi.org/10.1016/j.ebiom.2016.02.043
Lok, B. H., Gardner, E. E., Schneeberger, V. E., Ni, A., Desmeules, P., Rekhtman, N., et al. (2017). PARP inhibitor activity correlates with SLFN11 expression and demonstrates synergy with temozolomide in small cell lung cancer. Clinical Cancer Research, 23(2), 523–535. https://doi.org/10.1158/1078-0432.CCR-16-1040
Lord, C. J., & Ashworth, A. (2017). PARP inhibitors: synthetic lethality in the clinic. Science, 355(6330), 1152–1158. https://doi.org/10.1126/science.aam7344
Lou, Z., Minter-Dykhouse, K., Wu, X., & Chen, J. (2003). MDC1 is coupled to activated CHK2 in mammalian DNA damage response pathways. Nature, 421(6926), 957–961. https://doi.org/10.1038/nature01447
Lu, X. H., Mattis, V. B., Wang, N., Al-Ramahi, I., van den Berg, N., Fratantoni, S. A., et al. (2014). Targeting ATM ameliorates mutant Huntingtin toxicity in cell and animal models of Huntington’s disease. Science Translational Medicine, 6(268), 268ra178. https://doi.org/10.1126/scitranslmed.3010523
Lui, G. Y. L., Grandori, C., & Kemp, C. J. (2018). CDK12: an emerging therapeutic target for cancer. Journal of Clinical Pathology, 71(11), 957–962. https://doi.org/10.1136/jclinpath-2018-205356
Ma, Z., Jacobs, S. R., West, J. A., Stopford, C., Zhang, Z., Davis, Z., et al. (2015). Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proceedings of National Academy of Sciences, 112(31), E4306-4315. https://doi.org/10.1073/pnas.1503831112
Makharashvili, N., Arora, S., Yin, Y., Fu, Q., Wen, X., Lee, J. H., et al. (2018). Sae2/CtIP prevents R-loop accumulation in eukaryotic cells. Elife. https://doi.org/10.7554/eLife.42733
Marechal, A., & Zou, L. (2013). DNA damage sensing by the ATM and ATR kinases. Cold Spring Harbor Perspectives in Biology, 5, 9. https://doi.org/10.1101/cshperspect.a012716
Mariggio, G., Koch, S., Zhang, G., Weidner-Glunde, M., Ruckert, J., Kati, S., et al. (2017). Kaposi Sarcoma Herpesvirus (KSHV) Latency-Associated Nuclear Antigen (LANA) recruits components of the MRN (Mre11-Rad50-NBS1) repair complex to modulate an innate immune signaling pathway and viral latency. PLoS Pathogens, 13(4), e1006335. https://doi.org/10.1371/journal.ppat.1006335
Marteijn, J. A., Lans, H., Vermeulen, W., & Hoeijmakers, J. H. (2014). Understanding nucleotide excision repair and its roles in cancer and ageing. Nature Reviews Molecular Cell Biology, 15(7), 465–481. https://doi.org/10.1038/nrm3822
Matsuoka, S., Ballif, B. A., Smogorzewska, A., McDonald, E. R., 3rd., Hurov, K. E., Luo, J., et al. (2007). ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science, 316(5828), 1160–1166. https://doi.org/10.1126/science.1140321
Mattiroli, F., Vissers, J. H., van Dijk, W. J., Ikpa, P., Citterio, E., Vermeulen, W., et al. (2012). RNF168 ubiquitinates K13–15 on H2A/H2AX to drive DNA damage signaling. Cell, 150(6), 1182–1195. https://doi.org/10.1016/j.cell.2012.08.005
McKinnon, P. J. (2016). Topoisomerases and the regulation of neural function. Nature Reviews Neuroscience, 17(11), 673–679. https://doi.org/10.1038/nrn.2016.101
Menissier de Murcia, J., Ricoul, M., Tartier, L., Niedergang, C., Huber, A., Dantzer, F., et al. (2003). Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. EMBO Journal, 22(9), 2255–2263. https://doi.org/10.1093/emboj/cdg206
Miao, Z. H., Rao, V. A., Agama, K., Antony, S., Kohn, K. W., & Pommier, Y. (2006). 4-Nitroquinoline-1-oxide induces the formation of cellular topoisomerase I-DNA cleavage complexes. Cancer Research, 66(13), 6540–6545. https://doi.org/10.1158/0008-5472.CAN-05-4471
Min, A., Im, S. A., Jang, H., Kim, S., Lee, M., Kim, D. K., et al. (2017). AZD6738, a novel oral inhibitor of ATR, induces synthetic lethality with ATM deficiency in gastric cancer cells. Molecular Cancer Therapeutics, 16(4), 566–577. https://doi.org/10.1158/1535-7163.MCT-16-0378
Mordes, D. A., Nam, E. A., & Cortez, D. (2008). Dpb11 activates the Mec1-Ddc2 complex. Proceedings of National Academy of Sciences, 105(48), 18730–18734. https://doi.org/10.1073/pnas.0806621105
Moreau, S., Ferguson, J. R., & Symington, L. S. (1999). The nuclease activity of Mre11 is required for meiosis but not for mating type switching, end joining, or telomere maintenance. Molecular and Cellular Biology, 19(1), 556–566. https://doi.org/10.1128/mcb.19.1.556
Murai, J., Huang, S. Y., Das, B. B., Renaud, A., Zhang, Y., Doroshow, J. H., et al. (2012). Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Research, 72(21), 5588–5599. https://doi.org/10.1158/0008-5472.CAN-12-2753
Nalepa, G., & Clapp, D. W. (2018). Fanconi anaemia and cancer: an intricate relationship. Nature Reviews Cancer, 18(3), 168–185. https://doi.org/10.1038/nrc.2017.116
Navadgi-Patil, V. M., & Burgers, P. M. (2009). The unstructured C-terminal tail of the 9–1–1 clamp subunit Ddc1 activates Mec1/ATR via two distinct mechanisms. Molecular Cell, 36(5), 743–753. https://doi.org/10.1016/j.molcel.2009.10.014
Neale, M. J., Pan, J., & Keeney, S. (2005). Endonucleolytic processing of covalent protein-linked DNA double-strand breaks. Nature, 436(7053), 1053–1057. https://doi.org/10.1038/nature03872
Nicholson, J., Jevons, S. J., Groselj, B., Ellermann, S., Konietzny, R., Kerr, M., et al. (2017). E3 ligase cIAP2 mediates downregulation of MRE11 and radiosensitization in response to HDAC inhibition in bladder cancer. Cancer Research, 77(11), 3027–3039. https://doi.org/10.1158/0008-5472.CAN-16-3232
Nicolae, C. M., Aho, E. R., Choe, K. N., Constantin, D., Hu, H. J., Lee, D., et al. (2015). A novel role for the mono-ADP-ribosyltransferase PARP14/ARTD8 in promoting homologous recombination and protecting against replication stress. Nucleic Acids Research, 43(6), 3143–3153. https://doi.org/10.1093/nar/gkv147
Nicolae, C. M., Aho, E. R., Vlahos, A. H., Choe, K. N., De, S., Karras, G. I., et al. (2014). The ADP-ribosyltransferase PARP10/ARTD10 interacts with proliferating cell nuclear antigen (PCNA) and is required for DNA damage tolerance. Journal of Biological Chemistry, 289(19), 13627–13637. https://doi.org/10.1074/jbc.M114.556340
Nowsheen, S., Aziz, K., Aziz, A., Deng, M., Qin, B., Luo, K., et al. (2018). L3MBTL2 orchestrates ubiquitin signalling by dictating the sequential recruitment of RNF8 and RNF168 after DNA damage. Nature Cell Biology, 20(4), 455–464. https://doi.org/10.1038/s41556-018-0071-x
O’Connor, M. J. (2015). Targeting the DNA damage response in cancer. Molecular Cell, 60(4), 547–560. https://doi.org/10.1016/j.molcel.2015.10.040
Oh, J., & Symington, L. S. (2018). Role of the Mre11 complex in preserving genome integrity. Genes (Basel). https://doi.org/10.3390/genes9120589
Paull, T. T. (2015). Mechanisms of ATM activation. Annual Review of Biochemistry, 84, 711–738. https://doi.org/10.1146/annurev-biochem-060614-034335
Paull, T. T. (2018). 20 years of Mre11 biology: no end in sight. Molecular Cell, 71(3), 419–427. https://doi.org/10.1016/j.molcel.2018.06.033
Perry, J., & Kleckner, N. (2003). The ATRs, ATMs, and TORs are giant HEAT repeat proteins. Cell, 112(2), 151–155. https://doi.org/10.1016/s0092-8674(03)00033-3
Prevo, R., Fokas, E., Reaper, P. M., Charlton, P. A., Pollard, J. R., McKenna, W. G., et al. (2012). The novel ATR inhibitor VE-821 increases sensitivity of pancreatic cancer cells to radiation and chemotherapy. Cancer Biology and Therapy, 13(11), 1072–1081. https://doi.org/10.4161/cbt.21093
Przetocka, S., Porro, A., Bolck, H. A., Walker, C., Lezaja, A., Trenner, A., et al. (2018). CtIP-mediated fork protection synergizes with BRCA1 to suppress genomic instability upon DNA replication stress. Molecular Cell, 72(3), 568–582. https://doi.org/10.1016/j.molcel.2018.09.014
Qi, F., Meng, Q., Hayashi, I., & Kobayashi, J. (2020). FXR1 is a novel MRE11-binding partner and participates in oxidative stress responses. Journal of Radiation Research. https://doi.org/10.1093/jrr/rraa011
Quennet, V., Beucher, A., Barton, O., Takeda, S., & Lobrich, M. (2011). CtIP and MRN promote non-homologous end-joining of etoposide-induced DNA double-strand breaks in G1. Nucleic Acids Research, 39(6), 2144–2152. https://doi.org/10.1093/nar/gkq1175
Ray Chaudhuri, A., Callen, E., Ding, X., Gogola, E., Duarte, A. A., Lee, J. E., et al. (2016). Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature, 535(7612), 382–387. https://doi.org/10.1038/nature18325
Reale, A., Matteis, G. D., Galleazzi, G., Zampieri, M., & Caiafa, P. (2005). Modulation of DNMT1 activity by ADP-ribose polymers. Oncogene, 24(1), 13–19. https://doi.org/10.1038/sj.onc.1208005
Regairaz, M., Zhang, Y. W., Fu, H., Agama, K. K., Tata, N., Agrawal, S., et al. (2011). Mus81-mediated DNA cleavage resolves replication forks stalled by topoisomerase I-DNA complexes. Journal of Cell Biology, 195(5), 739–749. https://doi.org/10.1083/jcb.201104003
Rijkers, T., Van Den Ouweland, J., Morolli, B., Rolink, A. G., Baarends, W. M., Van Sloun, P. P., et al. (1998). Targeted inactivation of mouse RAD52 reduces homologous recombination but not resistance to ionizing radiation. Molecular and Cellular Biology, 18(11), 6423–6429. https://doi.org/10.1128/mcb.18.11.6423
Rodriguez-Vargas, J. M., Nguekeu-Zebaze, L., & Dantzer, F. (2019). PARP3 comes to light as a prime target in cancer therapy. Cell Cycle, 18(12), 1295–1301. https://doi.org/10.1080/15384101.2019.1617454
Sacho, E. J., & Maizels, N. (2011). DNA repair factor MRE11/RAD50 cleaves 3’-phosphotyrosyl bonds and resects DNA to repair damage caused by topoisomerase 1 poisons. Journal of Biological Chemistry, 286(52), 44945–44951. https://doi.org/10.1074/jbc.M111.299347
Sartori, A. A., Lukas, C., Coates, J., Mistrik, M., Fu, S., Bartek, J., et al. (2007). Human CtIP promotes DNA end resection. Nature, 450(7169), 509–514. https://doi.org/10.1038/nature06337
Sasanuma, H., Tsuda, M., Morimoto, S., Saha, L. K., Rahman, M. M., Kiyooka, Y., et al. (2018). BRCA1 ensures genome integrity by eliminating estrogen-induced pathological topoisomerase II-DNA complexes. Proceedings of National Academy of Sciences, 115(45), E10642–E10651. https://doi.org/10.1073/pnas.1803177115
Sawyer, D. B. (2013). Anthracyclines and heart failure. New England Journal of Medicine, 368(12), 1154–1156. https://doi.org/10.1056/NEJMcibr1214975
Schaue, D., & McBride, W. H. (2015). Opportunities and challenges of radiotherapy for treating cancer. Nature Reviews Clinical Oncology, 12(9), 527–540. https://doi.org/10.1038/nrclinonc.2015.120
Schellenberg, M. J., Lieberman, J. A., Herrero-Ruiz, A., Butler, L. R., Williams, J. G., Munoz-Cabello, A. M., et al. (2017). ZATT (ZNF451)-mediated resolution of topoisomerase 2 DNA-protein cross-links. Science, 357(6358), 1412–1416. https://doi.org/10.1126/science.aam6468
Schiller, C. B., Lammens, K., Guerini, I., Coordes, B., Feldmann, H., Schlauderer, F., et al. (2012). Structure of Mre11-Nbs1 complex yields insights into ataxia-telangiectasia-like disease mutations and DNA damage signaling. Nature Structural and Molecular Biology, 19(7), 693–700. https://doi.org/10.1038/nsmb.2323
Schlacher, K., Christ, N., Siaud, N., Egashira, A., Wu, H., & Jasin, M. (2011). Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell, 145(4), 529–542. https://doi.org/10.1016/j.cell.2011.03.041
Scully, R., Panday, A., Elango, R., & Willis, N. A. (2019). DNA double-strand break repair-pathway choice in somatic mammalian cells. Nature Reviews Molecular Cell Biology, 20(11), 698–714. https://doi.org/10.1038/s41580-019-0152-0
Shah, G. A., & O’Shea, C. C. (2015). Viral and cellular genomes activate distinct DNA damage responses. Cell, 162(5), 987–1002. https://doi.org/10.1016/j.cell.2015.07.058
Shen, J., Zhao, W., Ju, Z., Wang, L., Peng, Y., Labrie, M., et al. (2019). PARPi triggers the STING-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCAness. Cancer Research, 79(2), 311–319. https://doi.org/10.1158/0008-5472.CAN-18-1003
Shibata, A., Moiani, D., Arvai, A. S., Perry, J., Harding, S. M., Genois, M. M., et al. (2014). DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Molecular Cell, 53(1), 7–18. https://doi.org/10.1016/j.molcel.2013.11.003
Shiloh, Y., & Ziv, Y. (2013). The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nature Reviews Molecular Cell Biology, 14(4), 197–210. https://doi.org/10.1038/nrm3546
Smith, J., Tho, L. M., Xu, N., & Gillespie, D. A. (2010). The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Advances in Cancer Research, 108, 73–112. https://doi.org/10.1016/B978-0-12-380888-2.00003-0
Sotiriou, S. K., Kamileri, I., Lugli, N., Evangelou, K., Da-Re, C., Huber, F., et al. (2016). Mammalian RAD52 functions in break-induced replication repair of collapsed DNA Replication Forks. Molecular Cell, 64(6), 1127–1134. https://doi.org/10.1016/j.molcel.2016.10.038
Stewart, G. S., Wang, B., Bignell, C. R., Taylor, A. M., & Elledge, S. J. (2003). MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature, 421(6926), 961–966. https://doi.org/10.1038/nature01446
Sun, C., Yin, J., Fang, Y., Chen, J., Jeong, K. J., Chen, X., et al. (2018). BRD4 inhibition is synthetic lethal with PARP inhibitors through the induction of homologous recombination deficiency. Cancer Cell, 33(3), 401–416. https://doi.org/10.1016/j.ccell.2018.01.019
Terao, C., Suzuki, A., Momozawa, Y., Akiyama, M., Ishigaki, K., Yamamoto, K., et al. (2020). Chromosomal alterations among age-related haematopoietic clones in Japan. Nature, 584(7819), 130–135. https://doi.org/10.1038/s41586-020-2426-2
Thangavel, S., Berti, M., Levikova, M., Pinto, C., Gomathinayagam, S., Vujanovic, M., et al. (2015). DNA2 drives processing and restart of reversed replication forks in human cells. Journal of Cell Biology, 208(5), 545–562. https://doi.org/10.1083/jcb.201406100
Trenz, K., Smith, E., Smith, S., & Costanzo, V. (2006). ATM and ATR promote Mre11 dependent restart of collapsed replication forks and prevent accumulation of DNA breaks. EMBO Journal, 25(8), 1764–1774. https://doi.org/10.1038/sj.emboj.7601045
Unterholzner, L. (2013). The interferon response to intracellular DNA: why so many receptors? Immunobiology, 218(11), 1312–1321. https://doi.org/10.1016/j.imbio.2013.07.007
Vermeulen, W. (2011). Dynamics of mammalian NER proteins. DNA Repair, 10(7), 760–771. https://doi.org/10.1016/j.dnarep.2011.04.015
Wang, C., Guan, Y., Lv, M., Zhang, R., Guo, Z., Wei, X., et al. (2018). Manganese increases the sensitivity of the cGAS-STING pathway for double-stranded DNA and is required for the host defense against DNA viruses. Immunity, 48(4), 675–687. https://doi.org/10.1016/j.immuni.2018.03.017
Wang, C., Xu, W., Zhang, Y., Zhang, F., & Huang, K. (2018). PARP1 promote autophagy in cardiomyocytes via modulating FoxO3a transcription. Cell Death and Disease, 9(11), 1047. https://doi.org/10.1038/s41419-018-1108-6
Wang, J., Aroumougame, A., Lobrich, M., Li, Y., Chen, D., Chen, J., et al. (2014). PTIP associates with artemis to dictate DNA repair pathway choice. Genes and Development, 28(24), 2693–2698. https://doi.org/10.1101/gad.252478.114
Wang, W., Daley, J. M., Kwon, Y., Krasner, D. S., & Sung, P. (2017). Plasticity of the Mre11-Rad50-Xrs2-Sae2 nuclease ensemble in the processing of DNA-bound obstacles. Genes and Development, 31(23–24), 2331–2336. https://doi.org/10.1101/gad.307900.117
Wang, Z., Gong, Y., Peng, B., Shi, R., Fan, D., Zhao, H., et al. (2019). MRE11 UFMylation promotes ATM activation. Nucleic Acids Research, 47(8), 4124–4135. https://doi.org/10.1093/nar/gkz110
Wienert, B., Wyman, S. K., Richardson, C. D., Yeh, C. D., Akcakaya, P., Porritt, M. J., et al. (2019). Unbiased detection of CRISPR off-targets in vivo using DISCOVER-Seq. Science, 364(6437), 286–289. https://doi.org/10.1126/science.aav9023
Wright, W. D., Shah, S. S., & Heyer, W. D. (2018). Homologous recombination and the repair of DNA double-strand breaks. Journal of Biological Chemistry, 293(27), 10524–10535. https://doi.org/10.1074/jbc.TM118.000372
Ying, S., Hamdy, F. C., & Helleday, T. (2012). Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Research, 72(11), 2814–2821. https://doi.org/10.1158/0008-5472.CAN-11-3417
Yu, D., Liu, R., Yang, G., & Zhou, Q. (2018). The PARP1-Siah1 axis controls HIV-1 transcription and expression of Siah1 substrates. Cell Report, 23(13), 3741–3749. https://doi.org/10.1016/j.celrep.2018.05.084
Yu, W., Li, L., Wang, G., Zhang, W., Xu, J., & Liang, A. (2018). KU70 inhibition impairs both non-homologous end joining and homologous recombination DNA damage repair through SHP-1 induced dephosphorylation of SIRT1 in T-cell acute lymphoblastic leukemia (T-ALL) [corrected]. Cellular Physiology and Biochemistry, 49(6), 2111–2123. https://doi.org/10.1159/000493815
Zander, S. A., Kersbergen, A., van der Burg, E., de Water, N., van Tellingen, O., Gunnarsdottir, S., et al. (2010). Sensitivity and acquired resistance of BRCA1;p53-deficient mouse mammary tumors to the topoisomerase I inhibitor topotecan. Cancer Research, 70(4), 1700–1710. https://doi.org/10.1158/0008-5472.CAN-09-3367
Zhao, B., Liu, P., Fukumoto, T., Nacarelli, T., Fatkhutdinov, N., Wu, S., et al. (2020). Topoisomerase 1 cleavage complex enables pattern recognition and inflammation during senescence. Nature Communication, 11(1), 908. https://doi.org/10.1038/s41467-020-14652-y
Zhao, H., Sifakis, E. G., Sumida, N., Millan-Arino, L., Scholz, B. A., Svensson, J. P., et al. (2015). PARP1- and CTCF-mediated interactions between active and repressed chromatin at the lamina promote oscillating transcription. Molecular Cell, 59(6), 984–997. https://doi.org/10.1016/j.molcel.2015.07.019
Zhao, W., Steinfeld, J. B., Liang, F., Chen, X., Maranon, D. G., Jian Ma, C., et al. (2017). BRCA1-BARD1 promotes RAD51-mediated homologous DNA pairing. Nature, 550(7676), 360–365. https://doi.org/10.1038/nature24060
Zimmer, J., Tacconi, E. M. C., Folio, C., Badie, S., Porru, M., Klare, K., et al. (2016). Targeting BRCA1 and BRCA2 deficiencies with G-quadruplex-interacting compounds. Molecular Cell, 61(3), 449–460. https://doi.org/10.1016/j.molcel.2015.12.004
Zimmermann, M., Murina, O., Reijns, M. A. M., Agathanggelou, A., Challis, R., Tarnauskaite, Z., et al. (2018). CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature, 559(7713), 285–289. https://doi.org/10.1038/s41586-018-0291-z
Zoppoli, G., Regairaz, M., Leo, E., Reinhold, W. C., Varma, S., Ballestrero, A., et al. (2012). Putative DNA/RNA helicase Schlafen-11 (SLFN11) sensitizes cancer cells to DNA-damaging agents. Proceedings of National Academy of Sciences, 109(37), 15030–15035. https://doi.org/10.1073/pnas.1205943109
Zou, L. (2017). DNA replication checkpoint: new ATR activator identified. Current Biology, 27(1), R33–R35. https://doi.org/10.1016/j.cub.2016.11.025
Zou, L., & Elledge, S. J. (2003). Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science, 300(5625), 1542–1548. https://doi.org/10.1126/science.1083430
Acknowledgements
This review was supported by grants from the National Natural Science Foundation of China (81672981 and 81972608), and Innovation Fund for Outstanding Doctoral Candidates of Peking University Health Science Center.
Author information
Affiliations
Department of Radiation Medicine, Institute of Systems Biomedicine, School of Basic Medical Sciences, Peking University Health Science Center, Beijing, 100191, China
Yongtai Bai, Weibin Wang & Jiadong Wang
Corresponding authors
Correspondence to Weibin Wang or Jiadong Wang.
Ethics declarations
Conflict of interests
The authors declare that there are no competing interests associated with the manuscript.
Ethics approval
Not applicable.
Consent to participate
All the authors consent to participate.
Consent for publication
All the authors agreed to publish this review.
Availability of data and material
Not applicable.
Rights and permissions
About this article
Cite this article
Bai, Y., Wang, W. & Wang, J. Targeting DNA repair pathways: mechanisms and potential applications in cancer therapy. GENOME INSTAB. DIS. (2020). https://doi.org/10.1007/s42764-020-00026-7
Received10 September 2020
Revised13 October 2020
Accepted24 October 2020
Published05 November 2020
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
DNA damage response
PARP inhibitors
Topoisomerase
ATM
MRE11
Cancer therapy
用户登录
还没有账号?
立即注册