






TIP60 recruits SUV39H1 to chromatin to maintain heterochromatin genome stability and resist hydrogen peroxide-induced cytotoxicity
Original Research Paper
Bo Tu, Yantao Bao, Ming Tang, Qian Zhu, Xiaopeng Lu, Hui Wang, Tianyun Hou, Ying Zhao, Ping Zhang & Wei-Guo Zhu Genome Instability & Disease (2020)
Abstract
Tat interacting protein 60 (TIP60) is a histone acetyltransferase involved in chromatin remodeling and the DNA damage response. However, the role of TIP60 in maintaining genome stability is poorly understood. Here, we show that TIP60 can directly interact with suppressor of variegation 3–9 homologue 1 (SUV39H1), a methyltransferase that is responsible for histone H3 tri-methylation on Lys9 (H3K9me3). When we knocked down TIP60 or treated cells with hydrogen peroxide, a typical reactive oxygen species (ROS) generator that induces genome instability, SUV39H1 dissociated from chromatin but its acetylation levels remained unchanged. Consequently, H3K9me3 levels in the heterochromatin decreased, leading to a significant increase in the expression of satellite 2 (Sat2) and α-satellite (α-Sat), indicators of heterochromatin relaxation. Micrococcal nuclease sensitivity and colony formation assays further demonstrated that TIP60 knockdown or hydrogen peroxide treatment resulted in a relaxing of the heterochromatin and genome instability. Exogenous TIP60 could rescue the assembly of SUV39H1 on chromatin and ensure genome stability in response to hydrogen peroxide. This study is the first to describe a role for TIP60 in maintaining heterochromatin structure and genome stability by recruiting SUV39H1 to the chromatin upon oxidative stress, presenting TIP60 as a promising target to sensitize cancer cells to ROS-promoting therapies.
Introduction
Histone acetylation is one of the most important modifications in eukaryote: it affects the DNA binding capability, stability, and enzyme activities of proteins (Cao et al. 2016; Kim et al. 2019; Strahl and Allis, 2000). The histone acetyltransferase TIP60 belongs to the conserved histone acetyltransferase MYST (Moz, Ybf2/Sas3, Sas2, TIP60) family (Kamine et al. 1996). Recombinant TIP60 can directly acetylate H2A (Lys5), H3 (Lys14) and H4 (Lys5, 8, 12, 16), but not H2B in vitro (Yamamoto and Horikoshi 1997); however, TIP60 must form a complex with many other proteins such as TIP60-associated protein (TAP) 54α and β to acetylate the nucleosome in vivo (Ikura et al. 2000). TIP60 also interacts with various non-histone substrates, including aurora-B, p53 and ataxia telangiectasia mutated (ATM), to catalyze their acetylation (Ghobashi and Kamel 2018; Li et al. 2020; Mo et al. 2016; Sun et al. 2005; Tang et al. 2006). Unsurprisingly, TIP60 is closely associated with gene expression, DNA damage repair and tumor development (Kim et al. 2019), and its function is likely dependent on its enzymatic activity.
TIP60 might differentially affect cell fate in different contexts, such as oncogenesis and DNA damage. For example, in response to severe genotoxic stress, TIP60 can cooperate with multiple proteins, including p53 (Liu et al. 2019; Tang et al. 2006; Yang et al. 2020), programmed cell death 5 (PDCD5) (Li et al. 2016), or axin (Li et al. 2009), to induce apoptosis. In response to reparable DNA damage, TIP60 activates proteins including H4 (Tang et al. 2013), ATM (Ghobashi and Kamel 2018; Sun et al. 2005) and the ribonucleotide reductase catalytic subunit M1 (RRM1) (Niida et al. 2010) to arrest the cell cycle, recruit the DNA damage repair complex and facilitate cell survival. In this way, TIP60 has a dynamic role in protecting DNA from damage and maintaining genome stability during the DNA damage response (DDR). The underlying mechanisms by which TIP60 maintains genome stability, however, are not fully understood.
A key to the mechanisms of TIP60 function in genome stability might lie in its cooperation with suppressor of variegation 3–9 homolog 1 (SUV39H1), an H3K9 histone-lysine methyltransferase (Fritsch et al. 2010; Kaniskan et al. 2015; Vaquero et al. 2007). SUV39H1 is a heterochromatin-enriched enzyme that also transiently accumulates around centromeres during mitosis (Chiba et al. 2015): it has various well-documented functions, but importantly is critical for maintaining heterochromatin and genome stability (Jehanno et al. 2017; Shirai et al. 2017; Wang et al. 2013). Understanding how SUV39H1 is recruited to chromatin is critical for understanding of heterochromatin structure maintenance and genome stability. One study showed that treating cells with hydrogen peroxide, a reactive oxygen species (ROS) generator, can suppress Sp1 transcription factor (SP1)-dependent SUV39H1 recruitment to the Cyclin B1 promoter and perturb chromatin structure regulation (Chuang et al. 2011). ROS thus seems to regulate SUV39H1 recruitment and chromatin remodeling. Whether hydrogen peroxide (i.e. ROS) also regulates SUV39H1 recruitment to heterochromatin and subsequent genome stability is unknown.
In terms of the relationship between TIP60 and SUV39H1, cells lacking SUV39H1 are defective in TIP60 and ATM activation, which results in reduced DNA double-strand break (DSB) repair and enhanced radiosensitivity (Ayrapetov et al. 2014). Additionally, depleting TIP60 promotes heterochromatin dysfunction in SUV39H-deficient cells (Grezy et al. 2016), indicating that both SUV39H1 and TIP60 are crucial for maintaining heterochromatin functions. Moreover, a complex containing kinesin II associated protein (KAP-1), heterochromatin protein 1 (HP1), and SUV39H1 rapidly loads onto chromatin upon detecting DSBs and helps TIP60 to acetylate ATM and histone H4, providing a mechanism of cooperation between SUV39H1 and TIP60 in the DDR (Ayrapetov et al. 2014). Finally, the UFM1 specific ligase 1 (UFL1), which is recruited by the MRN complex, facilitates the recruitment of the SUV39H1 complex and TIP60 to DSBs (Qin et al. 2019). Despite these advances, a direct interaction between TIP60 and SUV39H1 has not yet been shown.
In this study, we aimed to dig deeper into a more close relationship between TIP60 and SUV39H1 in DDR response. To do so, we started by identifying if these two protein could directly interact with each other. Subsequently, we explored how this interaction may respond to different genotoxic stimulations and found that ROS could specifically induce a decreased TIP60-SUV39H1 interaction. Hence, we continued to investigate the alterations in SUV39H1 recruitment to chromatin, H3K9me3 levels in chromatin, heterochromatin structures and genome stability in response to oxidative stress. Altogether, we reveal a novel interaction between TIP60 and SUV39H1 that regulates SUV39H1 chromatin assembly. This assembly is necessary to maintain heterochromatin structure and genome stability in response to oxidative stress.
Materials and methods
Cell lines and cell culture
HEK293T, H1299, HeLa and HepG2 cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA). Wild-type TIP60 stably transfected and knocked down HepG2 cells were a gift from Professor Lin Shengcai (Xiamen University, China). All cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (Invitrogen) and penicillin–streptomycin (50 units/ml). The cells were cultured in a humidified incubator at 37 °C with 5% CO2 and passaged by trypsinization every 3 days.
Antibodies and reagents
The following antibodies and reagents were used in this study: Hydrogen peroxide (Sigma-Aldrich), anti-SUV39H1 (Cell Signaling Technology, #8729), anti-TIP60 (Santa Cruz, sc-5725), anti-Myc (MBL, M047-3), anti-HA (Santa Cruz, sc-805), anti-G9a (Sigma-Aldrich, G6919), anti-His (MBL PM032), anti-GST (Sigma-Aldrich, G1160), anti-H3K9 tri-methylation (Abcam ab8898), anti- Acetyl Histone H4K12 (Epigentek A-4029), anti-H3 (Bethyl A300-823A), anti-Flag (Sigma-Aldrich, F1804), Goat anti-mouse IgG horseradish peroxidase (HRP) (Santa Cruz), and goat anti-rabbit IgG HRP (Santa Cruz).
Plasmid construction and small interfering RNA
The human TIP60 full-length coding region was amplified by PCR from human TIP60 cDNA in the human brain library (Clontech) and was introduced to a pCMV5 vector with an HA and a Flag tag. The FLAG-TIP60 HAT-deficient mutant expression vector was a gift from Professor Lin Shengcai (Xiamen University, China). Full-length SUV39H1 cDNA was obtained from our own lab. TIP60-chromo, TIP60-Zn, TIP60-HAT, chromo-SUV39H1, Mid-SUV39H1 and SET-SUV39H1 fragments were amplified from TIP60 or SUV39H1 full-length sequences, and these PCR products were introduced into the pGEX4T-3 vector and then verified by Sanger sequencing. Site-mutated SUV39H1, zinc finger-deleted TIP60 and RNA interfering resistant TIP60 expressing plasmids were produced using a MutExpress II kit (Vazyme) following the manufacturer’s protocol. Small interfering RNA sequences were designed as follows: Non-silencing siRNA: sense: 5′-UUCUCCGAACGUGUCACGU-3′; TIP60 #1 siRNA: sense: 5′-ACGGAAGGUGGAGGUGGUU-3′; TIP60 #2 siRNA: sense: 5′-GUACGGCCGUAGUCUCAAG -3’.
Plasmids and siRNAs were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions.
GST pull-down assay
GST alone or TIP60/SUV39H1-GST fusion proteins (full-length or fragments) were induced by IPTG for 4 h at 28 °C in E. coli and purified using glutathione–Sepharose 4B beads (GE Healthcare). An equal amount of each individual fusion protein was incubated with His–SUV39H1 (from E. coli) in TEN buffer (10 mM Tris·HCl, pH8.0, 1 mM EDTA, 100 mM NaCl) for 4 h at 4 °C. After washing in TEN buffer, the precipitated components were analyzed by western blotting.
RNA preparation and quantitative real-time PCR
Total RNA was extracted using TRIzol solution (Invitrogen) according to the manufacturer's specifications. Genomic DNA was removed from 5 μg of total RNA by incubating the samples with 10 U RNase-free DNase I (New England Biolabs) and 2 U RNase inhibitor (New England Biolabs) in DEPC-treated water. The reaction mixture was incubated for 1 h at 37 °C and then for 10 min at 60 °C. RNA concentrations were determined by spectrophotometric analysis. All RNA isolates had an A260:A280 between 1.8 and 2.0, indicating that the isolated RNA was suitable for subsequent analyses. cDNAs were prepared from the isolated RNAs using an ImProm-II reverse transcription system (Promega). RT-PCR was performed in a Step One cycler (Applied Biosystems, Foster City, CA, USA) using an SYBR green premix (Applied Biosystems), according to the manufacturer’s instructions. The following primers were used for RT-PCR: SUV39H1 sense: 5-ATCCGCGAACAGGAATATTACC-3; SUV39H1 antisense: 5-GAGGATACGCACACACTTGAGATT-3; Sat2 sense: 5-CATCGAATGGAAATGAAAGGAGTC-3; Sat2 antisense: 5-ACCATTGGATGATTGCAGTCAA-3; α-Sat sense: 5-CTGCACTACCTGAAGAGGAC-3; α-Sat antisense: 5-GATGGTTCAACACTCTTACA-3;
GAPDH sense: 5-GAAGGTGAAGGTCGGAGTC-3; GAPDH antisense: 5-GAAGATGGTGATGGGATTTC-3.
GAPDH was used for normalization. The relative mRNA expression was calculated by the 2 − (ΔΔCt) method.
Immunoprecipitation and western blotting
H1299 and HepG2 cells were seeded in 60-mm plates at an initial density of 2 × 106 cells and allowed to grow for 12 h. The cells were transfected with the respective plasmids, further incubated for 24 h, and lyzed in buffer [1% Triton X-100, 150 mM NaCl, 50 mM Tris–HCl, pH 7.5, 0.1% sodium dodecyl sulfate (SDS), 1% Nonidet P-40, and 1 mM PMSF]. The cell suspensions were incubated on ice for 20 min and centrifuged at 14,000g at 4 °C for 20 min. For immunoprecipitation assays, the supernatants were pre-cleared with 20 μl protein A/G-agarose beads (50% slurry) and then incubated at 4 °C overnight with 40 μl fresh protein A/G beads and the appropriate antibodies. A normal IgG antibody was used as a negative control in each experiment. The beads were washed three times in PBS, resuspended in SDS loading buffer, and boiled for 10 min. The protein samples were electrophoresed on a 10% SDS–polyacrylamide gel and transferred to a nitrocellulose membrane (Whatman, PROTRAN). The membrane was blocked with 5% skimmed milk diluted in TBS-T (20 mM Tris–HCl, pH 7.6, 137 mM NaCl, and 0.1% Tween 20), then incubated with the indicated primary antibody at 4 °C overnight and ultimately the indicated secondary antibody at room temperature for 1 h. The membranes were exposed and visualized by the ECL method (Millipore).
Isolation of histone proteins
Histone proteins were isolated as previously described (Lu et al. 2019). Cells (2–5 × 107) were homogenized in 1 ml nuclear preparation buffer (10 mM Tris–HCl, pH 7.6; 150 mM NaCl; 1.5 mM MgCl2; 0.65% Nonidet P-40; and 1 mM PMSF) in the presence of protein phosphatase inhibitors (10 mM NaF, 1 mM sodium orthovanadate, and 25 mM β-glycerophosphate). The nuclei were recovered by centrifugation at 1500g for 10 min. All centrifugations were carried out at 4 °C. The nuclei were resuspended in 0.3 ml resuspension buffer (10 mM Tris–HCl, pH 7.6; 3 mM MgCl2; 10 mM NaCl; 1 mM PMSF; and protein phosphatase inhibitors). The nuclei were extracted with 0.4 nM H2SO4 to isolate total histones. The samples were precipitated with trichloroacetic acid (TCA) and then resuspended in double-distilled H2O.
ChIP assay
Cells were fixed with 1%formaldehyde for 10 min at room temperature and lyzed in ChIP lysis buffer (50 mM Tris·HCl, pH 8.0, 5 mM EDTA, 1% SDS) for 10 min at 4 °C. After sonication (5 s each time for nine times, separated by 10 s’ intervals), the lysate was centrifuged, and the supernatant was collected and pre-cleared in a dilution buffer (20 mM Tris·HCl, pH 8.0, 2 mM EDTA, 150 mM NaCl, 1% Triton X-100) with 45 μl protein G or A Sepharose and 2 μl salmon sperm DNA for 2 h at 4 °C. The pre-cleared samples were divided equally and incubated with the indicated antibody overnight at 4 °C. Then, protein G or A Sepharose was added to each sample and incubated for 2 h at 4 °C. The beads were washed with TSE I (20 mM Tris·HCl, pH 8.0, 2 mM EDTA, 150 mM NaCl, 0.1% SDS, 1% Triton X-100), TSE II (20 mM Tris·HCl, pH 8.0, 2 mM EDTA, 500 mM NaCl, 0.1% SDS, 1% Triton X-100), buffer III (10 mM Tris·HCl, pH 8.0, 1 mM EDTA, 0.25 M LiCl, 1% deoxycholate, 1% Nonidet P-40), and tris–EDTA buffer (TE) sequentially. The samples were heated at 65 °C for at least 6 h to reverse the cross-links. The DNA was purified using a PCR Cleaning Kit (Macherey–Nagel). Real-time PCR was performed to detect the relative occupancy with the same Sat2 and α-Sat primers described above.
Micrococcal nuclease (MNase) sensitivity assay
MNase sensitivity assay was carried out as previously described (Li et al. 2018). Briefly, cells were washed with cold PBS and lyzed with a nuclei extraction (NE) buffer (10 mM Tris–HCl pH 8.0, 0.1 mM EDTA, 2 mM MgCl2, 2 mM CaCl2, 1 mM DTT, 0.2% (v/v) NP-40) on ice. The resultant nuclei were washed twice with NE buffer, suspended in NE buffer and digested at 25 °C for 5 min with 1 U/ml MNase. The reaction was stopped upon addition of a stop buffer (50 mM Tris–HCl pH 8.0, 25 mM EDTA, 1% (w/v) SDS). The DNA was purified by incubating the samples with 200 μg/ml proteinase K for 1 h at 55 °C, followed by phenol–chloroform extraction and ethanol precipitation. The DNA was resuspended in TE buffer and separated by agarose gel electrophoresis and visualized using ethidium bromide. Each line of the ethidium bromide gels was scanned, and profiles representing band intensity were obtained using ImageJ software (https://rsbweb.nih.gov/ij/).
Soft agar colony formation assay
Transfected HepG2 cells were counted using a hemocytometer. Then, 5 × 103 cells in 1 ml growth medium containing 0.35% Noble agar (Difco) and G418 (800 μg/ml) were overlaid on 1.5 ml 0.5% base agar medium in a 60-mm culture dish. The cells were incubated at 37 °C in a humidified atmosphere of 95% air and 5% CO2, and the number of colonies was counted 2 weeks later, after 0.005% crystal violet staining.
Statistical analyses
Analysis of variances between two different experimental groups was conducted with Tukey’s post hoc comparison test using SPSS (Version K12). All experiments were repeated at least three times. A p < 0.05 was considered statistically significant.
Results
TIP60 interacts with SUV39H1 in vivo and in vitro
Given the close association reported between SUV39H1 and TIP60, we first investigated the nature of the interaction between these two proteins. To do so, we performed a co-immunoprecipitation (Co-IP) assay in HEK293T and HeLa cells, in which we over-expressed both TIP60 and SUV39H1. An interaction between exogenous TIP60 and SUV39H1 was readily detected (Fig. 1a). We also confirmed an endogenous interaction between SUV39 and TIP60 by Co-IP assay. While we observed an obvious interaction between TIP60 and SUV39H1 in the chromatin fraction (Fig. 1b), TIP60 did not interact with another H3K9 methyltransferase G9A, which is mainly that is responsible for H3K9 mono- and di-methylation in euchromatin (Fig. 1b).
Fig. 1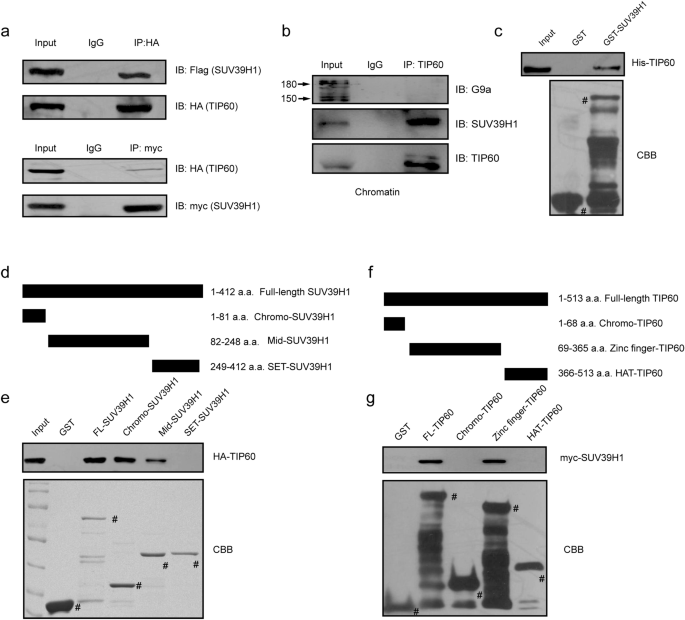
TIP60 interacts with SUV39H1 in vivo and in vitro. a Cell lysates of HEK293T cells over-expressing HA-Flag-TIP60 and Flag-SUV39H1 were subjected to immunoprecipitation (IP) with anti-HA (upper panel) or anti-myc antibodies (lower panel), followed by immunoblotting (IB) with anti-Flag or anti-HA antibodies. b chromatin fraction lysates of H1299 cells were subjected to IP with an anti-TIP60 antibody (N-17), followed by IB with an anti-SUV39H1 and an anti-G9a antibody. c GST-SUV39H1 and His-TIP60 were purified from E. coli respectively. After mixture, incubation and precipitation with glutathione-Sepharose beads, proteins were subject to IB with an anti-His antibody. The GST-fused proteins were stained with Coomassie blue. d, e Full-length SUV39H1 and various truncation mutants were purified from E.Coli. Whole-cell lysates derived from HEK293T cells expressing HA-TIP60 were incubated with glutathione-Sepharose beads containing GST, GST-SUV39H1-WT, -Middle region, or –SET domain, respectively. After being washed, the precipitated proteins were subjected to IB with anti-HA antibody. The GST-fused proteins were stained with Coomassie blue. f, g Full-length TIP60 and various truncation mutants were purified from E. coli. Whole-cell lysates derived from HEK293T cells expressing myc-TIP60 were incubated with glutathione-Sepharose beads containing GST, GST-TIP60-WT, -zinc finger, -chromo domain, or –HAT domain, respectively. After being washed, precipitated proteins were subjected to immunoblot analysis with anti-HA antibody. The GST-fused proteins were stained with Coomassie brilliant blue (CBB)
Full size imageImportantly, a GST pull-down assay confirmed a direct in vitro interaction between His-TIP60 and GST-SUV39H1, which we had purified from E. coli (Fig. 1c). Next we assessed the interactions between the full-length (FL) or fragments of GST–SUV39H1 (1–412 aa; chromodomain containing fragment, 1–81 aa; middle region fragment, 82–248 aa; and SET domain-containing fragment, 249–412 aa) and HA–TIP60, which was purified from the HEK293T cell line. Here, we found that HA–TIP60 interacted with the chromo domain-containing and middle region fragment of GST–SUV39H1, but not with the SET domain (Fig. 1d, e). Similarly, we purified FL or GST-TIP60 fragments (1–513 aa; Chromo domain fragment, 1–68 aa; zinc finger region fragment, 69–365 aa; and HAT domain-containing fragment, 366–513 aa.) and myc-SUV39H1 for GST pulldown assays. Here, only the zinc finger region fragments of TIP60 bound to SUV39H1 (Fig. 1f, g). Taken together, these data suggest that TIP60 directly interacts with SUV39H1 in vitro and in vivo.
Hydrogen peroxide treatment decreases the interaction between TIP60 and SUV39H1
To investigate whether SUV39H1 is acetylated by TIP60, we next transfected wild-type or mutant (catalytic defect) Flag-TIP60 plasmids into H1299 cells. Then, we monitored the acetylation levels of immunoprecipitated SUV39H1 using an anti-pan lysine acetylation antibody. Neither wild-type nor mutant TIP60 over-expression increased SUV39H1 acetylation levels in vivo (Fig. 2a). Consistently, knockdown of TIP60 did not decrease SUV39H1 acetylation levels (Fig. 2b). In addition, SUV39H1 protein levels were unchanged when TIP60 was overexpressed or knocked down in the H1299 cell line (Fig. 2c).
Fig. 2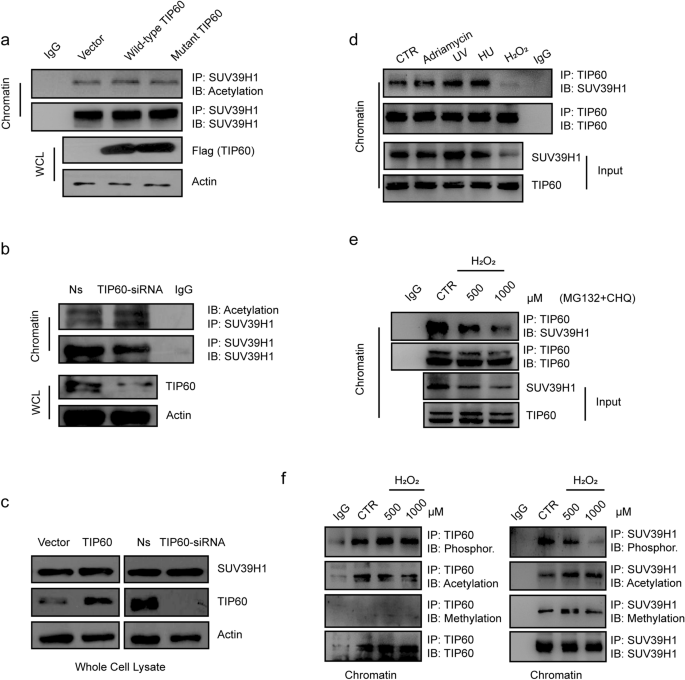
Decrease of the interaction between TIP60 and SUV39H1 in response to ROS. a The H1299 cells were transfected with wild-type or mutant TIP60 for 48 h. The chromatin fraction was immunoprecipitated (IP) with anti-SUV39H1 antibody, then the acetylation levels of SUV39H1 were detected by immunoblotting (IB) with an anti-lysine acetylation antibody. The lower panel showed the levels of over-expressed TIP60 detected by IB. b H1299 cells were pretreated with IP60 siRNA for 72 h. The chromatin fraction was IP with anti-SUV39H1 antibody, then the acetylation levels of SUV39H1 were detected by IB with an anti-lysine acetylation antibody. c In H1299 cells over-expressing TIP60 or those pretreated with the TIP60 siRNA for 72 h, the total protein was extracted and IB was performed with the indicated antibodies. d H1299 cells were treated with 1 μM adriamycin, 1 mM Hydroxyurea (HU) or 1 mM hydrogen peroxide for 6 h, or subject to UV radiation at a dosage of 10 J/m2. The chromatin fraction lysates were subjected to IP with anti-TIP60 antibody (N-17), followed by IB with an anti-SUV39H1 antibody. e H1299 cells were pre-treated with MG132 (1 μM) and CHQ (50 μM), and then treated with hydrogen peroxide at different concentrations for 6 h. The chromatin fraction lysates were subjected to IP with an anti-TIP60 antibody (N-17), followed by IB with an anti-SUV39H1 antibody. f TIP60 (left) and SUV39H1 (right) in the chromatin fraction were IP and tested for post-translational modifications with or without treatments of hydrogen peroxide at 0.5 mM or 1 mM. WCL, whole-cell lysate; CTR, control; Ns, non-silencing siRNA
Full size imageNext, we monitored the in vivo interactions between TIP60 and SUV39H1 in the chromatin fraction in response to different DNA damaging agents. The interaction between SUV39H1 and TIP60 and SUV39H1 protein levels in chromatin were both decreased upon treatment with hydrogen peroxide but not in the presence of other treatments, such as adriamycin, hydroxyurea (HU) or UV light (Fig. 2d). In addition, in the presence of a proteasome inhibitor, MG132, and a lysosome inhibitor, chloroquine (CHQ), we were still able to observe a dose-dependent decrease in the interaction between SUV39H1 and TIP60 and a loss of SUV39H1 protein levels in chromatin induced by hydrogen peroxide (Fig. 2e).
We also detected the common post-translational modifications (PTMs) of both TIP60 and SUV39H1, including phosphorylation, acetylation and methylation. We found that TIP60 phosphorylation and acetylation levels remained unchanged by hydrogen peroxide treatment, while TIP60 methylation was barely detected (Fig. 2f). For SUV39H1, its phosphorylation levels were remarkably decreased by hydrogen peroxide treatment in a dose-dependent manner, whereas its acetylation levels were only slightly increased and methylation levels were unchanged (Fig. 2f).
Altogether, these results suggest that the TIP60–SUV39H1 interaction does not affect the acetylation status or protein expression levels of SUV39H1 at chromatin. However, the affinity of this interaction is decreased in response to a ROS-inducing treatment, such as hydrogen peroxide, probably due to SUV39H1 dephosphorylation that results in decreased SUV39H1 recruitment to chromatin.
SUV39H1 de-phosphorylation at T146 suppresses its interaction with TIP60
We next determine whether SUV39H1 phosphorylation is prerequisite for TIP60-SUV39H1 interaction. An online phosphositeplus database (Hornbeck et al. 2015) predicted four potential phosphorylation sites of SUV39H1, i.e., Ser70, Thr146, Tyr279 and Ser391, which were tested in the following experiments (Fig. 3a). Plasmids of SUV39H1 with single-site mutation at each of these four sites (Serine/Threonine/Tyrosine mutated to Alanine which mimics a de-phosphorylated status at each of these sites, i.e. S70A, T146A, Y279A and S391A), were constructed and separately introduced in H1299 cells. In the meantime, wild-type SUV39H1 plasmids were also introduced as control. As shown in Fig. 3b and c, the TIP60-SUV39H1 interaction was remarkably decreased for SUV39H1 (T146A) compared with wild-type SUV39H1, whereas the interactions detected for other mutant SUV39H1 proteins were not different from that for wild-type SUV39H1. Consistently, within chromatin fraction, less TIP60 proteins interacted with SUV39H1 (T146A) mutant proteins, while more TIP60 proteins interacted with SUV39H1 (T146D), a phosphorylated-mimicry mutant, compared with wild-type SUV39H1 proteins (Fig. 3d). In addition, the affinity between TIP60 and mutant SUV39H1 (T146D) proteins remained strong after hydrogen peroxide treatment, whereas the interaction between TIP60 and wild-type SUV39H1 was obviously decreased, each compared with its corresponding non-treated control (Fig. 3e). Moreover, the recruitment of mutant SUV39H1 (T146D) to chromatin remained mostly unchanged, while the wild-type SUV39H1 recruitment to chromatin was decreased by treatment of hydrogen peroxide (Fig. 3f). These data suggested that de-phosphorylation of SUV39H1 at T146 is responsible for the decreased TIP60-SUV39H1 interaction and thus less SUV39H1 recruitment to chromatin after hydrogen peroxide-treatment.
Fig. 3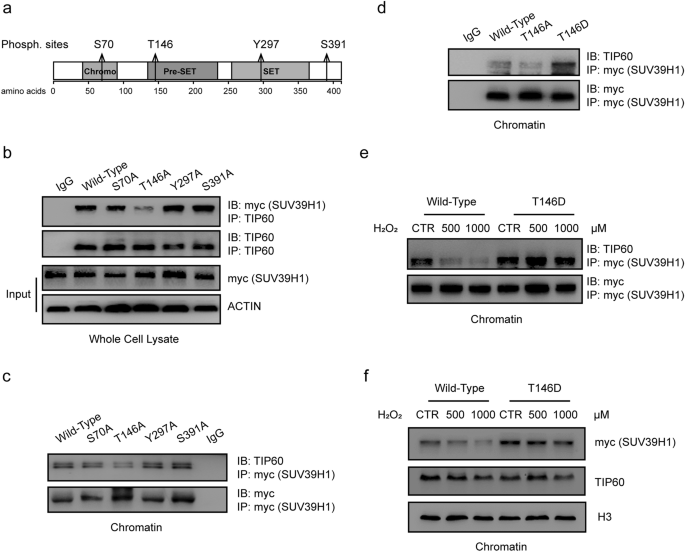
SUV39H1 de-phosphorylation at T146 suppresses TIP60-SUV39H1 interaction in response to hydrogen peroxide. a diagram showing four potential sites of SUV39H1 phosphorylation, i.e. S70, T146, Y297, S391, that were documented in phosphositePlus website (https://www.phosphosite.org/homeAction.action). b De-phosphorylated-mimicry mutant SUV39H1 plasmids with site mutations (S/T/Y mutated to A) at each of these four sites and wild-type SUV39H1 were separately introduced in H1299 cells. TIP60-SUV39H1 was co-immunoprecipitated by a TIP60 antibody from whole-cell lysates, and SUV39H1 was detected by Western Blotting using a myc-tag antibody. The expression levels of SUV39H1 proteins in whole-cell lysate was also detected. c Chromatin factions of the same above groups of cells were extracted. The introduced SUV39H1 proteins were immunoprecipitated by a myc-tag antibody. The TIP60 levels that were co-immunoprecipitated by the standardized same amount of SUV39H1 proteins of each group were detected by Western blotting. d Wild-type, T146A mutant and T146D mutant SUV39H1, a phosphorylated-mimicry mutation proteins, were separately introduced in H1299 cells. Chromatin fracture was extracted, and the TIP60 levels that were co-immunoprecipitated by the standardized same amount of SUV39H1 proteins of each group were detected by Western blotting. e Cells expressing wild-type, T146D mutant SUV39H1 were treated or not treated by hydrogen peroxide at concentrations of 500 μM, 1000 μM, chromatin factions were extracted. Similarly, the TIP60 levels that were co-immunoprecipitated by the standardized same amount of SUV39H1 proteins of each group were detected by Western blotting. f exogenous SUV39H1 and TIP60 protein levels in chromatin were detected by Western blotting in cells that overexpress wild-type, T146D mutant SUV39H1 and were treated or not treated by hydrogen peroxide at concentrations of 500 μM, 1000 μM
Full size imageTIP60 recruits SUV39H1 to chromatin
Based on the above observations, we hypothesized that the localization of SUV39H1 to chromatin might be regulated by TIP60. To prove our hypothesis, we knocked down TIP60 in HCT1299 cells to detect any alterations in SUV39H1 levels in the chromatin fraction. Indeed, we found that SUV39H1 levels in the chromatin fraction were remarkably decreased when TIP60 was depleted (Fig. 4a). To confirm this result, we transfected SUV39H1 plasmids into H1299 cells with or without TIP60 knockdown. Here, both the endogenous and exogenous SUV39H1 protein levels in the chromatin fraction were remarkably decreased when TIP60 was depleted (Fig. 4b). In addition, a wild-type TIP60 or a Zinc Finger domain deleted (ΔZNF) mutant TIP60 that loses the ability to interact with SUV39H1, was re-introduced in TIP60 knockdown cells to rescue the expression levels of TIP60, and then the SUV39H1 recruitment to chromatin was detected (Fig. 4c). As expected, the decreased SUV39H1 recruitment to chromatin in TIP60 knockdown cells was not rescued by ΔZNF TIP60 re-expressing cells, but was recovered in wild-type re-expressing cells (Fig. 4c).
Fig. 4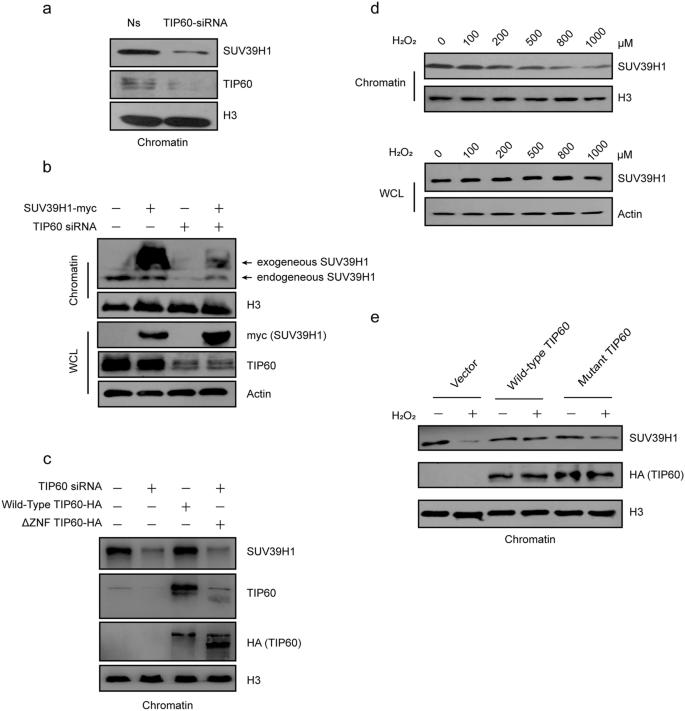
TIP60 is required for the recruitment of SUV39H1 to chromatin. a H1299 cells were treated with non-specific or TIP60 siRNA for 72 h. Then SUV39H1 and TIP60 protein levels in the chromatin fractions were checked by immunoblotting (IB). H3 served as a loading control. b H1299 cells were treated with non-specific or TIP60 siRNA for 36 h. Then vector or SUV39H1 were transfected for 36 h. The cells were harvested and the chromatin fractions were tested for SUV39H1 and TIP60 protein levels by IB. H3 served as a loading control. c TIP60 was knocked down by RNA interfering for 72 h in H1299 cells. An RNA interfering-resistant wild-type TIP60 or a ZNF-domain-deleted mutant TIP60, which loses its interaction with SUV39H1, was introduced into cells by transfection 24 h after TIP60 RNAi, and continue to incubate for 48 h. Chromatin fracture was obtained and SUV39H1 recruitment to heterochromatin is detected by Western Blot. d H1299 cells were treated with hydrogen peroxide at different concentrations for 6 h. Then, SUV39H1 or TIP60 protein levels in the chromatin fraction and total protein lysates were tested by IB, using H3 or actin as loading controls for chromatin or total cell lysates, respectively. e H1299 cells were transfected with wild-type TIP60 and mutant-TIP60 or vector for 42 h and then treated with 1 mM hydrogen peroxide for 6 h. SUV39H1 and TIP60 protein levels in the chromatin fractions were tested by IB. H3 served as a loading control. Ns, non-silencing siRNA; WCL, whole-cell lysate
Full size imageNext, we investigated whether SUV39H1 levels in chromatin were decreased by hydrogen peroxide treatment. As expected, the abundance of SUV39H1 in chromatin was reduced by hydrogen peroxide treatment in a dose-dependent manner (Fig. 4d); however, the total protein levels of SUV39H1 remained unchanged (Fig. 4d). Moreover, when we over-expressed wild-type or a catalytic inactive TIP60 mutant in H1299 cells, and then treated the cells with 1 mM hydrogen peroxide for 6 h, we found that the hydrogen peroxide-mediate decrease in SUV39H1 in chromatin levels were reversed (Fig. 4e).
These results suggest that hydrogen peroxide or TIP60 knockdown induces the detachment of SUV39H1 from chromatin due to an alleviation of the TIP60–SUV39H1 interaction. This detachment is not dependent on TIP60 enzymatic activity.
TIP60 regulates the H3K9me3 levels through SUV39H1
Because SUV39H1 is mainly responsible for catalyzing histone H3K9me3 levels in vivo, we investigated whether TIP60 modulates H3K9me3 levels through SUV39H1. To do so, we over-expressed SUV39H1 in TIP60 knockdown or untreated H1299 cells. We found that TIP60 knockdown resulted in decreased H3K9me3 levels in both SUV39H1 over-expressed and untreated cells (Fig. 5a). Consistently, HepG2 cells with stable TIP60 knockdown also exhibited lower H3K9me3 levels compared with TIP60 intact cells (Fig. 5b). Accordingly, the H3K9 mono-methylation (H3K9me1) and di-methylation levels (H3K9me2) increased when TIP60 was knocked down, as a result of decreased formation of H3K9me3 (Fig. 5a and b). In addition, re-expressing wild-type TIP60 rescued the H3K9me3 levels, whereas re-expressing ΔZNF mutant TIP60 that is unable to recruit SUV39H1 cannot restore the H3K9me3 levels (Fig. 5c), which is consistent with the levels of SUV39H1 recruitment shown in Fig. 4c under the same experimental conditions.
Fig. 5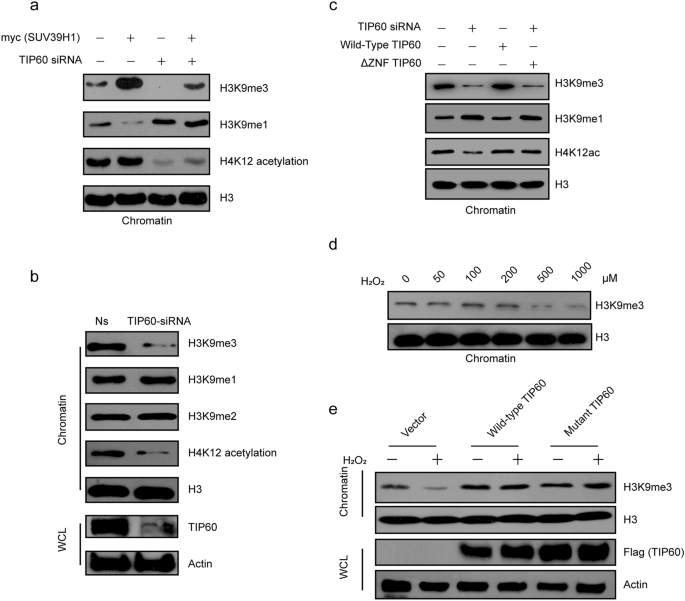
TIP60 is involved in regulating the H3k9me3 levels through SUV39H1. a H1299 cells were treated with non-specific or TIP60 siRNA for 36 h, and then transfected with vector or SUV39H1 for 36 h. Histones were extracted and detected by IB for H3K9 methylation levels. b Histones from TIP60 stably knocked down HepG2 cells and control cells were extracted and detected by Western blotting for H3K9 methylation levels. H3 served as a loading control and H4K12 acetylation as a positive control. c Chromatin fractions were separately extracted from control and TIP60 knockdown H1299 cells with or without re-expression of wild-type TIP60 or ZNF-domain-deleted mutant TIP60. Then H3K9me3 levels were detected accordingly in the non-treated, TIP60 knockdown, wildtype-TIP60-rescued and ZNF-domain-deleted mutant TIP60-rescued TIP60 knockdown cells by Western Blotting. d H1299 cells were treated with hydrogen peroxide at different concentrations for 6 h. Histones were extracted and H3K9me3 levels were detected by IB. H3 served as a loading control. e H1299 cells were transfected with wild-type or mutant TIP60 or vector only for 42 h and then treated with 1 mM hydrogen peroxide for 6 h. Histones were extracted and H3K9me3 levels were detected by IB. H3 served as a loading control. WCL, whole-cell lysate; Ns, non-silencing siRNA
Full size imageIn addition, we found that hydrogen peroxide could induce a dose-dependent decrease in H3K9me3 levels (Fig. 5d). Next, we over-expressed wild-type TIP60 or mutant TIP60 in H1299 cells and then treated the cells with 1 mM hydrogen peroxide for 6 h. Here, we found that TIP60 over-expression increased the H3K9me3 levels that were decreased by hydrogen peroxide treatments (Fig. 5e). The H3K9me3 levels were consistent with the TIP60-dependent recruitment of SUV39H1 to chromatin (Fig. 4e), which is also independent of the enzyme activity of TIP60 (Fig. 5e). In summary, TIP60 seems to promote H3K9me3 by recruiting SUV39H1 to chromatin.
TIP60 has a key role in maintaining heterochromatin stability
Based on the role of TIP60 in modulating SUV39H1 and H3K9me3, we next investigated the impact of TIP60 on heterochromatin structure. The expressions of centromeric satellite repeats, satellite 2 (Sat2) and α-satellite (α-Sat), are all well recognized as markers of heterochromatin relaxation and thus were evaluated in following experiments; these genes are transcribed when the heterochromatin region is relaxed (Wang et al. 2013). A qPCR analysis showed that Sat2 and α-Sat mRNA expression were both increased when TIP60 was knocked down (Fig. 6a). Accordingly, quantitative ChIP (qChIP) assay showed that H3K9me3 levels were down-regulated in both Sat2 and α-Sat loci when TIP60 was knocked down (Fig. 6b).
Fig. 6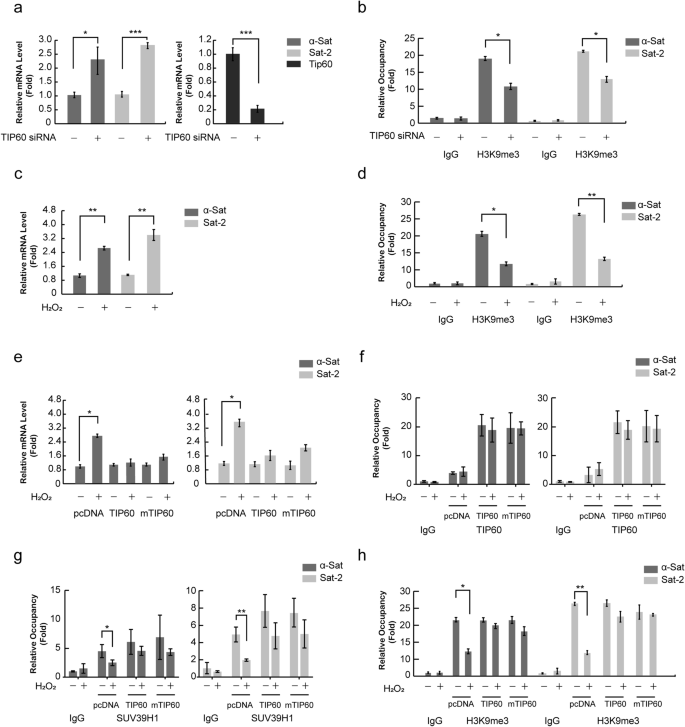
TIP60 participates in maintaining of the heterochromatin structure. a H1299 cells were treated with non-specific RNA or TIP60 siRNA for 72 h. Then mRNAs were extracted and Sat2 or α-Sat expression levels were detected by qPCR. GAPDH gene expression was determined for normalization. b H1299 cells were treated with non-specific RNA or TIP60 siRNA for 72 h. Then a ChIP assay using an H3K9me3 antibody was performed and the levels of precipitated Sat2 or α-Sat DNA fragments were tested by qPCR. c H1299 cells were treated with 1 mM H2O2 for 6 h. Then mRNA was extracted and tested for mRNA levels of Sat2 or α-Sat by qPCR. GAPDH gene expression was determined for normalization. d H1299 cells were treated with 1 mM H2O2 for 6 h. Then a ChIP assay using an H3K9me3 antibody was performed and the levels of precipitated Sat2 or α-Sat DNA fragments were tested by qPCR. e H1299 cells were transfected with wild-type or mutant TIP60 or vector for 42 h, and then treated with 1 mM hydrogen peroxide for 6 h. mRNA expression levels of Sat2 or α-Sat evaluated by qPCR. GAPDH gene expression was used for normalization. f, g, h H1299 cells were transfected with wild-type or mutant TIP60 or vector for 42 h and then treated with 1 mM hydrogen peroxide for 6 h. Levels of TIP60 (f), SUV39H1 (g) and H3K9me3 (h) antibody-precipitated Sat2 or α-Sat DNA fragments were tested by qPCR. The data represent the means ± SD (n = 3). The data represent the means ± SD (n = 3). * P < 0.05; **P < 0.01; ***P < 0.001 (ANOVA with Tukey’s post hoc test)
Full size imageWe also found that hydrogen peroxide treatment resulted in an increase in Sat2 and α-Sat expression (Fig. 6c). Accordingly, the H3K9me3 levels were down-regulated in both Sat2 and α-Sat loci in response to hydrogen peroxide treatment (Fig. 6d). To further investigate the role TIP60 plays during this ROS response, we over-expressed wild-type or mutant TIP60 in H1299 cells and subsequently treated them with hydrogen peroxide. We found that both wild-type and mutant TIP60 could decrease Sat2 and α-Sat expression that had been increased by ROS treatment (Fig. 6e). Moreover, TIP60 restored SUV39H1 and H3K9me3 levels that had been decreased in the Sat2 and α-Sat loci by ROS treatment (Fig. 6f–h). These findings support that TIP60 helps to maintain heterochromatin structure in an H3K9me3-dependent manner. Notably, the alterations of H3K9me3 levels were consistent with those of chromatin recruitment of SUV39H1 (Fig. 5) under the same experimental conditions, suggesting that TIP60-induced H3K9me3 is dependent of SUV39H1 recruitment to chromatin.
TIP60 maintains genome stability and preserves cell survival in response to ROS
The relaxation of heterochromatin may result in genome instability (Wang et al. 2013). Therefore, we investigated if TIP60 helps to maintain genome stability. We evaluated genome instability by performing a quantitative micrococcal nuclease (MNase) sensitivity assay in TIP60-overexpressing or TIP60 knockdown H1299 cells as well as their corresponding control cells, with or without hydrogen peroxide treatment. We found that hydrogen peroxide could markedly induce genome instability, which was reversed upon the over-expression of wild-type or mutant TIP60 (Fig. 7a, b). We obtained similar results when knocking down TIP60 in H1299 cells (Fig. 7a, b). In addition, we further confirmed that overexpression of wild-type SUV39H1 can reverse the hydrogen peroxide-induced genome instability (Fig. 7c, d). Moreover, overexpression of T146D mutant SUV39H1 that preserves its recruitment to chromatin led to a more remarkable effect in reversing the genome instability induced by hydrogen peroxide compared with overexpression of wild-type SUV39H1 (Fig. 7c, d).
Fig. 7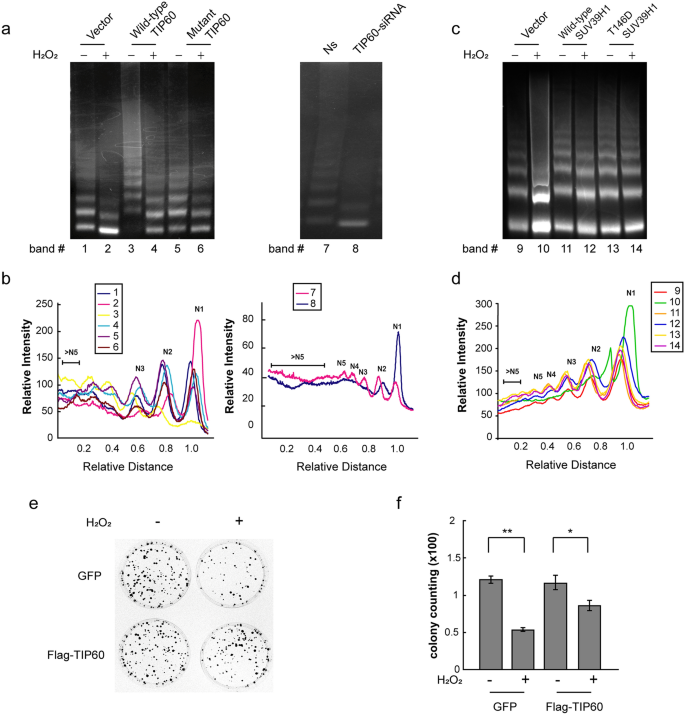
TIP60 is critical for genome stability maintenance and resistance to ROS cytotoxicity. a left panel, H1299 cells were transfected with wild-type or mutant TIP60 or vector only for 42 h and then treated with 1 mM hydrogen peroxide for 6 h. A micrococcal nuclease (MNase) sensitivity assay was performed. Indicated cells were digested with MNase for 2 min at 37 °C, and the genome DNA was subsequently extracted and separated on a 1.2% agarose gel. Right panel, the same experiments were performed with H1299 cells that were interfered with non-specific or TIP60 siRNA for 72 h. b The intensity of each lane in a was consecutively quantified using Quantity One software (Bio-Rad). N1–N5 indicate the number of nucleosomes in each oligonucleosome. c H1299 cells were transfected with wild-type SUV39H1 or T146D mutant SUV39H1 for 48 h and then treated with 1 mM hydrogen peroxide for 12 h. Indicated cells were digested with MNase for 2 min at 37 °C, and the genome DNA was subsequently extracted and separated on a 1.8% agarose gel. d The intensity of each lane in c was consecutively quantified using Quantity One software (Bio-Rad). e, f TIP60 stably over-expressing and control cells were treated with 1 mM H2O2. Then, 5 × 103 cells in 1 ml growth medium containing 0.35% Noble agar (Difco) and G418 (800 μg/ml) were overlaid on 1.5 ml 0.5% base agar medium in a 60-mm culture dish. The cells were incubated at 37 °C in a moist atmosphere of 95% air and 5% CO2; 2 weeks later, the colonies were enumerated after 0.005% crystal violet staining (f). The data represent the means ± SD (n = 3). * P < 0.05; **P < 0.01; (ANOVA with Tukey’s post hoc test). Ns, non-silencing siRNA
Full size imageWe next examined if TIP60 influences the colony-forming abilities of hydrogen peroxide-pretreated or non-treated HepG2 cells. We found that hydrogen peroxide could decrease the colony numbers from 121 to 54 per 5000 cells in average. However, over-expression of TIP60 resulted in an increase in colony formation ability in hydrogen peroxide-pretreated cells (Fig. 7e, f). These results demonstrate that TIP60 has a key role in preserving genome stability in response to ROS treatments, resulting in increased cell survival against ROS (Fig. 8).
Fig. 8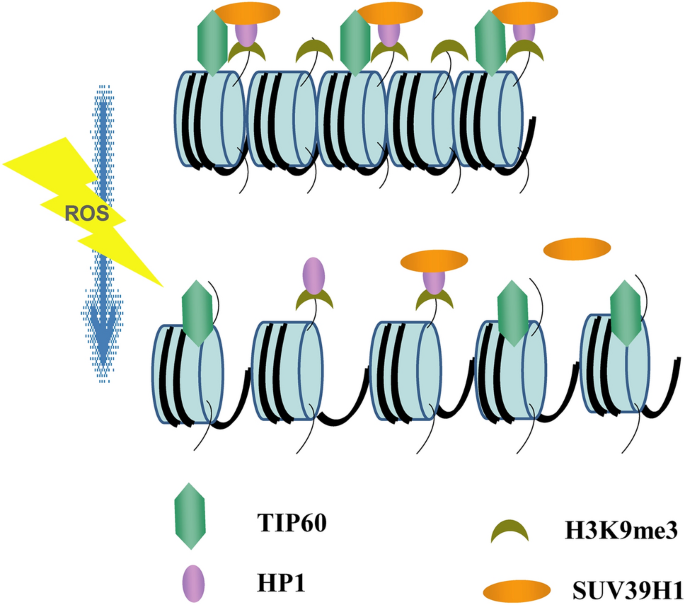
A proposed model for the role of TIP60 in regulating SUV39H1 protein and genome stability. TIP60 directly binds to SUV39H1 in vivo. This interplay recruits SUV39H1 to chromatin to catalyze H3K9me3, maintain compact heterochromatin structure and preserve genome stability. Oxidative stress compromises the TIP60–SUV39H1 interaction and promotes heterochromatin relaxation and genome instability
Full size imageDiscussion
Prior to our study, the detailed mechanisms underlying how TIP60 regulates heterochromatin and genome stability had not been fully explored. Here, we identified a direct TIP60–SUV39H1 interaction that permits TIP60 to recruit SUV39H1 to chromatin, independent of its enzyme activity. Hydrogen peroxide treatment inducing ROS interrupts this interaction and thus compromises SUV39H1 recruitment to chromatin. As a consequence, the heterochromatin H3K9me3 levels decrease, and the heterochromatin structure relaxes, resulting in genome instability and inhibited cell proliferation in response to oxidative stress (Fig. 8).
We identified that the TIP60 binding partner SUV39H1 frequently undergoes post-translational modifications. These PTMs are closely associated with the protein functions of SUV39H1. For example, SUV39H1 is acetylated within the SET domain, which alleviates its in vivo activity (Vaquero et al. 2007). Additionally, MDM2-dependent SUV39H1 ubiquitination mediates its degradation, resulting in genome instability (Mungamuri et al. 2016; Wienken et al. 2017). Moreover, our own previous work identified SUV39H1 methylations catalyzed by SET7/9 promotes genome instability and ultimately cell death as a result of DNA damage (Wang et al. 2013). Despite these important prior advances, our study is the first to identify a direct TIP60–SUV39H1 interaction that neither catalyzes SUV39H1 acetylation nor affects its stabilization, but rather mediates SUV39H1 recruitment to chromatin. This effect is likely because the TIP60 HAT domain is not involved in the TIP60–SUV39H1 interaction.
ROS can have a wide influence on the PTMs of various proteins. For example, ROS can induce S6K-mediated PTEN phosphorylation, which increases its interaction with USP7, causing a decrease in PTEN mono-ubiquitination (Wu et al. 2014). In addition, ROS induces the demethylation of transcriptional factor Sp1 and thereby increases the Sp1–SUV39H1 interaction and SUV39H1 recruitment to the promoter region of Cyclin B1, resulting in transcriptional silencing of Cyclin B1 (Chuang et al. 2011). We previously showed that ROS treatment-induced UNG2 deacetylation, resulting in its decreased interaction with UHRF1 (Bao et al. 2020). Here in this study, we not only discovered that SUV39H1 was de-phosphorylated in response to ROS treatment but also demonstrated, for the first time, that SUV39H phosphorylation at site T146 promotes the TIP60-SUV39H1 interaction and SUV39H1 recruitment to heterochromatin. Interestingly, the previous study reported that hydrogen peroxide may up-regulate Sirt1 that in turn deacetylate SUV39H1 at K266 (Vaquero et al. 2007) and stabilize the protein, although the acetylation levels of SUV39H1 were not directly investigated in their study (Bosch-Presegue et al. 2011). However, in our study, SUV39H1 acetylation levels in the chromatin were somehow increased at chromatin fraction in response to ROS treatment. In addition, our results suggest that the ROS-decreased SUV39H1 levels at chromatin were resulted from less SUV39H1 recruitment to chromatin rather than SUV39H1 degradation. Moreover, we observed that the overall SUV39H1 protein levels remained unchanged after ROS treatment, suggesting that the increased acetylation of SUV39H1 at chromatin that found in our study probably may not be associated with Sirt1 activity. Furthermore, these evidences might also suggest that SUV39H1 protein may react differently depending on different cell lines or tissues upon ROS stress. The functions and mechanisms of SUV39H1 acetylation as well as other modifications in response to ROS remain to be explored.
Both TIP60 and SUV39H1 are closely implicated in DDR pathways (Ikura et al. 2000; Wang et al. 2013). As such, the TIP60–SUV39H1 interaction might function in DNA repair processes. ROS induce various types of DNA damage, including base oxidization, nucleotide damage, inter-strand DNA cross-links and single-strand breakage (Hong and Greenberg, 2005; Pietrzak et al. 2018). As a result, multiple DDR pathways—including base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR) and ICL repair signaling—are profoundly activated in response to oxidative stress (Bao et al. 2020; Pietrzak et al. 2018; Shafirovich and Geacintov, 2017). This paradigm differs from the DDR pathways such as ATR–Chk1 signaling that specifically responds to replication stress, or homologous recombination (HR) and non-homologous end joining (NHEJ) that respond to DSBs. ROS can also directly modify lipids/proteins by oxidization, and impact multiple biological pathways including metabolism, inflammation and autophagy (Forrester et al. 2018). Therefore, the cellular response to ROS is especially complicated when compared with other genotoxic reagents. This situation might explain why decreased SUV39H1 recruitment to chromatin by TIP60 is induced only by hydrogen peroxide but not UV (inducing pyrimidine dimer), HU (inducing replication stress) or adriamycin (inducing DSB).
SUV39H1 recruitment to chromatin is essential for maintaining H3K9me3, a hallmark of mammalian heterochromatin (Hyun et al. 2017) and the docking site for heterochromatin protein 1α and β (HP1α and HP1β). HP1α/HP1β can interact with SUV39H1 and thereby recruit SUV39H1 to heterochromatin to maintain H3K9me3 levels (Grezy et al. 2016; Hyun et al. 2017). Upon activation of the DNA damage response, however, TIP60 is recruited by the MRN complex to H3K9me3 sites instead of HP1β. This event constitutes an essential step to activate TIP60 acetyltransferase activity necessary for DNA repair signaling. Heterochromatin-localized TIP60 is thus specifically important for DNA damage repair (Sun et al. 2009). We further propose that TIP60 mediates SUV39H1 recruitment to heterochromatin to maintain genome stability, representing a novel function of TIP60 during the DDR. In addition, because H3K9me3 is a dynamic docking site for TIP60 during the DDR (Sun et al. 2009), our data suggest that TIP60 will recruit more SUV39H1 to the heterochromatin as a positive feedback mechanism to ensure heterochromatin structure and preserve genome resilience against DNA damage (Wang et al. 2018).
The relationship between chromatin structure and genome stability is complex. For example, DNA damage increases H3K9 acetylation and accordingly decreases H3K9 methylation at damage sites due to mutual exclusivity between these two modifications (Hou et al. 2020; Nicolas et al. 2003). As a result, the chromatin structure is relaxed to facilitate the recruitment of DNA repair proteins and to ultimately ensure genome stability. Heterochromatin also relaxes to promote DNA repair protein recruitment and genome stability (Nair et al. 2017; Vargaweisz 2001). However, heterochromatic DNA is highly compacted by proteins such as HP1α/β in heterochromatin, which protect the DNA from damage (Luijsterburg et al. 2009). Therefore, the relaxation of the heterochromatin will result in increased genome instability and subsequently cell death (Wang et al. 2013). Now we demonstrate the existence of a similar paradox between ROS-modulated chromatin structure and genome stability. Previous studies already noticed that ROS treatments can result in chromatin relaxation, which facilitates the transcription of DNA repair proteins and ultimately ensures genome stability and cell survival against ROS (Wang et al. 2020b; Whongsiri et al. 2019). Our findings, however, have demonstrated a novel pathway by which hydrogen peroxide can interrupt the TIP60–SUV39H1 interaction, thus relaxing the heterochromatin and compromising genome instability. The overall result is an increase in cell death.
Because heterochromatin-dependent genome stability is vital for cell survival against oxidative stress (Choi et al. 2018), our mechanism highlights TIP60 as a specific therapeutic target to sensitize cancer cells to ROS-mediated heterochromatin relaxation, genome instability and cytotoxicity. Combined treatments involving ROS and DNA-damaging agents indeed have synergistic anti-cancer effects and thus hold promise as clinical cancer treatments (Cottini et al. 2015). Therefore, our data strengthen the biological foundation for the utility of these combined treatments of ROS-generating agents and DNA-damaging agents (such as TIP60 and SUV39H1 inhibitors) during cancer treatment. However, mice models are still needed to assess the therapeutic effects of TIP60-silencing or disrupted TIP60-SUV39H1 interaction. In addition, typical ROS-generating agents, such as hydrogen peroxide used in our study, cannot be directly used in clinical treatment. Fortunately, there are clinical chemicals that also promote ROS generation, such as piperlongumine (Huang et al. 2016), cisplatin and paclitaxel (Wang et al. 2020a), and thus could be candidates of substitute for hydrogen peroxide. Still, a screening of these ROS-generating therapeutic agents is needed to determine which drugs are best suitable for combined use with DNA damaging agents.
In summary, we have identified a novel function of TIP60 to recruit SUV39H1 to heterochromatin, to maintain heterochromatin structure, preserve genome stability and thus facilitate cell survival in response to oxidative stress, which inspires therapeutic strategy to inhibit TIP60 and exacerbate ROS-induced cancer cytotoxicity. Questions now remain as to whether potential inhibitor could be developed to selectively decrease the protein levels of TIP60 or to disrupt TIP60-SUV39H1 interaction, as our results indicated that the genome stability maintained by TIP60 is independent of its enzyme activity. As such, further work that investigates how the transcript or protein levels of TIP60 can be specifically targeted and inhibited is now warranted. Once we understand a detailed mechanism controlling the transcriptional inactivation or protein degradation of TIP60, we might be well positioned to design novel anticancer therapies based on targeting TIP60-dependent heterochromatin structure/genome stability and sensitizing cancer genome to ROS-generating agents.
References
Ayrapetov, M. K., Gursoyyuzugullu, O., Xu, C., Xu, Ye., & Price, B. D. (2014). DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proceedings of the National Academy of Sciences of the United States of America, 111, 9169–9174. https://doi.org/10.1073/pnas.1403565111
Bao, Y., Tong, L., Song, B., Liu, Ge., Zhu, Q., Lu, X., et al. (2020). UNG2 deacetylation confers cancer cell resistance to hydrogen peroxide-induced cytotoxicity. Free Radical Biology and Medicine. https://doi.org/10.1016/j.freeradbiomed.2020.06.010
Bosch-Presegue, L., Raurellvila, H., Marazueladuque, A., Kanegoldsmith, N., Valle, A., Oliver, J., et al. (2011). Stabilization of Suv39H1 by SirT1 is part of oxidative stress response and ensures genome protection. Molecular Cell, 42, 210–223. https://doi.org/10.1016/j.molcel.2011.02.034
Cao, L.-L., Shen, C., & Zhu, W.-G. (2016). Histone modifications in DNA damage response. Science China Life Sciences, 59, 257–270. https://doi.org/10.1007/s11427-016-5011-z
Chiba, T., Saito, T., Yuki, K., Zen, Y., Koide, S., Kanogawa, N., et al. (2015). Histone lysine methyltransferase SUV39H1 is a potent target for epigenetic therapy of hepatocellular carcinoma. International Journal of Cancer, 136, 289–298. https://doi.org/10.1002/ijc.28985
Choi, J. E., Heo, S.-H., Kim, M. J., & Chung, W.-H. (2018). Lack of superoxide dismutase in a rad51 mutant exacerbates genomic instability and oxidative stress-mediated cytotoxicity in Saccharomyces cerevisiae. Free Radical Biology and Medicine, 129, 97–106. https://doi.org/10.1016/j.freeradbiomed.2018.09.015
Chuang, J. Y., Chang, W. C., & Hung, J. J. (2011). Hydrogen peroxide induces Sp1 methylation and thereby suppresses cyclin B1 via recruitment of Suv39H1 and HDAC1 in cancer cells. Free Radical Biology and Medicine, 51, 2309–2318. https://doi.org/10.1016/j.freeradbiomed.2011.10.001
Cottini, F., Hideshima, T., Suzuki, R., Tai, Y.-T., Bianchini, G., Richardson, P. G., et al. (2015). Synthetic lethal approaches exploiting DNA damage in aggressive myeloma. Cancer Discovery, 5, 972. https://doi.org/10.1158/2159-8290.CD-14-0943
Forrester, S. J., Kikuchi, D. S., Hernandes, M. S., Xu, Q., & Griendling, K. K. (2018). Reactive oxygen species in metabolic and inflammatory signaling. Circulation Research, 122, 877–902. https://doi.org/10.1161/CIRCRESAHA.117.311401
Fritsch, L., Robin, P., Mathieu, J., Souidi, M., Hinaux, H., Rougeulle, C., et al. (2010). A subset of the histone H3 lysine 9 methyltransferases Suv39h1, G9a, GLP, and SETDB1 participate in a multimeric complex. Molecular Cell, 37, 46–56. https://doi.org/10.1016/j.molcel.2009.12.017
Ghobashi, A. H., & Kamel, M. A. (2018). Tip60: updates. Journal of Applied Genetics, 59, 161–168. https://doi.org/10.1007/s13353-018-0432-y
Grezy, A., Chevillardbriet, M., Trouche, D., & Escaffit, F. (2016). Control of genetic stability by a new heterochromatin compaction pathway involving the Tip60 histone acetyltransferase. Molecular Biology of the Cell, 27, 599–607. https://doi.org/10.1091/mbc.E15-05-0316
Hong, I. S., & Greenberg, M. M. (2005). DNA interstrand cross-link formation initiated by reaction between singlet oxygen and a modified nucleotide. Journal of the American Chemical Society, 127, 10510–10511. https://doi.org/10.1021/ja053493m
Hornbeck, P. V., Zhang, B., Murray, B., Kornhauser, J. M., Latham, V., & Skrzypek, E. (2015). PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Research, 43, D512–D520. https://doi.org/10.1093/nar/gku1267
Hou, T., Cao, Z., Zhang, J., Tang, M., Tian, Y., Li, Y., et al. (2020). SIRT6 coordinates with CHD4 to promote chromatin relaxation and DNA repair. Nucleic Acids Research, 48, 2982–3000. https://doi.org/10.1093/nar/gkaa006
Huang, B. K., Langford, T. F., & Sikes, H. D. (2016). Using sensors and generators of H2O2 to elucidate the toxicity mechanism of piperlongumine and phenethyl isothiocyanate. Antioxidants & Redox Signaling, 24, 924–938.
Hyun, K., Jeon, J., Park, K., & Kim, J. (2017). Writing, erasing and reading histone lysine methylations. Experimental Molecular Medicine, 49, 1–22. https://doi.org/10.1038/emm.2017.11
Ikura, T., Ogryzko, V., Grigoriev, M., Groisman, R., Wang, J., Horikoshi, M., et al. (2000). Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell, 102, 463–473. https://doi.org/10.1016/s0092-8674(00)00051-9
Jehanno, C., Flouriot, G., Le Goff, P., & Michel, D. (2017). A model of dynamic stability of H3K9me3 heterochromatin to explain the resistance to reprogramming of differentiated cells. Biochimica et Biophysica Acta (BBA): Gene Regulatory Mechanisms, 1860, 184–195. https://doi.org/10.1016/j.bbagrm.2016.11.006
Kamine, J., Elangovan, B., Subramanian, T., Coleman, D., & Chinnadurai, G. (1996). Identification of a cellular protein that specifically interacts with the essential cysteine region of the HIV-1 tat transactivator. Virology, 216, 357–366. https://doi.org/10.1006/viro.1996.0071
Kaniskan, H. U., Konze, K. D., & Jin, J. (2015). Selective inhibitors of protein methyltransferases. Journal of Medicinal Chemistry, 58, 1596–1629. https://doi.org/10.1021/jm501234a
Kim, J. J., Lee, S. Y., & Miller, K. M. (2019). Preserving genome integrity and function: the DNA damage response and histone modifications. Molecular Biology, 54, 208–241. https://doi.org/10.1080/10409238.2019.1620676
Li, Q., Lin, S., Wang, X., Lian, G., Lu, Z., Guo, H., et al. (2009). Axin determines cell fate by controlling the p53 activation threshold after DNA damage. Nature Cell Biology, 11, 1128–1134. https://doi.org/10.1038/ncb1927
Li, G., Ma, D., & Chen, Y. (2016). Cellular functions of programmed cell death 5. Biochimica et Biophysica Acta (BBA): Molecular Cell Research, 1863, 572–580. https://doi.org/10.1016/j.bbamcr.2015.12.021
Li, Y., Li, Z., Dong, L., Tang, M., Zhang, P., Zhang, C., et al. (2018). Histone H1 acetylation at lysine 85 regulates chromatin condensation and genome stability upon DNA damage. Nucleic Acids Research, 46, 7716–7730. https://doi.org/10.1093/nar/gky568
Li, Z., Chen, Y., Tang, M., Li, Y., & Zhu, W.-G. (2020). Regulation of DNA damage-induced ATM activation by histone modifications. Genome Instability & Disease, 1, 20–33. https://doi.org/10.1007/s42764-019-00004-8
Liu, Y., Tavana, O., & Gu, W. (2019). p53 modifications: exquisite decorations of the powerful guardian. Journal of Molecular Cell Biology, 11, 564–577. https://doi.org/10.1093/jmcb/mjz060
Lu, X., Tang, M., Zhu, Q., Yang, Q., Li, Z., Bao, Y., et al. (2019). GLP-catalyzed H4K16me1 promotes 53BP1 recruitment to permit DNA damage repair and cell survival. Nucleic Acids Research, 47, 10977–10993. https://doi.org/10.1093/nar/gkz897
Luijsterburg, M. S., Dinant, C., Lans, H., Stap, J., Wiernasz, E., Lagerwerf, S., et al. (2009). Heterochromatin protein 1 is recruited to various types of DNA damage. Journal of Cell Biology, 185, 577–586. https://doi.org/10.1083/jcb.200810035
Mo, F., Zhuang, X., Liu, X., Yao, P. Y., Qin, Bo., Su, Z., et al. (2016). Acetylation of Aurora B by TIP60 ensures accurate chromosomal segregation. Nature Chemical Biology, 12, 226–232. https://doi.org/10.1038/nchembio.2017
Mungamuri, S. K., Qiao, R. F., Yao, S., Manfredi, J. J., Gu, W., & Aaronson, S. A. (2016). USP7 enforces heterochromatinization of p53 target promoters by protecting SUV39H1 from MDM2-mediated degradation. Cell Reports, 14, 2528–2537. https://doi.org/10.1016/j.celrep.2016.02.049
Nair, N., Shoaib, M., & Sorensen, C. S. (2017). Chromatin dynamics in genome stability: Roles in suppressing endogenous DNA damage and facilitating DNA repair. International Journal of Molecular Sciences, 18, 1486. https://doi.org/10.3390/ijms18071486
Nicolas, E., Roumillac, C., & Trouche, D. (2003). Balance between acetylation and methylation of histone H3 lysine 9 on the E2F-responsive dihydrofolate reductase promoter. Molecular and Cellular Biology, 23, 1614–1622. https://doi.org/10.1128/Mcb.23.5.1614-1622.2003
Niida, H., Katsuno, Y., Sengoku, M., Shimada, M., Yukawa, M., Ikura, M., et al. (2010). Essential role of Tip60-dependent recruitment of ribonucleotide reductase at DNA damage sites in DNA repair during G1 phase. Genes & Development, 24, 333–338. https://doi.org/10.1101/gad.1863810
Pietrzak, J., Spickett, C. M., Ploszaj, T., Virag, L., & Robaszkiewicz, A. (2018). PARP1 promoter links cell cycle progression with adaptation to oxidative environment. Redox biology, 18, 1–5. https://doi.org/10.1016/j.redox.2018.05.017
Qin, Bo., Yu, J., Nowsheen, S., Wang, M., Tu, X., Liu, T., et al. (2019). UFL1 promotes histone H4 ufmylation and ATM activation. Nature Communications, 10, 1242. https://doi.org/10.1038/s41467-019-09175-0
Shafirovich, V., & Geacintov, N. E. (2017). Removal of oxidatively generated DNA damage by overlapping repair pathways. Free Radical Biology and Medicine, 107, 53–61. https://doi.org/10.1016/j.freeradbiomed.2016.10.507
Shirai, A., Kawaguchi, T., Shimojo, H., Muramatsu, D., Ishidayonetani, M., Nishimura, Y., et al. (2017). Impact of nucleic acid and methylated H3K9 binding activities of Suv39h1 on its heterochromatin assembly. eLife. https://doi.org/10.7554/eLife.25317
Strahl, B. D., & Allis, C. D. (2000). The language of covalent histone modifications. Nature, 403, 41–45. https://doi.org/10.1038/47412
Sun, Y., Jiang, X., Chen, S., Fernandes, N., & Price, B. D. (2005). A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proceedings of the National Academy of Sciences of the United States of America, 102, 13182–13187. https://doi.org/10.1073/pnas.0504211102
Sun, Y., Jiang, X., Xu, Ye., Ayrapetov, M. K., Moreau, L. A., Whetstine, J. R., & Price, B. D. (2009). Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nature Cell Biology, 11, 1376–1382. https://doi.org/10.1038/ncb1982
Tang, Y., Luo, J., Zhang, W., & Gu, W. (2006). Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Molecular Cell. https://doi.org/10.1016/j.molcel.2006.11.021
Tang, J., Cho, N. W., Cui, G., Manion, E. M., Shanbhag, N. M., Botuyan, M. V., et al. (2013). Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nature Structural & Molecular Biology, 20, 317–325. https://doi.org/10.1038/nsmb.2499
Vaquero, A., Scher, M., Erdjumentbromage, H., Tempst, P., Serrano, L., & Reinberg, D. (2007). SIRT1 regulates the histone methyl-transferase SUV39H1 during heterochromatin formation. Nature, 450, 440–444. https://doi.org/10.1038/nature06268
Vargaweisz, P. (2001). ATP-dependent chromatin remodeling factors: Nucleosome shufflers with many missions. Oncogene, 20, 3076–3085. https://doi.org/10.1038/sj.onc.1204332
Wang, D., Zhou, J., Liu, X., Lu, D., Shen, C., Du, Y., et al. (2013). Methylation of SUV39H1 by SET7/9 results in heterochromatin relaxation and genome instability. Proceedings of the National Academy of Sciences of the United States of America, 110, 5516–5521. https://doi.org/10.1073/pnas.1216596110
Wang, C., Zhu, B., & Xiong, J. (2018). Recruitment and reinforcement: maintaining epigenetic silencing. Science China Life Sciences, 61, 515–522. https://doi.org/10.1007/s11427-018-9276-7
Wang, K., Liu, X., Liu, Q., Ho, I., Wei, X., Yin, T., et al. (2020). Hederagenin potentiated cisplatin- and paclitaxel-mediated cytotoxicity by impairing autophagy in lung cancer cells. Cell Death & Disease, 11, 611. https://doi.org/10.1038/s41419-020-02880-5
Wang, S., Wu, X.-M., Liu, C.-H., Shang, J.-Y., Gao, F., & Guo, H.-S. (2020). Verticillium dahliae chromatin remodeling facilitates the DNA damage repair in response to plant ROS stress. PLoS Pathogens, 16, e1008481. https://doi.org/10.1371/journal.ppat.1008481
Whongsiri, P., Pimratana, C., Wijitsettakul, U., Sanpavat, A., Jindatip, D., Hoffmann, M. J., et al. (2019). Oxidative stress and LINE-1 reactivation in bladder cancer are epigenetically linked through active chromatin formation. Free Radical Biology and Medicine, 134, 419–428. https://doi.org/10.1016/j.freeradbiomed.2019.01.031
Wienken, M., Moll, U., & Dobbelstein, M. (2017). Mdm2 as a chromatin modifier. Journal of Molecular Cell Biology, 9, 74–80. https://doi.org/10.1093/jmcb/mjw046
Wu, Y., Zhou, H., Wu, Ke., Lee, S., Li, R., & Liu, X. (2014). PTEN phosphorylation and nuclear export mediate free fatty acid-induced oxidative stress. Antioxidants & Redox Signaling, 20, 1382–1395. https://doi.org/10.1089/ars.2013.5498
Yamamoto, T., & Horikoshi, M. (1997). Novel substrate specificity of the histone acetyltransferase activity of HIV-1-tat interactive protein Tip60. Journal of Biological Chemistry, 272, 30595–30598. https://doi.org/10.1074/jbc.272.49.30595
Yang, N., Sun, T., & Shen, P. (2020). Deciphering p53 dynamics and cell fate in DNA damage response using mathematical modeling. Genome Instability & Disease. https://doi.org/10.1007/s42764-020-00019-6
Acknowledgements
The authors would like to thank Professor Lin Shengcai for gifting the FLAG-TIP60 HAT-deficient mutant expression vector and HepG2 cells. We also thank Dr. Jessica Tamanini of ETediting, Shenzhen University for language editing. The authors would like to recognize that there are many other valuable papers that could have been included in this study, but space limitations prevented them all from being cited.
Funding
This work was supported by National Key R&D Program of China [2017YFA0503900]; National Natural Science Foundation of China [81720108027, 81530074]; Science and Technology Program of Guangdong Province in China [2017B030301016]; Shenzhen Municipal Commission of Science and Technology Innovation [JCYJ20170818092450901]; Discipline Construction Funding of Shenzhen [(2016)1452]; Shenzhen Bay Laboratory [SZBL2019062801011].
Author information
Bo Tu, Yantao Bao and Ming Tang contributed equally to this work.
Affiliations
Guangdong Key Laboratory of Genome Instability and Human Disease Prevention, Department of Biochemistry and Molecular Biology, Shenzhen University School of Medicine, Shenzhen, 518055, China
Bo Tu, Yantao Bao, Qian Zhu, Xiaopeng Lu, Hui Wang & Wei-Guo Zhu
Fred Hutchinson Cancer Research Center, Seattle, WA, 98101, USA
Bo Tu
Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education), Beijing Key Laboratory of Protein Posttranslational Modifications and Cell Function, Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Peking University Health Science Center, Beijing, 100191, China
Bo Tu, Ming Tang, Tianyun Hou, Ying Zhao, Ping Zhang & Wei-Guo Zhu
International Cancer Center, Shenzhen University School of Medicine, Room 901, Building A6, No. 1066, Xueyuan Avenue, Nanshan District, Shenzhen, 518055, Guangdong, China
Wei-Guo Zhu
Shenzhen Bay Laboratory, Shenzhen University School of Medicine, Shenzhen, 518055, China
Wei-Guo Zhu
Contributions
Experiments and data analysis were carried out by BT and YB; The study was conceived by W-GZ with intellectual contributions from QZ, XL, HW, TH, YZ and PZ; All authors contributed to writing and editing of the manuscript and have approved the final submission.
Corresponding authors
Correspondence to Ping Zhang or Wei-Guo Zhu.
Ethics declarations
Conflict of interest
The authors declare no competing financial interests.
Ethics approval
Not applicable.
Consent for publication
All authors have approved the final submission and consented to publication.
Rights and permissions
About this article
Cite this article
Tu, B., Bao, Y., Tang, M. et al. TIP60 recruits SUV39H1 to chromatin to maintain heterochromatin genome stability and resist hydrogen peroxide-induced cytotoxicity. GENOME INSTAB. DIS. (2020). https://doi.org/10.1007/s42764-020-00025-8
Received29 August 2020
Revised12 October 2020
Accepted22 October 2020
Published06 November 2020
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
TIP60
SUV39H1
ROS
H3K9 tri-methylation
Heterochromatin
Genome stability
用户登录
还没有账号?
立即注册