






End resection: a key step in homologous recombination and DNA double-strand break repair
Review Article Open Access
Sijie Liu & Daochun Kong
Genome Instability & Disease 2, 39–50(2021)
Abstract
DNA end resection in eukaryotes is a key step in DNA homologous recombination (HR) and HR-mediated DNA double-strand break (DSB) repair, in which DNA2, EXO1 and MRE11 endo- and exonucleases remove several kilobases from the 5′ terminus of the DNA with DSB, while the 3′ terminus remains intact. The end resection-generated 3′ single-stranded DNA (ssDNA) overhang is then coated by RAD51 for subsequent strand invasion. In the last two decades, great progress has been made in understanding the biochemical mechanisms of end resection, including the identification of various enzymes involved in this process. However, some important questions about this process remain to be resolved. In this review, we summarize the general process of end resection and discuss the implications of the most recent findings for understanding of the end resection machinery.
Introduction
Genomic DNA is frequently at risk of damages from a variety of physical or chemical hazards, such as ultraviolet (UV) radiation, ionizing radiation, and chemotherapy drugs, that trigger a diverse range of DNA lesions, including phosphodiester backbone breakage, inter-strand cross-linking, and base oxidation and reduction (Sancar et al. 2004; Tubbs and Nussenzweig 2017; Ciccia and Elledge 2010). These DNA lesions can severely affect genomic integrity and the survival of living organisms. Among the various types of DNA lesions, DNA double-strand breaks (DSBs) are the most lethal and can lead to chromosomal loss, genomic aberrations, carcinogenesis, or cell death, if not properly repaired (Featherstone and Jackson 1999). Efficient DNA repair pathways have evolved in eukaryotic cells to accurately repair DSBs induced by ionizing radiation or chemicals (Filippo et al. 2008; Mehta and Haber 2014; Jasin and Rothstein 2013). Among them, homologous recombination (HR) is one of the two major DNA DSB repair pathways. HR is also essential for DNA replication, genetic variation during the course of evolution, and production of gametes during meiosis (Hunter 2015; Mehta and Haber 2014; Lam and Keeney 2014). In humans, deficiencies in the HR pathway can result in serious diseases, including breast and ovarian cancer, ataxia–telangiectasia, Bloom syndrome, and Fanconi anemia (Jackson and Bartek 2009; Lord and Ashworth 2016).
The HR-mediated repair of DSBs predominantly occurs during the S and G2 phases of the cell cycle, as the process requires genetic information from a homologous DNA template, such as a sister chromatid (Filippo et al. 2008; Kadyk and Hartwell 1992). Programmed HR or HR-mediated DSB repair comprises three major steps: end resection, strand invasion, and resolution of Holliday junctions (Filippo et al. 2008). DNA DSB repair by HR requires many enzymes and protein complexes that effectively cooperate to perform each step of the repair process. In this review, we focus on the process of DNA end resection and its mechanism in eukaryotes.
DSB recognition
When DSBs occur in chromatin DNA, these breaks must be immediately recognized by the cell; otherwise, further DNA damage, such as genetic loss, may occur. Therefore, it is essential for the cell to quickly and efficiently recognize DNA breaks. In mammalian cells, DSB recognition in the HR pathway is performed by a complex known as the MRN (MRE11, RAD50, and NBS1) complex. The MRN complex is conserved among eukaryotes, from single-celled organisms such as yeast (where it is called the MRX complex) to multicellular organisms such as mammals (Haber 1998; D'Amours and Jackson 2002; Paull 2018; Stracker and Petrini 2011; Sung et al. 2014). A partial dysfunction of any of these MRN subunits in humans can result in severe genetic diseases, such as ataxia telangiectasia-like disorder (ATLD) and Nijmegen breakage syndrome (NBS). Patients with such genetic diseases exhibit developmental defects, genomic instability, hypersensitivity to ionizing radiation, and an increased risk of developing cancers (Petrini 2000; Carney et al. 1998; Varon et al. 1998). In mouse models, a deficiency in any of the MRN subunits leads to cell death and embryonic lethality (Bender et al. 2002; Stracker and Petrini 2011).
Immediately following DNA breakage, the MRN complex is recruited to the vicinity of DSBs (Lisby et al. 2004; Myler et al. 2017). However, the mechanism by which the MRN complex recognizes broken DNA ends is still unclear. Genetic and structural studies have revealed that two subunits each of MRE11 and RAD50 form the heterotetrameric (M2R2) core of the complex, with NBS1 at the periphery where it interacts with the MRE11 subunit (Williams et al. 2008; Dolganov et al. 1996; Park et al. 2011). Structurally, the MRN complex consists of three segments: A globular head, which is composed of the MRE11 dimer, RAD50 ATPase domains, and NBS1; a pair of elongated RAD50 coiled-coil domains; and a distal RAD50 hook domain (Williams et al. 2008). The MRN complex binds to DNA through its globular DNA-binding head, which primarily requires MRE11 and RAD50, although some studies suggest that NBS1 may also bind to DNA and influence the DNA-binding properties of MRE11 and RAD50 (Williams et al. 2008; Park et al. 2011; Paull and Gellert 1999). MRE11 has two conserved DNA-binding domains, one is located in its middle region and the other in the C terminus (Park et al. 2011). In vitro biochemical assays have shown that MRE11 has a higher affinity for single-stranded DNA (ssDNA) than for double-stranded DNA (dsDNA), and it is surprising that MRE11 binds the DNA backbone but not the broken ends of dsDNA, since one study has shown that MRE11 binds linear and circular DNA with the same efficiency (Hopfner et al. 2001). Recently, single molecular-imaging studies have revealed that MRN molecules localize to free DNA ends and internal DNA sites with similar efficiency. It was also found that MRN slides along a DNA strand to find free DNA ends via a homoduplex sliding activity, and its occupancy of DSB ends is more likely when free DSB ends are initially bound to the non-homologous end-joining (NHEJ) factor Ku70/80 (Myler et al. 2017) (Fig. 1). Currently, the mechanism by which the MRN complex recognizes DSB ends remains an open question. Further structural analysis of the entire MRN complex is necessary to elucidate the underlying mechanism by which MRN recognizes DNA ends. Additionally, whether there is an unknown factor that anchors MRN to DSB ends also remains to be determined (Fig. 1).
Fig. 1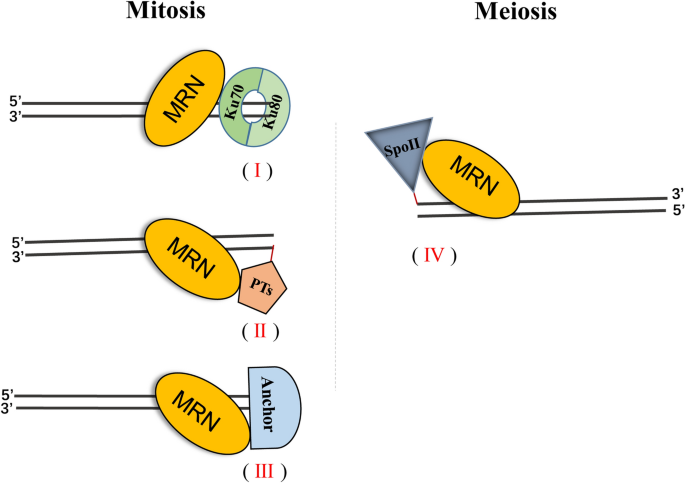
DSB end-binding or blocking factors facilitate MRN occupancy at DNA DSB ends. In mitotic cells, NHEJ factor Ku70/80 complex efficiently binds to DSBs and plays a key role as a scaffold to retain MRN at DNA break ends (I). The DSB ends conjugated with paused topoisomerases (PTs) are also appropriate substrates for MRN binding and retaining at DSB ends (II). Other unidentified DSB end-blocking factors (anchors) retain the MRN complex at the DSB end (III). During meiosis, Spo11generates DSBs in chromatin and conjugates with the 5′ end of DSBs, which promotes MRN binding to DSB ends (IV)
Full size imageProcessing DSB ends
The processing of DSB ends is a key step in HR, in which a region of few kilobases is resected from the 5′ strand of the DSB to form a 3′ ssDNA overhang. This 3′ ssDNA overhang is subsequently used to invade a homologous DNA strand. The end processing, also referred to as end resection, is executed by various factors under strict regulations (Daley et al. 2015). The process of end resection can be divided into three consecutive steps, namely initiation, processing, and termination. Next, we outline what is currently known about these three steps.
Initiation of DNA end resection
End resection is initiated when the MRE11 endonuclease nicks the 5′ strand of the DSB at a distance approximately 10 to 300 bases from the break end. Then, MRE11 digests the 5′ strand in the 3′ to 5′ direction (Garcia et al. 2011). MRE11 is part of the core of the MRN complex and interacts with itself, RAD50, NBS1, and the DNA substrate. In vitro biochemical studies have shown that MRE11 has three types of nuclease activity, namely ATP and Mn2+-dependent ssDNA endonuclease activity, dsDNA-dependent 3′–5′ exonuclease activity, and hairpin-opening activity (Paull and Gellert 1998; Haber 1998). The nuclease activities of MRE11 is critical for HR and cell survival. Genetic studies in both yeast and mammalian cells indicate that defects in the MRE11 nuclease activities result in severely aberrant HR phenotypes, such as recombination deficiency and hypersensitivity to ionizing radiation and chemotherapeutic drugs (Shibata et al. 2014a; Buis et al. 2008). The Mre11 nuclease-deficient homozygous allele in mice is embryonically lethal, similar to Mre11-null mice (Buis et al. 2008). However, for many years, it has been difficult to reconcile the polarity of the exonuclease activity of MRN with its known critical biological role in promoting the 5′ to 3′ resection of DSBs (Paull and Gellert 1998; Paull 2018). A major advance in unravelling this paradox was the observation that the processing of Spo11-induced breaks in Saccharomyces cerevisiae occurs through the endonucleolytic cleavage of the 5′ strand adjacent to the Spo11 covalent conjugate and subsequent 3′ to 5′ exonucleolytic degradation towards the conjugate (Paull 2018; Garcia et al. 2011) (Fig. 2). This endo-then-exo (bidirectional) end resection model explains the existence of the 3′ to 5′ exonuclease activity and suggests the existence of a protein block at the DSB end in HR (Garcia et al. 2011). Subsequent biochemical studies have confirmed the protein block-induced endonuclease activity of MRE11, and such protein block also displayed some inhibitory effect on the degradation of the 3′ strand by MRE11 (Cannavo and Cejka 2014; Anand et al. 2016, 2019; Cannavo et al. 2019). Furthermore, other studies have shown that this endonuclease activity is regulated by phosphorylated CtIP (or Sae2, the yeast Saccharomyces cerevisiae homolog of CtIP) and that CtIP itself is specifically modified by cell-cycle dependent kinase (CDK) in the S and G2 phases of the cell cycle (Huertas et al. 2008; Sartori et al. 2007; Cannavo and Cejka 2014; Anand et al. 2016). These findings not only demonstrate the existence of the protein-block-induced endonuclease activity of MRE11, but also suggest a mechanism for the coordination of HR with cell cycle progression, by regulating the initiation of DNA end resection or the endonuclease activity of MRE11 (G. Ira et al. 2004). Studies using endo- and exo-nuclease-specific inhibitors of MRE11 have also indicated that the MRE11 endonuclease activity determines the initiation of DNA end resection, thereby enabling MRE11 exonuclease activity and HR (Shibata et al. 2014b). During meiosis, Spo11, a member of the topoisomerase II family, is responsible for generating DSBs in chromatin and inducing meiotic recombination (Lam and Keeney 2014; Keeney 2001). However, unlike traditional type II topoisomerases, which break DNA by transiently forming a phosphodiester bond with the 5′ end and then re-ligating the ends immediately after topological changes, the Spo11 enzyme does not rejoin the two ends but instead covalently conjugates to the 5′ strand to form a protein-blockage, as shown in Fig. 1 (Keeney 2001). Although Spo11-blockage is sufficient to induce DNA end resection and HR in meiosis, it is only one way in which HR can be initiated, as the expression of Spo11 is limited to meiosis phase I and the Spo11-like blocking protein has not been found in mitotic cells (also see Box 1) (Keeney 2001; Paull 2018). HR commonly occurs in mitotic cells and is induced by certain cellular processes, such as endonuclease-mediated mating-type switches in yeast (Jasin and Rothstein 2013; Mehta and Haber 2014). Recent studies suggest that the NHEJ protein complex Ku70/80, as well as the DNA-dependent protein kinase catalytic subunit (DNA-PKcs), might play a role as a protein-block to induce the endonuclease activity of MRE11 (Langerak et al. 2011; Teixeira-Silva et al. 2017; Deshpande et al. 2020) (Fig. 2). This model appears to be excellent since Ku70/80 is highly abundant in cells and efficiently binds DSBs to promote NHEJ (Lieber 2010; Mimori and Hardin 1986). The endonuclease activity of MRE11 removes Ku70/80 from DSBs and facilitates the repair pathway to HR over NHEJ. However, there is some controversy surrounding this model, since genetic evidence from both yeast and mammalian cells indicates that Ku70/80-deficient cells exhibit defects in end-joining but HR is otherwise enhanced (Allen et al. 2002; Li et al. 2018; Langerak et al. 2011). Furthermore, the deletion of pku70 or pku80 (homologs of human Ku70 and Ku80, respectively) in Schizosaccharomyces pombe does not show any increased sensitivity to ionizing radiation or other DNA damaging agents like that exhibited by the HR factors (Manolis et al. 2001; Langerak et al. 2011). Therefore, further experimental evidence supporting this hypothesis is required to determine whether Ku70/80 or DNA-PK act as an end-blocking factor. Certainly, more studies are needed to more clearly understand how DNA end resection is initiated and how the MRN complex nuclease activity is regulated.
Fig. 2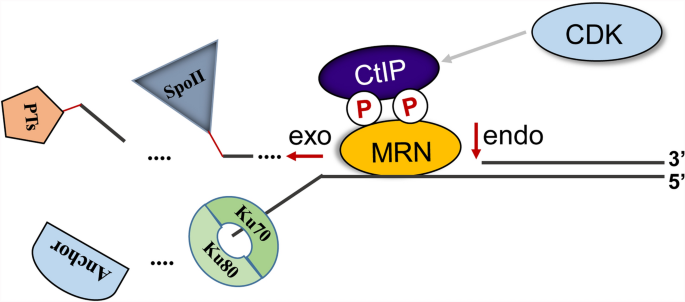
Regulation of MRE11 nuclease activity and DNA end resection. The nuclease activity of MRE11 is stimulated by CtIP and DSB end-blocking factors. CtIP is phosphorylated in the S and G2 cell phase by cell cycle-dependent Kinases (CDKs), which promotes CtIP to interact with MRE11 and stimulate the MRE11 5′-endonuclease activity. The presence of DSB end-blocking factors is also crucial for MRE11-catalyzed DNA end resection. After MRE11 endonuclease creates a nick on the 5′ strand near DSB ends, the 3′–5′ exonuclease activity of MRE11 begins to degrade the 5′ strand from the nick at 3′ to 5′ direction, leading to the removal of paused topoisomerases (PTs), 5′-conjugated Spo11 Ku70/80, or other anchors
Full size imageProcessing of DNA end resection
Following the initiation of DNA end resection, a few or more kilobases from the 5′ strand at the DSB end are further excised in the 5′–3′ direction. This step of DNA end resection generates a 3′ ssDNA overhang for subsequent strand invasion into a homologous DNA molecule. The length of the end resection depends on at least two nucleases, namely EXO1 and DNA2 (Zhu et al. 2008; Nimonkar et al. 2011) (Fig. 3). EXO1 is a 5′–3′ exonuclease that acts on dsDNA and is conserved in both prokaryotes and eukaryotes. The eukaryotic counterpart was first identified in S. pombe, where it plays a role in meiotic recombination (Szankasi and Smith 1992). Genetic analysis in yeast has revealed that exo1− cells are only slightly sensitive to DSB agents (e.g., camptothecin, bleomycin, and ionizing radiation). However, double-mutant cells of exo1− combined with sgs1− or dna2 temperature-sensitive mutations display severe synthetic sensitivity to such agents (Zhu et al. 2008; Zhang et al. 2016; Gravel et al. 2008). In vivo studies have shown that both short- and long-range end resection are affected in exo1 null cells (Zhu et al. 2008; Chen et al. 2013), suggesting that Exo1 may also play a role in the initiation of end resection. In mammalian cells, EXO1 is able to interact with CtIP and MRN, and such interaction may assist in the rapid recruitment of EXO1 to DNA DSB sites (Eid et al. 2010; Nimonkar et al. 2011). The level of EXO1 and its activity appears to be regulated by phosphorylation and ubiquitination in a cell cycle-dependent manner, which is correlated with the execution of the HR process (Xie et al. 2019; Tomimatsu et al. 2017, 2014). DNA2 nuclease is a multifunctional enzyme with 5′–3′ DNA helicase activity, DNA-dependent ATPase activity, 3′-exo/endonuclease activity, and 5′-exo/endonuclease activity (Masuda-Sasa et al. 2006b). It digests ssDNA or forked DNA structures with high efficiency but does not digest dsDNA (Hu et al. 2012; Kim et al. 2006). Dna2 plays an essential role in processing Okazaki fragments in yeast and thus is essential for cell growth (Liu et al. 2017). A temperature-sensitive mutant, dna2ts, is highly sensitive to DNA damaging agents and exhibits severe end-resection defects at non-permissive temperatures (Zhu et al. 2008; Zhang et al. 2016). A recent study revealed that in S. cerevisiae Dna2 inhibits the insertion of large and complex DNA fragments at chromosomal break sites (Yu et al. 2018). DNA2 homozygous knockout mice exhibit embryonic lethality before E7.5, whereas DNA2± mice are viable but exhibit shortened telomeres, chromosomal segregation errors, and increased tumor incidence associated with aneuploidy (Zheng et al. 2008). These results clearly indicate that both DNA2 and EXO1 are required for long-range end resection. Accordingly, considering that EXO1 alone may be sufficient given its 5′–3′ exonuclease activity (Zhu et al. 2008; Nimonkar et al. 2011), the question arises as to why cells need both DNA2 and EXO1 for end resection. A possible explanation is that some barriers can block EXO1, but DNA2 in complex with RECQ helicase can overcome those barriers, thus enabling end resection to continue (Masuda-Sasa et al. 2006b, 2006a). It has been demonstrated that DNA2 interacts with RecQ family helicases (Sgs1 in S. cerevisiae and five RECQ paralogs in humans: RECQL1, BLM, WRN, RECQL4, and RECQL5 (Bernstein et al. 2010)), DNA topoisomerase, and accessory protein RMI1 to form the end resection complex, in which each component is important for HR-mediated DSB repair (Gangloff et al. 1994; Mullen et al. 2005; Watt et al. 1995; Cejka et al. 2010; Gravel et al. 2008; Niu et al. 2010). In the fission yeast S. pombe, Dna2 is recruited to DNA break sites by the ssDNA binding complex RPA and the Cdc24 protein (Zhang et al. 2016). Cdc24 is essential for cell growth due to its role in DNA replication and processing of Okazaki fragments, but it is not a widely conserved protein, which makes it difficult to identify homologs in other species through database searches. Thus, it remains to be determined whether a homolog of Cdc24 exists in other species and acts to recruit Dna2 to DNA break sites. It has been suggested that DNA2 digests the 5′ strand of a DNA DSB through its interaction with RPA, which coats the relaxed 5′ ssDNA (Zhou et al. 2015; Stewart et al. 2008). Biochemical studies have also shown that the presence of RPA in the reaction promotes the DNA2-mediated 5′ strand digestion (Cejka et al. 2010; Niu et al. 2010). Due to the property of the DNA2 enzyme, which digest ssDNA, the generation of 5′ ssDNA should be a prerequisite for DNA2 digestion during DNA end resection. Following this logic, 3′ ssDNA strand should be simultaneously generated with the 5′ strand. This raises another question of why DNA2 digests the 5′ strand but not the 3′ strand, considering that DNA2 is able to digest both 5′ and 3′ ssDNA overhangs (Kim et al. 2006; Niu et al. 2010; Hu et al. 2012; Zhang et al. 2016). In vitro biochemical studies have provided some clues to the answer to this question. These studies showed that the binding of RPA on the 3′-strand reduced the degradation of this strand by DNA2 nuclease/helicase (Cejka et al. 2010; Niu et al. 2010; Nimonkar et al. 2011; Zhou et al. 2015). However, this finding has not been validated in vivo, as RPA depletion in eukaryotic cells renders them inviable. Conditional ablation of Rfa1 (the biggest subunit of RPA) in S. cerevisiae leads to defects in the long-range of DNA end resection, but the detailed mechanism underlying this effect has not been elucidated (Chen et al. 2013). Certainly, the RPA-mediated attenuation of 3′-ssDNA digestion by DNA2 does not meet the requirement of strict protection of the 3′-strand (see perspectives). Thus, the question of how the 3′ strand is protected remains an open question.
Fig. 3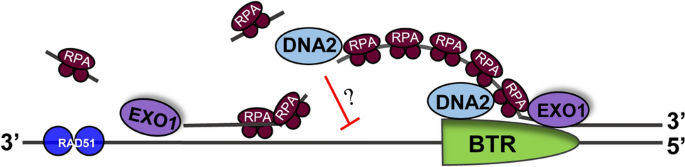
Multi-factors act in DNA end resection. A critical regulator for long-range end resection is the BTR (BLM-TOPIII-RMI1) complex, which is responsible for the unwinding of the broken DNA duplex along with the 3′-ended strand. The displaced 5′-ssDNA will quickly be coated by the single-stranded DNA binding protein complex RPA and subsequently degraded by DNA2 flap endonuclease. At the same time, the end resection-generated 3′-ssDNA is protected to avoid digestion by DNA2. The exonuclease EXO1 degrades the 5′-strand of duplexed DNA ahead of BTR or 5′ strand that re-anneals with the 3′-ssDNA strand. The 3′-ssDNA strand is eventually coated by RAD51 for the next step–strand invasion
Full size imageTermination of DNA end resection
Studies of the mechanism underlying the termination of the DNA end resection process very much lag behind those of the other HR processes. One of the main reasons for this is that there are no definite criteria for the extent of DNA end resection that is most suitable for subsequent HR processes. In most cases, the extent of DNA end resection is in the range of several hundred to a few thousand nucleotides (Zhou et al. 2014; Zierhut and Diffley 2008), although a break may sometimes be resected up to millions of nucleotides if a homologous template is absent (Zhu et al. 2008). In normally replicating cells or meiotic cells with sister chromatids available, the length of resection is even less (Mimitou et al. 2017; Symington 2016). Considering that both insufficient and excessive DNA end resection can affect the efficiency of HR or harm cells, it is highly possible that a cellular mechanism has evolved to control DNA end resection to a certain extent. Theoretically, such a regulatory mechanism could target both EXO1 and DNA2-RecQ, which are responsible for long-range DNA end resection (Mimitou and Symington 2008; Nimonkar et al. 2011; Zhu et al. 2008), by inhibiting their enzymatic activity. It has been reported that the nuclease activity of EXO1 is inhibited by post-translational modifications to ensure genomic stability (Keijzers et al. 2018; Tomimatsu et al. 2014; Xie et al. 2019). Another recent study indicated that EXO1 is phosphorylated and subsequently degraded via the ubiquitin–proteasome system immediately after DNA end resection, to limit the excessive resection of DNA ends and enable accurate DNA repair (Tomimatsu et al. 2017). However, compared with what is known about EXO1, much less is known about the regulation of the DNA2-RecQ activity during DNA end resection. Genetic and biochemical studies have revealed that the DNA2 end resection activity depends on the RecQ helicase complex (Zhu et al. 2008; Cejka et al. 2010; Gravel et al. 2008), suggesting that inhibition of this helicase may be an effective way to block helicase-mediated DNA unwinding and DNA2-catalyzed 5′-strand degradation. Additionally, in vitro biochemical analysis has shown that BLM, a human RecQ homolog, can interact with EXO1, thereby stimulating its nucleolytic activity (Nimonkar et al. 2008), further suggesting that RecQ helicase is a prominent target for regulating DNA end resection termination.
The RecQ helicase family is highly conserved, from bacteria to human cells (Bernstein et al. 2010; Croteau et al. 2014). In humans, mutations in BLM, WRN, and RTS/RECQ4 are associated with Bloom, Werner, and Rothmund–Thomson syndromes, respectively, while RECQL1 and RECQL5 are also critical for maintaining genomic stability (Bernstein et al. 2010; Hu et al. 2007). A prominent phenotype of Bloom syndrome involves markedly increased sister chromatid exchanges (SCEs) and hyper-crossover recombination products (Chaganti et al. 1974). This phenotype is also observed in S. cerevisiae Sgs1 deletion mutant (Watt et al. 1996,1995; Ira et al. 2003). In vitro studies suggest that BLM occurs in a complex with topoisomerase IIIɑ (hTOPIIIɑ) and BLAP75/RMI1 (BTR) to catalyze the dissolution of double Holliday junctions (dHJs) into non-crossover products (Leonard and Hickson 2003; Adams et al. 2003; Wu et al. 2006), which partially explains why a mutation in BLM leads to a hyper-crossover phenotype. However, BLM and Sgs1 are also critical for DNA DSB end resection in both human and yeast cells (Gravel et al. 2008; Mimitou and Symington 2008; Zhu et al. 2008), which suggests that the hyper-crossover phenotype in blm mutant cells may not be due to defects in the dissolution of Holliday junctions that are generated during HR. The reason for this is that, in blm mutants, DSBs rarely proceed to HR events due to a lack of DNA end-resection activity. Genetic studies in S. cerevisiae have revealed that DNA damage-induced SCEs are very low in sgs1-deleted cells (Gangloff et al. 2000), providing strong support for this hypothesis. Accordingly, the hyper-crossover between sister chromatids very likely results from abnormal processing of stalled DNA replication forks in blm mutant cells (Kaliraman et al. 2001), as the stalled DNA replication forks often form “chicken feet”-like structures that resemble Holliday junctions (Hu et al. 2012; Sogo et al. 2002). It is difficult to directly analyze how DSB-induced recombination intermediates are abnormally processed in RecQ mutants as they simultaneously affect the early step of HR-end resection and the late step of Holliday junction dissolution. However, some studies have been reported that mutations in proteins of the HR pathway (e.g., Rad51, Rad52, and Rad57) rescue the lethal effect of sgs1 deletion by reducing the accumulation of deleterious DNA structures (Gangloff et al. 2000; Oakley et al. 2002). Besides, BLM (Sgs1)-TOPIIIɑ-RMI1 (B/STR) has high affinities for forked DNA, HJ, and D-loop DNA structures (Bennett et al. 1998; Wu and Hickson 2003; Bachrati et al. 2006; Bussen et al. 2007). Therefore, B/STR is thought to inhibit the formation of HJs by disrupting the Rad51-ssDNA nucleofilament (Adams et al. 2003; Ira et al. 2003; Krejci et al. 2003; Wu and Hickson 2003; Wu et al. 2006).
Based on the findings of the above studies, we put forward a hypothesis related to the B/STR complex to explain how DNA end resection is terminated, as follows. At the beginning of HR, RECQ is recruited to DNA break sites and promotes extensive 5′-strand degradation. At the same time, RAD51 coordinately assembles on the exposed 3′ strand to form a nucleoprotein filament. Once the RAD51-coated 3′ filament is long enough to find a nearby homologous template, it invades the duplexed DNA template and immediately performs DNA synthesis, using the invaded strand as a primer. This process ultimately generates an HJ between the two DNA molecules. Based on the stronger affinity of B/STR for HJs and the extensive annealing of the invading strand to the template, the B/STR complex is unloaded from the 3′ strand of broken DNA and shifts onto the HJ structure, which results in the termination of DNA end resection (Fig. 4). The RECQ helicase, in turn, begins to unwind the invaded strand, which eventually results in a synthesis-dependent strand annealing (SDSA) product unless the invading strand ligates with the other end to form dHJs (Adams et al. 2003). In the other case, the B/STR helicase, together with nucleases MUS81-EME1, GEN1, and SLX1-SLX4, resolve the dHJs to complete the HR process (Fig. 4) (Kaliraman et al. 2001; Boddy et al. 2001; Ip et al. 2008).
Fig. 4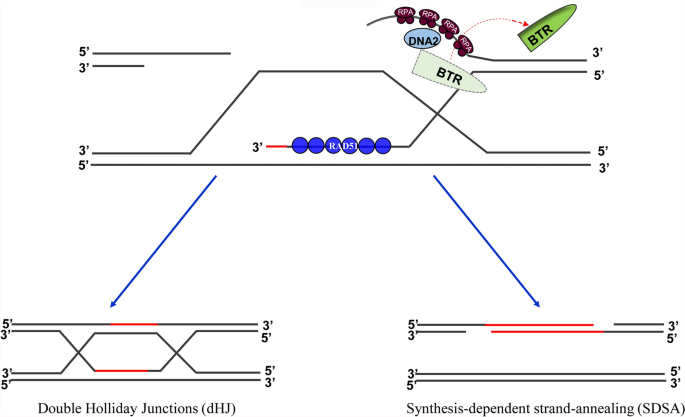
A putative model for the termination of DNA end resection. RAD51-nucleoprotein filament invading into a homologous dsDNA template will result in topological alteration at the BTR-bound junction. This may cause the BTR complex to fall off the DNA, and the DNA2-mediated nucleolytic degradation is terminated. After the strand invasion, DNA synthesis takes place immediately at the 3′-end of the invading strand. Subsequently, either synthesis-dependent strand-annealing (SDSA) or double Holliday junctions (dHJ) pathway occurs to repair DNA breaks
Full size imagePerspectives
First, DNA end resection is the primary step in HR and HR-mediated repair of DSBs. This step is also the critical point in cellular regulation to determine whether the NHEJ or HR pathway is used to repair DSBs. During the G1 phase, due to the lack of a sister chromatid, NHEJ is the only available choice for repairing a DNA break, and end resection or the HR pathway must be inhibited to avoid dire consequences. Since DNA end resection is a continuous process, cells can control the occurrence of end resection and HR by regulating the initiation step of end resection (Shibata et al. 2014b; Cannavo and Cejka 2014). Second, HR in eukaryotic cells occurs in chromatin, thus, chromatin structures must be loosened to enable the resection of the 5′ strand of DSB ends (Price and D'Andrea 2013). When a DSB occurs, histone modifications are changed in the chromatin region around the DSB (Clouaire et al. 2018). Chromatin remodeling factors, DNA damage response factors, and histone modification factors are recruited to DSB site to alter the chromatin structures to facilitate DSB repair (Price and D'Andrea 2013; Ciccia and Elledge 2010). During the S and G2 phases of the cell cycle, there are a number of chromatin-relaxing histone modifications that provide favorable conditions for DNA end resection. However, whether these relaxing histone modifications further contribute to recruit downstream HR-processing factors remains to be elucidated (Price and D'Andrea 2013). On the other hand, some DSB-induced histone modifications contribute to recruit the 53BP1-RIF1-Shieldin complex to DSBs (mostly in G1-phase cells), which spreads over megabases surrounding the DSB sites, to block access to resection nucleases and inhibit HR (Mailand et al. 2007; Doil et al. 2009; Mirman et al. 2018; Noordermeer et al. 2018; Bunting et al. 2010; Zimmermann et al. 2013). Third, a DNA broken end must be rapidly and adequately protected to avoid further damage to the end, such as unregulated DNA digestion; or prevent other types of DNA transactions, such as chromosomal translocations, insertions and deletions.
HR is a biological process relatively conserved from prokaryotes to eukaryotes. In eukaryotes, the process and principles of HR were first studied in yeast and subsequently extended to human cells. As a critical step in HR, DNA end resection is highly conserved from yeast to human cells. Most of the DNA end resection proteins in yeast cells can be found in human cells (Table 1). Besides the similarities in the basic principle of end resection, some new proteins have arisen in mammalian cells that are not present in yeast, such as the breast cancer type 1 susceptibility protein (BRCA1). BRCA1 is thought to be involved in DNA end resection, but its biochemical function is still unclear. Defects in BRCA1 are associated with high genetic susceptibility to breast and ovarian cancers in humans. Defects in other DNA end resection proteins in human also lead to severe diseases. For example, MRE11 mutations cause Ataxia-telangiectasia-like syndrome, mutations in RECQ-like helicases cause Bloom syndrome, Werner syndrome, Rothmund-Thomson syndrome, etc. Currently, research is focused on finding new proteins involved in DNA end resection in mammalian cells, and for many of the classical end resection proteins, their precise biological functions are still being extensively investigated. An in-depth biochemical analysis of all the DNA end resection proteins not only will improve our understanding of the HR process but will also help us gain a better insight into the mechanism by which defects in DNA end resection cause diseases, thus providing theoretical guidance for developing new therapy for these diseases.
Table 1 End resection factors in yeast and human cells
| Yeast (S. cerevisiae/S. pombe) | Human |
|---|---|
| Mre11 | MRE11 |
| Rad50 | RAD50 |
| Xrs2/Nbs1 | NBS1 |
| Sae2/Ctp1 | CtIP |
| Sgs1/Rqh1 | BLM (or RECQL1, WRN, RECQL4, and RECQL5) |
| Dna2 | DNA2 |
| Exo1 | EXO1 |
| – | BRCA1 |
Full size table
Although considerable progress has been made in understanding the general mechanism of HR and in particular the step of DNA end resection, some key questions remain to be answered in the near future. For instance, if a DNA break occurs in a cell during the G2 phase, does the cell prefer to use the HR pathway to repair the break? This question is raised because using the NHEJ pathway to repair a dirty break end will almost certainly result in a mutation in the genome, such as a deletion or insertion, but if the HR pathway is used, the repair of the break will most likely restore the original genome. For the HR pathway, the chromatin region surrounding a break must be relaxed through cellular regulation of end resection. Also, is chromatin also regulated for the NHEJ repair pathway? BRCA1 has been found to play a key role in promoting break repairs in the HR pathway, but its precise role in this process has not been biochemically determined. During DNA end resection, the 3′ strand must be carefully protected to avoid digestion by Dna2. It has been suggested that RPA binding may provide such protection. However, we believe that RPA may not play this role, for two reasons. First, it is unknown how the binding of RPA to the 3′ strand prior to DNA2 or other nuclease digestion can be ensured. Second, it is also unknown how the binding of RPA to every region on the 3′ strand can be ensured. The attenuated digestion of the 3′ strand in the presence of RPA does not signify strict protection of this strand. BRCA2 and RAD52 are essential for HR, but there are some controversies regarding the studies in yeast and mammals, suggesting that the functions of BRCA2 and RAD52 require further analysis. Finally, as stated above, the mechanism to terminate DNA end resection is poorly understood and requires further research. It is also highly likely that there exist as-yet-unidentified factors that play a role in the HR process. It is anticipated that future studies will uncover these factors.
Box 1. End resection in mitotic and meiotic DNA HR
The overall mechanisms underlying mitotic and meiotic recombination are very similar, particularly in DNA end resection processing. HR factors like MRE11, RAD50, NBS1 (Xrs2), CtIP (Sae2), EXO1, RPA, RAD51, and RECQ helicases are all required for meiotic recombination (Keeney 2001; Lam and Keeney 2014). However, the molecular function of DNA2 helicase/nuclease in meiotic recombination has been less studied, and its role in meiotic recombination remains to be elucidated (Manfrini et al. 2010). A prominent feature that distinguishes meiotic recombination is the programmed generation of DNA double-strand breaks by SPO11 and accessory factors, such as MEI1, MEI4, REC114, and HORMAD1 at the first meiosis phase (meiosis I) (Lam and Keeney 2014), while mitotic recombination resolves the accidental chromatin breaks and DNA replication problems.
References
Adams, M. D., McVey, M., & Sekelsky, J. J. (2003). Drosophila BLM in double-strand break repair by synthesis-dependent strand annealing. Science, 299(5604), 265–267.
Allen, C., Kurimasa, A., Brenneman, M. A., Chen, D. J., & Nickoloff, J. A. (2002). DNA-dependent protein kinase suppresses double-strand break-induced and spontaneous homologous recombination. Proceedings of the National Academy of Sciences of the United States of America, 99(6), 3758–3763.
Anand, R., Jasrotia, A., Bundschuh, D., Howard, S. M., Ranjha, L., Stucki, M., et al. (2019). NBS1 promotes the endonuclease activity of the MRE11-RAD50 complex by sensing CtIP phosphorylation. EMBO Journal. https://doi.org/10.15252/embj.2018101005.
Anand, R., Ranjha, L., Cannavo, E., & Cejka, P. (2016). Phosphorylated CtIP functions as a co-factor of the MRE11-RAD50-NBS1 endonuclease in DNA end resection. Molecular Cell, 64(5), 940–950. https://doi.org/10.1016/j.molcel.2016.10.017.
Bachrati, C. Z., Borts, R. H., & Hickson, I. D. (2006). Mobile D-loops are a preferred substrate for the Bloom’s syndrome helicase. Nucleic Acids Research, 34(8), 2269–2279. https://doi.org/10.1093/nar/gkl258.
Bender, C. F., Sikes, M. L., Sullivan, R., Huye, L. E., & Petrini, J. H. J. (2002). Cancer predisposition and hematopoietic failure in RAD50(S/S) mice. Genes & Development, 16(17), 2237–2251.
Bennett, R. J., Sharp, J. A., & Wang, J. C. (1998). Purification and characterization of the Sgs1 DNA helicase activity of Saccharomyces cerevisiae. Journal of Biological Chemistry, 273(16), 9644–9650.
Bernstein, K. A., Gangloff, S., & Rothstein, R. (2010). The RecQ DNA helicases in DNA repair. Annual Review of Genetics, 44, 393–417. https://doi.org/10.1146/annurev-genet-102209-163602.
Boddy, M. N., Gaillard, P. H., Mcdonald, W. H., Shanahan, P., Yates, J. R., & Russell, P. (2001). Mus81-Eme1 are essential components of a Holliday junction resolvase. Cell, 107(4), 537–548.
Buis, J., Wu, Y., Deng, Y., Leddon, J., Westfield, G., Eckersdorff, M., et al. (2008). Mre11 nuclease activity has essential roles in DNA repair and genomic stability distinct from ATM activation. Cell, 135(1), 85–96. https://doi.org/10.1016/j.cell.2008.08.015.
Bunting, S. F., Callen, E., Wong, N., Chen, H. T., Polato, F., Gunn, A., et al. (2010). 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell, 141(2), 243–254. https://doi.org/10.1016/j.cell.2010.03.012.
Bussen, W., Raynard, S., Busygina, V., Singh, A. K., & Sung, P. (2007). Holliday junction processing activity of the BLM-Topo IIIalpha-BLAP75 complex. Journal of Biological Chemistry, 282(43), 31484–31492. https://doi.org/10.1074/jbc.M706116200.
Cannavo, E., & Cejka, P. (2014). Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature, 514(7520), 122–125. https://doi.org/10.1038/nature13771.
Cannavo, E., Reginato, G., & Cejka, P. (2019). Stepwise 5’ DNA end-specific resection of DNA breaks by the Mre11-Rad50-Xrs2 and Sae2 nuclease ensemble. Proceedings of the National Academy of Sciences of the United States of America, 116(12), 5505–5513. https://doi.org/10.1073/pnas.1820157116.
Carney, J. P., Maser, R. S., Olivares, H., Davis, E. M., & Petrini, J. H. J. (1998). The hMre11/hRad50 protein complex and nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell, 93(3), 477–486.
Cejka, P., Cannavo, E., Polaczek, P., Masuda-Sasa, T., Pokharel, S., Campbell, J. L., et al. (2010). DNA end resection by Dna2-Sgs1-RPA and its stimulation by Top3-Rmi1 and Mre11-Rad50-Xrs2. Nature, 467(7311), 112–116. https://doi.org/10.1038/nature09355.
Chaganti, R., Schonberg, S., & German, J. (1974). A manyfold increase in sister chromatid exchanges in Bloom’s syndrome lymphocytes. Proceedings of the National Academy of Sciences, 71(11), 4508–4512.
Chen, H., Lisby, M., & Symington, L. S. (2013). RPA coordinates DNA end resection and prevents formation of DNA hairpins. Molecular Cell, 50(4), 589–600. https://doi.org/10.1016/j.molcel.2013.04.032.
Ciccia, A., & Elledge, S. J. (2010). The DNA damage response: making it safe to play with knives. Molecular Cell, 40(2), 179–204. https://doi.org/10.1016/j.molcel.2010.09.019.
Clouaire, T., Rocher, V., Lashgari, A., Arnould, C., Aguirrebengoa, M., Biernacka, A., et al. (2018). Comprehensive mapping of histone modifications at DNA double-strand breaks deciphers repair pathway chromatin signatures. Molecular Cell, 72(2), 250–262. https://doi.org/10.1016/j.molcel.2018.08.020.
Croteau, D. L., Popuri, V., Opresko, P. L., & Bohr, V. A. (2014). Human RecQ helicases in DNA repair, recombination, and replication. Annual Review of Biochemistry, 83, 519–552. https://doi.org/10.1146/annurev-biochem-060713-035428.
D’Amours, D., & Jackson, S. P. (2002). The Mre11 complex: at the crossroads of dna repair and checkpoint signalling. Nature Reviews Molecular Cell Biology, 3(5), 317–327. https://doi.org/10.1038/nrm805.
Daley, J. M., Niu, H., Miller, A. S., & Sung, P. (2015). Biochemical mechanism of DSB end resection and its regulation. DNA Repair (Amst), 32, 66–74. https://doi.org/10.1016/j.dnarep.2015.04.015.
Deshpande, R. A., Myler, L. R., Soniat, M. M., Makharashvili, N., Lee, L., Lees-Miller, S. P., et al. (2020). DNA-dependent protein kinase promotes DNA end processing by MRN and CtIP. Science Advances, 6(2), 922.
Doil, C., Mailand, N., Bekker-Jensen, S., Menard, P., Larsen, D. H., Pepperkok, R., et al. (2009). RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell, 136(3), 435–446. https://doi.org/10.1016/j.cell.2008.12.041.
Dolganov, G. M., Maser, R. S., Novikov, A., Tosto, L., Chong, S., Bressan, D. A., et al. (1996). Human Rad50 is physically associated with human Mre11: Identification of a conserved multiprotein complex implicated in recombinational DNA repair. Molecular and Cellular Biology, 16(9), 4832–4841.
Eid, W., Steger, M., El-Shemerly, M., Ferretti, L. P., Pena-Diaz, J., Konig, C., et al. (2010). DNA end resection by CtIP and exonuclease 1 prevents genomic instability. EMBO Reports, 11(12), 962–968. https://doi.org/10.1038/embor.2010.157.
Featherstone, C., & Jackson, S. P. (1999). DNA double-strand break repair. Current Biology, 9(20), R759–R761. https://doi.org/10.1016/s0960-9822(00)80005-6.
Filippo, J. S., Sung, P., & Klein, H. (2008). Mechanism of eukaryotic homologous recombination. Annual Review of Biochemistry, 77, 229–257. https://doi.org/10.1146/annurev.biochem.77.061306.125255.
Gangloff, S., McDonald, J. P., Bendixen, C., Arthur, L., & Rothstein, R. (1994). The yeast type I topoisomerase Top3 interacts with Sgs1, a DNA helicase homolog: A potential eukaryotic reverse gyrase. Molecular and Cellular Biology., 2, 2.
Gangloff, S., Soustelle, C., & Fabre, F. (2000). Homologous recombination is responsible for cell death in the absence of the Sgs1 and Srs2 helicases. Nature Genetics, 25(2), 192–194.
Garcia, V., Phelps, S. E., Gray, S., & Neale, M. J. (2011). Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature, 479(7372), 241–244. https://doi.org/10.1038/nature10515.
Gravel, S., Chapman, J. R., Magill, C., & Jackson, S. P. (2008). DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes & Development, 22(20), 2767–2772. https://doi.org/10.1101/gad.503108.
Haber, J. E. (1998). The many interfaces of Mre11. Cell, 95(5), 583–586.
Hopfner, K. P., Karcher, A., Craig, L., Woo, T. T., Carney, J. P., & Tainer, J. A. (2001). Structural biochemistry and interaction architecture of the DNA double-strand break repair Mre11 nuclease and RAD50-ATPase. Cell, 105(4), 473–485.
Hu, J., Sun, L., Shen, F., Chen, Y., Hua, Y., Liu, Y., et al. (2012). The intra-S phase checkpoint targets Dna2 to prevent stalled replication forks from reversing. Cell, 149(6), 1221–1232. https://doi.org/10.1016/j.cell.2012.04.030.
Hu, Y., Raynard, S., Sehorn, M. G., Lu, X., Bussen, W., Zheng, L., et al. (2007). RECQL5/Recql5 helicase regulates homologous recombination and suppresses tumor formation via disruption of Rad51 presynaptic filaments. Genes & Development, 21(23), 3073–3084. https://doi.org/10.1101/gad.1609107.
Huertas, P., Cortes-Ledesma, F., Sartori, A. A., Aguilera, A., & Jackson, S. P. (2008). CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature, 455(7213), 689–692. https://doi.org/10.1038/nature07215.
Hunter, N. (2015). Meiotic recombination: The essence of heredity. Cold Spring Harbor Perspectives in Biology. https://doi.org/10.1101/cshperspect.a016618.
Ip, S. C., Rass, U., Blanco, M. G., Flynn, H. R., Skehel, J. M., & West, S. C. (2008). Identification of Holliday junction resolvases from humans and yeast. Nature, 456(7220), 357–361. https://doi.org/10.1038/nature07470.
Ira, G., Malkova, A., Liberi, G., Foiani, M., & Haber, J. E. (2003). Srs2 and Sgs1–Top3 suppress crossovers during double-strand break repair in yeast. Cell, 115(4), 401–411.
Ira, G., Pellicioli, A., Balijja, A., Wang, X., Fiorani, S., Carotenuto, W., et al. (2004). DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature, 431(7011), 1011–1017. https://doi.org/10.1038/nature02964.
Jackson, S. P., & Bartek, J. (2009). The DNA-damage response in human biology and disease. Nature, 461(7267), 1071–1078. https://doi.org/10.1038/nature08467.
Jasin, M., & Rothstein, R. (2013). Repair of strand breaks by homologous recombination. Cold Spring Harbor Perspectives in Biology, 5(11), a012740. https://doi.org/10.1101/cshperspect.a012740.
Kadyk, L. C., & Hartwell, L. H. (1992). Sister chromatids are preferred over homologs as substrates for recombinational repair in saccharomyces-cerevisiae. Genetics, 132(2), 387–402.
Kaliraman, V., Mullen, J. R., Fricke, W. M., Bastin-Shanower, S. A., & Brill, S. J. (2001). Functional overlap between Sgs1–Top3 and the Mms4–Mus81 endonuclease. Genes & Development, 15(20), 2730–2740.
Keeney, S. (2001). Mechanism and control of meiotic recombination initiation. Current Topics in Developmental Biology, 52, 1–53. https://doi.org/10.1016/s0070-2153(01)52008-6.
Keijzers, G., Bakula, D., Petr, M. A., Madsen, N. G. K., Teklu, A., Mkrtchyan, G., et al. (2018). Human exonuclease 1 (EXO1) regulatory functions in DNA replication with putative roles in cancer. International Journal of Molecular Sciences. https://doi.org/10.3390/ijms20010074.
Kim, J. H., Kim, H. D., Ryu, G. H., Kim, D. H., Hurwitz, J., & Seo, Y. S. (2006). Isolation of human Dna2 endonuclease and characterization of its enzymatic properties. Nucleic Acids Research, 34(6), 1854–1864. https://doi.org/10.1093/nar/gkl102.
Krejci, L., Van Komen, S., Li, Y., Villemain, J., Reddy, M. S., Klein, H., et al. (2003). DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature, 423(6937), 305–309. https://doi.org/10.1038/nature01577.
Lam, I., & Keeney, S. (2014). Mechanism and regulation of meiotic recombination initiation. Cold Spring Harbor Perspectives in Biology, 7(1), a016634. https://doi.org/10.1101/cshperspect.a016634.
Langerak, P., Mejia-Ramirez, E., Limbo, O., & Russell, P. (2011). Release of Ku and MRN from DNA ends by Mre11 nuclease activity and Ctp1 is required for homologous recombination repair of double-strand breaks. PLoS Genetics, 7(9), e1002271. https://doi.org/10.1371/journal.pgen.1002271.
Leonard, W., & Hickson, I. D. (2003). The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature, 426(6968), 870–874.
Li, G., Liu, D., Zhang, X., Quan, R., Zhong, C., Mo, J., et al. (2018). Suppressing Ku70/Ku80 expression elevates homology-directed repair efficiency in primary fibroblasts. International Journal of Biochemistry & Cell Biology, 99, 154–160. https://doi.org/10.1016/j.biocel.2018.04.011.
Lieber, M. R. (2010). The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annual Review of Biochemistry, 79, 181–211. https://doi.org/10.1146/annurev.biochem.052308.093131.
Lisby, M., Barlow, J. H., Burgess, R. C., & Rothstein, R. (2004). Choreography of the DNA damage response: Spatiotemporal relationships among checkpoint and repair proteins. Cell, 118(6), 699–713. https://doi.org/10.1016/j.cell.2004.08.015.
Liu, B., Hu, J., Wang, J., & Kong, D. (2017). Direct Visualization of RNA-DNA primer removal from okazaki fragments provides support for flap cleavage and exonucleolytic pathways in eukaryotic cells. Journal of Biological Chemistry, 292(12), 4777–4788. https://doi.org/10.1074/jbc.M116.758599.
Lord, C. J., & Ashworth, A. (2016). BRCAness revisited. Nature Reviews Cancer, 16(2), 110–120. https://doi.org/10.1038/nrc.2015.21.
Mailand, N., Bekker-Jensen, S., Faustrup, H., Melander, F., Bartek, J., Lukas, C., et al. (2007). RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell, 131(5), 887–900. https://doi.org/10.1016/j.cell.2007.09.040.
Manfrini, N., Guerini, I., Citterio, A., Lucchini, G., & Longhese, M. P. (2010). Processing of meiotic DNA double strand breaks requires cyclin-dependent kinase and multiple nucleases. Journal of Biological Chemistry, 285(15), 11628–11637. https://doi.org/10.1074/jbc.M110.104083.
Manolis, K. G., Nimmo, E. R., Hartsuiker, E., Carr, A. M., Jeggo, P. A., & Allshire, R. C. (2001). Novel functional requirements for non-homologous DNA end joining in Schizosaccharomyces pombe. The EMBO journal, 20(1–2), 210–221.
Masuda-Sasa, T., Imamura, O., & Campbell, J. L. (2006). Biochemical analysis of human Dna2. Nucleic Acids Research, 34(6), 1865–1875. https://doi.org/10.1093/nar/gkl070.
Masuda-Sasa, T., Polaczek, P., & Campbell, J. L. (2006). Single strand annealing and ATP-independent strand exchange activities of yeast and human DNA2: Possible role in Okazaki fragment maturation. Journal of Biological Chemistry, 281(50), 38555–38564. https://doi.org/10.1074/jbc.M604925200.
Mehta, A., & Haber, J. E. (2014). Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harbor Perspectives in Biology, 6(9), a016428. https://doi.org/10.1101/cshperspect.a016428.
Mimitou, E. P., & Symington, L. S. (2008). Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature, 455(7214), 770–774. https://doi.org/10.1038/nature07312.
Mimitou, E. P., Yamada, S., & Keeney, S. (2017). A global view of meiotic double-strand break end resection. Science, 355(6320), 40–45. https://doi.org/10.1126/science.aak9704.
Mimori, T., & Hardin, J. A. (1986). Mechanism of interaction between Ku protein and DNA. Journal of Biological Chemistry, 261(22), 10375–10379.
Mirman, Z., Lottersberger, F., Takai, H., Kibe, T., Gong, Y., Takai, K., et al. (2018). 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polalpha-dependent fill-in. Nature, 560(7716), 112–116. https://doi.org/10.1038/s41586-018-0324-7.
Mullen, J. R., Nallaseth, F. S., Lan, Y. Q., Slagle, C. E., & Brill, S. J. (2005). Yeast Rmi1/Nce4 controls genome stability as a subunit of the Sgs1-Top3 complex. Molecular and Cellular Biology, 25(11), 4476–4487. https://doi.org/10.1128/MCB.25.11.4476-4487.2005.
Myler, L. R., Gallardo, I. F., Soniat, M. M., Deshpande, R. A., Gonzalez, X. B., Kim, Y., et al. (2017). Single-molecule imaging reveals how Mre11-Rad50-Nbs1 initiates DNA break repair. Molecular Cell, 67(5), 891–898. https://doi.org/10.1016/j.molcel.2017.08.002.
Nimonkar, A. V., Genschel, J., Kinoshita, E., Polaczek, P., Campbell, J. L., Wyman, C., et al. (2011). BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes & Development, 25(4), 350–362. https://doi.org/10.1101/gad.2003811.
Nimonkar, A. V., Oezsoy, A. Z., Genschel, J., Modrich, P., & Kowalczykowski, S. C. (2008). Human exonuclease 1 and Blm helicase interact to resect DNA and initiate DNA repair. Proceedings of the National Academy of Sciences of the United States of America, 105(44), 16906–16911.
Niu, H., Chung, W. H., Zhu, Z., Kwon, Y., Zhao, W., Chi, P., et al. (2010). Mechanism of the ATP-dependent DNA end-resection machinery from Saccharomyces cerevisiae. Nature, 467(7311), 108–111. https://doi.org/10.1038/nature09318.
Noordermeer, S. M., Adam, S., Setiaputra, D., Barazas, M., Pettitt, S. J., Ling, A. K., et al. (2018). The shieldin complex mediates 53BP1-dependent DNA repair. Nature, 560(7716), 117–121. https://doi.org/10.1038/s41586-018-0340-7.
Oakley, T. J., Goodwin, A., Chakraverty, R. K., & Hickson, I. D. (2002). Inactivation of homologous recombination suppresses defects in topoisomerase III-deficient mutants. DNA Repair, 1(6), 463–482.
Park, Y. B., Chae, J., Kim, Y. C., & Cho, Y. (2011). Crystal structure of human Mre11: Understanding tumorigenic mutations. Structure, 19(11), 1591–1602. https://doi.org/10.1016/j.str.2011.09.010.
Paull, T. T. (2018). 20 years of Mre11 biology: No end in sight. Molecular Cell, 71(3), 419–427. https://doi.org/10.1016/j.molcel.2018.06.033.
Paull, T. T., & Gellert, M. (1998). The 3′ to 5′ exonuclease activity of Mre11 facilitates repair of DNA double-strand breaks. Molecular Cell, 1(7), 969–979.
Paull, T. T., & Gellert, M. (1999). NBS1 potentiates ATP-driven DNA unwinding and endonuclease cleavage by the MRE11/RAD50 complex. Genes & Development, 13(10), 1276–1288.
Petrini, J. H. (2000). The Mre11 complex and ATM: Collaborating to navigate S phase. Current Opinion in Cell Biology, 12(3), 293–296.
Price, B. D., & D’Andrea, A. D. (2013). Chromatin remodeling at DNA double-strand breaks. Cell, 152(6), 1344–1354. https://doi.org/10.1016/j.cell.2013.02.011.
Sancar, A., Lindsey-Boltz, L. A., Unsal-Kacmaz, K., & Linn, S. (2004). Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annual Review of Biochemistry, 73, 39–85. https://doi.org/10.1146/annurev.biochem.73.011303.073723.
Sartori, A. A., Lukas, C., Coates, J., Mistrik, M., Fu, S., Bartek, J., et al. (2007). Human CtIP promotes DNA end resection. Nature, 450(7169), 509-U506. https://doi.org/10.1038/nature06337.
Shibata, A., Moiani, D., Arvai, A. S., Perry, J., Harding, S. M., Genois, M. M., et al. (2014a). DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Molecular Cell, 53(1), 7–18. https://doi.org/10.1016/j.molcel.2013.11.003.
Shibata, A., Moiani, D., Arvai, A. S., Perry, J., Harding, S. M., Genois, M. M., et al. (2014b). DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities (vol 53, pg 7, 2014). Molecular Cell, 53(2), 361–361. https://doi.org/10.1016/j.molcel.2014.01.008.
Sogo, J. M., Lopes, M., & Foiani, M. (2002). Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science, 297(5581), 599–602. https://doi.org/10.1126/science.1074023.
Stewart, J. A., Miller, A. S., Campbell, J. L., & Bambara, R. A. (2008). Dynamic removal of replication protein A by Dna2 facilitates primer cleavage during Okazaki fragment processing in Saccharomyces cerevisiae. Journal of Biological Chemistry, 283(46), 31356–31365. https://doi.org/10.1074/jbc.M805965200.
Stracker, T. H., & Petrini, J. H. (2011). The MRE11 complex: Starting from the ends. Nature Reviews Molecular Cell Biology, 12(2), 90–103. https://doi.org/10.1038/nrm3047.
Sung, S., Li, F., Park, Y. B., Kim, J. S., Kim, A. K., Song, O. K., et al. (2014). DNA end recognition by the Mre11 nuclease dimer: Insights into resection and repair of damaged DNA. EMBO Journal, 33(20), 2422–2435. https://doi.org/10.15252/embj.201488299.
Symington, L. S. (2016). Mechanism and regulation of DNA end resection in eukaryotes. Critical Reviews in Biochemistry and Molecular Biology, 51(3), 195–212. https://doi.org/10.3109/10409238.2016.1172552.
Szankasi, P., & Smith, G. R. (1992). A DNA exonuclease induced during meiosis of Schizosaccharomyces pombe. Journal of Biological Chemistry, 267(5), 3014–3023.
Teixeira-Silva, A., Ait Saada, A., Hardy, J., Iraqui, I., Nocente, M. C., Freon, K., et al. (2017). The end-joining factor Ku acts in the end-resection of double strand break-free arrested replication forks. Nature Communications, 8(1), 1982. https://doi.org/10.1038/s41467-017-02144-5.
Tomimatsu, N., Mukherjee, B., Catherine Hardebeck, M., Ilcheva, M., Vanessa Camacho, C., Louise Harris, J., et al. (2014). Phosphorylation of EXO1 by CDKs 1 and 2 regulates DNA end resection and repair pathway choice. Nature Communication, 5, 3561. https://doi.org/10.1038/ncomms4561.
Tomimatsu, N., Mukherjee, B., Harris, J. L., Boffo, F. L., Hardebeck, M. C., Potts, P. R., et al. (2017). DNA-damage-induced degradation of EXO1 exonuclease limits DNA end resection to ensure accurate DNA repair. Journal of Biological Chemistry, 292(26), 10779–10790. https://doi.org/10.1074/jbc.M116.772475.
Tubbs, A., & Nussenzweig, A. (2017). Endogenous DNA damage as a source of genomic instability in cancer. Cell, 168(4), 644–656. https://doi.org/10.1016/j.cell.2017.01.002.
Varon, R., Vissinga, C., Platzer, M., Cerosaletti, K. M., & Reis, A. (1998). Nibrin, a novel DNA double-strand break repair protein, is mutated in nijmegen breakage syndrome. Cell, 93(3), 467–476.
Watt, P., Louis, E., Borts, R., & Hickson, I. (1995). SGS1—a eukaryotic homolog of escherichia-coli recq that interacts with topoisomerase-II in-vivo and is required for faithful chromosome segregation. Cell, 2(2), 253–260.
Watt, P. M., Hickson, I. D., Borts, R. H., & Louis, E. J. (1996). SGS1, a homologue of the Bloom’s and Werner’s syndrome genes, Is required for maintenance of genome stability in Saccharomyces cerevisiae. Genetics, 144(3), 935.
Williams, R. S., Moncalian, G., Williams, J. S., Yamada, Y., Limbo, O., Shin, D. S., et al. (2008). Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell, 135(1), 97–109. https://doi.org/10.1016/j.cell.2008.08.017.
Wu, L., Bachrati, C. Z., Ou, J., Xu, C., & Hickson, I. D. (2006). BLAP75/RMI1 promotes the BLM-dependent dissolution of homologous recombination intermediates. Proceedings of the National Academy of Sciences of the United States of America, 103(11), 4068–4073.
Wu, L., & Hickson, I. D. (2003). The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature, 426(6968), 870–874.
Xie, Y., Liu, Y. K., Guo, Z. P., Guan, H., Liu, X. D., Xie, D. F., et al. (2019). RBX1 prompts degradation of EXO1 to limit the homologous recombination pathway of DNA double-strand break repair in G1 phase. Cell Death and Differentiation. https://doi.org/10.1038/s41418-019-0424-4.
Yu, Y., Pham, N., Xia, B., Papusha, A., Wang, G., Yan, Z., et al. (2018). Dna2 nuclease deficiency results in large and complex DNA insertions at chromosomal breaks. Nature, 564(7735), 287–290. https://doi.org/10.1038/s41586-018-0769-8.
Zhang, H., Hua, Y., Li, R., & Kong, D. (2016). Cdc24 is essential for long-range end resection in the repair of double-stranded DNA breaks. Journal of Biological Chemistry, 291(48), 24961–24973. https://doi.org/10.1074/jbc.M116.755991.
Zheng, L., Zhou, M., Guo, Z., Lu, H., Qian, L., Dai, H., et al. (2008). Human DNA2 is a mitochondrial nuclease/helicase for efficient processing of DNA replication and repair intermediates. Molecular Cell, 32(3), 325–336. https://doi.org/10.1016/j.molcel.2008.09.024.
Zhou, C., Pourmal, S., & Pavletich, N. P. (2015). Dna2 nuclease-helicase structure, mechanism and regulation by Rpa. Elife. https://doi.org/10.7554/eLife.09832.
Zhou, Y., Caron, P., Legube, G., & Paull, T. T. (2014). Quantitation of DNA double-strand break resection intermediates in human cells. Nucleic Acids Research, 42(3), e19. https://doi.org/10.1093/nar/gkt1309.
Zhu, Z., Chung, W. H., Shim, E. Y., Lee, S. E., & Ira, G. (2008). Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell, 134(6), 981–994. https://doi.org/10.1016/j.cell.2008.08.037.
Zierhut, C., & Diffley, J. F. X. (2008). Break dosage, cell cycle stage and DNA replication influence DNA double strand break response. Embo Journal, 27(13), 1875–1885.
Zimmermann, M., Lottersberger, F., Buonomo, S. B., Sfeir, A., & de Lange, T. (2013). 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science, 339(6120), 700–704.
Acknowledgements
We thank all members of the Kong Laboratory for discussion and support. This work was supported by grants from the National Natural Science Foundation of China (No. 31230021), the Ministry of Science and Technology of China (2013CB911000), the Peking-Tsinghua Center for Life Sciences, and the National Key Laboratory of Protein and Plant Gene Research.
Author information
Affiliations
Peking-Tsinghua Center for Life Sciences, The National Laboratory of Protein and Plant Gene Research, College of Life Sciences, Peking University, Beijing, 100871, China
Sijie Liu & Daochun Kong
Corresponding authors
Correspondence to Sijie Liu or Daochun Kong.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article
Cite this article
Liu, S., Kong, D. End resection: a key step in homologous recombination and DNA double-strand break repair. GENOME INSTAB. DIS. 2, 39–50 (2021). https://doi.org/10.1007/s42764-020-00028-5
Received04 September 2020
Revised21 October 2020
Accepted31 October 2020
Published27 November 2020
Issue DateFebruary 2021
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
Homologous recombination
DNA double-strand break repair
End resection
MRN complex
DNA2
用户登录
还没有账号?
立即注册