






USP11 suppresses CHK1 activation by deubiquitinating CLASPIN
Original Research Paper
Open Access
Genome Instability & Disease (2021)
Abstract
CLASPIN is an essential mediator of ATR-dependent CHK1 activation in the DNA replication checkpoint. K6-linked polyubiquitination of CLASPIN promotes its chromatin loading and subsequent CHK1 activation. Here, we found that ubiquitin-specific protease 11 (USP11) deubiquitinates the K6-linkage polyubiquitinated form of CLASPIN. Under steady-state conditions, USP11 interacts with CLASPIN, reducing CLASPIN K6-linked ubiquitination levels. In response to replication stress, USP11 is phosphorylated by ATR and subsequently disassociated from CLASPIN, promoting CLASPIN chromatin loading, CHK1 activation and ultimately genome stability. Taken together, our findings uncover a novel function of USP11 in negatively regulating CHK1 activation by suppressing CLASPIN chromatin loading.
Introduction
The ATR (ataxia telangiectasia and Rad3-related protein)–CHK1 (checkpoint kinase 1) pathway has key roles in maintaining genome stability and integrity during DNA replication (Berti et al. 2020; Menolfi and Zha 2020; Yazinski and Zou 2016). CLASPIN is a critical mediator of ATR-dependent CHK1 activation; both its stability and chromatin loading are essential for CHK1 activation (Smits et al. 2018). We and others have demonstrated that the E3 ligase HERC2 and the ubiquitin-specific protease 20 (USP20) coordinate CHK1 activation by modulating CLASPIN stability (Wang et al. 2017; Yuan et al. 2014; Zhu et al. 2014). In addition, BRCA1-mediated K6-linked polyubiquitination promotes CLASPIN loading onto chromatin, leading to CHK1 activation for homologous recombination (HR)-mediated DNA repair in response to DNA lesions, including camptothecin (CPT)-induced DNA damage (Sato et al. 2012; Wu-Baer et al. 2003). Despite these advances, the corresponding CLASPIN deubiquitinases have not yet been identified.
Ubiquitin-specific protease 11 (USP11) is a deubiquitinating enzyme that can remove conjugated ubiquitin chains from target proteins (Ideguchi et al. 2002); it resides in both the cytoplasm and nucleus, and associates with chromatin (Maertens et al. 2010). USP11 preferentially cleaves K63-, K6-, K33-, and K11-linked polyubiquitination chains over K27-, K29-, and K48-linked chains and linear chains (Harper et al. 2014). USP11 has been implicated in modulating different signaling pathways by deubiquitinating its substrates and antagonizing proteasome-mediated degradation, leading to either suppressed, or enhanced cell proliferation and tumorigenesis. USP11 deubiquitinates and stabilizes type I and type II TGF-β receptors to augment TGF-β signaling and the epithelial–mesenchymal transition (Al-Salihi et al. 2012; Garcia et al. 2018; Jacko et al. 2016). Eukaryotic initiation factor 4B (eIF4B), X-linked inhibitor of apoptosis protein (XIAP), and the target of retinoblastoma protein E2F1 are all stabilized by USP11 by direct deubiquitination; this stabilization results in oncogenic translation, apoptosis inhibition, cell proliferation and wound healing in lung epithelial cells, respectively (Kapadia et al. 2018; D. Wang et al. 2018; Zhou et al. 2017). Indeed, USP11 has been identified as a marker of poor prognosis and metastasis in hepatocellular carcinoma (Zhang et al. 2018) and estrogen receptor α-positive breast cancer (Dwane et al. 2020). These studies imply that USP11 plays an oncogenic role in tumorigenesis. On the other hand, USP11 negatively regulates TNFα-induced NF-κB activation by deubiquitinating IκBα (Sun et al. 2010). Furthermore, it deubiquitinates and stabilizes Mgl-1, a mammalian homologue of the Drosophila tumor suppressor protein Lgl, the transcriptional repressor VGLL4, and the promyelocytic leukemia protein (PML), suppressing cell proliferation (Lim et al. 2016; Wu et al. 2014; Zhang et al. 2016). Finally, USP11 targets p21 to protect it from degradation, thus activating the DNA damage-induced cell-cycle checkpoint (Deng et al. 2018). These reports support a tumor suppression role for USP11.
USP11 has been specifically implicated in the DNA damage response (DDR). In a subgenome-wide shRNA screen, USP11 was one of the 73 genes that, when silenced, caused DDR activation and defects in genome maintenance (Lovejoy et al. 2009). Inhibiting USP11 expression compromised HR-mediated DSB repair (Wiltshire et al. 2010). These findings are supported by a recent study that showed that USP11 antagonizes PALB2 ubiquitination, thus promoting PALB2 dissociation from BRCA1; this process is a plausible mechanism to limit HR in G1 phase (Orthwein et al. 2015). It was reported that USP11 interacts with and deubiquitinates xeroderma pigmentosum complementation group C (XPC), promoting its retention at the DNA damage site and nucleotide excision repair (Shah et al. 2017). USP11-mediated histone deubiquitination coordinates with NuRD-associated histone deacetylation to promote chromatin reorganization and ensure timely termination of DNA repair (Ting et al. 2019).
In this study, we found that USP11 elicits its non-proteasome-mediated degradation effects by enzymatically removing K6-linked polyubiquitin chains from CLASPIN. In this way, CLASPIN chromatin loading and subsequent CHK1 activation are negatively regulated in response to replication stress.
Results
USP11 suppresses CPT-induced CHK1 activation
In a candidate screen for ubiquitin-specific proteases (USPs) in regulating replication stress-induced CHK1 activation, we previously identified that USP20 promotes CHK1 activation by deubiquitinating CLASPIN and enhancing its protein stability (Zhu et al. 2014). In addition, we found that USP11 depletion in 293 T cells using two independent siRNAs had no effect on the total CLASPIN protein levels (Fig. 1a). By contrast, USP11 depletion enhanced CHK1 activation (as measured by phosphorylation of CHK1 at serine 345) in response to CPT treatment compared to mock inhibition (Fig. 1b). CPT is an inhibitor of DNA topoisomerase I (Topo I) that binds to the Topo I and DNA complex and stabilizes it, thereby triggering the replication stress response by activating the ATR–CHK1 signaling cascade. To exclude the possibility that enhanced CHK1 activation resulted from off-target effects by the siUSP11 oligonucleotide duplexes, we engineered a siUSP11-resistant form of FLAG-USP11, termed FLAG-USP11res. Here, FLAG-USP11res expression in 293 T cells depleted of endogenous USP11 restored normal activation kinetics of CPT-induced CHK1 (Fig. 1c). Furthermore, overexpression of wild-type FLAG-USP11, but not the catalytically inactive mutant FLAG-USP11(C318S), suppressed CPT-induced CHK1 activation in 293 T cells (Fig. 1d). Taken together, these results reveal a novel role for USP11 in negatively regulating CPT-induced CHK1 activation. This negative regulation by USP11 requires its catalytic activity. Notably, USP11 depletion had no effect on CLASPIN protein stability.
Fig. 1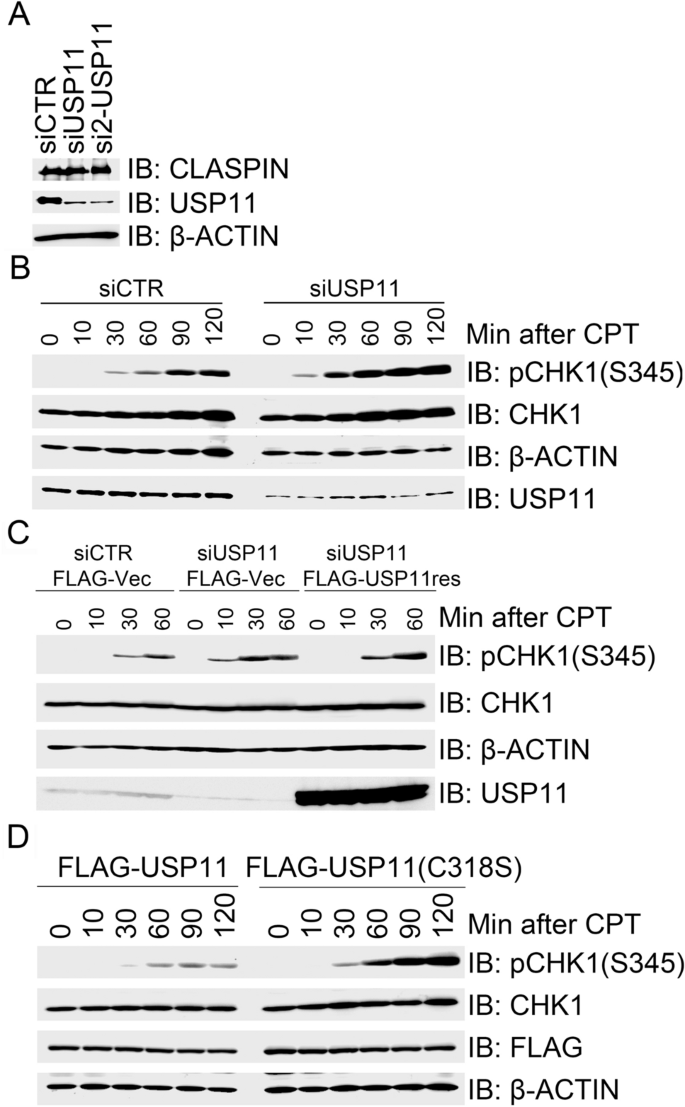
USP11 suppresses CHK1 activation in response to CPT-induced replication stress. a Depletion of USP11 has little impact on CLAPSIN protein levels. 293 T cells were transfected with control siRNA (siCTR) or USP11-specific siRNA oligos siUSP11 or si2-USP11, and total cell lysates were extracted 2 days after transfection for immunoblotting with the indicated antibodies. b Depletion of USP11 promoted CPT induced CHK1 activation. 293 T cells were transfected with siCTR or siUSP11, and the transfectants were exposed to CPT 2 days after transfection. Total cell lysates were extracted for immunoblotting at the indicated times, and analyzed with the indicated antibodies. c Re-expression of USP11 rescued its depletion-induced enhancement of CHK1 activation in response to CPT treatment. USP11-depleted 293 T cells were transfected with an empty vector (FLAG-Vec) or siRNA-resistant FLAG-USP11res. The transfectants were treated with CPT 2 days after transfection and total cell lysates were collected at the indicated time points. Total cell lysates were analyzed by immunoblotting with the indicated antibodies. d Over-expression of wild-type (WT) USP11, but not catalytically inactivated mutant USP11(C318S), suppressed CPT-induced CHK1 activation. 293 T cells were transfected with FLAG-USP11 or FLAG-USP11(C318S). Transfectants were treated with CPT 2 days after transfection and total cell lysates were then extracted at the indicated times for immunoblotting with the indicated antibodies
Full size imageATR-mediated phosphorylation of USP11 promotes CPT-induced CHK1 activation
Given that USP11 negatively regulates replication stress-induced CHK1 activation, we sought to determine if USP11 is a substrate of ATR. By co-immunoprecipitation assay, we detected endogenous USP11 in the ATR immunocomplex (Fig. 2a). To exclude the possibility of false-positive interaction caused by cross-reactivities of antibodies we used, we employed an anti-FLAG antibody to immunoprecipitated FLAG-ATR overexpressed in 293 T cells and found by immuonoblotting analysis with an anti-HA antibody that HA-USP11 co-overexpressed in 293 T cells was present in the FLAG-ATR immunocomplex (Fig. 2b).
Fig. 2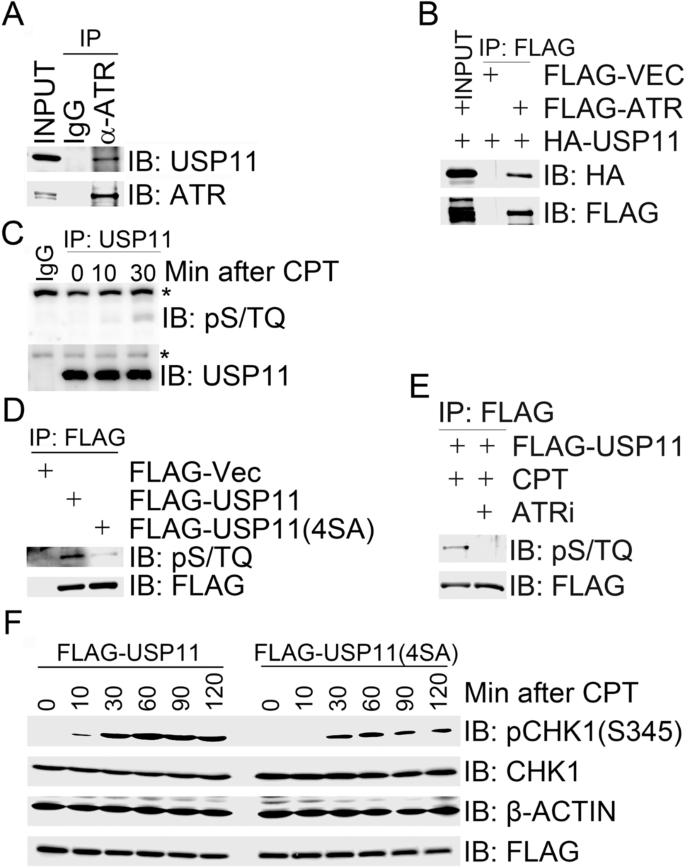
ATR-mediated USP11 phosphorylation promotes CPT-induced CHK1 activation. (A and B) USP11 interacted with ATR. Total cell lysates were extracted from 293 T cells (a) or 293 T cells co-transfected with HA-USP11 and FLAG-VEC or FLAG-ATR (b) and subjected to immunoprecipitation followed by immunoblotting with the indicated antibodies. c Endogenous USP11 is phosphorylated. Total cell lysates were extracted from 293 T cells mock-treated for 30 min or treated with CPT for 10 min or 30 min and subjected to immunoprecipitation followed by immunoblotting with the indicated antibodies. Asterisk indicates a non-specific signal. d HA-USP11 was phosphorylated on its S/TQ motifs. 293 T cells were transfected with FLAG-VEC, FLAG-USP11 or FLAG-USP11(4SA). Total cells lysates were harvested after 2 days and subjected to immunoprecipitation followed by immunoblotting with the indicated antibodies. e USP11 was phosphorylated by ATR in response to CPT treatment. 293 T cells expressing FLAG-USP11 were mocked-treated, treated with CPT for 1 h or pre-treated with the ATR inhibitor (ATRi) NU6027 for 1 h before CPT treatment. Total cells lysates were then harvested and subjected to immunoprecipitation followed by immunoblotting with the indicated antibodies. f Expression of phosphorylation-deficient mutant USP11(4SA) suppressed CHK1 activation. 293 T cells were transfected with FLAG-USP11 or FLAG-USP11(4SA) and the transfectants were treated with CPT (1 µM) 48 h after transfection at the indicated time points. Total cell lysates were analyzed by immunoblotting with the indicated antibodies
Full size imageATM/ATR kinase substrates typically contain a minimal phosphorylation motif S/TQ (Kim et al. 1999). USP11 encodes a polypeptide of 923 amino acids, which contains four SQ sites (S40, S351, S361, and S665) and no TQ site. We found that the ATM/ATR substrate pS/TQ antibody was almost unreactive with the immunoprecipitated endogenous USP11 under unperturbed conditions (Fig. 2c), and this reactivity increased upon CPT treatment (Fig. 2c). We also detected the pS/TQ reactivity in HA-USP11 when immunoprecipitated from 293 T cells overexpressing HA-USP11 (Fig. 2d). When cells were pretreated with the ATR-specific inhibitor (ATRi), NU6027, it was found that the pS/TQ reactivity in HA-USP11 after CPT treatment was diminished (Fig. 2e). However, we failed to attribute this phosphorylation signal to any individual or dual SQ sites by mutagenesis assays (data not shown). Furthermore, we failed to generate phospho-specific antibodies targeting individual SQ sites within USP11 (data not shown). Nevertheless, the pS/TQ antibody was not reactive with the immunoprecipitated phosphorylation-defective mutant FLAG-USP11 (4SA), in which all four SQ sites were mutated into AQ sites (Fig. 2d). Furthermore, over-expression of FLAG-USP11(4SA) in 293 T cells suppressed CPT-induced CHK1 activation (Fig. 2f). These results suggest that USP11 is a substrate of ATR and ATR-mediated USP11 phosphorylation promotes CPT-induced CHK1 activation.
USP11 dynamically interacts with CLASPIN
We next sought to uncover the molecular mechanisms by which USP11 suppresses ATR-dependent CHK1 activation in response to replication stress. Given that CLASPIN is an essential mediator of ATR-dependent CHK1 activation, we reasoned that USP11 might physically associate with CLASPIN. By co-immunoprecipitation assay, we found that endogenous CLASPIN was present in the USP11 immunocomplex (Fig. 3a), and vice versa (Fig. 3b). We then generated overlapping truncation mutants of CLASPIN (Fig. 3c). In vivo domain mapping analysis revealed that USP11 bound to the CLASPIN F1 fragment (1–330 aa) in the N terminus (Fig. 3d), where BRCA1-mediated K6-linked ubiquitination occurs (Sato et al. 2012; Wu-Baer et al. 2003).
Fig. 3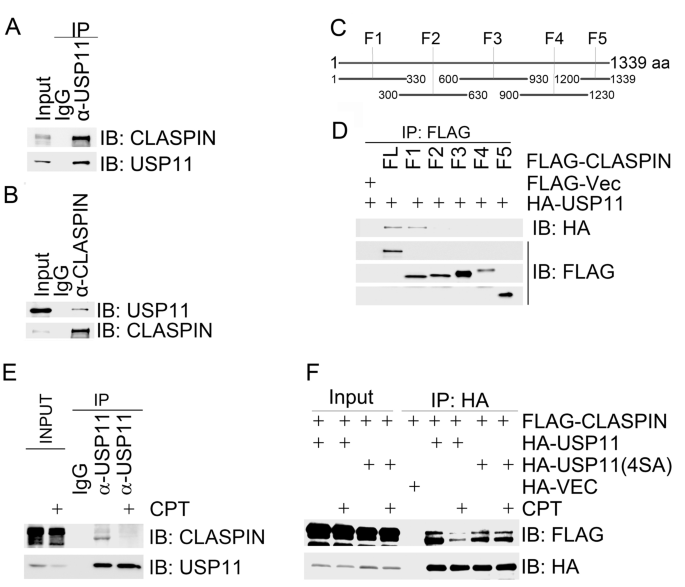
CPT-induced replication stress promotes USP11 dissociation from CLASPIN. a, b USP11 interacted with CLASPIN. Total cell lysates were extracted from 293 T cells and subjected to immunoprecipitation and immunoblotting with the indicated antibodies. c CLASPIN amino acid structure and fragmentation design. Full-length CLASPIN was divided into 5 overlapping fragments (F1–5). d The amino terminus of CLASPIN interacted with USP11. Total cell lysates extracted from 293 T cells co-transfected with the indicated constructs were immunoprecipitated with an anti-FLAG antibody and then analyzed by immunoblotting with an anti-HA antibody. e USP11 dissociated from CLASPIN upon CPT treatment. Total cell lysates were extracted from 293 T cells with mock treatment or CPT treatment for 1 h and subjected to immunoprecipitation with an anti-USP11 antibody. Immunoprecipitates were analyzed by immunoblotting with the indicated antibodies as indicated. f USP11(4SA) failed to dissociate from CLASPIN upon CPT treatment. 293 T cells co-expressing FLAG-CLASPIN with an empty HA vector (HA-Vec), HA-USP11 or HA-USP11(4SA) were mock-treated or treated with CPT for 1 h. Total cell lysates were extracted and subjected to immunoprecipitation with an HA antibody followed by immunoblotting with the indicated antibodies
Full size imageWe then asked if this interaction was modulated by replication stress. Indeed, we found that the majority of both endogenous and FLAG-tagged CLASPIN dissociated from endogenous and HA-tagged USP11 in 293 T cells treated with CPT for 1 h, respectively (Fig. 3e, f). FLAG-CLASPIN retained the association with the phosphorylation-defective mutant HA-USP11(4SA) after CPT treatment (Fig. 3f). These results demonstrate that ATR-mediated USP11 phosphorylation promotes dissociation of USP11 from CLASPIN.
USP11 deubiquitinates CLASPIN
Given that USP11 dynamically interacts with CLASPIN in response to replication stress, we decided to explore if CLASPIN is a substrate of USP11. We co-expressed MYC-USP11 or MYC-USP11(C318S) with FLAG-CLASPIN, and HA-tagged wild-type ubiquitin (UB) or K6/K48/K63-linkage-type-only UB mutants—UB(K6), UB(K48), and UB(K63), respectively. Here, UB or UB(K6)-linked polyubiquitination levels of FLAG-CLASPIN in USP11-expressing cells reduced compared to those in USP11(C318S)-expressing cells (Fig. 4a). We failed to detect obvious differences of UB(K48) or UB(K63)-linked polyubiquitination levels of FLAG-CLASPIN in USP11-expressing or USP11(C318S)-expressing cells (Fig. 4a). These results indicate that USP11 is enzymatically active towards CLASPIN K6-linkage-type polyubiquitination. This finding is consistent with a previous report that USP11 preferentially cleaves the K6 linkage over the K48 linkage or K63 linkage (Harper et al. 2014). In vivo deubiquitination assays using a similar strategy uncovered that both USP11 and USP11(4SA), but not USP11(C318S), efficiently reduce the K6-linked polyubiquitination levels of CLASPIN (Fig. 4b). Taken together, these results demonstrate that USP11 specifically deubiquitinates K6-linked polyubiquitinated CLASPIN. The phosphorylation-defective mutant USP11(4SA), in which all potential ATM/ATR phosphorylation sites are mutated, possesses similar deubiquitinating activity to wild-type USP11.
Fig. 4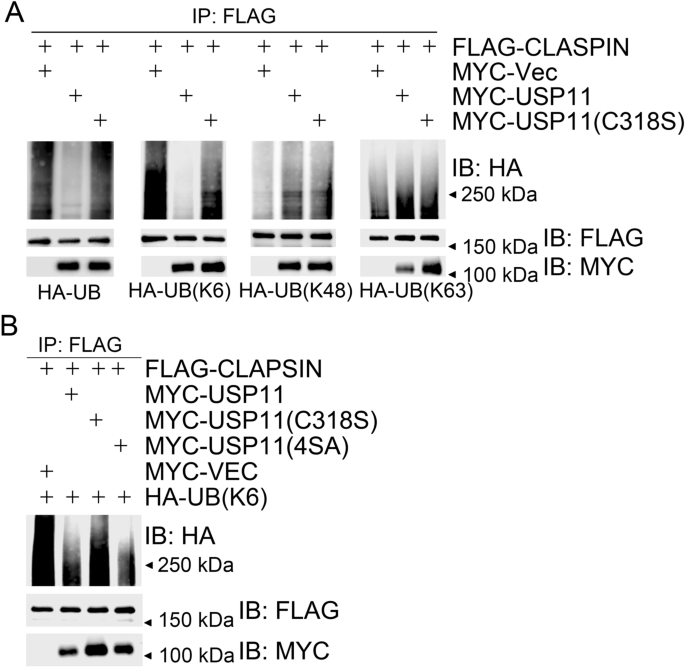
USP11 deubiquitinates CLASPIN. a USP11 deubiquitinated K6-linked polyubiquitination of CLASPIN. 293 T cells were co-transfected with FLAG-CLASPIN and HA-UB (K6/K48/K63) and other expression constructs as indicated. Total cell lysates were extracted 48 h after transfection and subjected to immunoprecipitation with an anti-FLAG antibody followed by immunoblotting with the indicated antibodies. b Over-expression of USP11 or USP11(4SA), but not USP11(C318S), decreased K6-linked ubiquitination of CLASPIN. 293 T cells were co-transfected with the expression constructs as indicated, and total cell lysates were harvested after 2 days and subjected to immunoprecipitation followed by immunoblotting with the indicated antibodies
Full size imageUSP11 suppresses CHK1 activation by reducing chromatin loading of CLASPIN upon replication stress
BRCA1-mediated K6-linked CLASPIN polyubiquitination promotes its chromatin loading and subsequent CHK1 activation in response to CPT treatment (Sato et al. 2012; Wu-Baer et al. 2003). We have found that USP11 specifically removes K6-linked polyubiquitin chains from CLASPIN (Fig. 4a). We, thus, attempted to determine if USP11 regulates CLASPIN chromatin loading. As expected, BRCA1 inhibition in 293 T cells by siRNA treatment reduced CLASPIN levels in the chromatin-enriched fraction (P3) after CPT treatment compared to mock inhibition. Depletion of USP11 increased CLASPIN levels in the chromatin-enriched fraction (Fig. 5a). Chromatin fractionation assays revealed that stable expression of catalytically inactive mutant USP11(C318S) in 293 T cells resulted in higher levels of CLASPIN in the P3 fraction than expression of wild-type USP11 after CPT treatment (Fig. 5b), indicating that it is USP11 enzymatic activity regulating CLASPIN chromatin loading. As expected, given that the phosphorylation-defective mutant USP11(4SA) retains its enzymatic activity (Fig. 4b) and fails to dissociate from CLASPIN in response to CPT treatment (Fig. 3f), USP11(4SA) over-expression decreased CLASPIN levels in the P3 fraction after CPT treatment compared to USP11 or USP11(C318S) over-expression (Fig. 5b). These results demonstrate that USP11 suppresses replication stress-induced CHK1 activation by chromatin loading of CLASPIN.
Fig. 5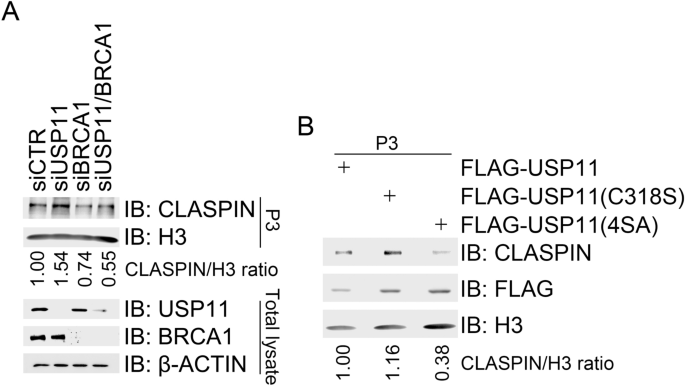
USP11 suppresses CHK1 activation by reducing CLASPIN chromatin loading. a Depletion of USP11 promoted chromatin loading of CLASPIN. 293 T cells were transfected with a control siCTR, siUSP11, a BRCA1-specific siRNA (siBRCA1), or siUSP11 plus with siBRCA1. Transfectants were treated 2 days after transfection with CPT for 1 h. Half of the transfectants were subjected to chromatin fractionation followed by immunoblotting with antibodies against H3 and CLASPIN in P3 (chromatin-enriched fraction). Total cell lysates were extracted from the other half and subjected to immunoblotting with the indicated antibodies. b Over-expression of USP11 or USP11(4SA) impaired CLASPIN chromatin. 293 T cells expressing FLAG-USP11, FLAG-USP11 (C318S), or FLAG-USP11(4SA) were treated with CPT (1 µM) for 1 h and then subjected to chromatin fractionation followed by immunoblotting of the P3 fraction with the indicated antibodies
Full size imageUSP11 promotes genome instability
We next hypothesized that over-expression of USP11 may confer drug resistance to CPT, abnormal S-phase checkpoint activation, and chromosomal instability. To test these parameters, we engineered U2OS cells to stably express FLAG-Vec, FLAG-USP11, FLAG-USP11(C318S), or FLAG-USP11(4SA) by lentiviral transduction. Upon treating cells with increasing concentrations of CPT (0–4.0 µM), clonogenic assays revealed that cells expressing USP11 or USP11(4SA) exhibited significantly higher survival rate than vector control cells or cells expressing catalytically inactive mutant USP11(C318S) (Fig. 6a). BrdU incorporation assays demonstrated that over-expression of wild-type USP11 or phosphorylation-defective mutant USP11(4SA), but not USP11(C318S), compromised UV-induced S-phase checkpoint activation (Fig. 6b). Furthermore, cells expressing USP11 or USP11(4SA), but not USP11(C318S), showed significantly enhanced premature chromosome condensation (PCC) under both unperturbed conditions and CPT treatment (Fig. 6c, d). Taken together, these results reveal a novel function of USP11 in promoting genome instability through suppressing replication stress-induced CHK1 activation.
Fig. 6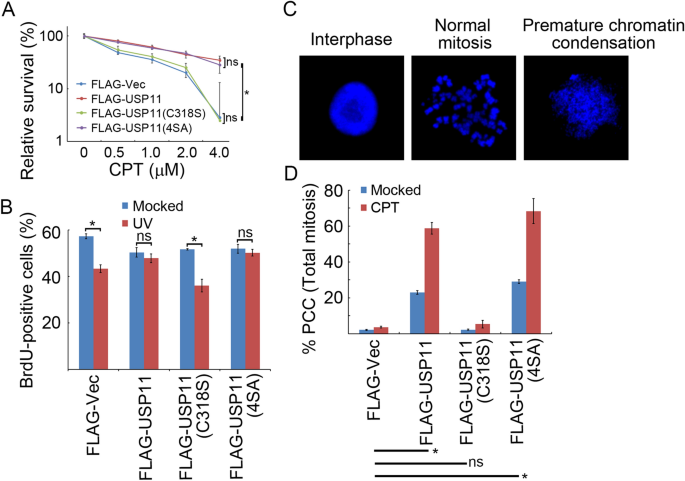
USP11 promotes genome instability. a Over-expression of USP11 or USP11(4SA), but not USP11(C318S), promoted cell survival after CPT treatment. U2OS cells stably expressing inducible FLAG-USP11, FLAG-USP11 (C318S) or FLAG-USP11(4SA) were incubated with doxycycline 2 days before exposure to the indicated dose of CPT for 1 h. Surviving colonies were counted in three independent experiments. The data represent the means ± SD. b Over-expression of USP11 or USP11(4SA), but not USP11(C318S), compromised the intra-S-phase checkpoint. U2OS cells stably expressing inducible FLAG-USP11, FLAG-USP11 (C318S) or FLAG-USP11(4SA) were incubated with doxycycline for 2 days before UV irradiation at 20 J/m2 followed by pulse labeling with BrdU. The percentage of BrdU-positive cells from three independent experiments is presented as the means ± SD. c, d Over-expression of USP11 or USP11(4SA), but not USP11(C318S), enhanced PCC. U2OS cells stably expressing inducible FLAG-USP11, FLAG-USP11 (C318S) or FLAG-USP11(4SA) were incubated with doxycycline for 2 days before exposure to CPT (1 µM) and nocodazole (200 ng/ml) for 20 h. Mitotic chromosome spreads were prepared, and the percentage of cells containing PCC was evaluated under a confocal microscope. The data represent the means ± SD of three independent experiments. *p < 0.05; ns not significant
Full size imageDiscussion
We and others have previously reported that USP20 activates CHK1 upon replication stress by deubiquitinating K48-linked polyubiquitinated CLASPIN and subsequently enhancing its protein stability (Yuan et al. 2014; Zhu et al. 2014). In this report, we describe that USP11 instead negatively regulates replication stress-induced CHK1 activation by deubiquitinating CLASPIN. Under basal conditions, USP11 binds to CLASPIN, antagonizing BRCA1-mediated CLASPIN K6-linkage polyubiquitination and maintaining normal CLASPIN levels on chromatin. In response to replication stress, ATR-mediated phosphorylation of USP11 promotes its dissociation from CLASPIN, increasing CLASPIN K6-linked polyubiquitination levels and its chromatin loading. Given that phosphorylated USP11 dissociates from CLASPIN, we speculate that this dissociation leads to an increase of the K6-linked polyubiquitination levels of CLASPIN (Fig. 4). Thus, USP11 serves as a negative regulator of ATR–CHK1-dependent replication checkpoint signaling. It would now be interesting to determine if and how USP11 communicates with USP20 in the CLASPIN-mediated replication stress response. It is highly possible that USP11 has an important role in replication checkpoint recovery, as it enzymatically clips away the polyubiquitin chains from CLASPIN. This possibility warrants further investigation.
BRCA1 is a ring E3 ligase that uniquely catalyzes K6-linkage-type polyubiquitin chain conjugation and elongation; its known substrates include BRAC1 itself and CLASPIN. In vitro, USP11 can directly remove K6-linked polyubiquitin chains during ubiquitin chain cleavage using artificial substrates (Harper et al. 2014). Here, we showed for the first time that USP11 efficiently cleaves the K6-linked polyubiquitin chain from CLASPIN, concluding that USP11 is a protease for K6-linkage-type polyubiquitin chains.
USP11 has been associated with tumorigenesis, but it is debated whether USP11 promotes or suppresses tumor formation. An unbiased DUB siRNA screen uncovered that USP11 deubiquitinates and stabilizes the promyelocytic leukemia (PML) protein to suppress Notch-induced malignancy in brain tumors (Wu et al. 2014). USP11 exhibits deubiquitinating activity against Mgl-1, a mammalian homologue of the Drosophila tumor suppressor protein Lgl, and prevents it from proteasome-mediated degradation, maintaining cell polarity and epithelial integrity (Lim et al. 2016). Both studies, thus, imply that USP11 may suppress tumorigenesis. However, mitoxantrone, a DNA intercalator and a potent inhibitor for USP11, blocked pancreatic ductal adenocarcinoma (PDA) cell survival with an IC50 of < 10 nM in an in vitro drug sensitivity assay (Burkhart et al. 2013). In addition, the cBioPortal for Cancer Genomics database (http://www.cbioportal.org/) indicates that USP11 is amplified in 30.8% neuroendocrine prostate cancers (33/107 cases) (Beltran et al. 2016), 9.8% metastatic prostate adenocarcinomas (6/61 cases), 2.7% metastatic prostate cancers (Robinson et al. 2015), 7.1% ovarian serous cystadenocarcinomas (22/311 cases), 7.1% uterine carcinosarcomas (4/51 cases) and 4.7% sarcomas (7/150 cases).
USP11 antagonizes E3 ubiquitin ligase complex-mediated PALB2 ubiquitination and subsequent dissociation from BRCA1. This mechanism suppresses HR in the G1 phase (Orthwein et al. 2015). Thus, it is conceivable that USP11 over-expression promotes chromosomal instability due to improper activation of the S-phase checkpoint and unscheduled DNA replication resulting from compromised HR suppression in G1 phase. These previous studies, and our latest findings in this report, argue for a role of USP11 in promoting tumorigenesis. The role of USP11 in tumorigenesis may be cell-type specific. We are now generating conditional knockout mouse models of Usp11 to produce a more definitive conclusion.
Experimental procedures
Reagents, antibodies and cell lines
CPT was used at a final concentration of 1 µM throughout this study. The ATR inhibitor NU6027 was used at a final concentration of 10 µM. Both reagents were purchased from Sigma. BrdU was used at a final concentration of 20 µM and was purchased from BD Biosciences.
Rabbit polyclonal antibodies used for immunoblotting and immunoprecipitation, including anti-MYC (A190–205A), anti-HA (A190–208A), anti-USP11 (A301–613A), anti-CLASPIN (A300–267A), anti-BRCA1 (A300–000A), and anti-ATR (A300–138A) were purchased from Bethyl Laboratories. Antibodies for Chk1 (sc-8408), and Ub (sc-8408) were obtained from Santa Cruz Biotechnology. The mouse monoclonal antibody against FLAG M2 (F1804) was from Sigma. Phosphorylation-specific antibodies against CHK1 (Ser345) (#2348), the (Ser/Thr) ATM/ATR substrate motif (#2851) and the anti-H3 antibody (#9715) were from Cell Signaling Technology. Anti-BrdU FITC (347583) was used for IF and was purchased from BD Biosciences.
All cultured cells were grown in high-glucose Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum, in a humidified 37 °C incubator with 5% CO2.
Expression constructs
Human open reading frames (ORFs) of USP11, CLASPIN, and Ub were generated by RT-PCR form total RNA extracted from HeLa cells and subcloned in frame into the pcDNA3.0 mammalian expression vector with a N-terminal epitope of 3 X HA or FLAG or a C-terminal MYC epitope. The phosphorylation-defective mutant USP11(4SA), the catalytically inactive mutant USP11(C318S), and the Ub mutants (K63-linked only mutant, K6-linked only mutant, and K48-linked only mutant) were generated using a QuikChange Site-Directed Mutagenesis Kit (Beijing SBS Genetech), according to the manufacturer conditions. To generate overlapping fragments covering the entire ORF of human CLASPIN, individual fragments were amplified by PCR from pcDNA-FLAG-CLASPIN. The rescue expression constructs that were siRNA resistant contained 4–6 silent point mutations within the siRNA targeting region. All the constructs were confirmed by DNA sequencing.
RNA interference
A mixture of four pre-designed OnTarget plus siRNA oligonucleotide duplexes specific for BRCA1 was purchased from Dharmacon, and the siRNA oligonucleotide duplexes against USP11 (si-USP11 sequence: CUGAAAUGAAAAAAAGGUA; si2-USP11 sequence: GAAATCAATTGGAGATTAA) were purchased from Guangzhou RiboBio. The gene-specific siRNA or the non-target siRNA control (siCTR) was transfected into cells at a final concentration of 20 nM using RNAimax (Invitrogen), according to the manufacturer’s instructions.
Immunoblotting and immunoprecipitation
Both immunoblotting and immunoprecipitation assays were performed with the indicated antibodies as previously described (Xu and Stern 2003).
BrdU incorporation assay
For BrdU incorporation assay, CTR-over-expression or USP11-over-expression cells were grown on cover slips and UV irradiated with a dose of 20 J/m2 and pulsed labeled 40 min later with 20 μM BrdU (BD Biosciences) for 20 min. Cells were then washed with PBS twice and fixed with ethanol (75%, vol/vol) at room temperature (RT) for 30 min. The fixed cells were washed with PBS, denatured by incubating with 2 M HCl at room temperature for 30 min, and then neutralized with Na2PO4 for 10 min. The samples were sequentially blocked with 2% BSA at room temperature for 30 min, incubated with a FITC BrdU antibody for 60 min, washed with PBS three times, and stained with DAPI for 2 min to visualize nuclear DNA. The cover slips were mounted onto glass slides with anti-fade solution and visualized under a fluorescence microscope.
Clonogenic survival assay
Clonogenic survival assays were performed essentially as previously described (Peng et al. 2015). In brief, U2OS cells stably expressing FLAG-VEC, FLAG-USP11, FLAG-USP11(C318S) or FLAG-USP11(4SA) were treated with CPT at the indicated doses. Colony formation occurred over the next 10–14 days under standard cell culture conditions. Colonies were counted after staining with 0.5% crystal violet/20% ethanol. The results were normalized to plating efficiencies. The colonies were counted and the results are presented as the mean of three independent experiments.
De-ubiquitination assay
De-ubiquitination assays were performed as described previously (Zhu et al. 2014). In brief, 293 T cells co-transfected with MYC-USP11, MYC-USP11 (C318S) or MYC-USP11 (4SA), FLAG-CLASPIN and HA-Ub (WT, K48 only, K63 only or K6 only) were harvested 48 h after transfection. Total cell lysates were extracted and subjected to immunoprecipitation with anti-FLAG agarose beads (Sigma) followed by immunoblotting with the indicated antibodies.
Chromatin fractionation
Chromatin fractionation was performed as previously described (Xu and Stern 2003).
Premature chromatin condensation assays
U2OS cells stably expressing FLAG-VEC, FLAG-USP11, FLAG-USP11(C318S) or FLAG-USP11(4SA) were treated with 1 µM CPT plus nocodazole (0.2 µg/ml) for 20 h. Preparation of mitotic chromosome spreads (Howe et al. 2014) and chromosome condensation was performed as previously described (Nghiem et al. 2001).
Availability of data and material
All data generated or analyzed during this study are included in this manuscript. Materials described in this manuscript are available upon request.
Code availability (software application or custom code)
Not applicable.
References
Al-Salihi, M. A., Herhaus, L., Macartney, T., & Sapkota, G. P. (2012). USP11 augments TGFbeta signalling by deubiquitylating ALK5. Open Biology, 2(6), 120063. https://doi.org/10.1098/rsob.120063
Beltran, H., Prandi, D., Mosquera, J. M., Benelli, M., Puca, L., Cyrta, J., et al. (2016). Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nature Medicine, 22(3), 298–305. https://doi.org/10.1038/nm.4045
Berti, M., Cortez, D., & Lopes, M. (2020). The plasticity of DNA replication forks in response to clinically relevant genotoxic stress. Nature Reviews Molecular Cell Biology, 21(10), 633–651. https://doi.org/10.1038/s41580-020-0257-5
Burkhart, R. A., Peng, Y., Norris, Z. A., Tholey, R. M., Talbott, V. A., Liang, Q., et al. (2013). Mitoxantrone targets human ubiquitin-specific peptidase 11 (USP11) and is a potent inhibitor of pancreatic cancer cell survival. Molecular Cancer Research, 11(8), 901–911. https://doi.org/10.1158/1541-7786.MCR-12-0699
Deng, T., Yan, G., Song, X., Xie, L., Zhou, Y., Li, J., et al. (2018). Deubiquitylation and stabilization of p21 by USP11 is critical for cell-cycle progression and DNA damage responses. Proceedings of the National Academy of Sciences of the United States of America, 115(18), 4678–4683. https://doi.org/10.1073/pnas.1714938115
Dwane, L., O’Connor, A. E., Das, S., Moran, B., Mulrane, L., Pinto-Fernandez, A., et al. (2020). A functional genomic screen identifies the deubiquitinase USP11 as a novel transcriptional regulator of ERalpha in Breast Cancer. Cancer Research, 80(22), 5076–5088. https://doi.org/10.1158/0008-5472.CAN-20-0214
Garcia, D. A., Baek, C., Estrada, M. V., Tysl, T., Bennett, E. J., Yang, J., et al. (2018). USP11 enhances TGFbeta-induced epithelial-mesenchymal plasticity and human breast cancer metastasis. Molecular Cancer Research, 16(7), 1172–1184. https://doi.org/10.1158/1541-7786.MCR-17-0723
Harper, S., Gratton, H. E., Cornaciu, I., Oberer, M., Scott, D. J., Emsley, J., et al. (2014). Structure and catalytic regulatory function of ubiquitin specific protease 11 N-terminal and ubiquitin-like domains. Biochemistry, 53(18), 2966–2978. https://doi.org/10.1021/bi500116x
Howe, B., Umrigar, A., & Tsien, F. (2014). Chromosome preparation from cultured cells. Journal of Visualized Experiments, 83, e50203. https://doi.org/10.3791/50203
Ideguchi, H., Ueda, A., Tanaka, M., Yang, J., Tsuji, T., Ohno, S., et al. (2002). Structural and functional characterization of the USP11 deubiquitinating enzyme, which interacts with the RanGTP-associated protein RanBPM. The Biochemical Journal, 367(Pt 1), 87–95. https://doi.org/10.1042/BJ20011851
Jacko, A. M., Nan, L., Li, S., Tan, J., Zhao, J., Kass, D. J., et al. (2016). De-ubiquitinating enzyme, USP11, promotes transforming growth factor beta-1 signaling through stabilization of transforming growth factor beta receptor II. Cell Death Dis, 7(11), e2474. https://doi.org/10.1038/cddis.2016.371
Kapadia, B., Nanaji, N. M., Bhalla, K., Bhandary, B., Lapidus, R., Beheshti, A., et al. (2018). Fatty acid synthase induced S6Kinase facilitates USP11-eIF4B complex formation for sustained oncogenic translation in DLBCL. Nature Communications, 9(1), 829. https://doi.org/10.1038/s41467-018-03028-y
Kim, S. T., Lim, D. S., Canman, C. E., & Kastan, M. B. (1999). Substrate specificities and identification of putative substrates of ATM kinase family members. Journal of Biological Chemistry, 274(53), 37538–37543.
Lim, K. H., Suresh, B., Park, J. H., Kim, Y. S., Ramakrishna, S., & Baek, K. H. (2016). Ubiquitin-specific protease 11 functions as a tumor suppressor by modulating Mgl-1 protein to regulate cancer cell growth. Oncotarget, 7(12), 14441–14457. https://doi.org/10.18632/oncotarget.7581
Lovejoy, C. A., Xu, X., Bansbach, C. E., Glick, G. G., Zhao, R., Ye, F., et al. (2009). Functional genomic screens identify CINP as a genome maintenance protein. Proceedings of the National Academy of Sciences of the United States of America, 106(46), 19304–19309. https://doi.org/10.1073/pnas.0909345106
Maertens, G. N., El Messaoudi-Aubert, S., Elderkin, S., Hiom, K., & Peters, G. (2010). Ubiquitin-specific proteases 7 and 11 modulate Polycomb regulation of the INK4a tumour suppressor. EMBO Journal, 29(15), 2553–2565. https://doi.org/10.1038/emboj.2010.129
Menolfi, D., & Zha, S. (2020). ATM, DNA-PKcs and ATR: shaping development through the regulation of the DNA damage responses [review]. Genome Instability & Disease, 1(2), 47–68. https://doi.org/10.1007/s42764-019-00003-9
Nghiem, P., Park, P. K., Kim, Y., Vaziri, C., & Schreiber, S. L. (2001). ATR inhibition selectively sensitizes G1 checkpoint-deficient cells to lethal premature chromatin condensation. Proceedings of the National Academy of Sciences of the United States of America, 98(16), 9092–9097. https://doi.org/10.1073/pnas.161281798
Orthwein, A., Noordermeer, S. M., Wilson, M. D., Landry, S., Enchev, R. I., Sherker, A., et al. (2015). A mechanism for the suppression of homologous recombination in G1 cells. Nature, 528(7582), 422–426. https://doi.org/10.1038/nature16142
Peng, B., Wang, J., Hu, Y., Zhao, H., Hou, W., Zhao, H., et al. (2015). Modulation of LSD1 phosphorylation by CK2/WIP1 regulates RNF168-dependent 53BP1 recruitment in response to DNA damage. Nucleic Acids Research, 43(12), 5936–5947. https://doi.org/10.1093/nar/gkv528
Robinson, D., Van Allen, E. M., Wu, Y. M., Schultz, N., Lonigro, R. J., Mosquera, J. M., et al. (2015). Integrative clinical genomics of advanced prostate cancer. Cell, 161(5), 1215–1228. https://doi.org/10.1016/j.cell.2015.05.001
Sato, K., Sundaramoorthy, E., Rajendra, E., Hattori, H., Jeyasekharan, A. D., Ayoub, N., et al. (2012). A DNA-damage selective role for BRCA1 E3 ligase in claspin ubiquitylation, CHK1 activation, and DNA repair. Current Biology, 22(18), 1659–1666. https://doi.org/10.1016/j.cub.2012.07.034
Shah, P., Qiang, L., Yang, S., Soltani, K., & He, Y. Y. (2017). Regulation of XPC deubiquitination by USP11 in repair of UV-induced DNA damage. Oncotarget, 8(57), 96522–96535. https://doi.org/10.18632/oncotarget.22105
Smits, V. A. J., Cabrera, E., Freire, R., & Gillespie, D. A. (2018). Claspin-checkpoint adaptor and DNA replication factor. FEBS Journal. https://doi.org/10.1111/febs.14594
Sun, W., Tan, X., Shi, Y., Xu, G., Mao, R., Gu, X., et al. (2010). USP11 negatively regulates TNFalpha-induced NF-kappaB activation by targeting on IkappaBalpha. Cellular Signalling, 22(3), 386–394. https://doi.org/10.1016/j.cellsig.2009.10.008
Ting, X., Xia, L., Yang, J., He, L., Si, W., Shang, Y., et al. (2019). USP11 acts as a histone deubiquitinase functioning in chromatin reorganization during DNA repair. Nucleic Acids Research, 47(18), 9721–9740. https://doi.org/10.1093/nar/gkz726
Wang, C., Yang, C., Ji, J., Jiang, J., Shi, M., Cai, Q., et al. (2017). Deubiquitinating enzyme USP20 is a positive regulator of Claspin and suppresses the malignant characteristics of gastric cancer cells. International Journal of Oncology. https://doi.org/10.3892/ijo.2017.3904
Wang, D., Zhao, J., Li, S., Wei, J., Nan, L., Mallampalli, R. K., et al. (2018). Phosphorylated E2F1 is stabilized by nuclear USP11 to drive Peg10 gene expression and activate lung epithelial cells. Journal of Molecular Cell Biology, 10(1), 60–73. https://doi.org/10.1093/jmcb/mjx034
Wiltshire, T. D., Lovejoy, C. A., Wang, T., Xia, F., O’Connor, M. J., & Cortez, D. (2010). Sensitivity to poly(ADP-ribose) polymerase (PARP) inhibition identifies ubiquitin-specific peptidase 11 (USP11) as a regulator of DNA double-strand break repair. Journal of Biological Chemistry, 285(19), 14565–14571. https://doi.org/10.1074/jbc.M110.104745
Wu, H. C., Lin, Y. C., Liu, C. H., Chung, H. C., Wang, Y. T., Lin, Y. W., et al. (2014). USP11 regulates PML stability to control Notch-induced malignancy in brain tumours. Nature Communications, 5, 3214. https://doi.org/10.1038/ncomms4214
Wu-Baer, F., Lagrazon, K., Yuan, W., & Baer, R. (2003). The BRCA1/BARD1 heterodimer assembles polyubiquitin chains through an unconventional linkage involving lysine residue K6 of ubiquitin. Journal of Biological Chemistry, 278(37), 34743–34746. https://doi.org/10.1074/jbc.C300249200
Xu, X., & Stern, D. F. (2003). NFBD1/KIAA0170 is a chromatin-associated protein involved in DNA damage signaling pathways. Journal of Biological Chemistry, 278(10), 8795–8803. https://doi.org/10.1074/jbc.M211392200
Yazinski, S. A., & Zou, L. (2016). Functions, regulation, and therapeutic implications of the ATR checkpoint pathway. Annual Review of Genetics, 50, 155–173. https://doi.org/10.1146/annurev-genet-121415-121658
Yuan, J., Luo, K., Deng, M., Li, Y., Yin, P., Gao, B., et al. (2014). HERC2-USP20 axis regulates DNA damage checkpoint through Claspin. Nucleic Acids Research, 42(21), 13110–13121. https://doi.org/10.1093/nar/gku1034
Zhang, E., Shen, B., Mu, X., Qin, Y., Zhang, F., Liu, Y., et al. (2016). Ubiquitin-specific protease 11 (USP11) functions as a tumor suppressor through deubiquitinating and stabilizing VGLL4 protein. Am J Cancer Res, 6(12), 2901–2909.
Zhang, S., Xie, C., Li, H., Zhang, K., Li, J., Wang, X., et al. (2018). Ubiquitin-specific protease 11 serves as a marker of poor prognosis and promotes metastasis in hepatocellular carcinoma. Laboratory Investigation, 98(7), 883–894. https://doi.org/10.1038/s41374-018-0050-7
Zhou, Z., Luo, A., Shrivastava, I., He, M., Huang, Y., Bahar, I., et al. (2017). Regulation of XIAP turnover reveals a role for USP11 in promotion of tumorigenesis. EBioMedicine, 15, 48–61. https://doi.org/10.1016/j.ebiom.2016.12.014
Zhu, M., Zhao, H., Liao, J., & Xu, X. (2014). HERC2/USP20 coordinates CHK1 activation by modulating CLASPIN stability. Nucleic Acids Research, 42(21), 13074–13081. https://doi.org/10.1093/nar/gku978
Funding
This work was supported by the National Natural Science Foundation of China (NSFC) (Grants 31530016, 32090031, 32000911, and 31761133012), the National Basic Research Program of China (Grant 2017YFA0503900), and the Shenzhen Science and Technology Innovation Commission (Grants JCYJ20180507182213033, and JCYJ20170412113009742), Young Scientists Fund (Grant 32000911).
Author information
Hongchang Zhao, Zhifeng Wang and Min Zhu contributed equally to this work.
Affiliations
College of Life Sciences, Capital Normal University, Beijing, 100048, China
Hongchang Zhao, Min Zhu & Ji Liao
Guangdong Key Laboratory for Genome Stability and Disease Control, Carson International Cancer Center, and Marshall Laboratory of Biomedical Engineering, Shenzhen University School of Medicine, Shenzhen, 518060, Guangdong, China
Zhifeng Wang & Xingzhi Xu
Corresponding author
Correspondence to Xingzhi Xu.
Ethics declarations
Conflict of interest
The authors declare no competing financial interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article
Cite this article
Zhao, H., Wang, Z., Zhu, M. et al. USP11 suppresses CHK1 activation by deubiquitinating CLASPIN. GENOME INSTAB. DIS. (2021). https://doi.org/10.1007/s42764-021-00034-1
Received21 January 2021
Revised18 February 2021
Accepted21 February 2021
Published07 March 2021
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
Deubiquitination
Replication stress
ATR–CHK1 signaling
用户登录
还没有账号?
立即注册