






Nuclear envelope integrity, DNA replication, damage repair and genome stability
Review Article
Genome Instability & Disease 2, 102–114(2021)
Abstract
The nuclear envelope (NE) not only shields the genetic material inside the nucleus, maintains the dynamic shapes of nucleus, and regulates nuclear exchange with cytosol, but also participates in DNA replication, damage repair and transcription regulation. The loss of NE integrity, as observed in various diseases, has been shown to cause genome instability as a result of genetic material leaking into the cytoplasm. An underestimated but critically important factor of genome integrity is the role of NE components that involve in DNA replication and damage repair. In this review, we summarize the triggers of NE loss and its cellular consequences by focusing on the interactions between NE components and DNA replication and repair factors. Studies on how NE mediates DNA replication and damage repair could shed light on the diagnosis and treatment of human diseases such as cancer and laminopathy.
Introduction
DNA replication is an essential biological process in mammalian cells. Cells duplicate themselves properly within a cell cycle requiring delicate checkpoint control. On the contrary, the retarded replication forks create a regionally unreplicated genome-double-strand breaks (DSBs), resulting in genome instability by the rearrangement and mis-segregation of chromosomes. Abnormally, the failed or faulty completion of DNA replication induces cell stress (Techer et al., 2017). At the genome level, “replication stress” refers to any biological processing or physical conditions that can obstruct the formation of DNA replication forks or interfere with replication dynamics (Wilhelm et al., 2020). However, the mechanism of genome instability caused by replication stress and the discovery of corresponding cellular markers remain elusive. The replication stress can be induced by a variety of exogenous and endogenous factors during cell cycle, such as ultraviolet, reactive oxygen species, DNA lesions and tumorigenesis (Zeman & Cimprich, 2014). Thus, systematic understanding of the mechanism that generates replication stress and how the resulting genome stability affects the fate of cell cycle will illuminate the potential therapeutic targets for various diseases.
The NE, a protective structure in eukaryotic cells, shields the genetic material inside the nucleus and maintains regular processes of DNA replication, transcription, and repair. In normal cells, loss of NE integrity was considered to occur only when chromatin becomes condensed and separated during mitosis. However, recent studies have shown that NE integrity and function can be disrupted and dismantled under various pathological conditions, including cancer, laminopathy, or cell migration within a confined space, thereby causing an irregular exchange of cytoplasm and the chromosomal DNA in the nucleus (De Vos et al., 2011). In addition to the safeguarding role of NE, the NE also participates in cellular processes and signal transduction, including DNA replication, chromosome organization, mitosis, and mechanosensing (Malhas & Vaux, 2011).
In this review, we focus on the diverse cause of compromised NE integrity and the related repair pathways. We will detail the major components of NE and their roles in DNA replication and its stress response, emphasizing the importance of NE integrity in the maintenance of genome stability.
Components of the NE and NE stress
Components of NE
A fully functional NE contains three components: the nuclear lamina, a double lipid bilayer encompassing an inner nuclear membrane (INM) and outer nuclear membrane (ONM), and nuclear pore complexes (NPC). The major components of the nuclear lamina are nuclear lamins, the filamentous proteins that form the meshwork underlying the INM for sustaining the shape and stability of the nucleus, whereas ONM constitutes a perinuclear space contiguous with endoplasmic reticulum (ER) (Fig. 1). In mammalian cells, lamins are classified into two groups based on their structures: A-type lamins (e.g., lamin A and its splice variant lamin C) and B-type lamins (e.g., lamin B1 and lamin B2). The ratio between different lamins depends on the cell type and the extracellular matrix (Swift et al., 2013). The lamins not only bind with chromatin, interact with NE transmembrane proteins, and link the genome to the INM, but also anchor the cytoskeleton via the nucleoskeleton and cytoskeleton (LINC) complex (Jahed & Mofrad, 2019). LINC complex consists of transmembrane proteins SUN and nesprin that penetrate the INM and ONM, respectively (Horn, 2014). In addition, proteins containing certain domains such as LEM (i.e., LAP2, emerin, and Man1) interact with lamin meshwork at the inner membrane (Berk et al., 2013). The lamin meshwork offers a buffering force for the nuclear membrane, serves as a backbone for chromatin anchoring, and regulates gene expression during cell cycle (Gruenbaum & Foisner, 2015). Thus, maintenance of NE function is pivotal for an effective cell cycle and stable nuclear dynamics.
Fig. 1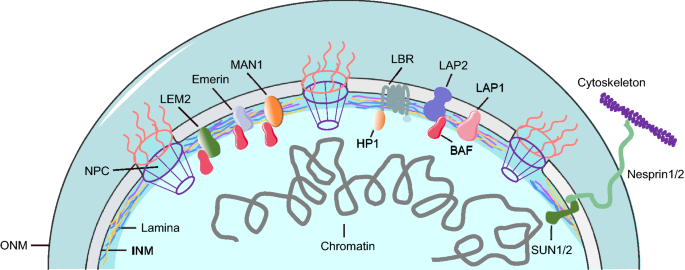
Schematic illustration of nuclear envelope structure. The lamina consists of different lamin filaments, including lamin A/C, lamin B1 and lamin B2. INM inner nuclear membrane, ONM outer nuclear membrane, NPC nuclear pore complex
Full size imageNE stress
Although compromised NE integrity was thought to occur only during mitosis, a process termed nuclear envelope breakdown (NEBD), studies have revealed a loss of NE integrity during interphase under degenerative conditions (Di Micco et al., 2016). Such compromised NE integrity is called “NE stress” that can be categorized into three types: NE erosion, NE shedding, and NE rupture (Robijns et al., 2018).
NE erosion is primarily caused by a disorder of NPC components, which prompts the slow loss of nuclear compartmentalization. Expression of nuclear import and transport factors decreased in the fibroblasts from older adults (Pujol et al., 2002). Clustered NPCs have been found in the cells of patients with Hutchinson–Gilford progeria syndrome (HGPS). Furthermore, overexpression of progerin, a permanently farnesylated, methylated, and truncated form of lamin A, is observed in HGPS (Goldman et al., 2004; Kelley et al., 2011). Taken together, the dysregulation of transport factors, the leakage of NPCs, and the mutation of nuclear lamins affect the formation of NE, leading to NE erosion.
Unlike NE erosion, NE shedding depends on the formation of localized blebs. NE shedding is accompanied by the budding of NE-derived vesicles, the creation of lamins via autophagy, the degradation of chromatin, and the fragmentation of cytoplasmic chromatin. During NE shedding, lamin B1 interacts with the autophagy protein LC3 (Dou et al., 2015; Lenain et al., 2015) and a defective A-type lamin that, along with emerin, cause vast quantities of LC3-positive autophagosomes to approach the NE (Park et al., 2009). NE shedding causes irreversible NE remodeling, increases nuclear permeability and intensifies genome instability (Ivanov et al., 2013).
NE rupture, first identified in HIV infection (de Noronha et al., 2001), shows the completely temporary loss of the NE’s barrier function. In general, a deficiency of lamins generates fragile sites on NE, which are vulnerable to mechanical forces (Cho et al., 2019). Close associations between NE rupture and the loss of lamin A/C and lamin B1 as well as the mutation of nuclear lamins exist in HGPS and cancer (Earle et al., 2020; Penfield et al., 2018). Cancer cells suffer more NE rupture, DNA damage and nuclear deformation through confining space (Denais et al., 2016; Kidiyoor et al., 2020; Shah et al., 2021). The confined migration and compression force increased replication stress and promoted replication fork stalling (Shah et al., 2021). In addition, the ATR defective cells generated NE rupture, perinuclear cGAS (cyclic GMP–AMP synthase) accumulation and aberrant perinuclear chromatin status during interstitial migration or mechanical force loading (Kidiyoor et al., 2020). Loss of NE integrity under mechanical stress also promoted senescence and invasive phenotype by increasing collagen degradation, activation of the ATM kinase and DNA damage. On the contrary, depletion of three-prime repair exonuclease 1 (TREX1) suppressed phenotypes associated DNA damage (Nader et al., 2020). Factors such as apoptosis and the phosphorylation of lamins may stimulate NE rupture (Hatch & Hetzer, 2014; Porwal et al., 2013). However, more evidence is needed to elucidate the dynamics and mechanism of those processes.
NE repair
Intriguingly, the cells will not die, but survive to continue dividing after NE rupture (Robijns et al., 2016), which implies the effective repairing mechanism for NE damage. The endosomal sorting complexes required for transport III (ESCRT-III) is first identified as a key factor in plasma membrane reseal (Jimenez et al., 2014). However, recent reports pointed out that NE rupture site recruited ESCRT-III during cell migration (Denais et al., 2016; Raab et al., 2016). The charged multivesicular body protein 4B (CHMP4B) relocated to rupture site upon NE rupture and depletion of CHMP4B slowed down rupture repair (Denais et al., 2016; Robijns et al., 2016). The LEM (Lap2-Emerin-Man1) family protein LEM2 served as a specific adaptor to recruit CHMP7 and ESCRT-III on the nuclear envelope (Gu et al., 2017). Furthermore, the DNA-binding protein barrier-to-autointegration factor (BAF) bound to exposed DNA, which further recruited LEM proteins to accumulate at rupture sites (Halfmann et al., 2019). The driving mechanism for the recruitment of ESCRT-III to rupture sites is still unknown. Since ESCRT-III as plasma membrane repair factor, other plasma membrane repair protein might involve with NE repair.
NE integrity and DNA replication
DNA replication and nuclear lamins
Although nuclear lamins have long been considered as safeguards that sustain regular cell dynamics, there is limited study on how nuclear lamins affect DNA replication. Early studies used truncated human lamins to disturb the organization of lamins in mammalian cells by microinjection or in vitro nuclear assembly from Xenopus extracts. In both cases, DNA replication decreased during elongation, without shifting the distribution of proteins required to initiate DNA replication, including xMCM3, xORC2, and DNA polymerase α. By contrast, the distribution of the replication factor complex (RFC) and proliferating cell nuclear antigen (PCNA) had colocalized with the lamins in aggregates at post-assembly disrupted nuclei, indicating the regulatory function of nuclear lamins in DNA replication elongation (Moir et al., 2000). Indeed, PCNA co-localizes with lamin B but not lamin A/C at replication sites in mid-late S phase (Moir et al., 1994). Replication proteins, nucleolus, and lamin A/C constitute the replication foci in the early G1 phase and the late S phase, while the DNA replication sites are distributed throughout the nucleus (Kennedy et al., 2000). Consistently, the lamins modulate PCNA’s position on chromatin, and mutations of LMNA decrease the binding affinity with PCNA compromising their interactions in vitro (Shumaker et al., 2008). Furthermore, the expression of prelamin A in U2OS cells can induce Polη and fork stalling during DNA replication with mono-ubiquitination of PCNA. The prelamin A intensifies the co-localization of PCNA and the DNA damaging marker γH2AX, thereby causing stalled DNA replication forks to collapse into DNA double-strand breaks, which competes with mature nuclear lamins to interact with PCNA (Cobb et al., 2016).
A study indirectly demonstrates that the mutated nuclear lamins can mitigate interaction between PCNA and lamins, which is based on mass spectrometric screening of proteins that interact with lamin A and progerin. The results showed that 110 proteins interact with both lamin A and progerin, in which 8 proteins preferentially interact with lamin A and 27 proteins preferentially interact with progerin. In particular, PCNA shows stronger interaction with progerin relative to lamin A (Kubben et al., 2010). Progerin-expressing cells cause the accumulation of xeroderma pigmentosum group A (XPA) at stalled or collapsed replication forks, accompanied by the reduction of PCNA. On the contrary, depletion of XPA or progerin could restore PCNA at the forks, which indicates the importance of nuclear lamins in DNA replication (Hilton et al., 2017). In addition, progerin induces replication stress that consequently causes the upregulation of the cGAS–STING cytosolic DNA-sensing pathway and interferon-like response (Kreienkamp et al., 2018). Despite the association between nuclear lamins and DNA replication factors, the absence of nuclear lamins enhances cellular sensitivity to replication stress. Lamin A/C-deficient cells are more sensitive to interstrand cross-links (ICL) and treatments for replication stress involving response-delayed γH2AX removal, increase chromosome aberrations, and reduce recruitment of the DNA repair factors Mre11, CtIP, RAD51, RPA, and FANCD2 (Singh et al., 2013).
Therefore, nuclear lamins play a key role in DNA replication and the response to replication stress: either directly, by binding to replisomes and facilitating the progression and stability of replication forks, or indirectly, by regulating PCNA and RFC. Mutant lamins, especially progerin, hinder the progress of replication by mislocalization of factors that associate with replisomes. Moreover, the dysfunction of lamins can obstruct the proper recruitment of the protective factors including RAD51, RPA and FANCD2 at replication forks upon fork stalling, thereby causing replication stress and genomic instability.
Lamina-associated domains and replication timing
In eukaryotes, DNA replication does not simply duplicate DNA, numerous proteins and molecules participate in and the entire genome needs to be stripped down and assembled at replication fork. To maintain faithful DNA replication, a spatiotemporal program known as replication timing is identified in eukaryotes (Vouzas & Gilbert, 2021). The complex microenvironment in nucleus can influence chromatin organization and activities. The lamina-associated domains (LADs) anchors to nuclear lamina which supports chromosome topology at interphase and overall genome spatial organization (van Steensel & Belmont, 2017). The LADs are firstly identified by DNA adenine methyltransferase identification (DamID) tethered to Lamin B1 (Pickersgill et al., 2006). Furthermore, LADs can be mapped by chromatin immunoprecipitation (ChIP) or visualized by microscopy (Handoko et al., 2011). Several thousands of genes have very low expression inside LADs suggesting the repression state of these genes (Leemans et al., 2019). The detachments or interactions of LADs and nuclear lamina lead to activating or reducing transcription (Robson et al., 2017). The contacts of LADs with nuclear lamina are rapidly established at G1 phase, where replication timing starts in the reassembled nucleus (van Schaik et al., 2020). The genes activated inside of the LADs reduce nuclear lamina interaction with transcription unit and shift the replication timing from late to early (Brueckner et al., 2020). Altogether, the LADs participate in transcription processing and affect replication timing. Nevertheless, the mechanisms of how the LADs affect transcription activation and whether more factors can interact with LADs need further exploration.
Other NE components in DNA replication
NE assembly deficiency on a lagging chromosome in mitosis may lead to the NE disruption and defects in nuclear import in the resulting micronuclei (Liu et al., 2018), which in turn causes genome instability and rearrangements, such as commonly observed chromothripsis in cancer (Zhang et al., 2015). The replication stress will be introduced in the S phase if NE rupture is not fully repaired. Many DNA replication factors need to be enriched in the nucleus for the repair. NE components such as LEM2, BAF and CHMP7 play important roles in NE dynamics and DNA replication. For example, the reformation of sealed nuclei requires ESCRTs (endosomal sorting complexes required for transport) and LEM2, a transmembrane ESCRT adaptor. LEM2 phase separation, together with CHMP7 promotes ESCRT-mediated nuclear envelope remodeling (von Appen et al., 2020). Furthermore, it has been demonstrated recently that K6-linked SUMOylation of BAF, which interacts with LEM2 and emerin, supports its binding to DNA and interaction with PCNA which regulates DNA replication. Disrupting the SUMOylation and de-SUMOylation cycle of BAF not only disturbs nuclear integrity, but also induces DNA replication failure (Lin et al., 2020). Interestingly, it has been shown that the BAF protein promotes the formation of a single nucleus via its DNA cross-bridging activity during nuclear assembly after mitosis (Samwer et al., 2017). It protects against a basal cGAS-STING response (Ma et al., 2020), and restricts cGAS on nuclear DNA to prevent innate immune activation (Guey et al., 2020). Despite their importance, the spatiotemporal regulation of NE rupture and repair, especially under stress and pathological conditions, remain poorly understood. It will be of interest to discover how the protein-protein interaction network which includes LEM2, CHMP7, BAF, emerin, cGAS, lamins and other unidentified factors operates to maintain NE stability, safeguard DNA replication and regulate immune response.
NE proteins and DNA repair
Maintaining regular DNA replication in the nucleus is essential for genomic stability and fundamental cellular functions, especially upon exposure to replication stress. Particular DNA repair systems or pathways have evolved to detect and repair various DNA lesions, although classical DNA repair proteins have been regarded as chief factors in different DNA repair pathways. NE and relevant proteins play essential roles in the response to DNA damage and its repair, including nuclear lamins, nuclear actin, and the LINC complex (Misteli & Soutoglou, 2009; Simon & Wilson, 2011). The significance of those proteins in DNA repair systems has been demonstrated in several pathological conditions, including cancer, aging, and neurological disorders such as Parkinson’s Disease and Alzheimer’s Disease (Brosh et al., 2017; Datta & Brosh, 2019; Worman et al., 2010). Therefore, elucidating their impact on DNA repair processes can clarify responses to DNA damage as well as its pathological consequences.
Nuclear lamins as key factors in DNA repair pathways
Nuclear lamins have both direct and indirect functions in DNA replication. During replication, the exogenous and endogenous factors that cause replication stress will induce additional DNA damage (De Vos et al., 2011); nuclear lamins play a unique role in response to DNA damage and repair (Gonzalo & Kreienkamp, 2015). In general, two major pathways for repairing DNA DSBs compose of the non-homologous end joining (NHEJ) and the homologous recombination (HR) pathways. The NHEJ is the primary pathway for DSBs not associated with replication and can function throughout the cell cycle, albeit chiefly in G0 and G1 phases (Kakarougkas & Jeggo, 2014; Pannunzio et al., 2018). HR predominantly occurs in the S and G2 phases using sister chromatids as a template for the repair process (Goodarzi & Jeggo, 2013; Wright et al., 2018). Aside from those two major DNA repair pathways, two other pathways can be used for DNA DSBs: alternative non-homologous end joining (A-NHEJ) and single-strand annealing (SSA) (Chang et al., 2017; Sallmyr & Tomkinson, 2018). To date, numerous studies have demonstrated that both types of lamins play critical roles in mediating the repair of DNA DSBs.
Although factors driving the choice of DNA repair pathway remain unclear, cell cycle may be a major one (Her & Bunting, 2018). A-type lamins (e.g., lamin A/C), considered to be essential in the repair response to DNA DSBs, can function in both the NHEJ and HR pathways, as well as mediate the cross talk between pathway choice and the factors involved (Gonzalo, 2014; Redwood, Gonzalez-Suarez, et al., 2011). Lamin A/C can bind 53BP1, a key modulator in the NHEJ repair pathway, and promote its nuclear retention and stability as results of recruiting relative proteins to the sites of DSBs (Gibbs-Seymour et al., 2015). Upon DNA damage, the heterodimer Ku70–Ku80 and DNA-dependent protein kinase (DNA-PK) are recruited to damage sites and form a complex during end-bridging processes. Beyond that, 53BP1 inhibits the recruitment of BRCA1 to DSB sites in the G1 phase, which reduces the HR repair pathway and the A-NHEJ pathway—both involving microhomology-mediated end joining (Brachner & Foisner, 2011; Gibbs-Seymour et al., 2015) (Fig. 2A). Meanwhile, the depletion of lamin A/C can destabilize 53BP1, which promotes 53BP1 to be degraded by the ubiquitin-conjugating enzyme H7 (UbcH7) or cleaved by an endosomal or lysosomal protease, cathepsin L, resulting in a deficiency in NHEJ (Gonzalo, 2014; Pozo et al., 2017).
Besides the role of lamins in NHEJ, lamin A/C is a dominant factor in regulating HR repair pathway, which associates with BRCA1 and RAD51 regulators (Redwood, Gonzalez-Suarez et al., 2011). Lamin A/C can inhibit p130-E2F4, the repressor complex that mediates the downregulation of BRCA1 and RAD51 (Redwood, Gonzalez-Suarez et al., 2011). The depletion of lamin A/C limits the inhibition of p130–E2F4 and represses the transcription of BRCA1 and RAD51, thereby causing the downregulated expression of BRCA1 and RAD51 (Redwood, Gonzalez-Suarez, et al., 2011). More recent evidence demonstrates the direct interaction between lamin A/C and RAD51 (Li et al., 2018). Lamin-binding ligand 1 bound directly with lamin A/C, which inhibited its interaction with RAD51 and accelerated the proteasome-mediated degradation of RAD51 (Li et al., 2018). Such findings suggest that lamin A/C can balance and shift between DNA DSB repair pathways (Fig. 2B). Understanding how lamin A/C supports the choice of DNA repair pathway and the interactions with proteins involved therein may elucidate the underlying mechanisms of those pathways and their roles in genome instability.
Fig. 2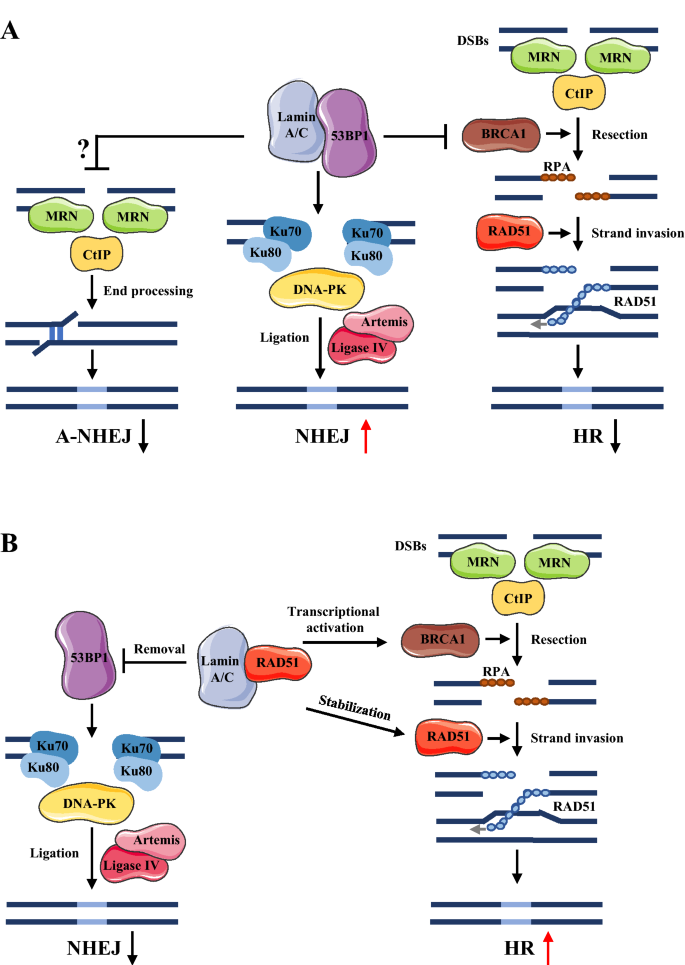
Lamin A/C plays important roles in DNA DSBs repair pathways. a Lamin A/C binding to 53BP to promote non-homologous end joining (NHEJ) DSBs repair pathway. b Lamin A/C binds to Rad51 and promotes homologous recombination (HR) repair pathway
Full size imageSimilar to A-type lamins, B-type lamins are also active in various functional processes, including DNA replication, repair, and transcription (Liu et al., 2015; Malhas et al., 2009; Tang et al., 2008). Lamin B1 has proven to adjust the HR DNA repair pathway and maintain the stability of RAD51 in DNA DSBs caused by replication fork collapse or ionizing radiation (Butin-Israeli et al., 2013; Liu et al., 2015). In addition, after the induction of DSBs, the elevated level of RAD51 can be attributed to the increase in lamin B1’s binding to RAD51 and stabilizing it by inhibiting its proteasome-mediated degradation (Liu et al., 2015) (Fig. 3). Lamin B1 regulates the expression of several essential genes in HR repair pathway (e.g., BRCA1, RAD51, and MRE11) and thus modulates the HR repair pathway (Butin-Israeli et al., 2013). In addition, lamin B1 also engages in nucleotide excision repair induced by ultraviolet damage. Lamin B1 depletion in cancer cells causes the downregulation of DNA damage binding protein 1, Cockayne syndrome B, PCNA, and Polη proteins that are required for NE repair (Butin-Israeli et al., 2013). The regulation of those proteins by lamin B1 has been attributed to transcription, mRNA-processing, and/or the stability of proteins (Butin-Israeli et al., 2015). Lamins can conjugate with each other and form networks within nuclei (Shimi et al., 2008), and the lamins’ interaction is critical for DNA repair pathways. For instance, lamin B1 is essential for maintaining the mobility and distribution of lamin A/C, which further affects DNA repair pathways (Shimi et al., 2008).
Fig. 3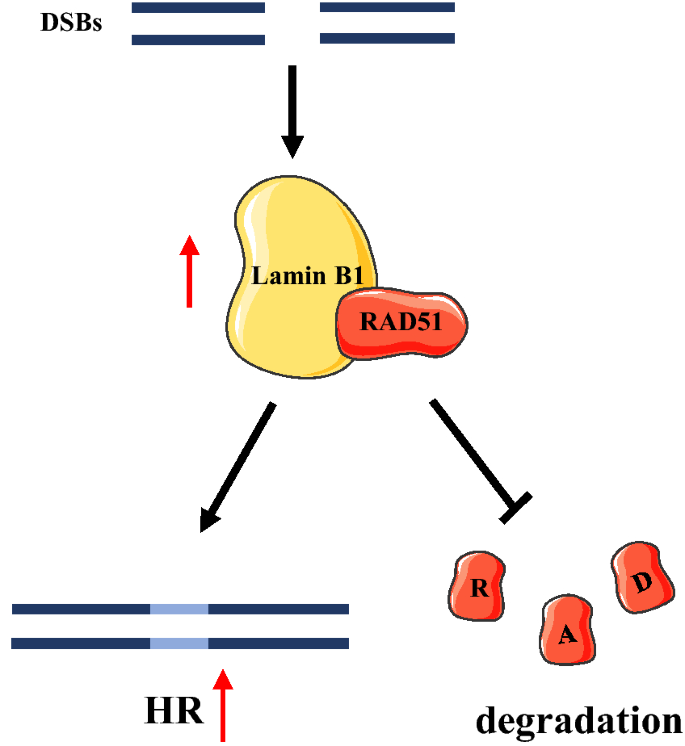
The role of lamin B1 in DNA DSBs repair pathway by HR. DSBs induced lamin B1 binding to RAD51 that increases HR repair pathway and prevents RAD51 degradation from proteasome
Full size imageAltogether, both types of lamins play pivotal roles in regulating specific DNA repair pathway genes. To expand such knowledge, further studies on the relationship between nuclear lamins and DNA repair mechanisms need to be conducted.
LINC complex in DNA damage repair
The LINC complex bridges the INM and ONM, which is the key link between the nucleoskeleton and cytoskeleton (Chang et al., 2015). The major proteins in the LINC complex are transmembrane SUN domain proteins SUN1-SUN5 in mammalian cells (Crisp et al., 2006). Among them, SUN1 and SUN2 interact with nuclear lamins, emerin, and INM proteins (Wilson & Foisner, 2010), as well as with nesprins, a KASH-domain protein located in the perinuclear space where it interacts with microtubules, microfilaments, and intermediate filaments in the cytoskeleton (Starr & Fridolfsson, 2010; Tapley & Starr, 2013). The LINC complex has been shown to participate in various functions, including mediating nuclear shape and position, nuclear migration, DNA damage repair, and mechano-transduction between the cytoplasm and nucleus (Gundersen & Worman, 2013; Hieda, 2017; Lawrence et al., 2016). The human (SUN1) and C. elegans (UNC-84) homologs inhibit the NHEJ pathway during DSB repair via interactions with both DNA-PK and Ku70-Ku80 and activate HR by promoting the binding of RAD51 to damage sites (Lawrence et al., 2016). Both SUN1 and UNC-84 also participate in DNA ICL repair by recruiting FAN-1 nuclease, the endonuclease shuttled to the nucleoplasm after treatment of ICL reagents (Lawrence et al., 2016). On top of that, SUN1 and SUN2 have been characterized as modulators in the 53BP1-dependent chromatin mobility pathway, which is associated with nesprin and in contact with microtubules, the transfer of cytoskeletal forces to nucleus, and the enhanced mobility of chromatin (Lottersberger et al., 2015). Interestingly, in mouse embryonic fibroblasts, the interaction between DNA-PK with SUN1 and SUN2 is not affected by DNA damaging reagents (Luxton et al., 2011). Thus, the LINC complex either promotes HR pathways by inhibiting NHEJ or plays an unclear role. As such, it continues to be a hotspot for researchers clarifying the relationship between the LINC complex and DNA repair pathways.
Nuclear pore complex and DNA damage repair
The nuclear pore complex (NPC) constitutes multiple copies of 30 different nucleoporins that associates as stable sub-complexes (D'Angelo & Hetzer, 2008). The largest mammalian NPC sub-complex is the Y-shaped Nup107-Nup160 complex (called Nup84 in budding yeast), which locates both at the cytoplasmic and nuclear side (Schwartz, 2016). Despite its role in molecule transport, nuclear organization and transcription, the function of NPC in DNA damage repair has also attracted more attention. In budding yeast, while the DNA damaging occurs, the NPCs or inner nuclear membrane SUN protein Mps3 act as binding site for DNA lesions, including persistent DSBs, collapsed forks and subtelomeric regions (Nagai et al., 2008; Therizols et al., 2006). The same phenomenon occurs on fission yeast homologue Mps3 and Sad1, indicating the role of NE in DSB repair (Swartz et al., 2014). Several studies have demonstrated binding DNA lesions to NPCs requiring SUMOylation processing (Churikov et al., 2016; Nagai et al., 2008). SUMOylated proteins as key substrates for the SUMO targeted ubiquitin ligase (STUbL) can target DNA damaging to NPCs, which further undergo degradation or direct SUMO removal by SENP proteases (Churikov et al., 2016; Hickey et al., 2012). In yeast, the SENP protease Ulp1 is constitutively attached to NPCs, while the mutation or depletion of Nup84 caused delocalization of Ulp1 resulting in DNA damage sensitivity (Palancade et al., 2007) and defective DNA repair and replication stress response (Gaillard et al., 2019). In addition, the E3 SUMO ligase Pli1 acted at arrested forks to secure nascent strands and promoted relocation to NPCs by producing poly-SUMOylation which further inhibited homologous recombination (HR)-dependent DNA synthesis pathway (Kramarz et al., 2020). Besides, another NPC protein Nup1 also regulated telomere relocalization to nuclear pores and reduced CAG/CTG triplet repeats relocated to the NPC (Aguilera et al., 2020). For instance, in eukaryotes, a SUMO-dependent manner effected on removing breaks within repeated sequences, such as heterochromatin, ribosomal DNA from their chromatin environment and facilitated RAD51 loading, further completed HR repair (Horigome et al., 2019; Tsouroula et al., 2016). Taken together, these studies indicate that SUMOylation-dependent NPCs participate in DNA damage repair pathway, which results in the transport of damage sites from their normal subnuclear position to the nuclear pore, a favored site for repairing.
The NE and diseases
Tumorigenesis and nuclear lamins
For decades, the mutation of nuclear lamin genes and the alternation of their structures have been known to be involved with cancer. Nevertheless, the role of nuclear lamins in tumorigenesis and in inducing NE rupture has not yet been elucidated. Because nuclear lamins can maintain the shape of the nucleus, changing expression levels of lamin proteins, especially lamin A/C, have long been detected in various types of cancer (Sakthivel & Sehgal, 2016). Studies have revealed the correlation between the expression of nuclear lamins and tumor malignance. One study showed positive association between upregulated expression of lamin A/C and better outcomes from therapy for colorectal, prostate, and breast cancers (Irianto et al., 2016). By contrast, other studies have indicated prostate, colon, ovarian, and gastric cancers, all of which exhibit a lower expression of nuclear lamins and poor survival outcomes (Denais & Lammerding, 2014; Irianto et al., 2016). In addition, depletion of lamin A/C in non-methylated neuroblastoma cells promotes tumorigenesis. Although the hypermethylation of CpG islands in the LMNA gene reduces the expression of lamin A/C, the effect can be reversed by a demethylating agent and thereby improving the outcomes of cancer treatment by inhibition of the migration and invasion of cancer cells, as well as inducing tumor senescence (Rauschert et al., 2017).
Apart from the association of the expression of nuclear lamins with tumoral properties, NE rupture has been found in several cancer cell lines. Two tumor suppressors, TP53 and RB1, have been reported to prevent NE rupture without affecting the expression of NE components and regulatory proteins, including lamin A/C, lamin B1, CHMP4B, and CHMP2A (Yang et al., 2017). Another study has shown varied responses to replication stress among different cancer cell lines expressing different levels of p53 and lamins. MCF7 cells with wild-type p53 and lamin A/C were senescent after replication stress due to the reduced expression of lamin B1. However, cells with p53 and lamin A/C mutations died during apoptosis. These findings demonstrate that both p53 and lamins play important roles in cells responding to replication stress with senescence or apoptosis (Lukasova et al., 2019).
Considering all of those findings, nuclear lamins clearly play a key role in tumorigenesis, although the effects of their expression in malignant transformation remain unknown. In response, the mechanism study by which tumor suppressor and lamin proteins react to tumorigenesis by inducing replication stress and NE rupture would be especially valuable.
Laminopathy and NE integrity
The laminopathy is a collection of 15 distinct diseases including muscular, neurologic, cardiac, metabolic and premature aging syndromes (Ho & Hegele, 2019), which are caused by gene mutations of lamins and NE proteins. The majority of the mutations occur in LMNA, LMNB1 and LMNB2, which encode lamin A/C, lamin B1 and lamin B2, respectively (Briand & Collas, 2018). Besides, several other NE genes also lead to laminopathy, such as EMD, LAP2, LBR, ZMPSTE24, SYNE-1, and NUP62 (Burke & Stewart, 2006). The altered NE proteins cause a reduction of mechanical stability of nucleus and increase of NE rupture in laminopathy. The normal NE proteins sustain the regular shape and stability of nucleus and repair the NE rupture in an efficiency way. On the contrary, the nuclei from laminopathic patients are more fragile and easily disrupted by mechanical stress compared with healthy controls (Zwerger et al., 2013). The nuclei of HGPS cells are stiffer than normal cells. For instance, Emery-Dreifuss muscular dystrophy mice cells have both deformed nuclei, decreased cytoskeletal stiffness and irregular gene in mechanosensing of the cellular environment (Broers et al., 2004). Taken together, the aberrant of NE integrity in laminopathy contributes to the frequency of NE rupture, which further leads to genome instability. Investigation of the molecular mechanisms of NE rupture underlying laminopathies, will provide guidance for novel therapeutic approach discovery.
Concluding remarks
The NE provides the fundamental environment for regular nuclear dynamics and nucleus-cytoplasm exchange. Under various diseases, conditions, and mechanical forces, the loss of NE integrity can occur and cause further disorder of transport involving the NE, NE rupture, nuclear remodeling, and, ultimately, genome instability. Although many studies have addressed the consequences of NE loss and NE stress, their catalyzing factors are critical to explore. For that reason, screening new interacting proteins and evaluating their roles in up- and downstream interaction with various degrees of NE integrity and NE functions can broaden perspectives on the regulation of NE. Besides, it is also essential to discover signaling pathways and new functions of key proteins in NE rupture and repair. This will address the importance and crosstalk between classical cell membrane regulation and novel NE dynamic pathways.
Aside from NE integrity’s role in modulating NE stress and repair, it actively participates in DNA replication and repair. However, the underlying molecular mechanism and function of those processes remain poorly understood. To shed light on the relationships among NE integrity and DNA replication and repair, it is important to determine what and how the upstream elements influence NE, which might affect DNA replication and repair. Whether single or multiple NEs factors are integrated into the same pathway and their cross-talk during DNA replication and repair require further investigation. Advanced techniques such as novel imaging platforms (e.g., multicolored super-resolution imaging) can record the dynamic movement of NE integrity and detect the interaction between NE proteins and DNA replication and repair proteins. Using such equipment to identify potential factors associated with NE proteins can be an efficient means to investigate the relevance and mechanisms of NE’s role in DNA replication and repair.
Current studies of NE focus on the effects of NE defects on DNA replication, damage repair and genome stability. It is possible that the components in these processes could regulate NE integrity via a feedback loop (Fig. 4). One important future direction is to understand how factors involved in DNA replication and DNA damage response modulate NE integrity. It would be of interest to investigate the role of DNA replication and damage repair factors in laminopathy. Altogether, thoroughly studying the underlying mechanisms of NE integrity in relation to genome stability can provide valuable insights into NE dynamics and help to identify new factors mediating DNA replication and repair. Such knowledge can afford clinical benefits, including the earlier diagnosis of severe diseases such as cancer and laminopathy, and, as a result, higher curative ratios and better outcomes for patients. These new insights will create testable hypotheses regarding potential future intervention strategies.
Fig. 4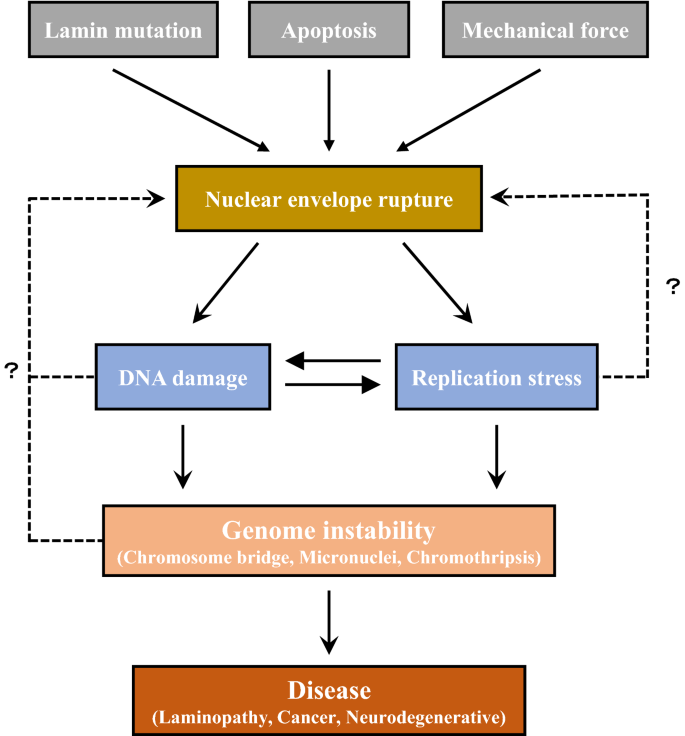
Relationship between NE rupture, DNA replication stress and genome instability. Many factors such as lamin mutation, apoptosis and mechanical stress could cause nuclear envelop rupture, followed by DNA damage and replication stress. This may in turn induce genome instability which could be a trigger for human diseases
Full size imageAbbreviations
A-NHEJ:
Alternative non-homologous end joining
ATM:
Ataxia-telangiectasia mutated protein
ATR:
Ataxia telangiectasia and Rad3-related protein
BAF:
Barrier-to-autointegration factor
cGAS:
Cyclic GMP-AMP synthase
ChIP:
Chromatin immunoprecipitation
CHMP:
Charged multivesicular body protein
DamID:
DNA adenine methyltransferase identification
DNA:
Deoxyribonucleic acid
DNA-PK:
DNA-dependent protein kinase
DSBs:
Double-strand breaks
ESCRT:
Endosomal sorting complexes required for transport
ER:
Endoplasmic reticulum
FANCD2:
Fanconi anemia group D2 protein
HGPS:
Hutchinson-Gilford progeria syndrome
HR:
Homologous recombination
ICL:
Interstrand cross-links
INM:
Inner nuclear membrane
LADs:
Lamina-associated domains
LEM:
Lap-Emerin-Man domain protein
LINC:
Linker of nucleoskeleton and cytoskeleton
NE:
Nuclear envelope
NEBD:
Nuclear envelope breakdown
NHEJ:
Non-homologous end joining
NPC:
Nuclear pore complexes
ONM:
Outer nuclear membrane
PCNA:
Proliferating cell nuclear antigen
RFC:
Replication factor complex
RPA:
Replication protein A
SSA:
Single-strand annealing
STING:
Stimulator of interferon genes
STUbL:
SUMO targeted ubiquitin ligase
SUMO:
Small ubiquitin-like modifier
TREX1:
Three-prime repair exonuclease 1
UbcH7:
Ubiquitin-conjugating enzyme H7
XPA:
Xeroderma pigmentosum group A
References
Aguilera, P., Whalen, J., Minguet, C., Churikov, D., Freudenreich, C., Simon, M. N., & Geli, V. (2020). The nuclear pore complex prevents sister chromatid recombination during replicative senescence. Nature Communications, 11, 160
Berk, J. M., Tifft, K. E., & Wilson, K. L. (2013). The nuclear envelope LEM-domain protein emerin. Nucleus, 4, 298–314
Brachner, A., & Foisner, R. (2011). Lamins reach out to novel functions in DNA damage repair Comment on: Redwood AB, et al. Cell Cycle, 2011(10), 2550–2561 (Cell Cycle 10, 3426–3427).
Briand, N., & Collas, P. (2018). Laminopathy-causing lamin A mutations reconfigure lamina-associated domains and local spatial chromatin conformation. Nucleus, 9, 216–226
Broers, J. L., Peeters, E. A., Kuijpers, H. J., Endert, J., Bouten, C. V., Oomens, C. W., Baaijens, F. P., & Ramaekers, F. C. (2004). Decreased mechanical stiffness in LMNA−/− cells is caused by defective nucleo-cytoskeletal integrity: implications for the development of laminopathies. Human Molecular Genetics, 13, 2567–2580
Brosh, R. M., Bellani, M., Liu, Y., & Seidman, M. M. (2017). Fanconi anemia: a DNA repair disorder characterized by accelerated decline of the hematopoietic stem cell compartment and other features of aging. Ageing Research Reviews, 33, 67–75
Brueckner, L., Zhao, P. A., van Schaik, T., Leemans, C., Sima, J., Peric-Hupkes, D., Gilbert, D. M., & van Steensel, B. (2020). Local rewiring of genome-nuclear lamina interactions by transcription. EMBO Journal, 39, e103159
Burke, B., & Stewart, C. L. (2006). The laminopathies: the functional architecture of the nucleus and its contribution to disease. Annual Review of Genomics and Human Genetics, 7, 369–405
Butin-Israeli, V., Adam, S. A., & Goldman, R. D. (2013). Regulation of nucleotide excision repair by nuclear lamin B1. PLoS ONE, 8 (1), e69169. https://doi.org/10.1371/journal.pone.0069169
Butin-Israeli, V., Adam, S. A., Jain, N., Otte, G. L., Neems, D., Wiesmuller, L., Berger, S. L., & Goldman, R. D. (2015). Role of lamin B1 in chromatin instability. Molecular and Cellular Biology, 35, 884–898
Chang, H. H. Y., Pannunzio, N. R., Adachi, N., & Lieber, M. R. (2017). Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nature Reviews Molecular Cell Biology, 18, 495–506
Chang, W., Worman, H. J., & Gundersen, G. G. (2015). Accessorizing and anchoring the LINC complex for multifunctionality. Journal of Cell Biology, 208, 11–22
Cho, S., Vashisth, M., Abbas, A., Majkut, S., Vogel, K., Xia, Y., Ivanovska, I. L., Irianto, J., Tewari, M., Zhu, K., et al. (2019). Mechanosensing by the lamina protects against nuclear rupture, DNA damage, and cell-cycle arrest. Developmental Cell, 49(920–935), e925
Churikov, D., Charifi, F., Eckert-Boulet, N., Silva, S., Simon, M. N., Lisby, M., & Geli, V. (2016). SUMO-dependent relocalization of eroded telomeres to nuclear pore complexes controls telomere recombination. Cell Reports, 15, 1242–1253
Cobb, A. M., Murray, T. V., Warren, D. T., Liu, Y., & Shanahan, C. M. (2016). Disruption of PCNA-lamins A/C interactions by prelamin A induces DNA replication fork stalling. Nucleus, 7, 498–511
Crisp, M., Liu, Q., Roux, K., Rattner, J. B., Shanahan, C., Burke, B., Stahl, P. D., & Hodzic, D. (2006). Coupling of the nucleus and cytoplasm: role of the LINC complex. Journal of Cell Biology, 172, 41–53
D’Angelo, M. A., & Hetzer, M. W. (2008). Structure, dynamics and function of nuclear pore complexes. Trends in Cell Biology, 18, 456–466
Datta, A., & Brosh, R. M. (2019). Holding All the cards-how Fanconi anemia proteins deal with replication stress and preserve genomic stability. Genes-Basel, 10, 170
de Noronha, C. M., Sherman, M. P., Lin, H. W., Cavrois, M. V., Moir, R. D., Goldman, R. D., & Greene, W. C. (2001). Dynamic disruptions in nuclear envelope architecture and integrity induced by HIV-1 Vpr. Science, 294, 1105–1108
De Vos, W. H., Houben, F., Kamps, M., Malhas, A., Verheyen, F., Cox, J., Manders, E. M., Verstraeten, V. L., van Steensel, M. A., Marcelis, C. L., et al. (2011). Repetitive disruptions of the nuclear envelope invoke temporary loss of cellular compartmentalization in laminopathies. Human Molecular Genetics, 20, 4175–4186
Denais, C., & Lammerding, J. (2014). Nuclear mechanics in cancer. Advances in Experimental Medicine and Biology, 773, 435–470
Denais, C. M., Gilbert, R. M., Isermann, P., McGregor, A. L., te Lindert, M., Weigelin, B., Davidson, P. M., Friedl, P., Wolf, K., & Lammerding, J. (2016). Nuclear envelope rupture and repair during cancer cell migration. Science, 352, 353–358
Di Micco, A., Frera, G., Lugrin, J., Jamilloux, Y., Hsu, E. T., Tardivel, A., De Gassart, A., Zaffalon, L., Bujisic, B., Siegert, S., et al. (2016). AIM2 inflammasome is activated by pharmacological disruption of nuclear envelope integrity. Proceedings of the National academy of Sciences of the United States of America, 113, E4671–E4680
Dou, Z., Xu, C., Donahue, G., Shimi, T., Pan, J. A., Zhu, J., Ivanov, A., Capell, B. C., Drake, A. M., Shah, P. P., et al. (2015). Autophagy mediates degradation of nuclear lamina. Nature, 527, 105–109
Earle, A. J., Kirby, T. J., Fedorchak, G. R., Isermann, P., Patel, J., Iruvanti, S., Moore, S. A., Bonne, G., Wallrath, L. L., & Lammerding, J. (2020). Mutant lamins cause nuclear envelope rupture and DNA damage in skeletal muscle cells. Nature Materials, 19, 464–473
Gaillard, H., Santos-Pereira, J. M., & Aguilera, A. (2019). The Nup84 complex coordinates the DNA damage response to warrant genome integrity. Nucleic Acids Research, 47, 4054–4067
Gibbs-Seymour, I., Markiewicz, E., Bekker-Jensen, S., Mailand, N., & Hutchison, C. J. (2015). Lamin A/C-dependent interaction with 53BP1 promotes cellular responses to DNA damage. Aging Cell, 14, 162–169
Goldman, R. D., Shumaker, D. K., Erdos, M. R., Eriksson, M., Goldman, A. E., Gordon, L. B., Gruenbaum, Y., Khuon, S., Mendez, M., Varga, R., et al. (2004). Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proceedings of the National academy of Sciences of the United States of America, 101, 8963–8968
Gonzalo, S. (2014). DNA damage and lamins. Cancer Biology and the Nuclear Envelope: Recent Advances May Elucidate Past Paradoxes, 773, 377–399
Gonzalo, S., & Kreienkamp, R. (2015). DNA repair defects and genome instability in Hutchinson-Gilford Progeria Syndrome. Current Opinion in Cell Biology, 34, 75–83
Goodarzi, A. A., & Jeggo, P. A. (2013). The repair and signaling responses to DNA double-strand breaks. Advances in Genetics, 82, 1–45
Gruenbaum, Y., & Foisner, R. (2015). Lamins: nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Annual Review of Biochemistry, 84, 131–164
Gu, M., LaJoie, D., Chen, O. S., von Appen, A., Ladinsky, M. S., Redd, M. J., Nikolova, L., Bjorkman, P. J., Sundquist, W. I., Ullman, K. S., et al. (2017). LEM2 recruits CHMP7 for ESCRT-mediated nuclear envelope closure in fission yeast and human cells. Proceedings of the National academy of Sciences of the United States of America, 114, E2166–E2175
Guey, B., Wischnewski, M., Decout, A., Makasheva, K., Kaynak, M., Sakar, M. S., Fierz, B., & Ablasser, A. (2020). BAF restricts cGAS on nuclear DNA to prevent innate immune activation. Science, 369, 823–828
Gundersen, G. G., & Worman, H. J. (2013). Nuclear positioning. Cell, 152, 1376–1389
Halfmann, C. T., Sears, R. M., Katiyar, A., Busselman, B. W., Aman, L. K., Zhang, Q., O’Bryan, C. S., Angelini, T. E., Lele, T. P., & Roux, K. J. (2019). Repair of nuclear ruptures requires barrier-to-autointegration factor. Journal of Cell Biology, 218, 2136–2149
Handoko, L., Xu, H., Li, G., Ngan, C. Y., Chew, E., Schnapp, M., Lee, C. W., Ye, C., Ping, J. L., Mulawadi, F., et al. (2011). CTCF-mediated functional chromatin interactome in pluripotent cells. Nature Genetics, 43, 630–638
Hatch, E., & Hetzer, M. (2014). Breaching the nuclear envelope in development and disease. Journal of Cell Biology, 205, 133–141
Her, J., & Bunting, S. F. (2018). How cells ensure correct repair of DNA double-strand breaks. Journal of Biological Chemistry, 293, 10502–10511
Hickey, C. M., Wilson, N. R., & Hochstrasser, M. (2012). Function and regulation of SUMO proteases. Nature Reviews Molecular Cell Biology, 13, 755–766
Hieda, M. (2017). Implications for diverse functions of the LINC complexes based on the structure. Cells, 6, 3. https://doi.org/10.3390/cells6010003
Hilton, B. A., Liu, J., Cartwright, B. M., Liu, Y., Breitman, M., Wang, Y., Jones, R., Tang, H., Rusinol, A., Musich, P. R., et al. (2017). Progerin sequestration of PCNA promotes replication fork collapse and mislocalization of XPA in laminopathy-related progeroid syndromes. The FASEB Journal, 31, 3882–3893
Ho, R., & Hegele, R. A. (2019). Complex effects of laminopathy mutations on nuclear structure and function. Clinical Genetics, 95, 199–209
Horigome, C., Unozawa, E., Ooki, T., & Kobayashi, T. (2019). Ribosomal RNA gene repeats associate with the nuclear pore complex for maintenance after DNA damage. PLoS Genetics, 15, e1008103
Horn, H. F. (2014). LINC complex proteins in development and disease. Current Topics in Developmental Biology, 109, 287–321
Irianto, J., Pfeifer, C. R., Ivanovska, I. L., Swift, J., & Discher, D. E. (2016). Nuclear lamins in cancer. Cellular and Molecular Bioengineering, 9, 258–267
Ivanov, A., Pawlikowski, J., Manoharan, I., van Tuyn, J., Nelson, D. M., Rai, T. S., Shah, P. P., Hewitt, G., Korolchuk, V. I., Passos, J. F., et al. (2013). Lysosome-mediated processing of chromatin in senescence. Journal of Cell Biology, 202, 129–143
Jahed, Z., & Mofrad, M. R. (2019). The nucleus feels the force, LINCed in or not! Current Opinion in Cell Biology, 58, 114–119
Jimenez, A. J., Maiuri, P., Lafaurie-Janvore, J., Divoux, S., Piel, M., & Perez, F. (2014). ESCRT machinery is required for plasma membrane repair. Science, 343, 1247436
Kakarougkas, A., & Jeggo, P. A. (2014). DNA DSB repair pathway choice: an orchestrated handover mechanism. British Journal of Radiology, 87, 20130685
Kelley, J. B., Datta, S., Snow, C. J., Chatterjee, M., Ni, L., Spencer, A., Yang, C. S., Cubenas-Potts, C., Matunis, M. J., & Paschal, B. M. (2011). The defective nuclear lamina in Hutchinson-Gilford progeria syndrome disrupts the nucleocytoplasmic Ran gradient and inhibits nuclear localization of Ubc9. Molecular and Cellular Biology, 31, 3378–3395
Kennedy, B. K., Barbie, D. A., Classon, M., Dyson, N., & Harlow, E. (2000). Nuclear organization of DNA replication in primary mammalian cells. Genes & Development, 14, 2855–2868
Kidiyoor, G. R., Li, Q., Bastianello, G., Bruhn, C., Giovannetti, I., Mohamood, A., Beznoussenko, G. V., Mironov, A., Raab, M., Piel, M., et al. (2020). ATR is essential for preservation of cell mechanics and nuclear integrity during interstitial migration. Nature Communications, 11, 4828
Kramarz, K., Schirmeisen, K., Boucherit, V., Ait Saada, A., Lovo, C., Palancade, B., Freudenreich, C., & Lambert, S. A. E. (2020). The nuclear pore primes recombination-dependent DNA synthesis at arrested forks by promoting SUMO removal. Nature Communications, 11, 5643
Kreienkamp, R., Graziano, S., Coll-Bonfill, N., Bedia-Diaz, G., Cybulla, E., Vindigni, A., Dorsett, D., Kubben, N., Batista, L. F. Z., & Gonzalo, S. (2018). A cell-intrinsic interferon-like response links replication stress to cellular aging caused by progerin. Cell Reports, 22, 2006–2015
Kubben, N., Voncken, J. W., Demmers, J., Calis, C., van Almen, G., Pinto, Y., & Misteli, T. (2010). Identification of differential protein interactors of lamin A and progerin. Nucleus, 1, 513–525
Lawrence, K. S., Tapley, E. C., Cruz, V. E., Li, Q., Aung, K., Hart, K. C., Schwartz, T. U., Starr, D. A., & Engebrecht, J. (2016). LINC complexes promote homologous recombination in part through inhibition of nonhomologous end joining. Journal of Cell Biology, 215, 801–821
Leemans, C., van der Zwalm, M. C. H., Brueckner, L., Comoglio, F., van Schaik, T., Pagie, L., van Arensbergen, J., & van Steensel, B. (2019). Promoter-intrinsic and local chromatin features determine gene repression in LADs. Cell, 177(852–864), e814
Lenain, C., Gusyatiner, O., Douma, S., van den Broek, B., & Peeper, D. S. (2015). Autophagy-mediated degradation of nuclear envelope proteins during oncogene-induced senescence. Carcinogenesis, 36, 1263–1274
Li, B. B. X., Chen, J. J., Chao, B., Zheng, Y. X., & Xiao, X. S. (2018). A Lamin-binding ligand inhibits homologous recombination repair of DNA double-strand breaks. ACS Central Sci, 4, 1201–1210
Lin, Q., Yu, B., Wang, X., Zhu, S., Zhao, G., Jia, M., Huang, F., Xu, N., Ren, H., Jiang, Q., et al. (2020). K6-linked SUMOylation of BAF regulates nuclear integrity and DNA replication in mammalian cells. Proceedings of the National academy of Sciences of the United States of America, 117, 10378–10387
Liu, N. A., Sun, J. Y., Kono, K., Horikoshi, Y., Ikura, T., Tong, X., Haraguchi, T., & Tashiro, S. (2015). Regulation of homologous recombinational repair by lamin B1 in radiation-induced DNA damage. Faseb Journal, 29, 2514–2525
Liu, S., Kwon, M., Mannino, M., Yang, N., Renda, F., Khodjakov, A., & Pellman, D. (2018). Nuclear envelope assembly defects link mitotic errors to chromothripsis. Nature, 561, 551–555
Lottersberger, F., Karssemeijer, R. A., Dimitrova, N., & de Lange, T. (2015). 53BP1 and the LINC complex promote microtubule-dependent DSB mobility and DNA repair. Cell, 163, 880–893
Lukasova, E., Rezacova, M., Bacikova, A., Sebejova, L., Vavrova, J., & Kozubek, S. (2019). Distinct cellular responses to replication stress leading to apoptosis or senescence. FEBS Open Bio, 9, 870–890
Luxton, G. W., Gomes, E. R., Folker, E. S., Worman, H. J., & Gundersen, G. G. (2011). TAN lines: a novel nuclear envelope structure involved in nuclear positioning. Nucleus, 2, 173–181
Ma, H., Qian, W., Bambouskova, M., Collins, P.L., Porter, S.I., Byrum, A.K., Zhang, R., Artyomov, M., Oltz, E.M., Mosammaparast, N., et al. (2020). Barrier-to-autointegration factor 1 protects against a basal cGAS-STING response. mBio, 11, e00136–20. https://doi.org/10.1128/mBio.00136-20
Malhas, A. N., Lee, C. F., & Vaux, D. J. (2009). Lamin B1 controls oxidative stress responses via Oct-1. Journal of Cell Biology, 184, 45–55
Malhas, A. N., & Vaux, D. J. (2011). The nuclear envelope and its involvement in cellular stress responses. Biochemical Society Transactions, 39, 1795–1798
Misteli, T., & Soutoglou, E. (2009). The emerging role of nuclear architecture in DNA repair and genome maintenance. Nat Rev Mol Cell Bio, 10, 243–254
Moir, R. D., Montag-Lowy, M., & Goldman, R. D. (1994). Dynamic properties of nuclear lamins: lamin B is associated with sites of DNA replication. Journal of Cell Biology, 125, 1201–1212
Moir, R. D., Spann, T. P., Herrmann, H., & Goldman, R. D. (2000). Disruption of nuclear lamin organization blocks the elongation phase of DNA replication. Journal of Cell Biology, 149, 1179–1192
Nader, G., Aguera-Gonzalez, S., Routet, F., Gratia, M., Maurin, M., Cancila, V., Cadart, C., Gentili, M., Yamada, A., & Lodillinsky, C. (2020). Compromised nuclear envelope integrity drives tumor cell invasion. bioRxiv, preprint. https://doi.org/10.1101/2020.05.22.110122
Nagai, S., Dubrana, K., Tsai-Pflugfelder, M., Davidson, M. B., Roberts, T. M., Brown, G. W., Varela, E., Hediger, F., Gasser, S. M., & Krogan, N. J. (2008). Functional targeting of DNA damage to a nuclear pore-associated SUMO-dependent ubiquitin ligase. Science, 322, 597–602
Palancade, B., Liu, X., Garcia-Rubio, M., Aguilera, A., Zhao, X., & Doye, V. (2007). Nucleoporins prevent DNA damage accumulation by modulating Ulp1-dependent sumoylation processes. Molecular Biology of the Cell, 18, 2912–2923
Pannunzio, N. R., Watanabe, G., & Lieber, M. R. (2018). Nonhomologous DNA end-joining for repair of DNA double-strand breaks. Journal of Biological Chemistry, 293, 10512–10523
Park, Y. E., Hayashi, Y. K., Bonne, G., Arimura, T., Noguchi, S., Nonaka, I., & Nishino, I. (2009). Autophagic degradation of nuclear components in mammalian cells. Autophagy, 5, 795–804
Penfield, L., Wysolmerski, B., Mauro, M., Farhadifar, R., Martinez, M. A., Biggs, R., Wu, H. Y., Broberg, C., Needleman, D., & Bahmanyar, S. (2018). Dynein pulling forces counteract lamin-mediated nuclear stability during nuclear envelope repair. Molecular Biology of the Cell, 29, 852–868
Pickersgill, H., Kalverda, B., de Wit, E., Talhout, W., Fornerod, M., & van Steensel, B. (2006). Characterization of the Drosophila melanogaster genome at the nuclear lamina. Nature Genetics, 38, 1005–1014
Porwal, M., Cohen, S., Snoussi, K., Popa-Wagner, R., Anderson, F., Dugot-Senant, N., Wodrich, H., Dinsart, C., Kleinschmidt, J. A., Pante, N., et al. (2013). Parvoviruses cause nuclear envelope breakdown by activating key enzymes of mitosis. PLoS Pathogens, 9, e1003671
Pozo, F. M., Tang, J. S., Bonk, K. W., Keri, R. A., Yao, X. S., & Zhang, Y. W. (2017). Regulatory cross-talk determines the cellular levels of 53BP1 protein, a critical factor in DNA repair. Journal of Biological Chemistry, 292, 5992–6003
Pujol, G., Soderqvist, H., & Radu, A. (2002). Age-associated reduction of nuclear protein import in human fibroblasts. Biochemical and Biophysical Research Communications, 294, 354–358
Raab, M., Gentili, M., de Belly, H., Thiam, H. R., Vargas, P., Jimenez, A. J., Lautenschlaeger, F., Voituriez, R., Lennon-Dumenil, A. M., Manel, N., et al. (2016). ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science, 352, 359–362
Rauschert, I., Aldunate, F., Preussner, J., Arocena-Sutz, M., Peraza, V., Looso, M., Benech, J. C., & Agrelo, R. (2017). Promoter hypermethylation as a mechanism for Lamin A/C silencing in a subset of neuroblastoma cells. PLoS ONE, 12 (4), e0175953. https://doi.org/10.1371/journal.pone.0175953
Redwood, A. B., Gonzalez-Suarez, I., & Gonzalo, S. (2011). Regulating the levels of key factors in cell cycle and DNA repair New pathways revealed by lamins. Cell Cycle, 10, 3652
Redwood, A. B., Perkins, S. M., Vanderwaal, R. P., Feng, Z. H., Biehl, K. J., Gonzalez-Suarez, I., Morgado-Palacin, L., Shi, W., Sage, J., Roti-Roti, J. L., et al. (2011). A dual role for A-type lamins in DNA double-strand break repair. Cell Cycle, 10, 2549–2560
Robijns, J., Houthaeve, G., Braeckmans, K., & De Vos, W. H. (2018). Loss of nuclear envelope integrity in aging and disease. Int Rev Cel Mol Bio, 336, 205–222
Robijns, J., Molenberghs, F., Sieprath, T., Corne, T. D., Verschuuren, M., & De Vos, W. H. (2016). In silico synchronization reveals regulators of nuclear ruptures in lamin A/C deficient model cells. Science and Reports, 6, 30325
Robson, M. I., de Las Heras, J. I., Czapiewski, R., Sivakumar, A., Kerr, A. R. W., & Schirmer, E. C. (2017). Constrained release of lamina-associated enhancers and genes from the nuclear envelope during T-cell activation facilitates their association in chromosome compartments. Genome Research, 27, 1126–1138
Sakthivel, K. M., & Sehgal, P. (2016). A novel role of lamins from genetic disease to cancer biomarkers. Oncology Reviews, 10, 309
Sallmyr, A., & Tomkinson, A. E. (2018). Repair of DNA double-strand breaks by mammalian alternative end-joining pathways. Journal of Biological Chemistry, 293, 10536–10546
Samwer, M., Schneider, M. W. G., Hoefler, R., Schmalhorst, P. S., Jude, J. G., Zuber, J., & Gerlich, D. W. (2017). DNA cross-bridging shapes a single nucleus from a set of mitotic chromosomes. Cell, 170(956–972), e923
Schwartz, T. U. (2016). The structure inventory of the nuclear pore complex. Journal of Molecular Biology, 428, 1986–2000
Shah, P., Hobson, C. M., Cheng, S., Colville, M. J., Paszek, M. J., Superfine, R., & Lammerding, J. (2021). Nuclear deformation causes DNA damage by increasing replication stress. Current Biology, 31(753–765), e756
Shimi, T., Pfleghaar, K., Kojima, S. I., Pack, C. G., Solovei, I., Goldman, A. E., Adam, S. A., Shumaker, D. K., Kinjo, M., Cremer, T., et al. (2008). The A- and B-type nuclear lamin networks: microdomains involved in chromatin organization and transcription. Gene Dev, 22, 3409–3421
Shumaker, D. K., Solimando, L., Sengupta, K., Shimi, T., Adam, S. A., Grunwald, A., Strelkov, S. V., Aebi, U., Cardoso, M. C., & Goldman, R. D. (2008). The highly conserved nuclear lamin Ig-fold binds to PCNA: its role in DNA replication. Journal of Cell Biology, 181, 269–280
Simon, D. N., & Wilson, K. L. (2011). The nucleoskeleton as a genome-associated dynamic “network of networks.” Nat Rev Mol Cell Bio, 12, 695–708
Singh, M., Hunt, C. R., Pandita, R. K., Kumar, R., Yang, C. R., Horikoshi, N., Bachoo, R., Serag, S., Story, M. D., Shay, J. W., et al. (2013). Lamin A/C depletion enhances DNA damage-induced stalled replication fork arrest. Molecular and Cellular Biology, 33, 1210–1222
Starr, D. A., & Fridolfsson, H. N. (2010). Interactions between nuclei and the cytoskeleton are mediated by SUN-KASH nuclear-envelope bridges. Annual Review of Cell and Developmental Biology, 26, 421–444
Swartz, R. K., Rodriguez, E. C., & King, M. C. (2014). A role for nuclear envelope-bridging complexes in homology-directed repair. Molecular Biology of the Cell, 25, 2461–2471
Swift, J., Ivanovska, I. L., Buxboim, A., Harada, T., Dingal, P. C., Pinter, J., Pajerowski, J. D., Spinler, K. R., Shin, J. W., Tewari, M., et al. (2013). Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science, 341, 1240104
Tang, C. W., Maya-Mendoza, A., Martin, C., Zeng, K., Chen, S. B., Feret, D., Wilson, S. A., & Jackson, D. A. (2008). The integrity of a lamin-B1-dependent nucleoskeleton is a fundamental determinant of RNA synthesis in human cells. Journal of Cell Science, 121, 1014–1024
Tapley, E. C., & Starr, D. A. (2013). Connecting the nucleus to the cytoskeleton by SUN-KASH bridges across the nuclear envelope. Current Opinion in Cell Biology, 25, 57–62
Techer, H., Koundrioukoff, S., Nicolas, A., & Debatisse, M. (2017). The impact of replication stress on replication dynamics and DNA damage in vertebrate cells. Nature Reviews Genetics, 18, 535–550
Therizols, P., Fairhead, C., Cabal, G. G., Genovesio, A., Olivo-Marin, J.-C., Dujon, B., & Fabre, E. (2006). Telomere tethering at the nuclear periphery is essential for efficient DNA double strand break repair in subtelomeric region. The Journal of cell biology, 172, 189–199
Tsouroula, K., Furst, A., Rogier, M., Heyer, V., Maglott-Roth, A., Ferrand, A., Reina-San-Martin, B., & Soutoglou, E. (2016). Temporal and spatial uncoupling of DNA double strand break repair pathways within mammalian heterochromatin. Molecular Cell, 63, 293–305
van Schaik, T., Vos, M., Peric-Hupkes, D., Hn Celie, P., & van Steensel, B. (2020). Cell cycle dynamics of lamina-associated DNA. EMBO Reports, 21, e50636
van Steensel, B., & Belmont, A. S. (2017). Lamina-associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell, 169, 780–791
von Appen, A., LaJoie, D., Johnson, I. E., Trnka, M. J., Pick, S. M., Burlingame, A. L., Ullman, K. S., & Frost, A. (2020). LEM2 phase separation promotes ESCRT-mediated nuclear envelope reformation. Nature, 582, 115–118
Vouzas, A.E., & Gilbert, D.M. (2021). Mammalian DNA replication timing. Cold Spring Harbor Perspectives Biology, a040162. https://doi.org/10.1101/cshperspect.a040162
Wilhelm, T., Said, M., & Naim, V. (2020). DNA replication stress and chromosomal instability: Dangerous liaisons. Genes (Basel), 11, 642
Wilson, K. L., & Foisner, R. (2010). Lamin-binding proteins. Cold Spring Harbor Perspectives in Biology, 2, a000554
Worman, H. J., Ostlund, C., & Wang, Y. X. (2010). Diseases of the nuclear envelope. Cold Spring Harbor Perspectives in Biology, 2, a000760
Wright, W. D., Shah, S. S., & Heyer, W. D. (2018). Homologous recombination and the repair of DNA double-strand breaks. Journal of Biological Chemistry, 293, 10524–10535
Yang, Z., Maciejowski, J., & de Lange, T. (2017). Nuclear envelope rupture is enhanced by loss of p53 or Rb. Molecular Cancer Research, 15, 1579–1586
Zeman, M. K., & Cimprich, K. A. (2014). Causes and consequences of replication stress. Nature Cell Biology, 16, 2–9
Zhang, C. Z., Spektor, A., Cornils, H., Francis, J. M., Jackson, E. K., Liu, S., Meyerson, M., & Pellman, D. (2015). Chromothripsis from DNA damage in micronuclei. Nature, 522, 179–184
Zwerger, M., Jaalouk, D. E., Lombardi, M. L., Isermann, P., Mauermann, M., Dialynas, G., Herrmann, H., Wallrath, L. L., & Lammerding, J. (2013). Myopathic lamin mutations impair nuclear stability in cells and tissue and disrupt nucleo-cytoskeletal coupling. Human Molecular Genetics, 22, 2335–2349
Acknowledgements
We would like to thank the members of the Deng and Qin laboratories for helpful discussions. We apologize to those whose excellent work could not be cited directly in this review due to space limitations. This work is supported by the Guangdong Basic and Applied Basic Research Foundation (2020A1515110542, to L.D.), National Natural Science Foundation of China (31970752, to P.Q.), Science, Technology and Innovation Commission of Shenzhen Municipality (JSGG20191129110812, to P.Q.), and Shenzhen Bay Laboratory Open Fund (SZBL2020090501004, to P.Q.).
Author information
Affiliations
Shenzhen Bay Laboratory, Shenzhen, China
Wenjun Pu & Lin Deng
Institute of Biopharmaceutical and Health Engineering, Tsinghua Shenzhen International Graduate School, Tsinghua University, Shenzhen, 518055, China
Haihui Zhang & Peiwu Qin
Corresponding author
Correspondence to Lin Deng.
Rights and permissions
About this article
Cite this article
Pu, W., Zhang, H., Qin, P. et al. Nuclear envelope integrity, DNA replication, damage repair and genome stability. GENOME INSTAB. DIS. 2, 102–114 (2021). https://doi.org/10.1007/s42764-021-00039-w
Received12 February 2021
Revised05 April 2021
Accepted07 April 2021
Published15 April 2021
Issue DateApril 2021
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
Nuclear envelope
DNA replication stress
Damage repair
Genome stability
用户登录
还没有账号?
立即注册