






Human papillomavirus-mediated carcinogenesis and tumor progression
Review Article
Fadi Abboodi, Nella C. Delva, Jennifer Emmel, Ariana Renrick, Phillip Buckhaults, Carolyn E. Banister, Kim E. Creek & Lucia Pirisi
Genome Instability & Disease 2, 71–91(2021)
Abstract
High-risk human papillomaviruses (HPV) cause 5% of all human cancers and are primary etiologic agents of cervical, anal, and oropharyngeal cancer. HPV infection is necessary, but not sufficient per se to produce cancer: additional changes must occur that transform HPV-infected cells to malignancy. The HPV oncoproteins E6 and E7 immortalize human keratinocytes, cervical cells, and fibroblasts in culture. Each oncoprotein interacts with a variety of cellular binding partners; most important for transformation are E6 and E7’s interactions with p53 and RB (respectively) which lead to degradation of p53 and RB through the ubiquitin pathway. Inactivation of p53 and RB leads to inactivation of pivotal cell cycle checkpoints, thereby stimulating cell proliferation and allowing cell division to occur independently of the presence of DNA damage, replicative stress, and other such insults, leading to genome instability. Continuous expression of E6/E7 drives the proliferation and progression of most HPV-mediated cancers of the cervix and a substantial fraction of those of the oropharynx. However, at both cancer sites, “HPV-inactive” tumors that contain HPV DNA, but do not express E6/E7 arise. We propose that these HPV-inactive cancers begin as HPV-driven lesions, but lose E6/E7 expression at some point during progression. We have recently shown that p53 deletion in HPV-immortalized, premalignant cells allows for the emergence of cell populations that no longer express E6/E7. These findings corroborate the notion of a pivotal role of p53 in the context of HPV-mediated transformation, both at the initiation and progression stages of cancer development.
Human Papillomaviruses (HPVs)
The Papillomaviridae family includes viruses that infect and cause warts (papillomas) on skin and/or mucosae across a large number of animal species, with marked species- and site-specificity of infection (Bernard et al. 2010; Doorbar et al. 2015). Papillomaviruses co-evolve with their hosts over long periods of time and tend to produce long-term asymptomatic infections (Antonsson et al. 2000, 2006; Griffith 1999). Human papillomaviruses (HPV) include about 200 genome types, subdivided into five genera: alpha, beta, gamma, mu, and nu, on the basis of their DNA sequence (Bernard et al 2010; Doorbar et al. 2015; Serrano et al. 2017). All five genera infect cutaneous tissues; HPV of the alpha genus also infect mucosal tissues (Doorbar et al. 2015). Papillomaviruses (PV) are small non-enveloped viral particles 50–60 nm in diameter, with icosahedral capsid containing a circular double-stranded DNA genome of about 8000 base-pairs (Zheng & Baker, 2006). HPV can be further classified into high risk (HR) or oncogenic, and low risk (LR): this classification depends on whether or not the individual HPV types are associated with cancer. As we will describe later, the ability to cause cancer is linked to the activities of E6 and E7, their respective affinities for p53 and Rb, and their ability to cause degradation of these tumor suppressor proteins (Doorbar et al. 2015). HR HPV include the HPV types 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, and 59 (Table 1). Among these, HPV16 and 18 are by far the most common, causing about 70% of all cervical cancer (Table 1). HPV16 is the type found in most of the HPV-positive oropharyngeal carcinoma and in about 61% of cervical cancer cases (Doorbar et al. 2015; Serrano et al. 2017). Other tumors have also been linked to HPV like cancers of the vagina, vulva, penis, and anal canal (Szymonowicz et al. 2020). Among the LR types, HPV6 and 11 are associated with mostly benign genital warts (condyloma acuminatum) and also cause recurrent respiratory papillomatosis. In addition, a whole series of cutaneous HPV have been isolated and identified (Doorbar et al. 2015). Among these are oncogenic types (HPV5, 8, 14) involved in the pathogenesis of epidermodysplasia verruciformis, a rare but severe condition that results in the formation of numerous wart-like lesions on sun-exposed skin. These lesions have considerable potential for malignant conversion into squamous cell carcinomas (reviewed in Przybyszewska et al. 2017).
Table 1 Classification and pathogenicity of selected HPV typesFull size tableCervical cancer
The incidence of cervical cancer has decreased sharply in the United States and Europe, since screening for early lesions by the Pap test was widely implemented. However, cervical cancer incidence remains high in developing countries in Sub-Saharan Africa, Asia, and Central and South America. Cervical cancer is the fourth most common cancer worldwide and the second most common in women, with approximately 570,000 new cases and 311,000 deaths in 2018 (Bray et al. 2018; Wild et al. 2020). In USA, 13,800 new cervical cancer cases and 4290 deaths were estimated for 2020 (Siegel et al. 2020). HPV infection of the cervical transformation zone frequently occurs during adolescence, with early onset of sexual activity. The lifetime risk of exposure to HPV for women is about 80% and the risk decreases with increasing age (Moscicki et al. 2012). Asymptomatic HPV infection of the cervix is also quite frequent (Hammer et al. 2019): in the absence of clinical lesions, HPV is detected in the cervix of 11–12% of women. The prevalence of HPV increases with increasing severity of cervical lesions, ranging from 50–70% in low-grade neoplasia to 90–100% in invasive cervical cancer, with HPV16 and 18 detected in the majority of cases alone, combined, or in combination with multiple HPV types (Bruni et al. 2010).
The natural history of HR HPV infection and cancer development in the uterine cervix is summarized in Fig. 1. HR HPVs produce subclinical transient infection that clear within (approximately) 2 years in about 80% of cases. Persistent HR HPV infection may give rise to cervical intraepithelial neoplasia (CIN-1 and CIN-2) within a couple of years. However, in the majority of cases, these lesions regress spontaneously as HPV infection resolves. If lesions and viral infection persist for longer periods of time, high-grade cervical intraepithelial neoplasia grade 3 (CIN-3) or adenocarcinoma in situ (AIS) may develop which can progress to cervical cancer in about half of cases (Moscicki et al. 2012). However, the ratio of HPV-induced lesions that progress to cancer to the total number of HPV-induced lesions is low and this process may take anywhere between 5 and 30 years (McCredie et al. 2008; Ostör, 1993).
Fig. 1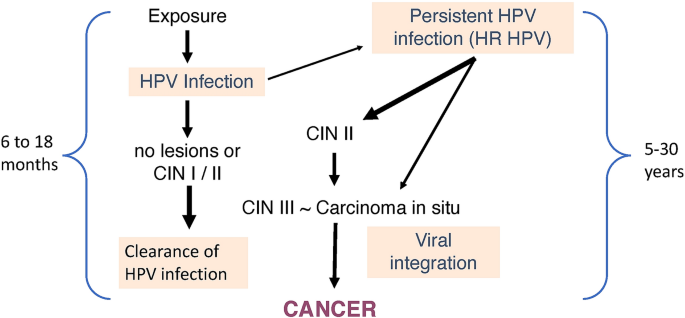
Natural history of HR HPV infection and cervical cancer development. CIN, cervical intraepithelial Neoplasia
Full size imageThe findings described above support the statement that HPV infection is common, but, in comparison, cervical cancer is quite rare, leading to the conclusion that HPV infection alone is not sufficient to produce cancer, as tumor development and progression require the contribution of multiple factors. Among the risk factors for cancer development and progression in women infected with HR HPV are the determinants of persistent infection, as it is well established that only women in whom HR HPV infection persists are at risk for cervical lesions that may progress to cancer (Banister et al. 2015 and references therein). HPV persistence has also been linked to HPV-mediated disease in men (Bettampadi et al. 2020). This is an important area of study, because in principle, if we were able to determine at a single visit whether or not an incident HR HPV infection will persist, we could target HPV-mediated cancer surveillance resources to the people who present with persistent infection. Our own (unpublished) findings support the concept that women with persistent HPV infection fail to mount a strong immune response to HPV. In turn, immune responses to HPV are likely to be influenced by HLA and SNP profiles, both of which have been linked with cervical cancer susceptibility (Chen et al. 2014; Das Gosh et al. 2017). Among the many SNPs that have relevance for cervical cancer development, the Arg/Pro TP53 polymorphism at codon 72 has received considerable attention, as the homozygote Arg/Arg phenotype is associated with a higher risk of developing cervical cancer, at least in certain populations (Ojeda et al. 2003; Chuery et al. 2017). TP53 codon 72 polymorphism has been connected with higher HPV E6/E7 expression, which appears to correlate with the Arg/Arg genotype (Chuery et al. 2017). Despite the continuing controversies in this area, there is evidence that this particular polymorphism plays a role in cervical cancer development, albeit with additional intervening factors that may modulate its impact in different populations.
In the USA, African American women have higher cervical cancer incidence and mortality rates than Caucasian women (Banister et al. 2015 and references therein). These findings are only partially explained by access to care and can be ascribed, in part, to a higher prevalence of persistent HR HPV infection in this population (Banister et al. 2015).
Therapeutic approaches to cervical cancer
Cervical cancer therapy often includes a combination of two or more therapeutic approaches. The feasibility of a surgical approach depends on the extent of the cancerous spread as well as other factors like the patient’s age and desire to preserve fertility. The standard surgical approach is radical hysterectomy (removing the whole uterus, tubes, upper vagina, and ovaries) with or without pelvic lymphadenectomy especially for early stages of cervical cancer (FIGO 2018 score IA1–IB2) (Goodman et al. 2019; Shuurman et al. 2021). Small and limited lesions can be treated by less invasive, fertility-preserving procedures like conization; LLETZ (Large Loop Excision of Transformation Zone); simple or radical trachelectomy (removing the cervix and upper vagina but not the body of the uterus) (Shuurman et al. 2021). The oncological prognosis after these surgical procedures is acceptable with low recurrence rate in stages lower that IB1 (Prodromidou et al. 2019). However, about 4% of the fertility-preserving procedures carry a higher risk of cancer development like positive resection margins, positive lymph nodes, or other features that necessitate a more invasive operation or the use of adjuvant chemo- and/or radiotherapy (Berman & Schiller, 2017; Bhatla et al. 2019).
When the cancer is at advanced stage at presentation, surgical intervention may no longer be an option. Hence, radiation therapy (RT) and/or chemotherapy (CT) become main treatment modalities. External beam radiation and brachytherapy are the radiation approaches most commonly utilized for cervical cancer (Goodman et al. 2019). Adjuvant chemotherapy may include cisplatin or 5-fluoruracil (Goodman et al. 2019). A combination of platinum-based chemotherapeutic agents like cisplatin, paclitaxel, and/or tropotecan is utilized for recurrent, or metastatic cancer (Tewari et al 2014). Additionally, cervical cancer is considered a good candidate for immunotherapy. For example, cervical cancer tissue often has a high activity in checkpoint-controlling immune targets such as programmed death ligand 1 and 2 (PD-L1 and PD-L2) and the non-coding RNA of BRCA-4 (Cancer Genome Atlas 2017). These tumors are usually infiltrated by immune cells like CD8 + cytotoxic T lymphocytes and macrophages that can be re-programmed to efficiently attack the tumor cells (Mandal et al 2016; Wang et al. 2019). Cervical cancer also has a high antigenic profile produced by the viral antigens or their interactions with cellular factors, and by the profile of somatic mutations characterizing this cancer (Alexandrov et al. 2013; Samstein et al. 2019). PD-1 receptor is expressed on T- and B-cells and also on cancer cells. In normal tissue, PD-1 activation by ligand 1 or 2 results in inhibition of immune cell function to protect the body from unnecessary lymphocytic stimulation, hence inducing tolerance to our body cells and avoiding the development of autoimmune diseases (Riely et al. 2019). However, tumor cells may use this receptor in an inappropriate way to induce T-cell tolerance to tumor cells, thereby the tumor can escape immune surveillance (Pauken et al. 2015; Diskin et al. 2020; Herbst et al. 2014). Therefore, targeting this immune inhibitory pathway by anti PD-L1 or anti PD-L2 medications is a very promising approach to cancer treatment. An example of an FDA-approved PD-1 antagonistic immunotherapeutic agent used in the treatment of cervical cancer is pembrolizumab (Verhoeven et al 2021).
Head and neck cancer (HNC)
Head and neck squamous cell carcinomas (HNSCC) represent more than 90% of HNC with a global incidence of 600,000 new cases per year, which translates into about 20 cases per 100,000 people (Jemal et al. 2011). HNSCC is responsible for 3.5% of cancer cases in the United States and Europe (Siegel et al. 2012). However, HNSCC is the sixth most common malignancy worldwide due to the high prevalence of the disease in India, Southeast Asia, and Latin America (Parkin et al. 2005). Although the overall incidence of HNSCC has decreased in the United States, the incidence of oropharyngeal squamous cell carcinoma (OPSCC) is on the rise mainly in younger adults and especially in regions of the tonsils and the base of the tongue, which together account for 90% of OPSCC cases (Chaturvedi et al. 2008; Licitra et al. 2002).
HPV is widely accepted as a causative agent of HNSCC, particularly of OPSCC, alongside with tobacco smoking and alcohol consumption. About 25% of HNSCC and 60–90% of OPSCC are positive for HPV (HPV +) with 90% of the cases being positive for HPV16 (Gillison et al. 2008). HNSCC is classified as HPV-positive (HPV +) and HPV-negative (HPV–) with differences in the molecular, etiological, clinical, and behavioral characteristics between the two groups (Chung & Gillison, 2009). Molecular analysis of HNSCC identified distinct gene expression profiles between HPV + and HPV– cancers, with higher expression of genes involved in cell cycle progression, mitosis, and proliferation in HPV + cancers, forming what can be described as the HPV signature (Schlecht et al. 2013; Tomar et al. 2016) while HPV- tumors lack this proliferative signature and have higher expression of genes involved in cell motility, epithelial-to-mesenchymal transition, and angiogenesis (Reviewed in Graves et al. 2014). HPV + tumors occur more commonly in younger patients who are usually non-smokers and do not consume alcohol in excess. HPV + OPSCC is also common in HIV infected individuals, marijuana users, and more in men than women, especially homosexual men. In contrast, HPV- tumors are more likely to affect older men who are smokers and/or alcoholics (Beachler et al. 2012; Del Mistro et al. 2012). HPV + HNSCC tend to be poorly differentiated, non-keratinizing, with basaloid morphology, without field cancerization and usually present with nodal metastases, features that are opposite to what is observed in HPV- HNSCC (Stanley, 2012). Several studies have shown a more favorable prognosis for HPV-induced HNSCC, with a 3-year disease-free survival and overall survival in HPV + compared to HPV– of 85% versus 49% and 90% versus 65%, respectively, leading to the definition of HPV positivity as an independent good prognostic factor (Nichols et al. 2013). Although some have pointed to the intensive treatment that patients with HPV + tumors receive as a reason for the more favorable outcome, others showed that HPV + tumors have favorable outcome regardless of the treatment (Guihard et al. 2012).
The better prognosis of HPV + HNSCC is thought to have a multifactorial explanation. HPV + tumors usually have wild-type p53 and Rb tumor suppressor gene products which, despite the downregulation by viral oncoproteins, still may allow for the apoptotic response of cancer cells to radio- and/or chemotherapy. These tumors lack field cancerization which means the absence of wide-spread genetic mutations (Adelstein et al. 2008). HPV + OPSCC patients are younger, non-smokers, and non-alcoholic, and therefore, they have less comorbidities and are less vulnerable to the negative effects of standard cancer therapeutic modalities (Guihard et al. 2012). Moreover, better prognosis could be owed to body immune surveillance against HPV as it was shown that there is activation of the adaptive immune system in the form of CD8 + T-cell lymphocytic tumor infiltration in HPV + cancers, and more extensive infiltration is associated with better outcome (King et al. 2014).
Therapeutic approaches to HNC Surgery alone is uncommonly a primary option for HNC treatment because most of these cancers present at late stage that requires a multimodality therapeutic approach. A common approach to inducing remission is cisplatin-induced chemoradiotherapy with or without surgery. This is usually followed by adjuvant radiotherapy or chemotherapy (Vokes et al. 2015; Wirth, 2016). Studies show that HPV-positive oropharyngeal squamous cell carcinoma cell lines (OPSCC) are more sensitive to radiation therapy than their HPV-negative counterpart (Arenz et al. 2014). This observation is in agreement with the data that support a better prognosis for HPV-positive OPSCC compared to HPV-negative OPSCC, leading to several clinical trials aiming at de-escalating the therapeutic protocols for HPV-positive OPSCC (Vokes et al. 2015). With the advancement in targeted and immune-modulatory agents like cetuximab, it is now possible to minimize the side effects of toxic chemotherapy in HPV-positive HNC. Cetuximab can be used as an adjuvant to give a more satisfactory local control with better survival without increasing the toxicity (Bonner et al. 2006). There are trials of administering definitive RT or transoral surgery alone in HPV-positive OPSCC (Vokes et al. 2015).
Similar to cervical cancer, immunotherapeutic agents targeting the PD-1/PD-L1 linkage have also been approved by the FDA for the treatment of HNSCC (Verhoeven et al. 2021). Nivolumab, pembrolizumab, and durvalumab have shown effectiveness in improving survival and controlling tumor microenvironment especially when there is marked infiltration by inflammatory cells (Vokes et al. 2015; Ferris 2016).
Therapeutic HPV vaccines and genome editing approaches in the treatment of cervical cancer and HPV + HNC Many trials have been directed to modify the natural history of HPV infection to progressive tumor development by the use of therapeutic vaccines and genome editing technologies. Therapeutic vaccines differ from the prophylactic vaccine by targeting E6/E7 rather than L1/L2. They aim at stimulating the cell-mediated immune system to kill the virus-loaded cells (reviewed in Chabeda et al 2018). Examples of these therapeutic vaccines are ADXS11-OO1 (Lm-LLo-E7), INO-3112, HPV16-SLP, and TA-CIN + GPI-0100 that target HPV16/18 E6/E7 oncoproteins in advanced cervical cancers. Others have been prepared to target persistent infection or intraepithelial neoplasia as PDS0101, ProCervix, GX-188E, pNGVL4a-CRT/E7 (detox), and others (reviewed in Yang et al. 2016). More recently, researchers are investigating the effectiveness of using genome editing techniques like CRISPR-Cas9, siRNA, shRNA, ribozymes, DNAzymes, zinc finger nucleases (ZFN), and transcription activator-like effector nucleases (TALENs) in treating cancers including HPV-associated cancers (reviewed in Pal & Kundu, 2020). The use of E6/E7-targeting agents in the treatment of HPV-mediated cancers is based on the notion that HPV + cancer cells are addicted to these viral oncoproteins, and their continuous expression is necessary for cancer cell growth and survival.
The HPV genome
The HPV genome consists of three regions: the noncoding upstream regulatory region (URR) and the early and late coding regions (Fig. 2) (de Villiers, 2013).
Fig. 2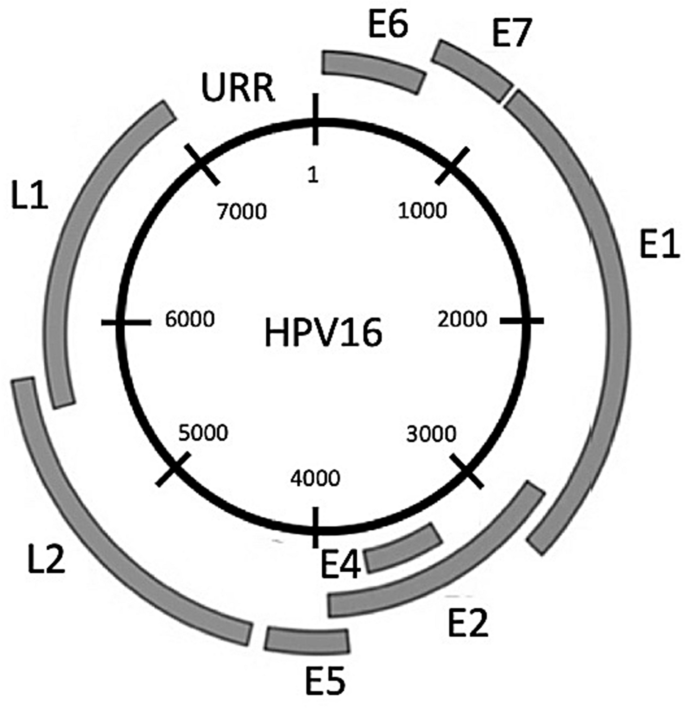
Genome map of HPV16. The viral genome can be divided into an Upstream Regulatory Region (URR) which is not translated, and an early (E) and a late (L) region, transcribed before and after the onset of DNA synthesis, respectively, during the virus life cycle
Full size imageThe URR contains the origin of replication and the promoter and transcription factor binding sites necessary to regulate early gene expression (Burley et al 2020). The early coding region contains seven open-reading frames (ORFs): E1, E2, E4, E5, E6, and E7 (shown in Fig. 2) and the lesser known E8, contained within the E1 ORF and expressed as a fusion E8^E2 protein that negatively regulates viral replication and transcription (Dreer et al. 2017). The E1 protein has DNA helicase activity and plays a key role in viral genome replication. E2 complexes with E1 to target it to the viral origin of replication and controls transcription of the viral genome (Doorbar et al. 2015). The E4 protein is thought to contribute to viral tropism, because different PV types contain marked variations in the E4 sequence (DiMaio & Petti, 2013). In oncogenic HPV, E4 disrupts the cytokeratin network, thus facilitating viral escape from infected epithelial surfaces (Doorbar, 2013). E5 enhances growth factor signaling pathways like EGFR activation which promotes cell proliferation and inhibits keratinocyte differentiation (DiMaio & Mattoon, 2001; Ilahi et al. 2020; McBride, 2017). The E6 and E7 proteins drive HPV-infected cells to re-enter the cell cycle and synthesize DNA. During the viral life cycle, E6 and E7 expression is carefully regulated both spatially and temporally. If expressed in an unregulated fashion, E6 and E7 promote abnormal proliferation, immortalization, and genomic instability, de facto pre-disposing the host cells to carcinogenesis (Doorbar et al. 2015; Moody & Laimins, 2010). The late region of the HPV genome codes for L1 and L2, the major and minor capsid proteins. In early phases of infection, L2 facilitates HPV transfer into the host cell nucleus by attaching it to Golgi apparatus, and then, the vesicle delivers the genome into the nucleus at time of nuclear envelop cleavage during mitosis (Aydin et al. 2017; Calton et al. 2017). HPV lacks polymerases essential for replication, and therefore employs the host cell replication proteins for viral DNA synthesis (Moody & Laimins, 2010).
Mode of transmission
HPV is the most common sexually transmitted virus (Bruni et al. 2010; Moscicki et al. 2012). Transmission occurs through vaginal, anal, and oral sexual contact, and the risk of infection increases with early onset of sexual activity and higher numbers of sexual partners (Bruni et al. 2010; Moscicki et al. 2012). In addition, HPV can be transmitted from an infected mother to her baby during birth and in utero transmission may also occur (Zouridis et al. 2018). Oncogenic HPV types have been detected in breast milk; however, the likelihood of transmission by this route is low (Donalisio et al. 2014). Non-sexual modes of transmission are also proposed, as HPV DNA can be found on fomites, surfaces, and even water (Petca et al. 2020).
HPV infection and life cycle
HPV have a very specific tropism for squamous epithelial tissues which contain a basal layer where progenitor cells reside, and supra-basal layers of differentiated cells that die as they approach the surface (Fig. 3). Basal layer cells periodically undergo asymmetric mitosis to produce two types of cells: one basal cell, to maintain a constant population of progenitor cells, and the other destined to differentiate, to replace the cells that slough off the upper layers (McLaughlin-Drubin & Münger, 2009). HPV infection usually occurs through minor abrasions of the cutaneous and mucosal surfaces that expose the basal layer cells which are particularly susceptible to infection (Ozbun et al. 2019). LR HPV take advantage of cell replication driven by the wound-healing processes to infect cells and transport the viral DNA to the nucleus by a process that involves cellular transport proteins and the viral L2 protein (Doorbar, 2006; Popa et al. 2015). In HR HPV, E6 and E7 drive cell proliferation independently of the wound-healing process (Doorbar, 2006). In both HR and LR HPV, E1 and E2 are essential to initiate viral replication.
Fig. 3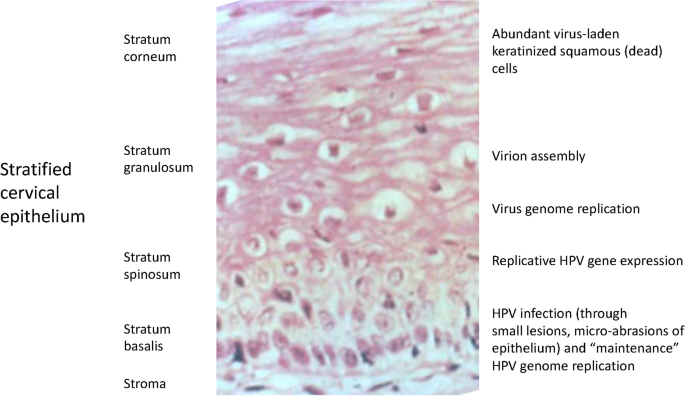
HPV life cycle and epithelial cell differentiation. The picture shows a normal cervical epithelium, the site of HPV infection, in correlation with the phases of the HPV life cycle. Details in the text
Full size imageInfection of the basal layer stem cell helps establish a long-term infection as the viral DNA is maintained in these cells in low copy number and these infected cells escape immune surveillance by a variety of virus-driven mechanisms, including inhibition of antigen presentation and interference with dendritic cell function (Doorbar et al. 2015; Yilmaz & Strati, 2019).
In this regard, our recent work demonstrates that epithelial stem cells are targeted by HPV16-mediated transformation: we demonstrated that keratinocyte stem cells, which form multicellular spheroids in 3-D culture and express, among other keratinocyte stem cell markers such as p63 and keratin 14, low epidermal growth factor receptor (EGFR), and high integrin-a6 protein levels (EGFRlo/ITG-a6hi) as detected by flow cytometry, are immortalized by HPV16 DNA about sixfold more efficiently than keratinocytes unable to form spheroids in suspension (Woappi et al. 2018). Furthermore, immortalized lines derived from EGFRlo/ITG-a6hi cells readily progress to the pre-malignant differentiation-resistant stage of HPV16-mediated transformation in vitro (Pirisi et al. 1987, 1988; Woappi et al. 2018). The density of EGFRlo/ITG-a6hi cells varies dramatically among foreskin keratinocyte cultures derived from different individuals, so that only about 40% of normal individual keratinocyte strains are able to form spheroids in suspension (Woappi et al. 2018, 2020). These findings provided a plausible explanation for the marked variability in immortalization responses we observed among individual normal human keratinocyte strains over the years, and for our previous finding that human keratinocyte strains with intrinsically high EGFR levels were refractory to HPV16-mediated immortalization (Akerman et al. 2001). Intrinsic interindividual differences such as the ones highlighted here may be added to immunological and genetic determinants of responses to HPV to explain why only relatively few people develop cancer, among the many infected with HPV.
Although early infection occurs in the basal layer of a mucosal epithelium, viral amplification occurs in the supra-basal terminally differentiated layers where cells have exited the cell cycle (Fig. 3) (reviewed in Lee & Laimins, 2007). HPV redirects these differentiating cells to re-enter the cell cycle (McLaughlin-Drubin & Münger, 2009). In the productive HPV life cycle, viral genome amplification occurs as the differentiating cells are pushed towards the surface of the epithelium and the viral genomes remain in the nucleus as episomes (De Leo et al. 2020; Parish et al. 2006). Then, as late viral genes L1 and L2 are expressed, capsids are assembled and HPV genomes packaged to produce new progeny virions that shed from the surface and spread to infect other cells (Doorbar et al. 2015). Productive HPV infection produces non-cancerous, often exuberantly proliferative and highly keratinizing lesions such as warts and condylomas. LR HPV almost never integrate their DNA into the host cell genome. However, HR HPV integrate their genome, often in tandem repeats, preferentially at chromosomal fragile sites, i.e., sites of active transcription where DNA is more accessible (Smith et al. 1992; Ziegert et al. 2003).
Integration is thought to be crucial for transformation of HR HPV-infected cells. In fact, most cervical cancers contain integrated HPV genomes (Moody & Laimins, 2010). HPV16 is integrated in about 74% of the associated cancers, while HPV18 is integrated in all the cases (Cancer Genome Atlas 2017). The HPV genome is less frequently integrated in HPV-positive OPSCC compared to cervical cancer (Morgan et al. 2017). In cervical cancer-derived cell lines, HPV DNA integration is often found within the E1–E2 region of the HPV genome, disrupting E2 expression. Since E2 is essential for the coordinated and precisely timed expression of E6/E7, in the absence of E2 the two oncoproteins are expressed in an unregulated fashion (Jeon et al. 1995; McBride & Warburton, 2017). E6 and E7 messenger RNAs produced by integrated genomes are more stable than those produced by episomal genomes (Jeon et al. 1995). However, integrated and episomal HPV DNA can coexist in infected cells. There is evidence that this may lead to enhanced carcinogenic potential, as episomal E1 and E2 can further promote the expression of integrated HPV gene expression (Kadaja et al. 2009). In the host cell genome, integration is at least in part mediated by areas of micro-homology between the viral DNA and the human genome (Hu et al. 2015).
The HPV replication cycle relies on the DNA Damage Response (DDR; reviewed in Blackford & Jackson, 2017) for crucial phases of virus genome amplification, early and late gene expression and virus production (reviewed in Spriggs & Laimins, 2017) and many molecular mechanisms of these interactions have been elucidated. For example, viral genome amplification in the suprabasal layers requires the activation of the ATM pathway; E7 binds to ATM through its LXCXE motif, thereby promoting CHK2 activation and activation of caspase needed for proper cleavage and activation of the E1 replication protein; HPV16E1/E2 mediate viral DNA replication in the presence of etoposide, which activates both the ATM and ATR pathways; HPV E1 and E2 co-localize with many components of the DDR pathway at integrated HPV18 genome replication centers (reviewed in Benedetti et al. 2021).
Genomic instability also characterizes HPV-immortalized cells and underlies the progression of HPV-immortalized cells toward malignancy (Gupta et al. 2018). HPV-transformed cells have deficient p53 and Rb function and often display severe chromosomal abnormalities, aneuploidy, and abnormal mitoses (reviewed in Korzeniewski et al. 2011). However, they incur relatively few mutations, in comparison with other solid tumors of non-viral origin (Litwin et al. 2017).
The HPV E6 and E7 oncoproteins and their interactions with cellular components
Among the many activities elicited by E6 and E7, their respective interactions with p53 and pRB are crucial for immortalization. In addition, these oncoproteins have many more activities and interact with a plethora of cellular factors with far reaching consequences not only for the virus life cycle, but also for transformation (see for example: Neveu et al. 2012; Poirson et al. 2017; Vats et al. 2021). Figure 4 shows some of the key interactions of E6 and E7 with cellular components that are important for immortalization and cancer development, highlighting in particular those that are most relevant to the areas discussed in this paper.
Fig. 4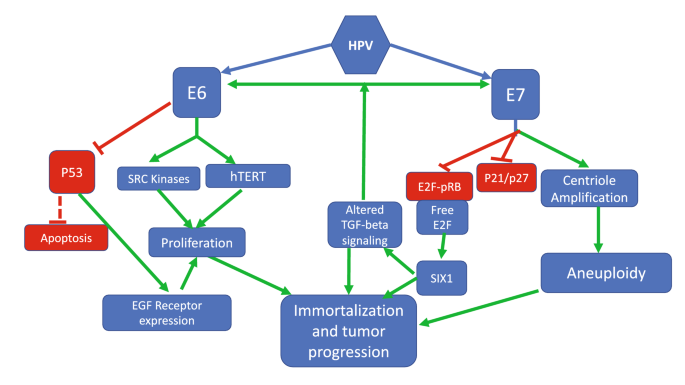
Selected pathways of E6 and E7 interactions with cellular components that contribute to immortalization and cancer progression. Details in the text
Full size imageHPV E6 and p53
HPV E6 is a small protein of 18 kDa that resides in the nucleus but also can be detected in the cytoplasm (McLaughlin-Drubin & Münger, 2009). E6 plays an important role in HPV pathogenesis, especially for HR HPV (Doorbar et al. 2015). The best known and probably most important activity of HR HPV E6 is its interaction with the p53 protein, discovered more than 30 years ago (Scheffner et al. 1990).
Tumor suppressor genes are inactivated before or during the carcinogenesis process, as their products act as negative regulators of proliferation and keepers of cell and tissue homeostasis. In normal cells, these genes become activated in response to various types of stress that de-regulate normal cellular activities. For example, as a consequence of DNA damage, tumor suppressor gene products prevent cell cycle progression and arrest cell proliferation until the DNA damage response can repair the DNA or, if the damage cannot be repaired, direct the cell to apoptosis (Vogelstein et al. 2000). Initially classified as an oncogene, and later defined as “the guardian of the genome” (Lane, 1992), p53 was the first tumor suppressor gene to be discovered (Carr, 2000; Vogelstein et al. 2000). p53 can be activated by three independent pathways: (1) by DNA damage through the ataxia telangiectasia mutated (ATM) gene product, activated by double-strand DNA breaks. ATM in turn activates the Checkpoint Kinase 2 (Chk2) (Carr, 2000) leading to cell cycle arrest and the recruitment of DNA repair complexes to the site of damage; (2) p14ARF-dependent activation in response to the expression of oncogenes (Lowe & Lin, 2000; Sherr & Weber, 2000). (3) The Ataxia Telangiectasia-Related (ATR) pathway that becomes activated by protein kinase inhibitors, UV radiation, and chemotherapeutic agents (Meek, 1999). p53 activation is mostly due to protein stabilization, rather than to increased expression (Vogelstein et al. 2000). In normal cells, p53 is rapidly degraded by ubiquitination facilitated by the MDM2 protein: a rise in p53 causes feedback activation of the MDM2 gene whose product binds to and ubiquitinates p53 for degradation (Vogelstein et al. 2000). All three pathways of p53 activation involve inhibition of MDM2-p53 binding through either modifications of the binding domains, or sequestration of MDM2 (Meek, 1999; Prives & Hall, 1999).
p53 functions through different pathways: (1) Inhibition of cyclin-dependent kinases (CDKs) by stimulation of the transcription of p21, resulting in inhibition of G1–S and G2-M transitions of the cell cycle (Meek, 1999). (2) Directing the cell to apoptosis by activation of the Bax gene whose product is the prototype of apoptosis-inducing proteins (Reed 1999) or by activating production of highly toxic reactive oxygen species in the mitochondria (Vogelstein et al. 2000). (3) Stimulation of DNA repair mechanism by the activation of repair genes (Vogelstein et al. 2000). (4) Inhibition of angiogenesis necessary for tumor development by activation of anti-angiogenesis genes (El-Deiry 1998).
Abnormalities in p53 pathways have been detected in well over half of the common tumors, and therefore, they are actively researched as potential therapeutic targets (Niazi et al. 2018). These abnormalities include mutations or deletions of the p53 gene; de-regulation of genes whose products interfere or interact with p53; or inactivation of p53 by viral proteins such as HR HPV E6, Adenovirus E1B, and the SV40 LT (Niazi et al. 2018; Vogelstein et al. 2000).
HR HPV E6 abolishes p53 functions by different mechanisms: (1) E6 forms a trimeric complex with the cellular E3 ubiquitin ligase E6-associated protein (E6AP) and p53 to direct the latter to ubiquitination and proteasomal degradation (Huibregtse et al. 1991). Both HR and LR HPV E6 can bind to the C-terminus of p53; however, HR HPV E6 can also bind the core site of p53 which is essential for its degradation. LR HPV E6 binding to p53 is weak and does not result in p53 degradation (Crook et al.1994). (2) Both HR and LR HPV E6 inhibit p53-mediated transcriptional activity by changing p53 attachment or inducing post-translational modifications (Crook et al. 1994). The degree of p53-DNA-binding inhibition depends on E6 affinity for p53 which is highest in HPV16, intermediate in HPV18, HPV31, and least in HPV11 (Lechner & Laimins, 1994). (3) Both HR and LR HPV E6 can sequester p53 in the cytoplasm by either exporting it outside the nucleus or by hiding the nuclear localization signal that is necessary to transport p53 to the nucleus (Mantovani & Banks, 2001). 4) HR HPV E6 interferes with p53 acetylation required for DNA binding (Patel et al. 1999).
HR HPV E6 has a PDZ (PSD95/Dlg/ZO-1)-binding motif (PBM) which enables the E6 protein to interact with variety of PDZ domain-containing proteins leading to proteasome-mediated degradation of these proteins (Kranjec & Banks, 2011). The PDZ-binding capacity of HR E6 is important for HPV-mediated carcinogenesis and abnormalities in this association result in abnormal genome amplification and S-phase re-entry (Delury et al 2013).
HR HPV E6 activates telomerase reverse transcriptase (TERT) transcription through a mechanism involving Myc. This activity is a strong contributor to cellular immortalization in conjunction with E7 inhibition of retinoblastoma protein (Rb) (Howie et al. 2009; Wise-Draper & Wells, 2008). E6 has additional targets that mediate modulation of the immune response, G-protein signaling, and disruption of cell adhesion and polarity (Doorbar et al. 2015).
HPV E7 and pRB
HPV E7 is a small 13 kDa protein localized in the nucleus and cytoplasm (MacLaughlin-Drubin and Münger 2009; Wise-Draper & Wells, 2008). During the virus life cycle, E7 plays a major role in activating DNA synthesis in differentiating cells (Cheng et al. 1995). When expressed in an unregulated fashion, such as when the HPV DNA is integrated into the host cell genome, E7 plays a comprehensive role in HPV pathogenesis as it functions on different levels to activate cell cycle progression. The hallmark role of E7 is mediated by its association with the tumor suppressor retinoblastoma protein (Rb) and other members of the same family p105, p107 and p130, and promoting their degradation. Rb represses G1–S transition by binding to and inactivating the transcription factor E2F (Münger et al. 1989). More specifically, when it is hypo- or de-phosphorylated, Rb binds E2F and prevents binding of E2F to specific binding sites in the promoter regions of genes that regulate cell cycle, differentiation, mitosis, and programmed cell death, thus impeding E2F action (DeGregori & Johnson, 2006). During the normal cell cycle, upon activation of growth factor receptor pathways, the cyclin-dependent kinases 4, 6, and 2 (CDK4/6 and CDK2) induce Rb phosphorylation (inactivation) releasing E2F which then can translocate to the nucleus to exert its transcriptional activity on S-phase entry genes (Stevaux & Dyson, 2002).
HPV E7 has no DNA-binding activity, but binds the Rb–E2F complex, causing the release of E2F, and promotes the ubiquitin-dependent proteasomal degradation of Rb (Boyer et al. 1996; Huang et al. 1993). HR HPV E7 has much higher binding affinity for Rb than LR HPV E7 (Gage et al. 1990).
The E2F family contains eight members (E2F1-E2F8). E2F1-E2F5 act as transcriptional activators and E2F6-E2F8 act as repressors (Lammens et al. 2009). Activation of E2F1 by E7 induces feedback E2F6 overexpression which aborts E2F1 action and directs the cell to exit the cell cycle and differentiate (Lyons et al. 2006). E7 can abolish this effect by preventing E2F6 repression of E2F1 which provides further cell cycle enhancement (McLaughlin-Drubin et al. 2008).
HR HPV E7 enhances CDK2 activity, which is important for G1–S-phase transition and progression, through different mechanisms: (1) HR HPV E7 proteins bind and inactivate p21 and p27 CDK inhibitors whose function is to repress cell cycle progression by targeting CDK2. Therefore, CDK2 levels remain elevated despite high levels of p21, which is inactive in HPV-infected cells; on the other hand, LR HPV E7 has low p21-binding affinity and less inhibitory effect (Funk et al. 1997; Jones et al. 1997); (2) Both HR and LR E7 bind directly to CDK2 maintaining constant activity (He et al. 2003). (3) HR E7 promotes CDK2 activity by enhancing its de-phosphorylation through the CDC25A tyrosine phosphatase enzyme (Blomberg & Hoffmann, 1999).
HR HPV E7 alone has transforming ability in cultured human keratinocytes albeit with a much lower efficiency than E6 and E7 combined (Halbert et al. 1991; Hawley-Nelson et al. 1989). HR E7 has telomere lengthening activity in the absence of E6 (Zhang et al. 2004) and produces marked mitotic aberrations which also contribute to major chromosomal alterations and aneuploidy (Duensing et al. 2009). The combined effects of HR HPV E6 and E7 synergize in mediating the transforming capacity of HR HPV (Hawley-Nelson 1989).
Sedman and colleagues (Sedman et al. 1992) showed that a dominant negative mutant of p53 cooperated with E7 to immortalize human keratinocytes (HKc). We utilized a p53 short hairpin RNA (shRNA) which we have extensively characterized (Bheda et al. 2008) in cooperation with E7 in immortalization assays of normal HKc to study E7-mediated immortalization across a panel of individual normal foreskin HKc strains, each derived from a different donor. Table 2 shows the results of these studies: out of a total of 12 individual HKc strains transfected with the retroviral vector LXSH (used as a control, LXSH); LXSH/HPV16 E7 (LXSHE7); p53shRNA + LXSH; or p53shRNA + LXSHE7, five failed to respond to any of the combinations. Among the seven HKc strains that exhibited extended life span after infection (i.e., grew for a greater number of passages than their respective LXSH-infected controls), six were immortalized by the combination of p53shRNA and E7 (marked by a + in the table), while only 2 strains were immortalized by E7 alone. p53shRNA alone did not immortalize any of the HKc strains. Interestingly, the combination of E7 and p53shRNA failed to immortalize 6 of the 12 individual HKc strains. In previous experiments, the full-length HPV16 genome immortalized eight out of ten individual HKc strains transfected (Pirisi et al. 1988). Taken all together, these results help us make some important points: (1) p53shRNA effectively cooperates with E7 in the immortalization of human keratinocyte strains; (2) p53shRNA alone does not immortalize human keratinocytes; (3) as we discussed above, individual normal HKc strains differ considerably in their susceptibility to HPV16-mediated immortalization.
Table 2 Results of immortalization assays with 12 normal HKc strains, derived from 12 different individual donors, transfected with retroviral vectors of the LXSH series, either empty (LXSH) or expressing the HPV16 E7 sequences (LXSHE7) with or without a pSuper vector expressing an shRNA against p53 which we have extensively characterized (Bheda et al. 2008)Full size tableThe data shown above again underscore the central role of p53 for the initial immortalization stages of HPV-mediated transformation.
HPV-mediated carcinogenesis and tumor progression in vivo and in vitro
Malignant progression of HPV-transformed cells is characterized in many cases by continuous expression of E6/E7, associated with genomic instability leading to chromosomal copy number variations and large rearrangements; mutations found in HPV-driven cancer are mainly driven by apolipoprotein B mRNA editing catalytic polypeptide-like (APOBEC) (reviewed in Litwin et al. 2017) which in turn is activated by E6 (Vieira et al. 2014) again pointing to the fact that the activities of the viral oncoproteins determine the pathways utilized for continuous cancer growth and survival in HPV + cancers. Common sites of mutation in HPV-mediated cancers include activating mutations of PIK3CA, particularly frequent in head and neck cancer, and inactivating mutations of SMAD4, found in 28% of HPV + cervical cancers (Litwin et al. 2017). A constant feature of HPV + cancers is the relative lack of inactivating mutations of p53, compared with the high prevalence of p53 mutations in solid tumors (Litwin et al. 2017). Another indication that HPV drives progression in these cancers is the fact that similar mutation patterns and to some extent also chromosomal sites of damage are found in HPV + cancers at different sites (Litwin et al. 2017).
To study the molecular details of HPV-mediated transformation and progression toward malignancy in vitro, we established a model system of normal HKc transfected with a plasmid carrying two copies of the full-length HPV16 genome in a head-to-tail configuration, in the vector pdMMTneo (pMHPV16d, Pirisi et al. 1987, 1988). Transfection of normal HKc with pMHPV16d readily resulted in the establishment of immortalized lines (HKc/HPV16) (Pirisi et al. 1987, 1988) (Fig. 5). Growth factor-independent (HKc/GFI) were obtained from HPV16-immortalized human keratinocytes (HKc/HPV16) by selection in media devoid of EGF and BPE. Differentiation-resistant, premalignant cell lines (HKc/DR) were selected from both HKc/HPV16 and, more readily, from HKc/GFI in media containing 5% fetal bovine serum and 1 mM CaCl2 (Pirisi et al. 1987, 1988; Zyzak et al. 1994) (Fig. 5). Differentiation-resistant HPV16-immortalized human cervical cells developed according to the same experimental protocols we utilized can be fully transformed by v-ras, or HSV2 to form squamous cell carcinomas in nude mice (DiPaolo et al. 1988, 1989). HKc/DR were fully transformed to malignancy by transfection with SIX1 (Xu et al. 2014). Progressive increases in the levels of SIX1 and EGFR mRNA and protein were observed during progression of HKc/HPV16 cells in vitro (Fig. 5) (Xu et al. 2014; Zyzak et al. 1994). In addition, while normal HKc and early-passage HKc/HPV16 were extremely sensitive to growth inhibition by TGF-beta, this sensitivity was completely lost with in vitro progression (Fig. 5) (Borger et al. 2000; Creek et al. 1995; Mi et al. 2000). This work uncovered important connections between HPV E7, the TGF-beta signaling, and SIX1.
Fig. 5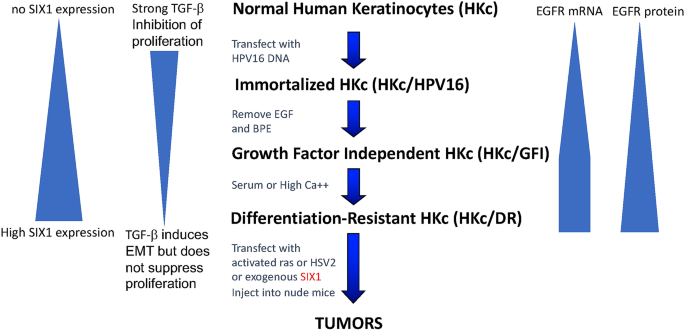
Flow chart of the in vitro model system for HPV16-mediated multistage carcinogenesis utilized in the studies described in this paper. The flow chart includes the various steps in the progression from normal human keratinocytes (HKc) to differentiation-resistant, pre-malignant keratinocytes (HKc/DR) to tumor cells. In the course of this process, epidermal growth factor receptor (EGFR) and SIX1 mRNA and protein levels increase progressively, while sensitivity to growth inhibition by transforming growth factor-beta (TGF-β) progressively decreases
Full size imageThe homeobox gene SIX1 is essential in development, particularly for the auditory and olfactory systems and the kidney and is silenced in normal adult tissue (Christensen et al. 2008). However, like other developmentally relevant genes, SIX1 is re-expressed in cancers of the breast, ovaries, uterine cervix, and other tissues (Coletta et al. 2010; Wan et al. 2008). SIX1 overexpression in cancer correlates with metastatic behavior and decreased survival (Tan et al. 2011; Zheng 2010). SIX1 has also been shown to transform immortalized mammary cell lines to malignancy, and to promote epithelial–mesenchymal transition (EMT) and invasive behavior when overexpressed in cancer cells (Coletta et al. 2008; Micalizzi et al. 2009; Xu et al. 2014). We became interested in the role of SIX1 in HPV16-mediated carcinogenesis when we identified SIX1 as one of the genes with robust overexpression in all HKc/DR versus their HKc/HPV16 line of origin, and in HKc/HPV16 versus normal HKc in our model system (Wan et al. 2008). When we explored the expression of SIX1 protein in cervical tissue microarrays with paired cancer and surrounding normal tissue samples, we found that about 23% of cervical cancers stained strongly for SIX1, while none of the normal tissue samples had strong staining for this protein (Wan et al. 2008).
In subsequent work, we identified SIX1 as one of the main “drivers” of progression and a primary cause of malignant conversion of HPV16-transformed human cells in vitro. We also uncovered the involvement of the TGF-β signaling pathway in this process (Xu et al. 2014). We determined that SIX1 increases HPV16 E6/E7 expression and, in turn, E7 increases SIX1 levels, very likely by mediating RB degradation and release of E2F, as SIX1 expression is under the control of E2F (Young et al. 2003). SIX1 induces E7 expression indirectly, by modifying TGF-β signaling (Xu et al. 2014, 2015). In short, when SIX1 is expressed, the levels of TGF-β receptors I, II, and III increase. However, signaling through the canonical Smad pathway is markedly decreased, and much of the TGF-β signaling occurs through non-canonical pathways, in particular MAPK pathways such as ERK1/ERK2, p38 MAPK, and JNK (Xu et al. 2014). Phenotypically, this translates into progressive and ultimately complete loss of sensitivity to growth inhibition by TGF-β that HKc/HPV16 cells exhibit as they progress to HKc/DR in vitro (Borger et al. 2000; Creek et al. 1995; Mi et al. 2000).
We have found that, in HKc/HPV16, growth inhibition by TGF-β is mediated through Smad signaling, while the induction of epithelial–mesenchymal transition (EMT) is driven primarily by non-canonical pathways of TGF-β signaling (Xu et al. 2014, 2015). We also determined that SIX1 overexpression induces EMT and differentiation resistance in HKc/HPV16, and further EMT and malignant conversion in HKc/DR (Xu et al. 2014, 2015). Canonical TGF-β signaling through the Smad pathway also decreases transcription from the p97 promoter, effectively inhibiting E6/E7 expression (Baldwin et al. 2004).
In summary, in HPV-immortalized cells at early stages of transformation in vitro, TGF-β and E7 exist in a balance: E7, through SIX1, modifies TGF-β signaling in such a way as to decrease the Smad-mediated signals that inhibit transcription from the p97 promoter (Baldwin et al. 2004; Kowli et al. 2013; Xu et al. 2014, 2015). As the cells progress, E7 levels increase and so do SIX1 levels, driving TGF-β signaling to non-canonical pathways, freeing the cells from growth inhibition by TGF-β, promoting differentiation resistance and EMT. When we attempted to reduce SIX1 levels in HKc/HPV16 and HKc/DR with siRNA approaches, we found that these cells did not tolerate a loss of SIX1 and either overcame the siRNA effects or died (Hosseinipour et al. 2019).
Taken all together, our results identify SIX1 and TGF-β as major players in at least one pathway of malignant progression of HPV-transformed cells that depend on E6/E7 expression for proliferation and survival. It is important to verify these pathways in human tumors, because SIX1 is an attractive, albeit difficult, therapeutic target frequently overexpressed in cancer cells but not expressed in normal adult cells (Blevins et al. 2015; Zhou et al. 2020). Importantly, we have previously shown that many of the gene expression changes that characterize progression in our in vitro model system correspond to changes detected in HPV + cancer, and that SIX1 is overexpressed in at least 23% of cervical cancers (Wan et al. 2008).
Evolving concepts on the role of HPV in head and neck and cervical cancer progression
About 25% of all head and neck squamous cell carcinomas (HNSCC) and up to 90% of oropharyngeal cancers are positive for HPV DNA (Chung & Gillison, 2009). As we have discussed above, HPV-positive HNSCC present at a younger age than do HPV-negative tumors, are not associated with smoking or alcohol consumption, and have a better response to standard chemo/radiation therapy and a better prognosis (Chung & Gillison, 2009; Weinberger et al. 2010). African American (AA) patients, especially AA males, present primarily with HPV-negative HNSCC (Weinberger et al. 2010). We found that, even when positive for HPV DNA, oral, and oropharyngeal cancers in most AA patients (and in some Caucasian patients as well) were HPV-inactive: these cancers were positive for viral genomic DNA, but did not express the transcripts encoding the E6 and E7 oncoproteins (Tomar et al. 2016). Interestingly, primary HNSCC that are HPV-positive tend to be HPV-active (express E6/E7 transcripts), whereas recurring HPV-positive cancers of the head and neck are more often inactive (Weinberger et al. 2010). Gene expression profiling identified a gene expression signature of HPV-inactive tumors that is intermediate between HPV-active and HPV-negative cancers (Tomar et al. 2016). In addition, HPV-inactive cancers have survival rates similar to those of HPV-negative tumors, completely distinct from those of HPV-active tumors (Tomar et al. 2016). Last, but not least, p16 positivity (a marker of E7 activity) in HPV + tumor masses is not uniformly distributed across every HPV + cancer: in many cases, different components can be observed in a tumor mass, some strongly positive for p16 staining, others virtually negative and behave more aggressively than the p16 expressing tumors (Nicolas et al. 2020; Ryu et al. 2017), suggesting that populations of cells that no longer express E7 can arise during tumor progression in vivo.
Recently, we analyzed the mutation and gene expression profile data from 264 cases of cervical cancers in the TCGA database. We determined that 7.7% of cervical cancers are also HPV-inactive (Banister et al. 2017). Similar to what has been shown in head and neck cancer, HPV-inactive cervical cancers were distinct clinical entities: they occurred in older women (median age 54 vs. 45 years, p = 0.02) and had worse survival (median 715 vs 3046 days, p = 0.0003). The mutational, gene expression and methylation profiles of these tumors were also distinct from those of HPV-active cervical cancers (Banister et al. 2017).
The existence of HPV-inactive cancers in the head and neck had been previously documented (Weinberger et al. 2006, 2010). However, the widely accepted interpretation was that HPV was a mere “passenger” with no causal role in the development of those neoplasms. The existence of HPV-inactive cervical cancers was a completely novel finding which, together with the observations made by us and others in cancers of the head and neck, challenged the widespread notion that continuous expression of E6/E7 is necessary to maintain cancerous growth in HPV-mediated cancers. Taken all together, these observations strongly suggest that one of the possible pathways of malignant progression for HPV-mediated tumors involves the acquisition of independence from the continuous expression of E6/E7 for growth and survival of HPV-transformed cells.
To investigate the pathways that may lead to independence from E6/E7 expression in HPV-transformed cells, we compared mutation patterns between HPV-negative and HPV-positive cancers of the cervix and the head and neck included in the TCGA database. Similar to head and neck cancer (Westra et al. 2008), p53 mutations were rare among HPV-active cancers; however, they were much more common in HPV-inactive tumors of the cervix (Banister et al. 2017). Along the same lines, among the somatic mutations found most commonly in HPV-negative and HPV-positive cancers of the head and neck in the TCGA database, TP53 mutations and CDKN2A mutations show the highest differential distribution (Table 3).
Table 3 examples of genes mutated with different frequencies between HPV-positive and HPV-negative head and neck cancersFull size tableBased on these findings, we set out to determine whether deletion of p53 by CRISPR-Cas 9 in HKc/DR cells decreased E6/E7 expression. Loss of p53 did not immediately cause a complete loss of E7 expression in p53 knockout cells. Rather, we observed only about a 50% decrease in E7 mRNA levels, measured by real-time qPCR, in the p53 knockout cells soon after selection and clonal expansion. However, over time and with repeated low-density passaging, p53 deletion allowed for the development of distinct cell populations that no longer expressed HPV16 E6/E7, as demonstrated by RNAScope in situ hybridization for HPV16 E6/E7 in the clonal p53 knockout lines (Abboodi et al. 2021). Importantly, cell populations devoid of E6/E7 expression did not develop in the control HKc/DR with intact p53 that had been subjected to the same transfection, selection, clonal expansion, and number of passages as the p53 knockout cells (Abboodi et al. 2021). Treatment with 5-aza-cytidine restored E7 mRNA levels in the p53 knockout lines, indicating that methylation events were likely responsible for suppressing E7 expression, although we do not yet know exactly what methylation target(s) are relevant in this regard, whether viral or cellular, or both (Abboodi et al. 2021).
We also asked whether expression of activated ras (HrasV12, Addgene) would influence E7 expression in HKc/HPV16: E7 mRNA levels decreased by about 60% in HrasV12-transfected cells. Conversely, Rb protein levels measured by ELISA increased by about sixfold in the HrasV12-transfected cells (Renrick, unpublished data), indicating that E7 function was indeed depressed. In a cognate line of work, we had determined that the growth factor-independent lines (HKc/GFI) derived from our HKc/HPV16 lines (Pirisi et al. 1988) express high levels of EGFR, much of which is active in the absence of exogenous growth factors (Zyzak et al. 1994). We have also explored the expression of autocrine/paracrine growth factors in HPV16-transformed HKc and cancer-derived cell lines and identified alternatively spliced forms of transforming growth factor-alpha (TGFa) lacking the C-terminal valine residues necessary for processing of the transmembrane pro-TGFa precursor and release of mature TGFa in the medium (Xu et al. 1999). These variants, when overexpressed in CHO cells, supported proliferation in the absence of exogenous growth factors; in addition, we determined that the TGFa variants C-terminal sequences mediated specific interactions with ErbB2 and ErbB4 (Xu et al. 1999, 2000).
Taken all together, these findings provide proof of principle for a causal role of HPV in the development of HPV-inactive cancers of the cervix and the head and neck and suggest some possible mechanisms by which cancer cells may lose dependence from the continued expression of E6 and E7.
We postulate that HPV-inactive cancers begin as HPV-active lesions (cancerous or pre-malignant) and progress to HPV-independence through a mechanism involving loss of p53, in conjunction with other changes that support continuous proliferation and survival in the absence of E7. If confirmed in human cancers in vivo, these findings have considerable ramifications for our understanding of the mechanisms of progression of HPV-driven cancers, and for the treatment of both HPV-active and HPV-inactive cancers.
We have evidence of some residual p53 activity in HPV-transformed human epithelial cells, despite E6-mediated degradation of p53. At any given time, HKc/HPV16 express about 30% levels of p53 protein found in normal HKc (Delva, 2015). Yet, HKc/HPV16 and to some extent HKc/DR respond to a brief (30 s) UV irradiation treatment with about a twofold increase in p53 protein levels. Maximum increase in p53 protein level was observed 12 h after UV treatment in normal HKc and HKc/HPV16, and much earlier (as soon as 1 h after treatment) in HKc/DR (Delva, 2015). Interestingly, the magnitude and time course of p53 responses to UV treatment vary dramatically among different individual normal HKc strains (Abboodi, 2016) again underscoring differences that may contribute to interindividual variability in susceptibility to HPV-mediated immortalization and transformation.
The fact that HPV-positive cancer cells may still mount a p53 response to radiation and chemotherapy, in addition to the proliferative nature of these cancers, is believed to contribute to the relative radio- and chemo-sensitivity of these tumors and therefore to their overall better response to therapy. However, the same therapeutic means have the potential to produce mutations, including p53 mutations and ras mutations. If our observations in vitro translate to what happens in tumors, we can reasonably expect that cells in which p53 is mutated as a consequence of cytotoxic therapy may give rise to secondary populations of HPV-inactive cells, which are now much more similar to HPV-negative cancers: radio- and chemo-resistant and prone to EMT, invasion, and angiogenesis according to their gene expression profiles (Tomar et al. 2016). Whether or not additional growth stimuli contribute to the loss of E6/E7 expression (i.e., activation of ras or other growth signaling pathways) should be investigated. Along the same lines, therapeutic vaccines developed against the E6/E7 antigens may kill off HPV-active cells in the context of a tumor but allow subpopulations of HPV-inactive cells to take over leading to recurrence of a more aggressive tumor. A demonstration that these mechanisms actually come into play in vivo would add to our understanding of the dynamics of progression of HPV-driven cancers and allow for the development of specific therapeutic approaches that take these mechanisms into account and perhaps even take advantage of them for better outcomes.
The fact that the origin of HPV-inactive cancers, which are relatively rare in the cervix but common among head and neck cancers, can also be attributed to HPV broadens the reach of the HPV vaccines in the prevention of cancer, particularly of the head and neck.
Summary and prospective
E6 and E7 are the major agents of HPV-mediated immortalization, primarily because of their respective interactions with p53 and Rb and their many and diverse accessory functions that, taken all together, contribute to promoting uncontrolled proliferation, mitotic abnormalities, chromosomal aberrations, and genomic instability. Progression of HPV16-immortalized cells toward malignancy occurs with different mechanisms, depending at least in part on p53 status: in cells with intact p53, continuous expression of E6/E7 is paramount to maintain tumor cell growth and survival. If p53 function is permanently lost, continuous expression of E6/E7 becomes unnecessary and in time is suppressed, giving rise to HPV-inactive cells. Among the many questions that remain to be investigated is whether or not the same mechanisms operate in vivo to give origin to HPV-inactive tumors; what additional changes are necessary for a complete loss of E6/E7 expression to occur; and which of these changes provide opportunities for novel therapeutic interventions.
References
Abboodi, F., Buckhaults, P., Altomare, D., Liu, C., Hosseinipour, M., Banister, C. E., Creek, K. E., & Pirisi, L. (2021). HPV-inactive cell populations arise from HPV16-transformed human keratinocytes after p53 knockout. Virology, 554, 9–16. https://doi.org/10.1016/j.virol.2020.12.005
Abboodi, F. F. (2016). Tumor Suppressor p53 Response To UV Light In Normal Human Keratinocyte Strains From Different Individuals. (Master's thesis). Retrieved from https://scholarcommons.sc.edu/etd/3427
Adelstein, D. J., Ridge, J. A., Gillison, M. L., Chaturvedi, A. K., D'Souza, G., Gravitt, P. et al. (2009). Head and neck squamous cell cancer and the human papillomavirus: summary of a National Cancer Institute State of the Science Meeting, November 9–10, 2008, Washington, D.C. Head & neck, 31(11), 1393–1422. https://doi.org/10.1002/hed.21269
Akerman, G. S., Tolleson, W. H., Brown, K. L., Zyzak, L. L., Mourateva, E., Engin, T. S., Basaraba, A., Coker, A. L., Creek, K. E., & Pirisi, L. (2001). Human papillomavirus type 16 E6 and E7 cooperate to increase epidermal growth factor receptor (EGFR) mRNA levels, overcoming mechanisms by which excessive EGFR signaling shortens the life span of normal human keratinocytes. Cancer Research, 61, 3837–3843
Alexandrov, L. B., Nik-Zainal, S., Wedge, D. C., Aparicio, S. A., Behjati, S., Biankin, A. V., Bignell, G. R., Bolli, N., Borg, A., Børresen-Dale, A. L., Boyault, S., Burkhardt, B., Butler, A. P., Caldas, C., Davies, H. R., Desmedt, C., Eils, R., Eyfjörd, J. E., Foekens, J. A., … Stratton, M. R. (2013). Signatures of mutational processes in human cancer. Nature, 500(7463), 415–421. https://doi.org/10.1038/nature12477
Antonsson, A., Forslund, O., Ekberg, H., Sterner, G., & Hansson, B. G. (2000). The ubiquity and impressive genomic diversity of human skin papillomaviruses suggest a commensalic nature of these viruses. Journal of Virology, 74(24), 11636–11641. https://doi.org/10.1128/jvi.74.24.11636-11641.2000
Antonsson, A., & McMillan, N. A. (2006). Papillomavirus in healthy skin of Australian animals. Journal of General Virology, 87(Pt 11), 3195–3200
Arenz, A., Ziemann, F., Mayer, C., Wittig, A., Dreffke, K., Preising, S., Wagner, S., Klussmann, J. P., Engenhart-Cabillic, R., & Wittekindt, C. (2014). Increased radiosensitivity of HPV-positive head and neck cancer cell lines due to cell cycle dysregulation and induction of apoptosis. Strahlentherapie und Onkologie : Organ der Deutschen Rontgengesellschaft ... [et al], 190(9), 839–846. https://doi.org/10.1007/s00066-014-0605-5
Aydin, I., Villalonga-Planells, R., Greune, L., Bronnimann, M. P., Calton, C. M., Becker, M., Lai, K. Y., Campos, S. K., Schmidt, M. A., & Schelhaas, M. (2017). A central region in the minor capsid protein of papillomaviruses facilitates viral genome tethering and membrane penetration for mitotic nuclear entry. PLoS Pathogens, 13, e1006308
Baldwin, A., Pirisi, L., & Creek, K. E. (2004). NFI-Ski interactions mediate transforming growth factor beta modulation of human papillomavirus type 16 early gene expression. Journal of Virology, 78(8), 3953–3964. https://doi.org/10.1128/jvi.78.8.3953-3964.2004
Banister, C. E., Liu, C., Pirisi, L., Creek, K. E., & Buckhaults, P. J. (2017). Identification and characterization of HPV-independent cervical cancers. Oncotarget, 8(8), 13375–13386. https://doi.org/10.18632/oncotarget.14533
Beachler, D. C., Weber, K. M., Margolick, J. B., Strickler, H. D., Cranston, R. D., Burk, R. D., Wiley, D. J., Minkoff, H., Reddy, S., Stammer, E. E., Gillison, M. L., & D’Souza, G. (2012). Risk factors for oral HPV infection among a high prevalence population of HIV-positive and at-risk HIV-negative adults. Cancer Epidemiology, Biomarkers & Prevention, 21(1), 122–133. https://doi.org/10.1158/1055-9965.EPI-11-0734
Benedetti, F., Curreli, S., Gallo, R. C., & Zella, D. (2021). Tampering of viruses and bacteria with host DNA repair: Implications for cellular transformation. Cancers, 13(2), 241. https://doi.org/10.3390/cancers13020241
Berman, T. A., & Schiller, J. T. (2017). Human papillomavirus in cervical cancer and oropharyngeal cancer: One cause, two diseases. Cancer, 123(12), 2219–2229. https://doi.org/10.1002/cncr.30588
Bernard, H. U., Burk, R. D., Chen, Z., van Doorslaer, K., & zur Hausen, H., de Villiers, E. M. . (2010). Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology, 401(1), 70–79. https://doi.org/10.1016/j.virol.2010.02.002
Bettampadi, D., Sirak, B. A., Abrahamsen, M. E., Reich, R. R., Villa, L. L., Ponce, E. L., & Giuliano, A. R. (2020). Factors associated with persistence and clearance of high-risk oral HPV among participants in the HPV Infection in Men (HIM) study. Clinical Infectious Diseases: an Official Publication of the Infectious Diseases Society of America, ciaa1701. Advance online publication. https://doi.org/10.1093/cid/ciaa1701
Bhatla, N., Berek, J. S., Cuello Fredes, M., Denny, L. A., Grenman, S., Karunaratne, K., et al. (2019). Revised FIGO staging for carcinoma of the cervix uteri. International Journal of Gynaecology and Obstetrics: The Official Organ of the International Federation of Gynaecology and Obstetrics, 145(1), 129–135. https://doi.org/10.1002/ijgo.12749
Bheda, A., Creek, K. E., & Pirisi, L. (2008). Loss of p53 induces epidermal growth factor receptor promoter activity in normal human keratinocytes. Oncogene, 27(31), 4315–4323. https://doi.org/10.1038/onc.2008.65
Blackford, A. N., & Jackson, S. P. (2017). ATM, ATR, and DNA-PK: The trinity at the heart of the DNA damage response. Molecular Cell, 66(6), 801–817. https://doi.org/10.1016/j.molcel.2017.05.015
Blevins, M. A., Towers, C. G., Patrick, A. N., Zhao, R., & Ford, H. L. (2015). The SIX1-EYA transcriptional complex as a therapeutic target in cancer. Expert Opinion on Therapeutic Targets, 19(2), 213–225. https://doi.org/10.1517/14728222.2014.978860
Blomberg, I., & Hoffmann, I. (1999). Ectopic expression of Cdc25A accelerates the G(1)/S transition and leads to premature activation of cyclin E- and cyclin A-dependent kinases. Molecular and Cellular Biology, 19(9), 6183–6194. https://doi.org/10.1128/mcb.19.9.6183
Bonner, J. A., Harari, P. M., Giralt, J., Azarnia, N., Shin, D. M., Cohen, , et al. (2006). Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. The New England Journal of Medicine, 354(6), 567–578. https://doi.org/10.1056/NEJMoa053422
Borger, D. R., Mi, Y., Geslani, G., Zyzak, L. L., Batova, A., Engin, T. S., Pirisi, L., & Creek, K. E. (2000). Retinoic acid resistance at late stages of human papillomavirus type 16-mediated transformation of human keratinocytes arises despite intact retinoid signaling and is due to a loss of sensitivity to transforming growth factor-beta. Virology, 270(2), 397–407. https://doi.org/10.1006/viro.2000.0282
Boyer, S. N., Wazer, D. E., & Band, V. (1996). E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Research, 56(20), 4620–4624
Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A., & Jemal, A. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a Cancer Journal for Clinicians, 68(6), 394–424. https://doi.org/https://doi.org/10.3322/caac.21492
Bruni, L., Diaz, M., Castellsagué, X., Ferrer, E., Bosch, F. X., & de Sanjosé, S. (2010). Cervical human papillomavirus prevalence in 5 continents: Meta-analysis of 1 million women with normal cytological findings. The Journal of Infectious Diseases, 202(12), 1789–1799. https://doi.org/10.1086/657321
Burley, M., Roberts, S., & Parish, J. L. (2020). Epigenetic regulation of human papillomavirus transcription in the productive virus life cycle. Semin Immunopathol, 42, 159–171. https://doi.org/10.1007/s00281-019-00773-0
Calton, C. M., Bronnimann, M. P., Manson, A. R., Li, S., Chapman, J. A., Suarez-Berumen, M., Williamson, T. R., Molugu, S. K., Bernal, R. A., & Campos, S. K. (2017). Translocation of the papillomavirus L2/vDNA complex across the limiting membrane requires the onset of mitosis. PLoS Pathogens, 13, e1006200
Cancer Genome Atlas Research Network. (2017). Integrated genomic and molecular characterization of cervical cancer. Nature, 543(7645), 378–384. https://doi.org/10.1038/nature21386
Carr A. M. (2000). Cell cycle. Piecing together the p53 puzzle. Science (New York, N.Y.), 287(5459), 1765–1766. https://doi.org/https://doi.org/10.1126/science.287.5459.1765
Chabeda, A., Yanez, R., Lamprecht, R., Meyers, A. E., Rybicki, E. P., & Hitzeroth, I. I. (2018). Therapeutic vaccines for high-risk HPV-associated diseases. Papillomavirus Research (Amsterdam, Netherlands), 5, 46–58. https://doi.org/10.1016/j.pvr.2017.12.006
Chaturvedi, A. K., Engels, E. A., Anderson, W. F., & Gillison, M. L. (2008). Incidence trends for human papillomavirus-related and -unrelated oral squamous cell carcinomas in the United States. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology, 26(4), 612–619. https://doi.org/10.1200/JCO.2007.14.1713
Chen, D., Juko-Pecirep, I., Hammer, J., Ivansson, E., Enroth, S., Gustavsson, I., Feuk, L., Magnusson, P. K., McKay, J. D., Wilander, E., & Gyllensten, U. (2013). Genome-wide association study of susceptibility loci for cervical cancer. Journal of the National Cancer Institute, 105(9), 624–633. https://doi.org/10.1093/jnci/djt051
Cheng, S., Schmidt-Grimminger, D. C., Murant, T., Broker, T. R., & Chow, L. T. (1995). Differentiation-dependent up-regulation of the human papillomavirus E7 gene reactivates cellular DNA replication in suprabasal differentiated keratinocytes. Genes & Development, 9(19), 2335–2349. https://doi.org/10.1101/gad.9.19.2335
Christensen, K. L., Patrick, A. N., McCoy, E. L., & Ford, H. L. (2008). The six family of homeobox genes in development and cancer. Advances in Cancer Research, 101, 93–126. https://doi.org/10.1016/S0065-230X(08)00405-3
Chung, C., & Gillison, M. (2009). Human papillomavirus in head and neck cancer: Its role in pathogenesis and clinical implications. Clinical Cancer Research, 15, 6758–6762. https://doi.org/10.1158/1078-0432.CCR-09-0784
Coletta, R. D., Christensen, K. L., Micalizzi, D. S., Jedlicka, P., Varella-Garcia, M., & Ford, H. L. (2008). Six1 overexpression in mammary cells induces genomic instability and is sufficient for malignant transformation. Cancer Research, 68(7), 2204–2213. https://doi.org/10.1158/0008-5472.CAN-07-3141
Coletta, R. D., McCoy, E. L., Burns, V., Kawakami, K., McManaman, J. L., Wysolmerski, J. J., & Ford, H. L. (2010). Characterization of the Six1 homeobox gene in normal mammary gland morphogenesis. BMC Developmental Biology, 10, 4. https://doi.org/10.1186/1471-213X-10-4
Creek, K. E., Geslani, G., Batova, A., & Pirisi, L. (1995). Progressive loss of sensitivity to growth control by retinoic acid and transforming growth factor-beta at late stages of human papillomavirus type 16-initiated transformation of human keratinocytes. Advances in Experimental Medicine and Biology, 375, 117–135. https://doi.org/10.1007/978-1-4899-0949-7_11
Crook, T., Fisher, C., Masterson, P., & Vousden, K. (1994). Modulation of transcriptional regulatory properties of p53 by HPV E6. Oncogene, 9, 1225–1230
Das Ghosh, D., Mukhopadhyay, I., Bhattacharya, A., Roy Chowdhury, R., Mandal, N. R., Roy, S., & Sengupta, S. (2017). Impact of genetic variations and transcriptional alterations of HLA class I genes on cervical cancer pathogenesis. International Journal of Cancer, 140(11), 2498–2508. https://doi.org/10.1002/ijc.30681
de Villiers, E. M. (2013). Cross-roads in the classification of papillomaviruses. Virology, 445(1–2), 2–10. https://doi.org/10.1016/j.virol.2013.04.023
DeGregori, J., & Johnson, D. G. (2006). Distinct and Overlapping Roles for E2F Family Members in Transcription, Proliferation and Apoptosis. Current Molecular Medicine, 6(7), 739–748. https://doi.org/10.2174/1566524010606070739
Delury, C. P., Marsh, E. K., James, C. D., Boon, S. S., Banks, L., Knight, G. L., & Roberts, S. (2013). The role of protein kinase A regulation of the E6 PDZ-binding domain during the differentiation-dependent life cycle of human papillomavirus type 18. Journal of Virology, 87, 9463–9472. https://doi.org/10.1128/JVI.01234-13
Delva, N. C. (2015). Tp53 and Hras Influence on HPV16 E7 Expression in HPV16-Transformed Human Keratinocytes. (Master's thesis). Retrieved from https://scholarcommons.sc.edu/etd/3090
DiMaio, D., & Mattoon, D. (2001). Mechanisms of cell transformation by papillomavirus E5 proteins. Oncogene, 20(54), 7866–7873. https://doi.org/10.1038/sj.onc.1204915
DiMaio, D., & Petti, L. M. (2013). The E5 proteins. Virology, 445(1–2), 99–114. https://doi.org/10.1016/j.virol.2013.05.006
DiPaolo, J. A., Popescu, N. C., Ablashi, D. V., Lusso, P., Zimonjic, D. B., & Woodworth, C. D. (1994). Multistage carcinogenesis utilizing human genital cells and human papillomaviruses. Toxicology Letters, 72(1–3), 7–11. https://doi.org/10.1016/0378-4274(94)90004-3
DiPaolo, J. A., Woodworth, C. D., Popescu, N. C., Koval, D. L., Lopez, J. V., & Doniger, J. (1990). HSV-2-induced tumorigenicity in HPV16-immortalized human genital keratinocytes. Virology, 177, 777–779. https://doi.org/10.1016/0042-6822(90)90548-6
DiPaolo, J. A., Woodworth, C. D., Popescu, N. C., Notario, V., & Doniger, J. (1989). Induction of human cervical squamous cell carcinoma by sequential transfection with human papillomavirus 16 DNA and viral Harvey ras. Oncogene, 4, 395–399
Diskin, B., Adam, S., Cassini, M. F., Sanchez, G., Liria, M., Aykut, B., Buttar, C., Li, E., Sundberg, B., Salas, R. D., Chen, R., Wang, J., Kim, M., Farooq, M. S., Nguy, S., Fedele, C., Tang, K. H., Chen, T., Wang, W., … Miller, G. (2020). PD-L1 engagement on T cells promotes self-tolerance and suppression of neighboring macrophages and effector T cells in cancer. Nature Immunology, 21(4), 442–454. https://doi.org/10.1038/s41590-020-0620-x
Donalisio, M., Cagno, V., Vallino, M., Moro, G. E., Arslanoglu, S., Tonetto, P., et al. (2014). Inactivation of high-risk human papillomaviruses by Holder pasteurization: Implications for donor human milk banking. Journal of Perinatal Medicine, 42(1), 1–8. https://doi.org/10.1515/jpm-2013-0200
Doorbar, J. (2013). The E4 protein; structure, function and patterns of expression. Virology, 445(1–2), 80–98. https://doi.org/10.1016/j.virol.2013.07.008
Doorbar, J., Egawa, N., Griffin, H., Kranjec, C., Murakami, I. (2015). Human papillomavirus molecular biology and disease association. Reviews in Medical Virology, 25 Suppl 1(Suppl Suppl 1), 2–23. https://doi.org/10.1002/rmv.1822
Doorbar J. (2006). Molecular biology of human papillomavirus infection and cervical cancer. Clinical Science (London, England: 1979), 110(5), 525–541. https://doi.org/10.1042/CS20050369
Dreer, M., van de Poel, S., & Stubenrauch, F. (2017). Control of viral replication and transcription by the papillomavirus E8^E2 protein. Virus Research, 231, 96–102. https://doi.org/10.1016/j.virusres.2016.11.005
Duensing, A., Spardy, N., Chatterjee, P., Zheng, L., Parry, J., Cuevas, R., Korzeniewski, N., & Duensing, S. (2009). Centrosome overduplication, chromosomal instability, and human papillomavirus oncoproteins. Environmental and Molecular Mutagenesis, 50, 741–747. https://doi.org/10.1002/em.20478
el-Deiry, W. S. (1998). Regulation of p53 downstream genes. Seminars in Cancer Biology, 8(5), 345–357. https://doi.org/10.1006/scbi.1998.0097
Ferris, R. L., Blumenschein, G., Jr., Fayette, J., Guigay, J., Colevas, A. D., Licitra, L., et al. (2016). Nivolumab for recurrent squamous-cell carcinoma of the head and neck. The New England Journal of Medicine, 375(19), 1856–1867. https://doi.org/10.1056/NEJMoa1602252
Funk, J. O., Waga, S., Harry, J. B., Espling, E., Stillman, B., & Galloway, D. A. (1997). Inhibition of CDK activity and PCNA-dependent DNA replication by p21 is blocked by interaction with the HPV-16 E7 oncoprotein. Genes & Development, 11, 2090–2100. https://doi.org/10.1101/gad.11.16.2090
Gage, J. R., Meyers, C., & Wettstein, F. O. (1990). The E7 proteins of the nononcogenic human papillomavirus type 6b (HPV-6b) and of the oncogenic HPV-16 differ in retinoblastoma protein binding and other properties. Journal of Virology, 64(2), 723–730. https://doi.org/10.1128/JVI.64.2.723-730.1990
Gillison, M. L., Chaturvedi, A. K., & Lowy, D. R. (2008). HPV prophylactic vaccines and the potential prevention of noncervical cancers in both men and women. Cancer, 113(10 Suppl), 3036–3046. https://doi.org/10.1002/cncr.23764
Goodman, A. M., Kato, S., Chattopadhyay, R., Okamura, R., Saunders, I. M., Montesion, M., Frampton, G. M., Miller, V. A., Daniels, G. A., & Kurzrock, R. (2019). Phenotypic and genomic determinants of immunotherapy response associated with squamousness. Cancer immunology research, 7(6), 866–873. https://doi.org/10.1158/2326-6066.CIR-18-0716
Graves, C., Abboodi, F., Tomar, S., Wells, J., & Pirisi, L. (2014). The translational significance of epithelial mesenchymal transition in head and neck cancer. Clinical and Translational Medicine, 3, 39–52. https://doi.org/10.1186/s40169-014-0039
Griffiths, P. (1999). Time to consider the concept of a commensal virus? Reviews in medical virology, 9(2), 73–74. https://doi.org/10.1002/(sici)1099-1654(199904/06)9:2%3c73::aid-rmv254%3e3.0.co;2-5
Guihard, S., Ramolu, L., Macabre, C., Wasylyk, B., Noël, G., Abecassis, J., & Jung, A. C. (2012). The NEDD8 conjugation pathway regulates p53 transcriptional activity and head and neck cancer cell sensitivity to ionizing radiation. International Journal of Oncology, 41(4), 1531–1540. https://doi.org/10.3892/ijo.2012.1584
Gupta, S., Kumar, P., & Das, B. C. (2018). HPV: Molecular pathways and targets. Current Problems in Cancer, 42(2), 161–174. https://doi.org/10.1016/j.currproblcancer.2018.03.003
Halbert, C. L., Demers, G. W., & Galloway, D. A. (1991). The E7 gene of human papillomavirus type 16 is sufficient for immortalization of human epithelial cells. Journal of Virology, 65(1), 473–478. https://doi.org/10.1128/JVI.65.1.473-478.1991
Hammer, A., de Koning, M. N., Blaakaer, J., Steiniche, T., Doorbar, J., Griffin, H., Mejlgaard, E., Svanholm, H., Quint, W. G., & Gravitt, P. E. (2019). Whole tissue cervical mapping of HPV infection: Molecular evidence for focal latent HPV infection in humans. Papillomavirus Res., 7, 82–87
Hawley-Nelson, P., Vousden, K. H., Hubbert, N. L., Lowy, D. R., & Schiller, J. T. (1989). HPV16 E6 and E7 proteins cooperate to immortalize human foreskin keratinocytes. The EMBO Journal, 8(12), 3905–3910
He, W., Staples, D., Smith, C., & Fisher, C. (2003). Direct activation of cyclin-dependent kinase 2 by human papillomavirus E7. Journal of Virology, 77(19), 10566–10574. https://doi.org/10.1128/jvi.77.19.10566-10574.2003
Herbst, R. S., Soria, J. C., Kowanetz, M., Fine, G. D., Hamid, O., Gordon, M. S., Sosman, J. A., McDermott, D. F., Powderly, J. D., Gettinger, S. N., Kohrt, H. E., Horn, L., Lawrence, D. P., Rost, S., Leabman, M., Xiao, Y., Mokatrin, A., Koeppen, H., Hegde, P. S., … Hodi, F. S. (2014). Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature, 515(7528), 563–567. https://doi.org/10.1038/nature14011
Hosseinipour, M., Wan, F., Altomare, D., Creek, K. E., & Pirisi, L. (2019). HPV16-transformed human keratinocytes depend on SIX1 expression for proliferation and HPV E6/E7 gene expression. Virology, 537, 20–30. https://doi.org/10.1016/j.virol.2019.08.009
Howie, H. L., Katzenellenbogen, R. A., & Galloway, D. A. (2009). Papillomavirus E6 proteins. Virology, 384(2), 324–334. https://doi.org/10.1016/j.virol.2008.11.017
Hu, Z., Zhu, D., Wang, W., Li, W., Jia, W., Zeng, X., et al. (2015). Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nature Genetics, 47(2), 158–163. https://doi.org/10.1038/ng.3178
Huang, P. S., Patrick, D. R., Edwards, G., Goodhart, P. J., Huber, H. E., Miles, L., Garsky, V. M., Oliff, A., & Heimbrook, D. C. (1993). Protein domains governing interactions between E2F, the retinoblastoma gene product, and human papillomavirus type 16 E7 protein. Molecular and Cellular Biology, 13(2), 953–960. https://doi.org/10.1128/mcb.13.2.953
Huibregtse, J. M., Scheffner, M., & Howley, P. M. (1991). A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. The EMBO Journal, 10(13), 4129–4135
Ilahi, N. E., & Bhatti, A. (2020). Impact of HPV E5 on viral life cycle via EGFR signaling. Microbial Pathogenesis, 139, 103923. https://doi.org/10.1016/j.micpath.2019.103923
Jemal, A., Bray, F., Center, M. M., Ferlay, J., Ward, E., Forman, D. (2011). Global cancer statistics. CA: a Cancer Journal for Clinicians, 61(2), 69–90. https://doi.org/10.3322/caac.20107
Jeon, S., Allen-Hoffmann, B. L., & Lambert, P. F. (1995). Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. Journal of Virology, 69(5), 2989–2997. https://doi.org/10.1128/JVI.69.5.2989-2997.1995
Jones, D. L., Alani, R. M., & Münger, K. (1997). The human papillomavirus E7 oncoprotein can uncouple cellular differentiation and proliferation in human keratinocytes by abrogating p21Cip1-mediated inhibition of cdk2. Genes & Development, 11(16), 2101–2111. https://doi.org/10.1101/gad.11.16.2101
Kadaja, M., Isok-Paas, H., Laos, T., Ustav, E., & Ustav, M. (2009). Mechanism of genomic instability in cells infected with the high-risk human papillomaviruses. PLoS Pathogens, 5(4), e1000397. https://doi.org/10.1371/journal.ppat.1000397
King, E., Ottensmeier, C., & Thomas, G. (2014). The immune response in HPV+ oropharyngeal cancer. OncoImmunology, 3, e27254
Korzeniewski, N., Spardy, N., Duensing, A., & Duensing, S. (2011). Genomic instability and cancer: lessons learned from human papillomaviruses. Cancer Letters, 305(2), 113–122. https://doi.org/10.1016/j.canlet.2010.10.013
Kowli, S., Velidandla, R., Creek, K. E., & Pirisi, L. (2013). TGF-β regulation of gene expression at early and late stages of HPV16-mediated transformation of human keratinocytes. Virology, 447(1–2), 63–73. https://doi.org/10.1016/j.virol.2013.08.034
Kranjec, C., & Banks, L. (2011). A systematic analysis of human papillomavirus (HPV) E6 PDZ substrates identifies MAGI-1 as a major target of HPV type 16 (HPV-16) and HPV-18 whose loss accompanies disruption of tight junctions. Journal of Virology, 85(4), 1757–1764. https://doi.org/10.1128/JVI.01756-10
Lammens, T., Li, J., Leone, G., & De Veylder, L. (2009). Atypical E2Fs: new players in the E2F transcription factor family. Trends in Cell Biology, 19(3), 111–118. https://doi.org/10.1016/j.tcb.2009.01.002
Lane, D. P. C. (1992). p53, guardian of the genome. Nature, 358(6381), 15–16. https://doi.org/10.1038/358015a0
Lechner, M. S., & Laimins, L. A. (1994). Inhibition of p53 DNA binding by human papillomavirus E6 proteins. Journal of Virology, 68(7), 4262–4273. https://doi.org/10.1128/JVI.68.7.4262-4273.1994
Lee C., Laimins L.A. (2007) The Differentiation-Dependent Life Cycle of Human Papillomaviruses in Keratinocytes. In: Garcea R.L., DiMaio D. (eds) The Papillomaviruses. Springer, Boston, MA. https://doi.org/10.1007/978-0-387-36523-7_4
De Leo, A., Calderon, A., & Lieberman, P. M. (2020). Control of viral latency by episome maintenance proteins. Trends in Microbiology, 28, 150–162. https://doi.org/10.1016/j.tim.2019.09.002
Licitra, L., Bernier, J., Grandi, C., Merlano, M., Bruzzi, P., & Lefebvre, J. L. (2002). Cancer of the oropharynx. Critical Reviews in Oncology Hematology, 41, 107–122. https://doi.org/10.1016/S1040-8428(01)00129-9
Litwin, T. R., Clarke, M. A., Dean, M., & Wentzensen, N. (2017). Somatic host cell alterations in HPV carcinogenesis. Viruses, 9(8), 206. https://doi.org/10.3390/v9080206
Lowe, S. W., & Lin, A. W. (2000). Apoptosis in cancer. Carcinogenesis, 21(3), 485–495. https://doi.org/10.1093/carcin/21.3.485
Lyons, T., Salih, M., & Tuana, B. (2006). Activating E2Fs mediate transcriptional regulation of human E2F6 repressor. American Journal of Physiology. Cell Physiology, 290, C189-199. https://doi.org/10.1152/ajpcell.00630.2004
Mandal, R., Şenbabaoğlu, Y., Desrichard, A., Havel, J. J., Dalin, M. G., Riaz, N., Lee, K. W., Ganly, I., Hakimi, A. A., Chan, T. A., & Morris, L. G. (2016). The head and neck cancer immune landscape and its immunotherapeutic implications. JCI Insight, 1(17), e89829. https://doi.org/10.1172/jci.insight.89829
Mantovani, F., & Banks, L. (2001). The human papillomavirus E6 protein and its contribution to malignant progression. Oncogene, 20(54), 7874–7887. https://doi.org/10.1038/sj.onc.1204869
McBride, A. A. (2017). Mechanisms and strategies of papillomavirus replication. Biological Chemistry, 398, 919–927
McBride, A. A., & Warburton, A. (2017). The role of integration in oncogenic progression of HPV-associated cancers. PLoS Pathogens, 13, e1006211
McCredie, M. R., Sharples, K. J., Paul, C., Baranyai, J., Medley, G., Jones, R. W., & Skegg, D. C. (2008). Natural history of cervical neoplasia and risk of invasive cancer in women with cervical intraepithelial neoplasia 3: A retrospective cohort study. The Lancet. Oncology, 9(5), 425–434. https://doi.org/10.1016/S1470-2045(08)70103-7
McLaughlin-Drubin, M. E., Huh, K. W., & Münger, K. (2008). Human papillomavirus type 16 E7 oncoprotein associates with E2F6. Journal of Virology, 82(17), 8695–8705. https://doi.org/10.1128/JVI.00579-08
McLaughlin-Drubin, M. E., & Münger, K. (2009). The human papillomavirus E7 oncoprotein. Virology, 384(2), 335–344. https://doi.org/10.1016/j.virol.2008.10.006
Meek, D. W. (1999). Mechanisms of switching on p53: A role for covalent modification? Oncogene, 18(53), 7666–7675. https://doi.org/10.1038/sj.onc.1202951
Mi, Y., Borger, D. R., Fernandes, P. R., Pirisi, L., & Creek, K. E. (2000). Loss of transforming growth factor-beta (TGF-beta) receptor type I mediates TGF-beta resistance in human papillomavirus type 16-transformed human keratinocytes at late stages of in vitro progression. Virology, 270(2), 408–416. https://doi.org/10.1006/viro.2000.0283
Micalizzi, D. S., Christensen, K. L., Jedlicka, P., Coletta, R. D., Barón, A. E., Harrell, J. C., Horwitz, K. B., Billheimer, D., Heichman, K. A., Welm, A. L., Schiemann, W. P., & Ford, H. L. (2009). The Six1 homeoprotein induces human mammary carcinoma cells to undergo epithelial-mesenchymal transition and metastasis in mice through increasing TGF-beta signaling. The Journal of Clinical Investigation, 119(9), 2678–2690. https://doi.org/10.1172/JCI37815
Del Mistro, A., Baboci, L., Frayle-Salamanca, H., Trevisan, R., Bergamo, E., Lignitto, L., et al. (2012). Oral human papillomavirus and human herpesvirus-8 infections among human immunodeficiency virus type 1-infected men and women in Italy. Sexually Transmitted Diseases, 39(11), 894–898. https://doi.org/10.1097/OLQ.0b013e31826ef2da
Moody, C. A., & Laimins, L. A. (2010). Human papillomavirus oncoproteins: pathways to transformation. Nature Reviews. Cancer, 10(8), 550–560. https://doi.org/10.1038/nrc2886
Morgan, I. M., DiNardo, L. J., & Windle, B. (2017). Integration of human papillomavirus genomes in head and neck cancer: Is it time to consider a paradigm shift? Viruses, 9, 208
Moscicki, A. B., Schiffman, M., Burchell, A., Albero, G., Giuliano, A. R., Goodman, M. T., Kjaer, S. K., & Palefsky, J. (2012). Updating the natural history of human papillomavirus and anogenital cancers. Vaccine, 30 Suppl 5(0 5), F24–F33. https://doi.org/10.1016/j.vaccine.2012.05.089
Münger, K., Werness, B. A., Dyson, N., Phelps, W. C., Harlow, E., & Howley, P. M. (1989). Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. The EMBO Journal, 8(13), 4099–4105
Neveu, G., Cassonnet, P., Vidalain, P. O., et al. (2012). Comparative analysis of virus-host interactomes with a mammalian high-throughput protein complementation assay based on Gaussia princeps luciferase. Methods (San Diego, Calif.), 58(4), 349–359. https://doi.org/10.1016/j.ymeth.2012.07.029
Niazi, S., Purohit, M., & Niazi, J. H. (2018). Role of p53 circuitry in tumorigenesis: A brief review. European Journal of Medicinal Chemistry, 158, 7–24. https://doi.org/10.1016/j.ejmech.2018.08.099
Nichols, A. C., Dhaliwal, S. S., Palma, D. A., Basmaji, J., Chapeskie, C., Dowthwaite, S., et al. (2013). Does HPV type affect outcome in oropharyngeal cancer?. Journal of otolaryngology - head & neck surgery = Le Journal d'oto-rhino-laryngologie et de chirurgie cervico-faciale, 42(1), 9. https://doi.org/10.1186/1916-0216-42-9
Nicolás, I., Saco, A., Barnadas, E., Marimon, L., Rakislova, N., Fusté, P., Rovirosa, A., Gaba, L., Buñesch, L., Gil-Ibañez, B., Pahisa, J., Díaz-Feijoo, B., Torne, A., Ordi, J., & Del Pino, M. (2020). Prognostic implications of genotyping and p16 immunostaining in HPV-positive tumors of the uterine cervix. Modern Pathology: An Official Journal of the United States and Canadian Academy of Pathology, Inc, 33(1), 128–137. https://doi.org/10.1038/s41379-019-0360-3
Ostör, A. G. (1993). Natural history of cervical intraepithelial neoplasia: a critical review. International Journal of Gynecological Pathology: Official Journal of the International Society of Gynecological Pathologists, 12(2), 186–192
Ozbun, M. A. (2019). Extracellular events impacting human papillomavirus infections: Epithelial wounding to cell signaling involved in virus entry. Papillomavirus Research (Amsterdam, Netherlands), 7, 188–192. https://doi.org/10.1016/j.pvr.2019.04.009
Pal, A., & Kundu, R. (2020). Human papillomavirus E6 and E7: The cervical cancer hallmarks and targets for therapy. Frontiers in Microbiology, 10, 3116. https://doi.org/10.3389/fmicb.2019.03116
Parish, J. L., Bean, A. M., Park, R. B., & Androphy, E. J. (2006). ChlR1 is required for loading papillomavirus E2 onto mitotic chromosomes and viral genome maintenance. Molecular Cell, 24(6), 867–876. https://doi.org/10.1016/j.molcel.2006.11.005
Parkin, D., Bray, F., Ferlay, J., & Pisani, P. (2005). Global cancer statistics, 2002. CA: A Cancer Journal for Clinicians, 55, 74–108. https://doi.org/10.3322/canjclin.55.2.74
Patel, D., Huang, S. M., Baglia, L. A., & McCance, D. J. (1999). The E6 protein of human papillomavirus type 16 binds to and inhibits co-activation by CBP and p300. The EMBO Journal, 18(18), 5061–5072. https://doi.org/10.1093/emboj/18.18.5061
Pauken, K. E., & Wherry, E. J. (2015). Overcoming T-cell exhaustion in infection and cancer. Trends in Immunology, 36(4), 265–276. https://doi.org/10.1016/j.it.2015.02.008
Petca, A., Borislavschi, A., Zvanca, M. E., Petca, R. C., Sandru, F., & Dumitrascu, M. C. (2020). Non-sexual HPV transmission and role of vaccination for a better future (Review). Experimental and Therapeutic Medicine, 20(6), 186. https://doi.org/10.3892/etm.2020.9316
Pirisi, L., Creek, K. E., Doniger, J., & DiPaolo, J. A. (1988). Continuous cell lines with altered growth and differentiation properties originate after transfection of human keratinocytes with human papillomavirus type 16 DNA. Carcinogenesis, 9, 1573–1579. https://doi.org/10.1093/carcin/9.9.1573
Pirisi, L., Yasumoto, S., Feller, M., Doniger, J., & DiPaolo, J. A. (1987). Transformation of human fibroblasts and keratinocytes with human papillomavirus type 16 DNA. Journal of Virology, 61, 1061–1066. https://doi.org/10.1128/JVI.61.4.1061-1066.1987
Poirson, J., Biquand, E., Straub, M. L., et al. (2017). Mapping the interactome of HPV E6 and E7 oncoproteins with the ubiquitin-proteasome system. The FEBS Journal, 284(19), 3171–3201. https://doi.org/10.1111/febs.14193
Popa, A., Zhang, W., Harrison, M. S., Goodner, K., Kazakov, T., Goodwin, E. C., Lipovsky, A., Burd, C. G., & DiMaio, D. (2015). Direct binding of retromer to human papillomavirus type 16 minor capsid protein L2 mediates endosome exit during viral infection. PLoS Pathogens, 11, e1004699. https://doi.org/10.1371/journal.ppat.1004699
Prives, C., & Hall, P. A. (1999). The p53 pathway. The Journal of Pathology, 187(1), 112–126. https://doi.org/10.1002/(SICI)1096-9896(199901)187:1%3c112::AID-PATH250%3e3.0.CO;2-3
Prodromidou, A., Iavazzo, C., Fotiou, A., Psomiadou, V., Douligeris, A., Vorgias, G., & Kalinoglou, N. (2019). Short- and long-term outcomes after abdominal radical trachelectomy versus radical hysterectomy for early stage cervical cancer: A systematic review of the literature and meta-analysis. Archives of Gynecology and Obstetrics, 300(1), 25–31. https://doi.org/10.1007/s00404-019-05176-y
Przybyszewska, J., Zlotogorski, A., & Ramot, Y. (2017). Re-evaluation of epidermodysplasia verruciformis: Reconciling more than 90 years of debate. Journal of the American Academy of Dermatology, 76(6), 1161–1175. https://doi.org/10.1016/j.jaad.2016.12.035
Riley, R. S., June, C. H., Langer, R., & Mitchell, M. J. (2019). Delivery technologies for cancer immunotherapy. Nature reviews. Drug Discovery, 18(3), 175–196. https://doi.org/10.1038/s41573-018-0006-z
Ryu, H. J., Kim, E. K., Heo, S. J., Cho, B. C., Kim, H. R., & Yoon, S. O. (2017). Architectural patterns of p16 immunohistochemical expression associated with cancer immunity and prognosis of head and neck squamous cell carcinoma. APMIS: Acta Pathologica, Microbiologica, et Immunologica Scandinavica, 125(11), 974–984. https://doi.org/10.1111/apm.12744
Samstein, R. M., Lee, C. H., Shoushtari, A. N., Hellmann, M. D., Shen, R., Janjigian, Y. Y., Barron, D. A., Zehir, A., Jordan, E. J., Omuro, A., Kaley, T. J., Kendall, S. M., Motzer, R. J., Hakimi, A. A., Voss, M. H., Russo, P., Rosenberg, J., Iyer, G., Bochner, B. H., … Morris, L. (2019). Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nature Genetics, 51(2), 202–206. https://doi.org/10.1038/s41588-018-0312-8
Scheffner, M., Werness, B. A., Huibregtse, J. M., Levine, A. J., & Howley, P. M. (1990). The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell, 63(6), 1129–1136. https://doi.org/10.1016/0092-8674(90)90409-8
Schlecht, N. F., Burk, R. D., Adrien, L., Dunne, A., Kawachi, N., Sarta, C., et al. (2007). Gene expression profiles in HPV-infected head and neck cancer. The Journal of Pathology, 213(3), 283–293. https://doi.org/10.1002/path.2227
Schuurman, T., Zilver, S., Samuels, S., Schats, W., Amant, F., van Trommel, N., & Lok, C. (2021). Fertility-sparing surgery in gynecologic cancer: a systematic review. Cancers, 13(5), 1008. https://doi.org/10.3390/cancers13051008
Sedman, S. A., Hubbert, N. L., Vass, W. C., Lowy, D. R., & Schiller, J. T. (1992). Mutant p53 can substitute for human papillomavirus type 16 E6 in immortalization of human keratinocytes but does not have E6-associated trans-activation or transforming activity. Journal of Virology, 66, 4201–4208. https://doi.org/10.1128/JVI.66.7.4201-4208.1992
Serrano, B., Brotons, M., Bosch, F. X., & Bruni, L. (2017). Epidemiology and burden of HPV related disease. Best Practice & Research. Clinical Obstetrics & Gynaecology, 47, 14–26. https://doi.org/10.1016/j.bpobgyn.2017.08.006
Sherr, C. J., & Weber, J. D. (2000). The ARF/p53 pathway. Current Opinion in Genetics & Development, 10(1), 94–99. https://doi.org/10.1016/s0959-437x(99)00038-6
Siegel, R., Naishadham, D., & Jemal, A. (2012). Cancer statistics, 2012. CA: a Cancer Journal for Clinicians, 62(1), 10–29. https://doi.org/10.3322/caac.20138
Siegel, R. L., Miller, K. D., & Jemal, A. (2020). Cancer statistics, 2020. CA: a Cancer Journal for Clinicians, 70(1), 7–30. https://doi.org/10.3322/caac.21590
Smith, P. P., Friedman, C. L., Bryant, E. M., & McDougall, J. K. (1992). Viral integration and fragile sites in human papillomavirus-immortalized human keratinocyte cell lines. Genes, Chromosomes & Cancer, 5(2), 150–157. https://doi.org/10.1002/gcc.2870050209
Spriggs, C. C., & Laimins, L. A. (2017). Human papillomavirus and the DNA damage response: Exploiting host repair pathways for viral replication. Viruses, 9, 232. https://doi.org/10.3390/v9080232
Stanley, M. A. (2012). Epithelial cell responses to infection with human papillomavirus. Clinical Microbiology Reviews, 25(2), 215–222. https://doi.org/10.1128/CMR.05028-11
Stevaux, O., & Dyson, N. J. (2002). A revised picture of the E2F transcriptional network and RB function. Current Opinion in Cell Biology, 14(6), 684–691. https://doi.org/10.1016/s0955-0674(02)00388-5
Szymonowicz, K. A., & Chen, J. (2020). Biological and clinical aspects of HPV-related cancers. Cancer Biology & Medicine, 17(4), 864–878. https://doi.org/10.20892/j.issn.2095-3941.2020.0370
Tan, J., Zhang, C., & Qian, J. (2011). Expression and significance of Six1 and Ezrin in cervical cancer tissue. Tumour Biology, 32(6), 1241–1247. https://doi.org/10.1007/s13277-011-0228-8
Tewari, K. S., Sill, M. W., Long, H. J., 3rd., Penson, R. T., Huang, H., Ramondetta, L. M., Landrum, L. M., Oaknin, A., Reid, T. J., Leitao, M. M., Michael, H. E., & Monk, B. J. (2014). Improved survival with bevacizumab in advanced cervical cancer. The New England Journal of Medicine, 370(8), 734–743. https://doi.org/10.1056/NEJMoa1309748
Tomar, S., Graves, C. A., Altomare, D., Kowli, S., Kassler, S., Sutkowski, N., Gillespie, M. B., Creek, K. E., & Pirisi, L. (2016). Human papillomavirus status and gene expression profiles of oropharyngeal and oral cancers from European American and African American patients. Head Neck, 38. Suppl, 1, E694-704. https://doi.org/10.1002/hed.24072
Vats, A., Trejo-Cerro, O., Thomas, M., & Banks, L. (2021). Human papillomavirus E6 and E7: What remains?. Tumour virus research, 11, 200213. Advance online publication. https://doi.org/https://doi.org/10.1016/j.tvr.2021.200213
Verhoeven, Y., Quatannens, D., Trinh, X. B., Wouters, A., Smits, E., Lardon, F., De Waele, J., & van Dam, P. A. (2021). Targeting the PD-1 axis with pembrolizumab for recurrent or metastatic cancer of the uterine cervix: A brief update. International Journal of Molecular Sciences, 22(4), 1807. https://doi.org/10.3390/ijms22041807
Vieira, V. C., Leonard, B., White, E. A., Starrett, G. J., Temiz, N. A., Lorenz, L. D., Lee, D., Soares, M. A., Lambert, P. F., Howley, P. M., Harris, R. S. (2014). Human papillomavirus E6 triggers upregulation of the antiviral and cancer genomic DNA deaminase APOBEC3B. mBio, 5(6), e02234–14. https://doi.org/10.1128/mBio.02234-14
Vogelstein, B., Lane, D., & Levine, A. J. (2000). Surfing the p53 network. Nature, 408(6810), 307–310. https://doi.org/10.1038/35042675
Vokes, E. E., Agrawal, N., & Seiwert, T. Y. (2015). HPV-Associated Head and Neck Cancer. Journal of the National Cancer Institute, 107(12), djv344. https://doi.org/10.1093/jnci/djv344
Wan, F., Miao, X., Quraishi, I., Kennedy, V., Creek, K. E., & Pirisi, L. (2008). Gene expression changes during HPV-mediated carcinogenesis: A comparison between an in vitro cell model and cervical cancer. International Journal of Cancer, 123(1), 32–40. https://doi.org/10.1002/ijc.23463
Wang, J., Li, Z., Gao, A., Wen, Q., & Sun, Y. (2019). The prognostic landscape of tumor-infiltrating immune cells in cervical cancer. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie, 120, 109444. https://doi.org/10.1016/j.biopha.2019.109444
Weinberger, P. M., Merkley, M. A., Khichi, S. S., Lee, J. R., Psyrri, A., Jackson, L. L., & Dynan, W. S. (2010). Human papillomavirus-active head and neck cancer and ethnic health disparities. The Laryngoscope, 120(8), 1531–1537. https://doi.org/10.1002/lary.20984
Weinberger, P. M., Yu, Z., Haffty, B. G., Kowalski, D., Harigopal, M., Brandsma, J., Sasaki, C., Joe, J., Camp, R. L., Rimm, D. L., & Psyrri, A. (2006). Molecular classification identifies a subset of human papillomavirus–associated oropharyngeal cancers with favorable prognosis. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology, 24(5), 736–747. https://doi.org/10.1200/JCO.2004.00.3335
Westra, W. H., Taube, J. M., Poeta, M. L., Begum, S., Sidransky, D., & Koch, W. M. (2008). Inverse relationship between human papillomavirus-16 infection and disruptive p53 gene mutations in squamous cell carcinoma of the head and neck. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 14(2), 366–369. https://doi.org/10.1158/1078-0432.CCR-07-1402
Wild, C.P., Weiderpass, E., Stewart, B.W. editors (2020). World Cancer Report: Cancer Research for Cancer Prevention. Lyon, France: International Agency for Research on Cancer. Available from: http://publications.iarc.fr/586. Licence: CC BY-NC-ND 3.0 IGO.
Wirth, L. J. (2016). Cetuximab in human papillomavirus-positive oropharynx carcinoma. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology, 34(12), 1289–1291. https://doi.org/10.1200/JCO.2015.65.1414
Wise-Draper, T. M., & Wells, S. I. (2008). Papillomavirus E6 and E7 proteins and their cellular targets. Frontiers in Bioscience: A Journal and Virtual Library, 13, 1003–1017. https://doi.org/10.2741/2739
Woappi, Y., Altomare, D., Creek, K. E., & Pirisi, L. (2020). Self-assembling 3D spheroid cultures of human neonatal keratinocytes have enhanced regenerative properties. Stem Cell Research, 49, 102048. https://doi.org/10.1016/j.scr.2020.102048
Woappi, Y., Hosseinipour, M., Creek, K. E., & Pirisi, L. (2018). Stem cell properties of normal human keratinocytes determine transformation responses to human papillomavirus 16 DNA. Journal of Virology, 92(11), e00331-e418. https://doi.org/10.1128/JVI.00331-18
Xu, X., Kelleher, K. F., Liao, J., Creek, K. E., & Pirisi, L. (2000). Unique carboxyl-terminal sequences of wild type and alternatively spliced variant forms of transforming growth factor-alpha precursors mediate specific interactions with ErbB4 and ErbB2. Oncogene, 19(28), 3172–3181. https://doi.org/10.1038/sj.onc.1203645
Xu, X., Liao, J., Creek, K. E., & Pirisi, L. (1999). Human keratinocytes and tumor-derived cell lines express alternatively spliced forms of transforming growth factor-alpha mRNA, encoding precursors lacking carboxyl-terminal valine residues. Oncogene, 18(40), 5554–5562. https://doi.org/10.1038/sj.onc.1203091
Xu, H., Pirisi, L., & Creek, K. E. (2015). Six1 overexpression at early stages of HPV16-mediated transformation of human keratinocytes promotes differentiation resistance and EMT. Virology, 474, 144–153. https://doi.org/10.1016/j.virol.2014.10.010
Xu, H., Zhang, Y., Altomare, D., Peña, M. M., Wan, F., Pirisi, L., & Creek, K. E. (2014). Six1 promotes epithelial-mesenchymal transition and malignant conversion in human papillomavirus type 16-immortalized human keratinocytes. Carcinogenesis, 35, 1379–1388. https://doi.org/10.1093/carcin/bgu050
Yang, A., Jeang, J., Cheng, K., Cheng, T., Yang, B., Wu, T. C., & Hung, C. F. (2016). Current state in the development of candidate therapeutic HPV vaccines. Expert Review of Vaccines, 15(8), 989–1007. https://doi.org/10.1586/14760584.2016.1157477
Yilmaz, V., & Strati, K. (2019). Regulating cellular plasticity to persist: A way for tumor viruses to triumph. Current Opinion in Virology, 39, 1–7. https://doi.org/10.1016/j.coviro.2019.06.007
Young, A. P., Nagarajan, R., & Longmore, G. D. (2003). Mechanisms of transcriptional regulation by Rb–E2F segregate by biological pathway. Oncogene, 22(46), 7209–7217. https://doi.org/10.1038/sj.onc.1206804
Zhang, A., Wang, J., Zheng, B., Fang, X., Angström, T., Liu, C., et al. (2004). Telomere attrition predominantly occurs in precursor lesions during in vivo carcinogenic process of the uterine cervix. Oncogene, 23(44), 7441–7447. https://doi.org/10.1038/sj.onc.1207527
Zheng, Z. M., & Baker, C. C. (2006). Papillomavirus genome structure, expression, and post-transcriptional regulation. Frontiers in Bioscience: A Journal and Virtual Library, 11, 2286–2302. https://doi.org/10.2741/1971
Zheng, X. H., Liang, P. H., Guo, J. X., Zheng, Y. R., Han, J., Yu, L. L., Zhou, Y. G., & Li, L. (2010). Expression and clinical implications of homeobox gene Six1 in cervical cancer cell lines and cervical epithelial tissues. International Journal of Gynecological Cancer, 20(9), 1587–1592
Zhou, H., Blevins, M. A., Hsu, J. Y., Kong, D., Galbraith, M. D., Goodspeed, A., et al. (2020). Identification of a small-molecule inhibitor that disrupts the SIX1/EYA2 Complex, EMT, and metastasis. Cancer Research, 80(12), 2689–2702. https://doi.org/10.1158/0008-5472.CAN-20-0435
Ziegert, C., Wentzensen, N., Vinokurova, S., Kisseljov, F., Einenkel, J., Hoeckel, M., & von Knebel, D. M. (2003). A comprehensive analysis of HPV integration loci in anogenital lesions combining transcript and genome-based amplification techniques. Oncogene, 22, 3977–3984. https://doi.org/10.1038/sj.onc.1206629
Zouridis, A., Kalampokas, T., Panoulis, K., Salakos, N., & Deligeoroglou, E. (2018). Intrauterine HPV transmission: A systematic review of the literature. Archives of Gynecology and Obstetrics, 298, 35–44. https://doi.org/10.1007/s00404-018-4787-4
Zyzak, L. L., MacDonald, L. M., Batova, A., Forand, R., Creek, K. E., & Pirisi, L. (1994). Increased levels and constitutive tyrosine phosphorylation of the epidermal growth factor receptor contribute to autonomous growth of human papillomavirus type 16 immortalized human keratinocytes. Cell Growth & Differentiation: The Molecular Biology Journal of the American Association for Cancer Research, 5(5), 537–547
Funding
This paper was made possible in part by Grant 1R21CA201853 from the National Institutes of Health, National Cancer Institute, USA to LP.
Author information
Nella C. Delva
Present address: Department of Biomedical Sciences, School of Medicine, Florida State University, Tallahassee, FL, USA
Ariana Renrick
Present address: Meharry Medical College Biomedical Sciences Program, Nashville, TN, USA
Affiliations
Department of Pathology, Microbiology and Immunology, University of South Carolina School of Medicine, Building 3, Room 226, 6311 Garners Ferry Road, Columbia, SC, 29208, USA
Fadi Abboodi, Nella C. Delva, Jennifer Emmel, Ariana Renrick & Lucia Pirisi
Department of Drug Discovery and Biomedical Sciences, University of South Carolina College of Pharmacy, Columbia, SC, 29208, USA
Fadi Abboodi, Phillip Buckhaults, Carolyn E. Banister & Kim E. Creek
Corresponding author
Correspondence to Lucia Pirisi.
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Rights and permissions
About this article
Cite this article
Abboodi, F., Delva, N.C., Emmel, J. et al. Human papillomavirus-mediated carcinogenesis and tumor progression. GENOME INSTAB. DIS. 2, 71–91 (2021). https://doi.org/10.1007/s42764-021-00038-x
Received23 February 2021
Revised02 April 2021
Accepted05 April 2021
Published15 April 2021
Issue DateApril 2021
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
Human papillomavirus
HPV
Genomic instability
Immortalization
用户登录
还没有账号?
立即注册