






Role of NEDD8 and neddylation dynamics in DNA damage response
Review Article
Genome Instability & Disease 2, 139–149 (2021)
Abstract
Every single cell in human body is suffering ~ 7000 DNA lesions of various types every day. Different kinds of DNA damage response (DDR) are evolved to overcome these threats to maintain genome stability. Failure in the DDR system leads to various diseases ranging from cancer to neurodevelopmental defects. Past years’ studies have unraveled the close relationship between neddylation, an ubiquitin-like modification, and DDR. This functional interplay involves the neddylation of Cullin RING E3 ligases, the prototype neddylation substrates, and non-Cullin-based neddylation targets, both of which have intimate links to DDR. More recently, unconjugated NEDD8 polymers are implicated in DDR as well. Here we summarize these findings to better understand the role that neddylation and deneddylation plays in DDR.
Introduction
DNA damage response (DDR) and neddylation are two essential biological processes that both involve many players. The intimate functional link between DDR and Neddylation is best illustrated by the early identification of damaged DNA binding proteins DDB1 and DDB2 (Chu & Chang, 1988), where were quickly found to be essential nucleotide excision repair genes mutated in certain Xeroderma Pigmentosum patients, and later also discovered as the adaptor and substrate receptor components of CRL4, a major neddylation target (Scrima et al., 2008;). In this review, we will briefly introduce the various DDR choices, i.e., cell cycle arrest, damage repair and/or tolerance, with an emphasis on the different repair pathways. We then focus on introducing Neddylation and NEDD8 related processes, followed by discussing their intersection with DDR, including the latest findings. We conclude with a summary and future perspective.
The DNA damage response
Every single cell in human body is facing tens of thousands of DNA lesions every day (Jackson & Bartek, 2009). These DNA lesions can originate from exogenous insults (physical and chemical, e.g., radiation), or from endogenous reactions (hydrolysis, alkylation, oxidation and DNA mismatches). DNA lesions can disrupt DNA replication and gene expression if they are not fixed correctly and timely. To keep the genetic material intact and unchanged, several strong systems have been evolved in lives. DNA damage response (DDR) is one of such system to protect life from endogenous and exogenous damage to DNA (Giglia-Mari et al., 2011; Harper & Elledge, 2007). Defects in DDR often result in enhanced sensitivity to DNA damage, cell death or mutations that could eventually leads to debilitating diseases such as cancer and neurodevelopmental defect/degeneration. DDR includes three key processes: damage repair, damage tolerance and cell-cycle checkpoint signaling. DDR has been extensively reviewed (Ciccia & Elledge, 2010; Harper & Elledge, 2007; Jackson & Bartek, 2009). We will briefly introduce DDR and then detail examples of neddylation–DDR interplay.
Different types of repair processes are defined by the types of DNA damage they repair. These include Base Excision Repair (BER), Nucleotide Excision Repair (NER), DNA Mismatch Repair (NMR) and Double Strand Breaks Repair (DSBR). Briefly, BER repairs a few bases with slight alterations by chemicals (Krokan & Bjoras, 2013). The single damaged base is detected and removed by specific DNA glycosylases, leaving an abasic site for further incision, end processing, and repair synthesis. NER mainly repairs bulky DNA lesions which are normally caused by ultraviolet (UV) radiation or environmental chemicals and cytotoxic drugs (Scharer, 2013). NER can be divided into two types according to their mode of damage detection: global genomic NER (GG-NER) for whole genome, and transcription-coupled NER (TC-NER) for actively transcribing genes (Scrima et al., 2008; Spivak, 2015; Sugasawa et al., 2005). Although GG-NER and TC-NER detect lesions in totally different ways, the following process converges into common steps. After lesion detection, basal transcription factor TFIIH stretches the damaged DNA section, while the undamaged bases are stabilized (de Laat et al., 1998). Finally, the gap between incisions is repaired by DNA replication proteins and the nick is filled by DNA ligase I or III. MMR recognizes and corrects replication errors (Li et al., 2016). Similar to BER and NER, MMR also involves excision of damaged bases and synthesis of new nucleotides based on the other strand, while the repair proteins including replication factors are recruited to the lesion sites for further lesion repair. DSBR deals with Double Strand Breaks, which normally caused by irradiation, certain radiomimetic drugs and DNA replication. These severe lesions could lead to DNA deletions, translocations and fusions and can cause cell death if they are not correctly repaired in time. There are two major repair mechanisms for DSBs: non-homologous end-joining (NHEJ) and homologous recombination (HR) (Aleksandrov et al., 2020; Wu et al., 2012). NHEJ repairs two DSB ends by direct ligation and thus possibly generates new mutations. The other mechanism HR always requires homologous sister chromatid to produce ssDNA for DSB repair. Therefore, NHEJ can initiate in all phases of cell cycle, whereas HR only operates in S and G2.
Before DNA lesions are repaired, or in the event of repair failure, cells can use DNA damage tolerance pathways to facilitate translesion polymerase for temporary DNA synthesis without actual damage repair (Giglia-Mari et al., 2011). Translesion synthesis (TLS) and recombination-dependent daughter-strand gap repair (DSGR) are two major processes for DNA tolerance. Cells also developed damage signaling programs to arrest cell cycle to extend the time window before the damage is totally removed. The cell cycle can be arrested at G1/S checkpoint, at the G2/M checkpoint or within the S-phase by dedicated checkpoint signaling pathways. To repair DNA lesions, dramatic responses also occur in nuclear domains, forming unclear segregations with the accumulation of functional factors (e.g., p53) (Dellaire & Bazett-Jones, 2007; Jackson & Bartek, 2009). Finally, in the case of unbearable DNA damage, cells undergo death pathways as part of the damage response, which is often p53-dependent. In sum, to safeguard our genome integrity, an array of DDR programs exist, which are mostly protein-based machineries. Timely accumulation and removal of these DDR protein machineries are essential for damage response and for homeostasis. Indeed, many DDR proteins are modified by ubiquitin and ubiquitin-like proteins (UBLs) to orchestrate the processes.
Neddylation
Neddylation is an enzymatic post-translational modification, whereby the ubiquitin-like protein NEDD8 (Neural Precursor Cell Expressed, Developmentally Down-regulated 8) is attached to the substrate protein. NEDD8 was first discovered as one of the 10 genes (neural precursor cell expressed, developmentally down-regulated 1–10) developmental down-regulated in the mouse brain (Rabut & Peter, 2008). Similar to the ubiquitylation pathway, E1 activating enzymes, E2 conjugating enzymes, and E3 ligases work together to conjugate NEDD8, via its carboxy-terminal glycine (Gly76), to a lysine residue on the substrate protein (Fig. 1) (Chung & Dellaire, 2015; Schwechheimer, 2018). Unlike ubiquitylation, however, the NEDD8 gene encodes a non-conjugatable precursor which requires a C-terminal hydrolase, NEDP1 (deneddylase 1, also known as DEN1 or SENP8), to cleave its additional residues beyond Gly76. Mature NEDD8 could then be activated by the NEDD8 E1 activating enzyme (heterodimer of APPBP1/NAE1 and UBA3), and transferred to E2 (Ubc12/UBE2M) before being conjugated to substrate proteins with the assistance of E3s (Rabut & Peter, 2008). Canonically, this neddylation pathway leads to substrate mono-neddylation. In addition, there is also an atypical neddylation pathway through the ubiquitin system rather than canonical NEDD8 pathway (Lobato-Gil et al., 2021). Thus, a recent proteomes study shows atypical neddylation occurs in ribosome and proteasome, while canonical NEDD8 neddylation is mainly found in DNA replication, mRNA surveillance, and spliceosome proteins (Lobato-Gil et al., 2021). This study also found different types of neddylation, which are poly-NEDD8, hybrid NEDD8-ubiquitin, and NEDD8-SUMO-2 chains, underscoring the complexity of neddylation pathways and the mixing of neddylation with other ubiquitin and ubiquitin-like modifications.
Fig. 1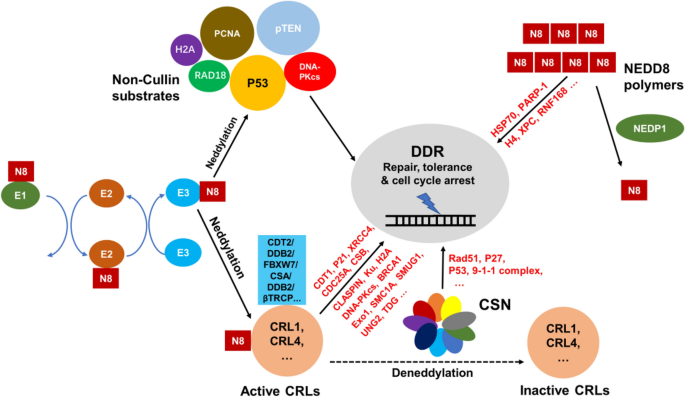
Regulation of DDR by neddylation. The neddylation of CRLs, which is negatively controlled by CSN, is a key regulator of DDR through CRL substrates (e.g., CDT1 by CRL4CDT2 and CLASPIN by SCFbTRCP). Meanwhile, CSN can also mediate DDR without CRLs involved. There are also many Non-Cullin substrates that regulate DDR directly such as p53, PCNA and pTEN. In addition, NEDD8 polymers as novel discovered DDR mediators can also be functional in DNA sensing, tolerance and repair. For reference on original studies, see Table 1
Full size imageCompared to that of ubiquitylation, only a limited number of neddylation substrates have been reported up to date. The first and by far the best characterized neddylation substrates are CRLs (Cullin–RING ubiquitin Ligases), which plays versatile roles during DDR. Neddylation is also reversible, similar to ubiquitylation. CRLs are specifically deneddylated by CSN (COP9 signalosome), which is only activated upon CRL binding and is known to be important in DDR (Chung & Dellaire, 2015). In addition, there are several other reported non-Cullin neddylation substrates. Among them, p53, PCNA and pTEN are proved to play key role in DDR as well. The deneddylase for non-Cullin-based neddylation substrate are not very clear, with NEDP1 being a potential candidate, at least in the case of PCNA (Guan et al., 2018). Moreover, NEDD8 polymers has recently been discovered to be functional in DDR (Ohki et al., 2009). The link between these neddylation events and DDR are shown in Fig. 1 and explained below.
Neddylation is an important clinical therapeutic target. Thus, MLN4924, is a general inhibitor of neddylation that acts by covalently inhibiting UBA3 activity (Mao & Sun, 2020; Soucy et al., 2009), has been wildly reported to display broad antitumor activity (Oladghaffari et al., 2016; Soucy et al., 2009). More than 30 completed or ongoing clinical studies have been taken to evaluate its antitumor activity, culminating in its approval by the FDA to treat Myelodysplastic Syndrome as a breakthrough therapy.
CRL neddylation and DNA damage response
The multi-subunit Cullin–RING ubiquitin Ligases (CRLs) as the largest family of E3 enzymes function with E1 and E2 for substrates ubiquitylation and degradation. About 20% of proteasomal degradation is mediated by CRL ubiquitylation (Petroski & Deshaies, 2005). In addition, CRL-mediated ubiquitylation also activates, represses or relocalizes proteins other than degradation. Normally, CRLs are comprised of a Cullin protein (Cul1, 2,-3, 4A/B, 5 or 7) as scaffold for E3 assembly, an RING protein Rbx1/2 (also called Roc1/2) for E2 recruiting, and Cullin-specific adaptors (e.g., DDB1 for CRL4) and/or substrate receptors for substrate recognition (e.g., DDB2 for CRL4). Cullins are modified by NEDD8 on a conserved lysine in the C-terminus (Rabut & Peter, 2008). NEDD8 stimulates CRL recruitment of E2 or partner E3 and blocks its binding from the native CRL inhibitor CAND1 (Cullin-associated NEDD8-dissociated 1) (Baek et al., 2020a, 2020b; Duda et al., 2008; Schwechheimer, 2018; Wu et al., 2013), thereby activating CRLs. The treatment of MLN4924 can lead to the deneddylation of CRLs and the accumulation of CRL substrates, many of which are involved in DDR (e.g., CDT1, p27 and p21) (Brown & Jackson, 2015; Hannss & Dubiel, 2011).
CRLs are reportedly essential in regulating cell cycle arrest during DDR for their function of restricting DNA replication, cell cycle checkpoint responses and G1–S transition (Hannss & Dubiel, 2011; Petroski & Deshaies, 2005) (as shown in Fig. 1 and summarized in Table 1). For examples, once DNA damage occurred, by binding to chromatin with PCNA, CRL4CDT2 degrades CDT1 and p21. p21 and CDT1 are cell cycle regulators, recruiting MCM complex to start DNA synthesis during S-phase (Rizzardi & Cook, 2012). Thus, the degradation of these two factors leads to recovery of DNA replication and promotion of damage repair by NER. In addition, p21 can also be ubiquitylated and degraded by CRL1Skp2 during lesions repair (Nishitani et al., 2008). Unlike p21 and CDT1, p53, the master regulator of DNA damage, is dynamically regulated by different E3 ligases. In response to ionizing radiation (IR) exposure, phosphorylation of p53 at S33 and S37, promoted by the activated ATM, disrupts the binding of p53 to Mdm2 but enhances the binding of p53 to SKP1–Cullin–F-box (SCF) containing FBXW7 (Cui et al., 2020; Marine & Lozano, 2010; Zhao et al., 2018). Thus, p53 is accumulated at the early stage to promote cell-cycle arrest and DNA repair (Brooks & Gu, 2006). Once the damage DNA is repaired, p53 will be quickly ubiquitylated by SCFFBXW7 for proteasomal degradation, which facilitates p53 recovery. Moreover, upon IR exposure, CDC25A and CLASPIN, two proteins which are phosphorylated during CHEK1/CHEK2 signaling, are ubiquitylated by SCFβTRCP (Mailand et al., 2006; Sancar et al., 2004). Then, CDC25A and CLASPIN are degraded for cell cycle arrest and checkpoint recovery, respectively. Moreover, When UBE2M, the E2 NEDD8 conjugating enzyme, is knocked-down, Cullins with defective neddylation leads to arrest of G1/S transition and impaired DNA damage response (Brown et al., 2015; Cukras et al., 2014).
Table 1 Neddylation targets and their role in DDR

CRLs also play key roles in various types of DNA damage repair pathways, including but not limited to NER, DSBR, and BER (Brown & Jackson, 2015; Zhou & Yan, 2020) (Fig. 1 and Table 1). As mentioned in the introduction, CRLs are key players in initiating NER, both genome-wide (GG-NER) and transcription-coupled (TC-NER). In GG-NER, upon UV-induced DNA damage, DDB2, which binds to DDB1 to form heterodimer, brings CRL4 to the chromatin. Meanwhile, CSA also helps CRL4 to associate with chromatin in this process (Fousteri & Mullenders, 2008). CRL4ADDB2 complex recognize and degrade substrates including histone H2A and possibly XPC after UV stimulation (Kapetanaki et al., 2006). In TC-NER, after UV-induced DNA damage is repaired, CSB is to be rapidly degraded by CRL4CSA to promote the resumption of transcription elongation (Fousteri & Mullenders, 2008; Groisman et al., 2006; Mahon et al., 2014).
CRL activity orchestrates both branches of DSBR: NHEJ and HR. XRCC4, an essential NHEJ factor, is polyubiquitylated by SCFFBXW7 (Zhang et al., 2016). During NHEJ repair, FBXW7 is phosphorylated by ATM upon radiation. The phosphorylated FBXW7 binds to poly(ADP-ribose) (PAR) and is recruited to damage sites (Zhang et al., 2019). Meanwhile, XRCC4 is phosphorylated by DNA-PKcs, facilitating its recognition and polyubiquitylation by SCFFBXW7 via K63-linked polyubiquitylation (Lan & Sun, 2019). Then, ubiquitylated XRCC4 promotes NHEJ repair by enhancing the assembly of NHEJ complex and its binding to DNA lesions. In addition, MLN4924 can abrogate the ubiquitylation of XRCC4 and NHEJ repair. Another study from Brown et al. also found the important role of neddylation for NHEJ initiation (Brown et al., 2015). They showed that the ubiquitylation and release of Ku, the key protein of NHEJ, can be promoted by Cullin neddylation for damage repair, although the precise CRL complex responsible for this Ku ubiquitylation remains unknown. EXO1, another important repair protein of both NHEJ and HR, is also ubiquitylated by SCF when it is phosphorylated by ATR upon the induction of DSBs by topoisomerase I poison camptothecin (CPT) (Tomimatsu et al., 2017). In addition, Dar et al. showed how Cul4B mediates CD4+ T cell activity changes in response to DSBs induced by etoposide (Dar et al., 2021). After T cell receptor receiving DNA damage signals, the expression and neddylation of Cul4B are promoted. Then, Cul4B with its adaptor DDB1 and substrate receptor DCAF1 direct binds to DNA sensing and repairing proteins (e.g., Rad50 and Mre11a, key components of Mre11-Rad50-Nbs1 (MRN) complex), promoting phosphorylation of SMC1A, a protein working with MRN to facilitate lesion HR repair. However, after DSBs induction by X-ray irradiation, CRL4ADTL is reported to degrade nuclear DNA-PKcs protein to inhibit NHEJ pathway and enhance the instability of genome. This may affect the repair choices of DSBR pathways (Feng et al., 2021).
The role of CRLs in BER is relatively understudied. However, Thymine DNA glycosylase (TDG), a based excision repair enzyme involved in DNA demethylation, needs to be degraded during S phase, a task triggered by CRL4CDT2-mediated ubiquitylation. The physical interaction of TDG and DNA polymerase clamp PCNA is required for its degradation by CRL4CDT2 (Slenn et al., 2014). Similarly, UV treatment also induces CRL4CDT2-mediated TDG ubiquitylation and degradation, which promotes NER and cell survival. (Nakamura et al., 2017). SMUG1 and UNG2, two Uracil–DNA glycosylases mediating the initial steps of BER, are ubiquitylated by CRL4DCAF1 for proteasomal degradation with the assistance of the HIV-1 Vpr protein, whose direct interaction with UNG2 is required for the later’s degradation (Fousteri & Mullenders, 2008; Pettersen et al., 2007; Wen et al., 2012), whether SMUG1 and UNG2 degradation occurs during any DNA damage process remains to be studied.
CSN-mediated CRL deneddylation in DNA damage response
CRLs undergo cycles of neddylation and deneddylation. The most famous isopeptidase of deneddylation is COP9 signalosome (CSN), which deneddylates Cullins (Rabut & Peter, 2008). CSN is a highly evolutionarily conserved complex initially thought to consist of eight subunits (CSN1–8) (Kato & Yoneda-Kato, 2009; Wei et al., 2008) (Rozen et al., 2015; Schwechheimer, 2004; Wei & Deng, 2003). CSNAP, the ninth subunit of CSN, is also recently identified (Rozen et al., 2015; Schwechheimer, 2004; Wei & Deng, 2003). Mutations of CSN lead to constitutive photomorphogenesis in plants and profound disorders in mammals, including cancer (Leal et al., 2008). CSN1 is essential for the assembly of the whole CSN complex (Wang et al., 2002). CSN2 is required for direct interaction with Cullin/Rbx1 (Lin et al., 2020; Yang et al., 2002). CSN5 (also named Jab1) is the well-studied subunit of CSN complex and carries the catalytic activity of CSN deneddylation (Chamovitz & Segal, 2001). In addition, CSN5 has been proved to bind to various proteins independent of the CSN complex, such as cell cycle regulator p27 and AP-1 transcription regulators c-jun. CSN5 can also function as proteasome regulators and translation initiation factor without the involvement of CSN complex (Echalier et al., 2013). Apart from typically catalyzing CRL deneddylation, CSN can also regulate CRLs by structural sequestration associated with physically binding (Bosu et al., 2010). Since the isopeptidase activity of CSN was first found to cleave NEDD8 from CRL1 (Cope & Deshaies, 2003), Ample amount of work showed that CSN regulation of CRL deneddylation extends CSN functions to DNA repair, embryonic development, cytokine signaling, regulation of nuclear transport, cell-cycle progression, angiogenesis, and antigen-induced responses (Schwechheimer, 2004; Wei & Deng, 2003). Given the pivotal roles of CRL-mediated ubiquitylation in DDR-related processes such as lesions sensing and damaged DNA binding, it is no surprise that, CSN is also a crucial mechanism for mediating DDR.
In response to DNA damage, CSN is involved in multiple processes including DNA repair, cell cycle progression and checkpoint control (Brown & Jackson, 2015) (Fig. 1). Using an increasing dose of DNA damage inducing drug mitomycin C (MMC), a dose-dependent accumulation of CSN subunits (CSN1, CSN3, CSN5 and CSN8) is observed (Tomasz & Palom, 1997). p27 and p53 are directly induced by knocking down CSN subunits, suggesting the important role of CSN responding to DNA damage. Shiloh and associates showed that neocarzinostatin, a DSB-inducing agent, elicits CSN phosphorylation and recruitment to damage site for balancing the two major DSB repair pathways: HR vs NHEJ, thereby mediating timely DSB repair (Meir et al., 2015). Groisman R et al. found that both DDB2 and CSA bind to DDB1, to form two different integrated complexes with CRL4 (Groisman et al., 2003). CSN is also the part of the two complexes. Strikingly, in response to UV irradiation, DDB2 and CSA are differently regulated by CRL neddylation status through CSN. After UV irradiation, DDB2 shuffles from the nuclear extract fraction to the chromatin fraction with the increased appearance of CRL neddylation for substrates degradation, while CSA complex still remains in chromatin with stronger CSN interaction. Füzesi-Levi et al. observed a significant shuffling for CSN within cytoplasm, nucleoplasm, and chromatin-bound fractions (Fuzesi-Levi et al., 2014). The proportion of CSN in each subcellular units remains steady until DNA lesions formed by UV damage. The whole CSN complex then shuffles into nucleus and the three subunits of CSN (CSN1, CSN3 and CSN8) are increasingly phosphorylated. Therefore, the activity of CSN is also regulated by both phosphorylation and translocation. It was also found that the loss of CSNAP, the ninth subunit of CSN, causes attenuated DNA damage response after treatment with UV irradiation (Fuzesi-Levi et al., 2020; Rozen et al., 2015). Although CSNAP does not regulate the neddylation of CRLs, it has steric effects on CRLs so that the affinity of CSN for CRLs is promoted when CSNAP is abolished. Therefore, CSNAP is also required for effective DNA damage response.
Work from our lab has elucidated how CSN responds to UV damage to mediate DDR (Fig. 2). Thus, Rao et al. first identified IP6K1 (inositol hexakisphosphate kinase 1) as a key regulator in this process (Rao et al., 2014). Under normal conditions, IP6K1, the enzyme that phosphorylates inositol hexakisphosphate (IP6) to generate the pyrophosphate-containing IP7, is catalytically inert but binds to CRL4 complex together with CSN, leading to CRL4 deneddylation and inactivation. Upon UV damage, IP6K1 is activated, leading to IP7 generation and CRL–CSN disassembly. IP6 but not IP7, maintains the assembly of CSN and CRL4 as “molecular glue”, thus CSN dissociates from CRL4 when IP6 is converted to IP7, which binds CSN2 less tightly (Lin et al., 2020; Scherer et al., 2016). Subsequently, CRL4 is neddylated with increased activity of ubiquitylation for substrate degradation, such as p53, CDT1, p27, p21, and maybe XPC and CSB (Hannss & Dubiel, 2011). Moreover, according to the recent study, CSN and the CRL4 substrate receptors competitively bind to CRLs, therefore, after DNA lesion detected, the dissociation of CSN not only enhances CRL neddylation but also promotes the assembly of CRL and its substrate receptor for substrate degradation (Lin et al., 2021). It will be important to determine whether other DNA damages stimuli activates CRL by dissociating CSN in a manner similar to UV radiation.
Fig. 2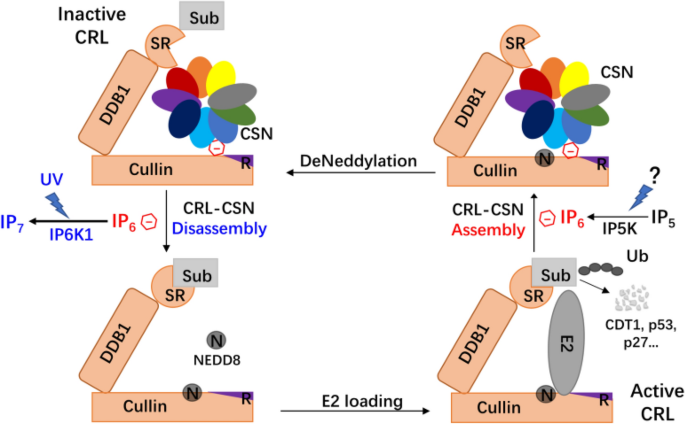
Role of inositol polyphosphate metabolism in CRL-deneddylation during DDR. IP6, as an intermolecular glue, assists CSN binding to deneddylated CRL, sequestering CRL from receiving the right substrates. UV radiation, via yet unknown mechanisms, induces IP6K1 activation and IP6 conversion to IP7, a process facilitating CRL–CSN dissociation and CRL-mediated substrate ubiquitylation. It remains unknown how widespread is this mechanism for the many CRL subcomplexes, or whether DNA damages other than UVR activates CRL in a similar manner. Whether IP6-mediated CRL–CSN assembly is also signaling dependent also requires further study
Full size imageCSN subcomplex can have isoform specific functions during DDR (Wang et al., 2021). Thus, mammalian COP9 signalosome (CSN) exists as two variant complexes containing either CSN7A or CSN7B. Nevertheless, CSNCSN7B, but not CSNCSN7A, has a unique function in DNA double-strand break (DSB) sensing, being required for ATM-dependent formation of γH2AX foci and DNA-damage-induced apoptosis triggered. CSN7B knockout cells are resistant to DNA damage by causing a switch in DNA repair pathway choice with increased utilization of non-homologous end joining over homologous recombination.
CSN subunits can also participate in DDR without the involvement of CRL (Chung & Dellaire, 2015). Thus, CSN5 is required for efficient HR (Meir et al., 2015; Pan et al., 2013). The disruption of CSN5 (also known as JAB1) results in increased spontaneous DNA damage and loss of function of HR. CSN5 overexpression upregulates Rad51 and 9-1-1 complex, enhancing the tolerance to cisplatin, IR and UV, and knocking down CSN5 leads to conferred sensitivity. Mechanistically, p53 accumulates in CSN5 knock-out cells, which directly block the transcription of DNA repair protein Rad51. Similarly, studies on CSN6 has revealed its intimate link to the regulation of DDR via p27 and p53 (Choi et al., 2015; Xue et al., 2012). Since CSN subunit expression is used as a key indicating for their complex-independent function, it would be essential to exclude whether there was dominant negative effects interfering with endogenous holo-CSN during DDR.
Non-Cullin-mediated neddylation in DNA damage response
Cullins are not the only proteins modified by NEDD8. More and more proteins, including but not limited to p53, the DNA polymerase processivity proliferating cell nuclear antigen (PCNA), the tumor suppressor pTEN, the ribosomal protein L11, caspase 7, breast cancer-associated gene 3 (BCA3), von Hippel–Lindau protein (VHL), hypoxia-inducible factor 1α (HIF1α) and histone H4 are reported to be neddylated as non-Cullin NEDD8 substrates (Gao et al., 2006; Guan et al., 2018; Ma et al., 2013; Oved et al., 2006; Ryu et al., 2011; Stickle et al., 2004; Sundqvist et al., 2009; Watson et al., 2006; Xie et al., 2021; Xirodimas et al., 2004). Some of these non-Cullin neddylation substrates are well-known mediators of DNA damage response (Fig. 1, Table 1). In response to DNA damage, p53 as a transcriptional factor is accumulated and activated to regulate transcription of target genes (Mijit et al., 2020). However, its neddylation by Mdm2 RING finger E3 ubiquitin ligase inhibits its function (Xirodimas et al., 2004). Strikingly, Mdm2 itself can also be modified by NEDD8 and is negatively regulated by neddylation. The first 4 h-UV treatment leads to accumulation of p53 level as well as its neddylation form in cells; however, the neddylation of p53 seems to disappear after 8 h UV stimulation. Therefore, the transcriptional activity of p53 is controlled by temporal neddylation upon DNA damage. PCNA can be neddylated by the E3 ligase RAD18 while deneddylated by NEDP1 (Guan et al., 2018). Besides the well-known role of PCNA in DNA repair, it also facilitates the recruitment of DNA polymerase η in TLS for tolerance (Guan et al., 2018). Moreover, upon oxidative DNA damage and NEDP1 dissociation, neddylation of both RAD18 and PCNA are increased to promote polymerase η recruitment after oxidative stress (Guan & Zheng, 2019). Another stimuli, glucose, was found to induce the neddylation of the membrane-bound protein phosphatase pTEN (Xie et al., 2021). The loss of pTEN causes over-activation of CHK1, which in turn generated double strand DNA breaks and inhibition of XPC (xeroderma pigmentosum C), one of the key GG-NER proteins (Ming et al., 2011; Ogino et al., 2016; Puc & Parsons, 2005). However, there is few direct evidence of pTEN neddylation regulating DDR. NEDD8 can binds to the N-terminal lysine residues of histone H4 for polyneddylation in DSBR, while the loss function of NEDD8 fails to recruit RNF168 and other DNA repair partners (Stiff et al., 2004; Stucki & Jackson, 2006).
E2 and E3s that neddylate non-Cullin substrates are also involved in DDR. Histone H4 poly-neddylation, catalyzed by RNF111 (E3) and Ube2M (E2), could determine the choice between NHEJ and HR (Jimeno et al., 2015). Neddylation catalyzed by RNF111 and UBE2M promotes NHEJ, while it plays an inhibitory role in BRCA1 and CtIP-mediated DNA end resection, the key process of HR. Moreover, after IR induction, NEDD8 accumulates and localizes with the markers of DSBs, γH2AX and 53BP1, at the DNA damage sites in E2 enzyme UBE2M and E3 ubiquitin ligase RNF111-dependent manner (Ma et al., 2013). Future studies would possibly reveal more neddylation targets involved in DDR.
The emerging role of NEDD8 polymer in DDR
Although poly-ubiquitin is well established, polymeric NEDD8 was not known to exist until very recently. It was found that Ubc12 catalyzes the formation of poly-NEDD8, which can be transferred to Cullins as well as non-Cullin neddylation substrates (Ohki et al., 2009). The monomer of NEDD8 is essential for DDR initiation, while the polymer inhibits some key genes of lesions repair. The conversion of unanchored ploy-to-mono-neddylation is reported to stimulate the HSP70-mediated lesion repair (Bailly et al., 2019). Upon DNA damage, this conversion is catalyzed by NEDP1 and HSP70 chaperone acts as an effector of this process to facilitate damage resistance and promote single strand DNA damage repair (Dubrez et al., 2020). Another related study also proved the importance of poly-NEDD8 (tri-NEDD8) in DDR by targeting PARP-1. PARP-1 is an early stage sensor of DNA strand breaks and can initiate DNA repair, control cell cycle and contribute to cell death or survive decision (Bouchard et al., 2003; Pascal, 2018). When NEDP1 is silenced or negatively regulated, unanchored NEDD8 trimers are formed and bind to PARP-1, resulting in the inhibition of PARP-1 (Keuss et al., 2019). The inhibition of PARP-1 often results in defect strand displacement DNA synthesis during BER, altered checkpoint proteins functions (eg. p53) and delay of oxidative stress-induced cell death (Bouchard et al., 2003; Keuss et al., 2019). Further work is required to understand the role of NEDD8 polymers, standalone or conjugated, in DDR.
Summary and future perspectives
Timely DNA damage response is essential for survival. The neddylation modification regulates this complex process through different aspects. Neddylation of Cullins, which is negatively controlled by CSN complex, regulates the ubiquitylation of core DDR proteins. Consistently, the neddylation inhibitor NLN4924 is an effective anti-cancer drug, primarily by targeting DDR. Given that CSN binds tightly to sequester CRL, it remained to be studied how different types of DNA-damage signaling leads to dynamic CRL–CSN complex assembly and disassembly to orchestrate DRR-related protein ubiquitylation by CRL. In addition, whether CSN mediates DDR as a whole complex or via specific subunits is incompletely understood. These outstanding questions can possibly be answered by a combination of biochemical reconstitution and cutting-edge high-resolution cryo-electron microscopy analysis.
Enough evidences have been found to demonstrate the importance of Non-Cullin neddylation. For example, the number of non-Cullin neddylation substrates is still limited and their functions still need to be further characterized, especially given the potential caveat that exogenous overexpressed NEDD8 can readily hijack the ubiquitylation pathway. In addition, the deneddylase for non-Cullin-based neddylation substrates is largely unknown. Nevertheless, neddylation of non-Cullin-based substrates can directly mediate the activity of DSBR and other types of DDR as well (Brown & Jackson, 2015). In view of recently efforts to detect endogenous neddylation targets by making NEDD8 R74K knock-in cells (Vogl et al., 2020) and by generating mice with endogenous tagged Nedd8 alleles (Zhang et al., 2020), more non-Cullin neddylation targets involved in DDR are likely to be identified in future work.
Last but not least, the newly discovered polymeric forms of NEDD8 draws attentions, since they mediate DNA damage tolerance and promote single strand DNA repair. Polymers of NEDD8 are a novel pathway; therefore, a lot more work, especially proteomic studies, are required for better understanding of the function and mechanism of them in response to DNA damage.
References
Aleksandrov, R., Hristova, R., Stoynov, S., & Gospodinov, A. (2020). The chromatin response to double-strand dna breaks and their repair. Cells, 9, 1853.
Baek, K., Krist, D. T., Prabu, J. R., Hill, S., Klugel, M., Neumaier, L. M., von Gronau, S., Kleiger, G., & Schulman, B. A. (2020a). NEDD8 nucleates a multivalent cullin-RING-UBE2D ubiquitin ligation assembly. Nature, 578, 461–466.
Baek, K., Scott, D. C., & Schulman, B. A. (2020b). NEDD8 and ubiquitin ligation by cullin-RING E3 ligases. Current Opinion in Structural Biology, 67, 101–109.
Bailly, A. P., Perrin, A., Serrano-Macia, M., Maghames, C., Leidecker, O., Trauchessec, H., Martinez-Chantar, M. L., Gartner, A., & Xirodimas, D. P. (2019). The balance between mono- and NEDD8-chains controlled by NEDP1 upon DNA damage is a regulatory module of the HSP70 ATPase activity. Cell Reports, 29, 212-224 e218.
Bosu, D. R., Feng, H., Min, K., Kim, Y., Wallenfang, M. R., & Kipreos, E. T. (2010). C. elegans CAND-1 regulates cullin neddylation, cell proliferation and morphogenesis in specific tissues. Developmental Biology, 346, 113–126.
Bouchard, V. J., Rouleau, M., & Poirier, G. G. (2003). PARP-1, a determinant of cell survival in response to DNA damage. Experimental Hematology, 31, 446–454.
Brooks, C. L., & Gu, W. (2006). p53 ubiquitination: Mdm2 and beyond. Molecular Cell, 21, 307–315.
Brown, J. S., & Jackson, S. P. (2015). Ubiquitylation, neddylation and the DNA damage response. Open Biology, 5, 150018.
Brown, J. S., Lukashchuk, N., Sczaniecka-Clift, M., Britton, S., le Sage, C., Calsou, P., Beli, P., Galanty, Y., & Jackson, S. P. (2015). Neddylation promotes ubiquitylation and release of Ku from DNA-damage sites. Cell Reports, 11, 704–714.
Chamovitz, D. A., & Segal, D. (2001). JAB1/CSN5 and the COP9 signalosome. A Complex Situation. EMBO Rep, 2, 96–101.
Choi, H. H., Guma, S., Fang, L., Phan, L., Ivan, C., Baggerly, K., Sood, A., & Lee, M. H. (2015). Regulating the stability and localization of CDK inhibitor p27(Kip1) via CSN6-COP1 axis. Cell Cycle, 14, 2265–2273.
Chu, G., & Chang, E. (1988). Xeroderma pigmentosum group E cells lack a nuclear factor that binds to damaged DNA. Science, 242, 564–567.
Chung, D., & Dellaire, G. (2015). The role of the COP9 signalosome and neddylation in DNA damage signaling and repair. Biomolecules, 5, 2388–2416.
Ciccia, A., & Elledge, S. J. (2010). The DNA damage response: Making it safe to play with knives. Molecular Cell, 40, 179–204.
Cope, G. A., & Deshaies, R. J. (2003). COP9 signalosome: A multifunctional regulator of SCF and other cullin-based ubiquitin ligases. Cell, 114, 663–671.
Cui, D., Xiong, X., Shu, J., Dai, X., Sun, Y., & Zhao, Y. (2020). FBXW7 confers radiation survival by targeting p53 for degradation. Cell Reports, 30, 497-509 e494.
Cukras, S., Morffy, N., Ohn, T., & Kee, Y. (2014). Inactivating UBE2M impacts the DNA damage response and genome integrity involving multiple cullin ligases. PLoS ONE, 9, e101844.
Dar, A. A., Sawada, K., Dybas, J. M., Moser, E. K., Lewis, E. L., Park, E., Fazelinia, H., Spruce, L. A., Ding, H., Seeholzer, S. H., and Oliver, P.M. (2021). The E3 ubiquitin ligase Cul4b promotes CD4+ T cell expansion by aiding the repair of damaged DNA. PLoS Biology, 19, e300101.
de Laat, W. L., Appeldoorn, E., Sugasawa, K., Weterings, E., Jaspers, N. G., & Hoeijmakers, J. H. (1998). DNA-binding polarity of human replication protein A positions nucleases in nucleotide excision repair. Genes & Development, 12, 2598–2609.
Dellaire, G., & Bazett-Jones, D. P. (2007). Beyond repair foci: Subnuclear domains and the cellular response to DNA damage. Cell Cycle, 6, 1864–1872.
Dubrez, L., Causse, S., Borges Bonan, N., Dumetier, B., & Garrido, C. (2020). Heat-shock proteins: Chaperoning DNA repair. Oncogene, 39, 516–529.
Duda, D. M., Borg, L. A., Scott, D. C., Hunt, H. W., Hammel, M., & Schulman, B. A. (2008). Structural insights into NEDD8 activation of cullin-RING ligases: Conformational control of conjugation. Cell, 134, 995–1006.
Echalier, A., Pan, Y., Birol, M., Tavernier, N., Pintard, L., Hoh, F., Ebel, C., Galophe, N., Claret, F. X., & Dumas, C. (2013). Insights into the regulation of the human COP9 signalosome catalytic subunit, CSN5/Jab1. Proc Natl Acad Sci U S A, 110, 1273–1278.
Essers, J., Theil, A. F., Baldeyron, C., van Cappellen, W. A., Houtsmuller, A. B., Kanaar, R., & Vermeulen, W. (2005). Nuclear dynamics of PCNA in DNA replication and repair. Molecular and Cellular Biology, 25, 9350–9359.
Feng, M., Wang, Y., Bi, L., Zhang, P., Wang, H., Zhao, Z., Mao, J. H., & Wei, G. (2021). CRL4A(DTL) degrades DNA-PKcs to modulate NHEJ repair and induce genomic instability and subsequent malignant transformation. Oncogene, 40, 2096–2111.
Fousteri, M., & Mullenders, L. H. (2008). Transcription-coupled nucleotide excision repair in mammalian cells: Molecular mechanisms and biological effects. Cell Research, 18, 73–84.
Fuzesi-Levi, M. G., Ben-Nissan, G., Bianchi, E., Zhou, H., Deery, M. J., Lilley, K. S., Levin, Y., & Sharon, M. (2014). Dynamic regulation of the COP9 signalosome in response to DNA damage. Molecular and Cellular Biology, 34, 1066–1076.
Fuzesi-Levi, M. G., Fainer, I., Ivanov Enchev, R., Ben-Nissan, G., Levin, Y., Kupervaser, M., Friedlander, G., Salame, T. M., Nevo, R., Peter, M., and Sharon, M. (2020). CSNAP, the smallest CSN subunit, modulates proteostasis through cullin-RING ubiquitin ligases. Cell Death and Differentiation, 27, 984–998.
Gao, F., Cheng, J., Shi, T., & Yeh, E. T. (2006). Neddylation of a breast cancer-associated protein recruits a class III histone deacetylase that represses NFkappaB-dependent transcription. Nature Cell Biology, 8, 1171–1177.
Giglia-Mari, G., Zotter, A., & Vermeulen, W. (2011). DNA damage response. Cold Spring Harbor Perspectives in Biology, 3, 745.
Groisman, R., Kuraoka, I., Chevallier, O., Gaye, N., Magnaldo, T., Tanaka, K., Kisselev, A. F., Harel-Bellan, A., & Nakatani, Y. (2006). CSA-dependent degradation of CSB by the ubiquitin-proteasome pathway establishes a link between complementation factors of the Cockayne syndrome. Genes & Development, 20, 1429–1434.
Groisman, R., Polanowska, J., Kuraoka, I., Sawada, J., Saijo, M., Drapkin, R., Kisselev, A. F., Tanaka, K., & Nakatani, Y. (2003). The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell, 113, 357–367.
Guan, J., & Zheng, X. (2019). NEDDylation regulates RAD18 ubiquitination and localization in response to oxidative DNA damage. Biochemical and Biophysical Research Communications, 508, 1240–1244.
Guan, J., Yu, S., & Zheng, X. (2018). NEDDylation antagonizes ubiquitination of proliferating cell nuclear antigen and regulates the recruitment of polymerase eta in response to oxidative DNA damage. Protein & Cell, 9, 365–379.
Guo, Z., Wang, S., Xie, Y., Han, Y., Hu, S., Guan, H., Xie, D., Bai, C., Liu, X., Gu, Y., Zhou, P.K., and Ma, T. (2020). HUWE1-dependent DNA-PKcs neddylation modulates its autophosphorylation in DNA damage response. Cell Death & Disease, 11, 400.
Hannss, R., & Dubiel, W. (2011). COP9 signalosome function in the DDR. FEBS Letters, 585, 2845–2852.
Harper, J. W., & Elledge, S. J. (2007). The DNA damage response: Ten years after. Molecular Cell, 28, 739–745.
Jackson, S. P., & Bartek, J. (2009). The DNA-damage response in human biology and disease. Nature, 461, 1071–1078.
Jimeno, S., Fernandez-Avila, M. J., Cruz-Garcia, A., Cepeda-Garcia, C., Gomez-Cabello, D., & Huertas, P. (2015). Neddylation inhibits CtIP-mediated resection and regulates DNA double strand break repair pathway choice. Nucleic Acids Research, 43, 987–999.
Kapetanaki, M. G., Guerrero-Santoro, J., Bisi, D. C., Hsieh, C. L., Rapic-Otrin, V., & Levine, A. S. (2006). The DDB1-CUL4ADDB2 ubiquitin ligase is deficient in xeroderma pigmentosum group E and targets histone H2A at UV-damaged DNA sites. Proceedings of the National Academy of Sciences U S A, 103, 2588–2593.
Kato, J. Y., & Yoneda-Kato, N. (2009). Mammalian COP9 signalosome. Genes to Cells, 14, 1209–1225.
Keuss, M. J., Hjerpe, R., Hsia, O., Gourlay, R., Burchmore, R., Trost, M., & Kurz, T. (2019). Unanchored tri-NEDD8 inhibits PARP-1 to protect from oxidative stress-induced cell death. EMBO Journal, 38, e100024.
Krokan, H. E., & Bjoras, M. (2013). Base excision repair. Cold Spring Harbor Perspectives in Biology, 5, a012583.
Lan, H., & Sun, Y. (2019). FBXW7 E3 ubiquitin ligase: Degrading, not degrading, or being degraded. Protein & Cell, 10, 861–863.
Leal, J. F., Fominaya, J., Cascon, A., Guijarro, M. V., Blanco-Aparicio, C., Lleonart, M., Castro, M. E., Ramon, Y. C. S., Robledo, M., Beach, D. H., and Carnero, A. (2008). Cellular senescence bypass screen identifies new putative tumor suppressor genes. Oncogene, 27, 1961–1970.
Li, T., Guan, J., Huang, Z., Hu, X., & Zheng, X. (2014). RNF168-mediated H2A neddylation antagonizes ubiquitylation of H2A and regulates DNA damage repair. Journal of Cell Science, 127, 2238–2248.
Li, Z., Pearlman, A. H., & Hsieh, P. (2016). DNA mismatch repair and the DNA damage response. DNA Repair (amst), 38, 94–101.
Lin, H. Y., Luo, Y., So, W. Y., Rao, F., et al. (2021). IP6-assisted CSN-COP1 competition regulates a CRL4-ETV5 proteolytic checkpoint to safeguard glucose-induced insulin secretion against hyperinsulinemia. Nature Communications, 12, 2461.
Lin, H., Zhang, X., Liu, L., Fu, Q., Zang, C., Ding, Y., Su, Y., Xu, Z., He, S., Yang, X., Wei, X., Mao, H., Cui, Y., Wei, Y., Zhou, C., Du, L., Huang, N., Zheng, N., Wang, T., and Rao, F. (2020). Basis for metabolite-dependent Cullin-RING ligase deneddylation by the COP9 signalosome. Proceedings of the National Academy of Sciences U S A, 117, 4117–4124.
Lobato-Gil, S., Heidelberger, J. B., Maghames, C., Bailly, A., Brunello, L., Rodriguez, M. S., Beli, P., & Xirodimas, D. P. (2021). Proteome-wide identification of NEDD8 modification sites reveals distinct proteomes for canonical and atypical NEDDylation. Cell Reports, 34, 108635.
Ma, T., Chen, Y., Zhang, F., Yang, C. Y., Wang, S., & Yu, X. (2013). RNF111-dependent neddylation activates DNA damage-induced ubiquitination. Molecular Cell, 49, 897–907.
Mahon, C., Krogan, N. J., Craik, C. S., & Pick, E. (2014). Cullin E3 ligases and their rewiring by viral factors. Biomolecules, 4, 897–930.
Mailand, N., Bekker-Jensen, S., Bartek, J., & Lukas, J. (2006). Destruction of Claspin by SCFbetaTrCP restrains Chk1 activation and facilitates recovery from genotoxic stress. Molecular Cell, 23, 307–318.
Mao, H., & Sun, Y. (2020). Neddylation-Independent Activities of MLN4924. Advances in Experimental Medicine and Biology, 1217, 363–372.
Marine, J. C., & Lozano, G. (2010). Mdm2-mediated ubiquitylation: P53 and beyond. Cell Death and Differentiation, 17, 93–102.
Meir, M., Galanty, Y., Kashani, L., Blank, M., Khosravi, R., Fernandez-Avila, M. J., Cruz-Garcia, A., Star, A., Shochot, L., Thomas, Y., Garrett, L.J., Chamovitz, D.A., Bodine, D.M., Kurz, T., Huertas, P., Ziv, Y., and Shiloh, Y. (2015). The COP9 signalosome is vital for timely repair of DNA double-strand breaks. Nucleic Acids Research, 43, 4517–4530.
Mijit, M., Caracciolo, V., Melillo, A., Amicarelli, F., & Giordano, A. (2020). Role of p53 in the regulation of cellular senescence. Biomolecules, 10, 420.
Ming, M., Feng, L., Shea, C. R., Soltani, K., Zhao, B., Han, W., Smart, R. C., Trempus, C. S., & He, Y. Y. (2011). PTEN positively regulates UVB-induced DNA damage repair. Cancer Research, 71, 5287–5295.
Nakamura, T., Murakami, K., Tada, H., Uehara, Y., Nogami, J., Maehara, K., Ohkawa, Y., Saitoh, H., Nishitani, H., Ono, T., et al. (2017). Thymine DNA glycosylase modulates DNA damage response and gene expression by base excision repair-dependent and independent mechanisms. Genes to Cells, 22, 392–405.
Nishitani, H., Shiomi, Y., Iida, H., Michishita, M., Takami, T., & Tsurimoto, T. (2008). CDK inhibitor p21 is degraded by a proliferating cell nuclear antigen-coupled Cul4-DDB1Cdt2 pathway during S phase and after UV irradiation. Journal of Biological Chemistry, 283, 29045–29052.
Ogino, M., Ichimura, M., Nakano, N., Minami, A., Kitagishi, Y., & Matsuda, S. (2016). Roles of PTEN with DNA repair in Parkinson’s disease. International Journal of Molecular Sciences, 17, 954.
Ohki, Y., Funatsu, N., Konishi, N., & Chiba, T. (2009). The mechanism of poly-NEDD8 chain formation in vitro. Biochemical and Biophysical Research Communications, 381, 443–447.
Oladghaffari, M., Islamian, J. P., Baradaran, B., & Monfared, A. S. (2016). MLN4924 therapy as a novel approach in cancer treatment modalities. Journal of Chemotherapy, 28, 74–82.
Oved, S., Mosesson, Y., Zwang, Y., Santonico, E., Shtiegman, K., Marmor, M. D., Kochupurakkal, B. S., Katz, M., Lavi, S., Cesareni, G., et al. (2006). Conjugation to Nedd8 instigates ubiquitylation and down-regulation of activated receptor tyrosine kinases. Journal of Biological Chemistry, 281, 21640–21651.
Pan, Y., Zhang, Q., Atsaves, V., Yang, H., & Claret, F. X. (2013). Suppression of Jab1/CSN5 induces radio- and chemo-sensitivity in nasopharyngeal carcinoma through changes to the DNA damage and repair pathways. Oncogene, 32, 2756–2766.
Pascal, J. M. (2018). The comings and goings of PARP-1 in response to DNA damage. DNA Repair (amst), 71, 177–182.
Petroski, M. D., & Deshaies, R. J. (2005). Function and regulation of cullin-RING ubiquitin ligases. Nature Reviews Molecular Cell Biology, 6, 9–20.
Pettersen, H. S., Sundheim, O., Gilljam, K. M., Slupphaug, G., Krokan, H. E., & Kavli, B. (2007). Uracil-DNA glycosylases SMUG1 and UNG2 coordinate the initial steps of base excision repair by distinct mechanisms. Nucleic Acids Research, 35, 3879–3892.
Puc, J., & Parsons, R. (2005). PTEN loss inhibits CHK1 to cause double stranded-DNA breaks in cells. Cell Cycle, 4, 927–929.
Rabut, G., & Peter, M. (2008). Function and regulation of protein neddylation. “Protein modifications: Beyond the usual suspects” review series. EMBO Reports, 9, 969–976.
Rao, F., Xu, J., Khan, A. B., Gadalla, M. M., Cha, J. Y., Xu, R., Tyagi, R., Dang, Y., Chakraborty, A., & Snyder, S. H. (2014). Inositol hexakisphosphate kinase-1 mediates assembly/disassembly of the CRL4-signalosome complex to regulate DNA repair and cell death. Proc Natl Acad Sci U S A, 111, 16005–16010.
Rizzardi, L. F., & Cook, J. G. (2012). Flipping the switch from g1 to s phase with e3 ubiquitin ligases. Genes & Cancer, 3, 634–648.
Rozen, S., Fuzesi-Levi, M. G., Ben-Nissan, G., Mizrachi, L., Gabashvili, A., Levin, Y., Ben-Dor, S., Eisenstein, M., & Sharon, M. (2015). CSNAP Is a Stoichiometric Subunit of the COP9 Signalosome. Cell Reports, 13, 585–598.
Ryu, J. H., Li, S. H., Park, H. S., Park, J. W., Lee, B., & Chun, Y. S. (2011). Hypoxia-inducible factor alpha subunit stabilization by NEDD8 conjugation is reactive oxygen species-dependent. Journal of Biological Chemistry, 286, 6963–6970.
Sancar, A., Lindsey-Boltz, L. A., Unsal-Kacmaz, K., & Linn, S. (2004). Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annual Review of Biochemistry, 73, 39–85.
Scharer, O. D. (2013). Nucleotide excision repair in eukaryotes. Cold Spring Harbor Perspectives in Biology, 5, a012609.
Scherer, P. C., Ding, Y., Liu, Z., Xu, J., Mao, H., Barrow, J. C., Wei, N., Zheng, N., Snyder, S. H., & Rao, F. (2016). Inositol hexakisphosphate (IP6) generated by IP5K mediates cullin-COP9 signalosome interactions and CRL function. Proc Natl Acad Sci U S A, 113, 3503–3508.
Schwechheimer, C. (2004). The COP9 signalosome (CSN): An evolutionary conserved proteolysis regulator in eukaryotic development. Biochimica Et Biophysica Acta, 1695, 45–54.
Schwechheimer, C. (2018). NEDD8-its role in the regulation of Cullin-RING ligases. Current Opinion in Plant Biology, 45, 112–119.
Scrima, A., Konickova, R., Czyzewski, B. K., Kawasaki, Y., Jeffrey, P. D., Groisman, R., Nakatani, Y., Iwai, S., Pavletich, N. P., & Thoma, N. H. (2008). Structural basis of UV DNA-damage recognition by the DDB1-DDB2 complex. Cell, 135, 1213–1223.
Slenn, T. J., Morris, B., Havens, C. G., Freeman, R. M., Jr., Takahashi, T. S., & Walter, J. C. (2014). Thymine DNA glycosylase is a CRL4Cdt2 substrate. Journal of Biological Chemistry, 289, 23043–23055.
Soucy, T. A., Smith, P. G., Milhollen, M. A., Berger, A. J., Gavin, J. M., Adhikari, S., Brownell, J. E., Burke, K. E., Cardin, D. P., Critchley, S., Cullis, C.A., Doucette, A., Garnsey, J.J., Gaulin, J.L., Gershman, R.E., Lublinsky, A.R., McDonald, A., Mizutani, H., Narayanan, U., Olhava, E.J., Peluso, S., Rezaei, M., Sintchak, M.D., Talreja, T., Thomas, M.P., Traore, T., Vyskocil, S., Weatherhead, G.S., Yu, J., Zhang, J., Dick, L.R., Claiborne, C.F., Rolfe, M., Bolen, J.B., and Langston, S.P. (2009). An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature, 458, 732–736.
Spivak, G. (2015). Nucleotide excision repair in humans. DNA Repair (amst), 36, 13–18.
Stickle, N. H., Chung, J., Klco, J. M., Hill, R. P., Kaelin, W. G., Jr., & Ohh, M. (2004). pVHL modification by NEDD8 is required for fibronectin matrix assembly and suppression of tumor development. Molecular and Cellular Biology, 24, 3251–3261.
Stiff, T., O’Driscoll, M., Rief, N., Iwabuchi, K., Lobrich, M., & Jeggo, P. A. (2004). ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Research, 64, 2390–2396.
Stucki, M., & Jackson, S. P. (2006). gammaH2AX and MDC1: Anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair (amst), 5, 534–543.
Sugasawa, K., Okuda, Y., Saijo, M., Nishi, R., Matsuda, N., Chu, G., Mori, T., Iwai, S., Tanaka, K., Tanaka, K., et al. (2005). UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell, 121, 387–400.
Sundqvist, A., Liu, G., Mirsaliotis, A., & Xirodimas, D. P. (2009). Regulation of nucleolar signalling to p53 through NEDDylation of L11. EMBO Reports, 10, 1132–1139.
Tomasz, M., & Palom, Y. (1997). The mitomycin bioreductive antitumor agents: Cross-linking and alkylation of DNA as the molecular basis of their activity. Pharmacology & Therapeutics, 76, 73–87.
Tomimatsu, N., Mukherjee, B., Harris, J. L., Boffo, F. L., Hardebeck, M. C., Potts, P. R., Khanna, K. K., & Burma, S. (2017). DNA-damage-induced degradation of EXO1 exonuclease limits DNA end resection to ensure accurate DNA repair. Journal of Biological Chemistry, 292, 10779–10790.
Vogl, A. M., Phu, L., Becerra, R., Giusti, S. A., Verschueren, E., Hinkle, T. B., Bordenave, M. D., Adrian, M., Heidersbach, A., Yankilevich, P., Stefani, F.D., Wurst, W., Hoogenraad, C.C., Kirkpatrick, D.S., Refojo, D., and Sheng, M. (2020). Global site-specific neddylation profiling reveals that NEDDylated cofilin regulates actin dynamics. Nature Structural & Molecular Biology, 27, 210–220.
Wang, J., Dubiel, D., Wu, Y., Cheng, Y., Wolf, D. A., & Dubiel, W. (2021). CSN7B defines a variant COP9 signalosome complex with distinct function in DNA damage response. Cell Reports, 34, 108662.
Wang, X., Kang, D., Feng, S., Serino, G., Schwechheimer, C., & Wei, N. (2002). CSN1 N-terminal-dependent activity is required for Arabidopsis development but not for Rub1/Nedd8 deconjugation of cullins: A structure-function study of CSN1 subunit of COP9 signalosome. Molecular Biology of the Cell, 13, 646–655.
Watson, I. R., Blanch, A., Lin, D. C., Ohh, M., & Irwin, M. S. (2006). Mdm2-mediated NEDD8 modification of TAp73 regulates its transactivation function. Journal of Biological Chemistry, 281, 34096–34103.
Wei, N., & Deng, X. W. (2003). The COP9 signalosome. Annual Review of Cell and Developmental Biology, 19, 261–286.
Wei, N., Serino, G., & Deng, X. W. (2008). The COP9 signalosome: more than a protease. Trends in Biochemical Sciences, 33, 592–600.
Wen, X., Casey Klockow, L., Nekorchuk, M., Sharifi, H. J., & de Noronha, C. M. (2012). The HIV1 protein Vpr acts to enhance constitutive DCAF1-dependent UNG2 turnover. PLoS ONE, 7, e30939.
Wu, J., Zhang, X., Zhang, L., Wu, C. Y., Rezaeian, A. H., Chan, C. H., Li, J. M., Wang, J., Gao, Y., Han, F., et al. (2012). Skp2 E3 ligase integrates ATM activation and homologous recombination repair by ubiquitinating NBS1. Molecular Cell, 46, 351–361.
Wu, S., Zhu, W., Nhan, T., Toth, J. I., Petroski, M. D., & Wolf, D. A. (2013). CAND1 controls in vivo dynamics of the cullin 1-RING ubiquitin ligase repertoire. Nature Communications, 4, 1642.
Xie, P., Peng, Z., Chen, Y., Li, H., Du, M., Tan, Y., Zhang, X., Lu, Z., Cui, C. P., Liu, C. H., et al. (2021). Neddylation of PTEN regulates its nuclear import and promotes tumor development. Cell Research, 31, 291–311.
Xirodimas, D. P., Saville, M. K., Bourdon, J. C., Hay, R. T., & Lane, D. P. (2004). Mdm2-mediated NEDD8 conjugation of p53 inhibits its transcriptional activity. Cell, 118, 83–97.
Xue, Y., Chen, J., Choi, H. H., Phan, L., Chou, P. C., Zhao, R., Yang, H., Santiago, J., Liu, M., Yeung, G. E., et al. (2012). HER2-Akt signaling in regulating COP9 signalsome subunit 6 and p53. Cell Cycle, 11, 4181–4190.
Yang, X., Menon, S., Lykke-Andersen, K., Tsuge, T., Di, X., Wang, X., Rodriguez-Suarez, R. J., Zhang, H., & Wei, N. (2002). The COP9 signalosome inhibits p27(kip1) degradation and impedes G1-S phase progression via deneddylation of SCF Cul1. Current Biology, 12, 667–672.
Zhang, Q., Karnak, D., Tan, M., Lawrence, T. S., Morgan, M. A., & Sun, Y. (2016). FBXW7 facilitates nonhomologous end-joining via K63-linked polyubiquitylation of XRCC4. Molecular Cell, 61, 419–433.
Zhang, Q., Mady, A. S. A., Ma, Y., Ryan, C., Lawrence, T. S., Nikolovska-Coleska, Z., Sun, Y., & Morgan, M. A. (2019). The WD40 domain of FBXW7 is a poly(ADP-ribose)-binding domain that mediates the early DNA damage response. Nucleic Acids Research, 47, 4039–4053.
Zhang, X., Zhang, Y. L., Qiu, G., Pian, L., Guo, L., Cao, H., Liu, J., Zhao, Y., Li, X., Xu, Z., Huang, X., Huang, J., Dong, J., Shen, B., Wang, H.X., Ying, X., Zhang, W.J., Cao, X., and Zhang, J. (2020). Hepatic neddylation targets and stabilizes electron transfer flavoproteins to facilitate fatty acid beta-oxidation. Proceedings of the National Academy of Sciences U S A, 117, 2473–2483.
Zhao, Y., Tan, M., Liu, X., Xiong, X., & Sun, Y. (2018). Inactivation of ribosomal protein S27-like confers radiosensitivity via the Mdm2-p53 and Mdm2-MRN-ATM axes. Cell Death & Disease, 9, 145.
Zhou, P., & Yan, F. (2020). CRL4 ubiquitin pathway and DNA damage response. Advances in Experimental Medicine and Biology, 1217, 225–239.
Acknowledgements
We thank members of the Rao Laboratory for discussion and support, and apologize to colleagues whose work were unable to be cited here. This work was supported by grants from the Science and Technology Innovation Commission of Shenzhen Municipal Government (KQJSCX20180322152418316, JCYJ20170412153517422, and JCYJ20170817104311912 to F.R.), the National Science Foundation of China (31872798 and 91853129 to F.R.;), and the Department of Science and Technology of Guangdong Province (2018A030313207, to F.R.).
Author information
Affiliations
Department of Biology, School of Life Sciences, Southern University of Science and Technology, Shenzhen, Guangdong, China
Yifan Luo, Yang Su & Feng Rao
School of Biological Sciences, The University of Hong Kong, Pokfulam, Hong Kong
Yifan Luo
Contributions
YL and FR wrote the paper, with the assistance from YS.
Corresponding author
Correspondence to Feng Rao.
Ethics declarations
Competing interests
The authors declare that they have no competing interest.
Rights and permissions
About this article
Cite this article
Luo, Y., Su, Y. & Rao, F. Role of NEDD8 and neddylation dynamics in DNA damage response. GENOME INSTAB. DIS. 2, 139–149 (2021). https://doi.org/10.1007/s42764-021-00044-z
Received02 April 2021
Revised27 May 2021
Accepted29 May 2021
Published07 June 2021
Issue DateJune 2021
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
Neddylation
COP9 Signalosome
NEDD8
DNA damage response
Cullin–RING ubiquitin ligase
用户登录
还没有账号?
立即注册