






BRCA1: a key player at multiple stages of homologous recombination in DNA double-strand break repair
Review Article
Genome Instability & Disease 2, 164–174 (2021)
Abstract
As a key tumor suppressor in several cancers, BRCA1 is required for the maintenance of genomic stability. Extensive studies have revealed multiple functions of BRCA1 that contribute to its role in tumor suppression, but the most recognized one is DNA damage repair, especially DNA double-strand break (DSB) repair. Once recruited to DSB in S/G2 phase of the cell cycle, BRCA1 promotes DSB repair through homologous recombination (HR). Mechanistically, it is well known that BRCA1 is required for the efficient loading of RAD51 recombinase to DSB. Studies in the past decade reveal that BRCA1 also regulates DNA end resection to generate single-stranded DNA (ssDNA) required for RAD51 loading and promotes RAD51-mediated synaptic complex assembly. In this review, we will summarize the underlying mechanisms of the recruitment BRCA1 to DSB and BRCA1’s functions at multiple stages of HR in DSB repair. We will also discuss how HR defects caused by BRCA1 deficiency can be rescued by modulating DSB repair pathway choice.
Introduction
Being the most deleterious form of DNA damage, DNA double-strand breaks (DSB) are disruptive to nearly all activities on DNA and are the major sources of large DNA structural variations including intra- and inter-chromosomal translocations. DSB can arise from either exogenous source such as ionizing radiation (IR) or endogenous source such as collapsed DNA replication forks. In response to such enormous threat to genomic stability, cells are equipped with a plethora of mechanisms that are coordinated to ensure timely DSB repair (Kass et al., 2016).
BRCA1 is first identified as a breast cancer susceptibility gene and is frequently mutated in familial breast and ovarian cancers. Mutations and transcriptional silencing of BRCA1 gene are also found in several types of sporadic cancers. BRCA1 is involved in multiple aspects of DNA damage response, establishing itself as a bona fide genomic caretaker and tumor suppressor (Li & Greenberg, 2012). BRCA1 is most characterized and appreciated for its pivotal role in promoting homologous recombination (HR) in DSB repair (Chen et al., 2018; Tarsounas & Sung, 2020; Zhao et al., 2019). BRCA1 mutated cancer cells are HR deficient and are sensitive to a number of DNA damaging agents, especially poly(ADP-ribose) polymerase inhibitors (PARPi) (Lord & Ashworth, 2017). Such characteristics have been exploited for targeted therapy, and PARPi have been approved for clinical use in multiple countries (Curtin & Szabo, 2020). In addition to DSB, BRCA1 repairs other types of DNA damage (Michl et al., 2016; Paull, 2019). BRCA1 is also required for protection, repair, and restart of stalled replication forks (Tarsounas & Sung, 2020). In this review, we will focus on BRCA1’s function in promoting HR in DSB repair. The audience are encouraged to read the aforementioned reviews on other aspects of BRCA1 in DNA damage repair.
Due to the requirement of sister chromatid as a repair template, HR is restricted to S/G2 phase of the cell cycle (Hustedt & Durocher, 2016). BRCA1 localizes to DSB specifically in S/G2 phase of the cell cycle and promotes HR at multiple stages. BRCA1 directs DSB repair pathway choice towards HR by promoting DNA end resection that generates single-stranded DNA (ssDNA). BRCA1 also facilitates the formation of RAD51-ssDNA presynaptic filaments, important intermediates that are required for RAD51-mediated strand invasion. In addition, BRCA1 and its partner BARD1 directly bind RAD51 and promote RAD51-mediated homologous DNA pairing. In the following paragraphs, we will discuss BRCA1’s functions in these steps of HR in detail.
Recruitment of BRCA1 to DSB
Studies over the past two decades have established that histone ubiquitination establishes a platform for the recruitment of a variety of DNA damage response proteins including BRCA1 to DSB (Mattiroli & Penengo, 2021; Uckelmann & Sixma, 2017). Histone ubiquitination is initiated by E3 ubiquitin ligase RNF8, which is recruited to DSB by γH2AX-MDC1 signaling. Together with UBC13, RNF8 generates K63-linked poly-ubiquitin chains on histone H1 and L3MBTL2 (Huen et al., 2007; Kolas et al., 2007; Mailand et al., 2007; Nowsheen et al., 2018; Thorslund et al., 2015; Wang & Elledge, 2007). Another E3 ubiquitin ligase RNF168 recognizes these K63-linked poly-ubiquitin chains using its MIU domains, relocates to DSB, and further mono-ubiquitinates histone H2A on lysine 13/15 (Doil et al., 2009; Horn et al., 2019; Mattiroli et al., 2012; Stewart et al., 2009). The activities of both RNF8 and RNF168 are required for the efficient recruitment of BRCA1 to DSB (Fig. 1).
Fig. 1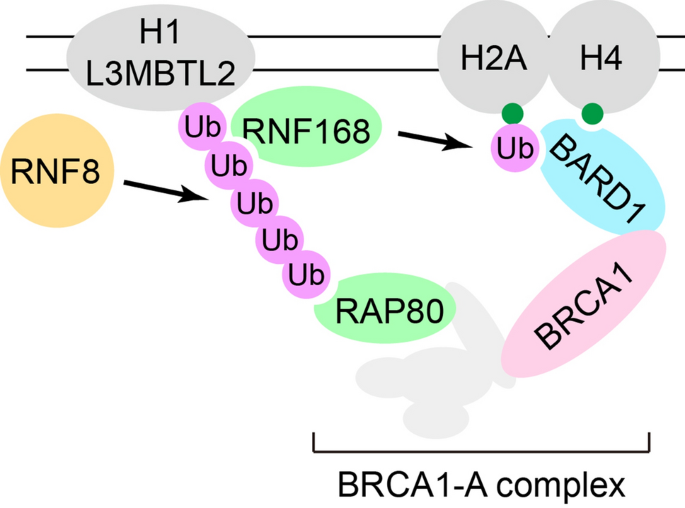
Recruitment of BRCA1 to DSB. RNF8 establishes K63-linked poly-ubiquitin chains on histones, especially on histone H1 and L3MBTL2. RNF168 recognizes these K63-linked poly-ubiquitin chains and further mono-ubiquitinates histone H2A on lysine 13/15. RAP80 recognizes K63-linked ubiquitin chains established by RNF8 and mediates the BRCA1-A complex to DSB. BARD1 recognizes both H4K20me0 specifically present in S/G2 phase and H2AK15Ub catalyzed by RNF168, both of which are important for BRCA1’s localization at DSB. The coordination between BRCA1-A complex and BARD1 is not clear
Full size imageBRCA1 is a large protein with multiple functional domains. The C-terminus of the protein contains tandem BRCT domains that bind phosphorylated proteins (Manke et al., 2003; Yu et al., 2003). Using these domains, BRCA1 is assembled into three major complexes through different binding partners, including phosphorylated ABRAXAS1/CCDC98 in BRCA1-A complex, phosphorylated FANCJ/BRIP1/BACH1 in BRCA1-B complex, and phosphorylated CTIP/RBBP8 in BRCA1-C complex (Huen et al., 2010). BRCA1-A complex is important for the recruitment of BRCA1 to DSB. In addition to CCDC98, BRCA1-A complex also contains UIMC1/RAP80, BRCC3/BRCC36, BABAM1/MERIT40/NBA1, and BABAM2/BRCC45/BRE (Huen et al., 2010). The UIM domains of RAP80 recognize K63-linked ubiquitin chains established by RNF8 and mediate the BRCA1-A complex to DSB (Fig. 1) (Kim et al., 2007; Sobhian et al., 2007; Wang et al., 2007).
In addition to BRCA1-A/B/C complex, BRCA1 forms a constitutive heterodimer with BARD1 through the N-terminal RING domains on both proteins (Tarsounas & Sung, 2020). Accumulating evidence has suggested that BARD1 also contributes to the recruitment of BRCA1 to DSB. Within seconds after DSB generation, massive poly(ADP)ribosylation (PAR) is generated at DSB to initiate the DNA damage response (Pandey & Black, 2021). BARD1 recognizes PAR through its C-terminal tandem BRCT domains and mediates the rapid recruitment of BRCA1 to DSB (Johnson et al., 2013). BARD1 is also required for retaining BRCA1 at DSB. BARD1’s ankyrin repeats recognize H4K20me0 while its tandem BRCT domains recognize H2AK15Ub catalyzed by RNF168, both of which are important for BRCA1’s localization at DSB (Fig. 1) (Becker et al., 2020; Nakamura et al., 2019). It is still not clear how BARD1 and BRCA1-A complex coordinate the recruitment of BRCA1 to DSB.
Since HR repair only operates in S/G2 phase of the cell cycle, the recruitment of BRCA1 to DSB is cell cycle regulated. BRCA1 is recruited to DSB and forms foci only at S/G2 phase, and BRCA1 foci can hardly be observed in G1 phase (Escribano-Diaz et al., 2013). Since H4K20 is unmethylated only when histone H4 is newly synthesized and incorporated into chromatin during replication (Saredi et al., 2016), the preference of BARD1’s ankyrin repeat for H4K20me0 explains S/G2 phase-specific recruitment of BRCA1 to DSB (Nakamura et al., 2019). In G1 phase, BRCA1 foci are suppressed by a mechanism that is not fully understood, but 53BP1 and its downstream effector RIF1 are involved since depletion of either of them significantly increases BRCA1 foci in G1 phase (Escribano-Diaz et al., 2013). On the contrary, BRCA1 localizes to DSB in S/G2 phase and promotes HR by counteracting 53BP1/RIF1’s localization and functions at DSB (discussed below).
BRCA1 lifts the barriers on DNA end resection and directs DSB repair pathway choice towards HR
In S/G2 phase, DSB can be repaired by either accurate HR or error-prone pathways including non-homologous end joining (NHEJ), microhomology-mediated end joining (MMEJ), and single-strand annealing (SSA) (Fig. 2). The DSB repair pathway choice is mainly determined by DNA end resection (Aparicio et al., 2014; Ceccaldi et al., 2016; Ronato et al., 2020; Scully et al., 2019). While DNA end resection inhibits NHEJ, it is required for generating 3’ ssDNA essential for HR. Studies over the past decade has revealed the antagonistic role of BRCA1 and 53BP1 complex in DSB repair pathway choice through regulation of DNA end resection (Mirman & Lange, 2020; Panier & Boulton, 2014; Zimmermann & Lange, 2014). 53BP1 localizes to DSB during all phases of the cell cycle and is hyper-phosphorylated by ATM/ATR at 28 N-terminal SQ/TQ sites (Bothmer et al., 2011). Several downstream factors are recruited to DSB by phosphorylated 53BP1 (Mirman & Lange, 2020; Shibata & Jeggo, 2020). These include PTIP/Artemis (Callen et al., 2013; Munoz et al., 2007; Wang et al., 2014) and RIF1/Shieldin complex/CST complex (Boersma et al., 2015; Chapman et al., 2013; Dev et al., 2018; Escribano-Diaz et al., 2013; Findlay et al., 2018; Gao et al., 2018; Ghezraoui et al., 2018; Gupta et al., 2018; Mirman et al., 2018; Noordermeer et al., 2018; Tomida et al., 2018; Virgilio et al., 2013; Xu et al., 2015; Zimmermann et al., 2013). DYNLL1 and ATMIN also associate with 53BP1 at DSB in a phosphorylation-independent manner (Becker et al., 2018; He et al., 2018; West et al., 2019). Members of the 53BP1 complex are not core components of NHEJ machinery. Instead, they promote NHEJ by suppressing HR through blocking DNA end resection or blunting the resected DSB (Mirman & Lange, 2020; Shibata & Jeggo, 2020). A recent study suggests that different downstream effectors of 53BP1 suppress different nucleases in DNA end resection. While RIF1/Shieldin complex suppresses the activity of EXO1, PTIP/Artemis suppresses the activity of DNA2/BLM (Callen et al., 2020).
Fig. 2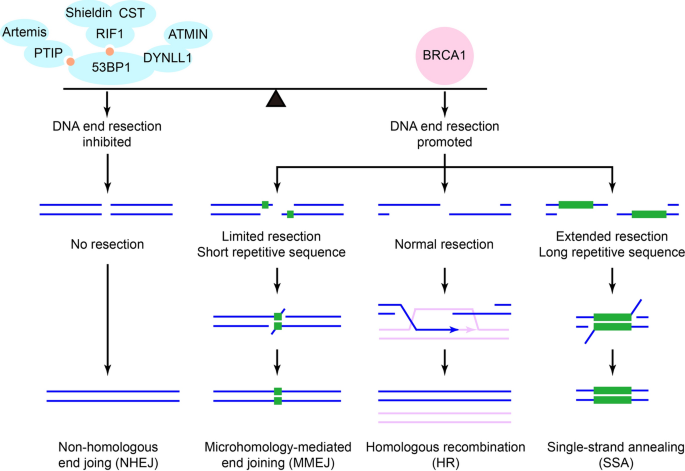
Antagonism between BRCA1 and 53BP1 in DSB repair pathway choice. In S/G2 phase, DSB can be repaired by either accurate homologous recombination (HR) or error-prone pathways including non-homologous end joining (NHEJ), microhomology-mediated end joining (MMEJ), and single-strand annealing (SSA). The DSB repair pathway choice is mainly determined by DNA end resection. 53BP1 recruits PTIP/Artemis and RIF1/Shieldin complex/CST complex through its N-terminal phosphorylation and recruits DYNLL1 and ATMIN in a phosphorylation-independent manner. Members of the 53BP1 complex block DNA end resection and promote NHEJ. BRCA1 antagonizes 53BP1 complex and promotes DNA end resection, but the mechanism remains elusive
Full size imageOnce recruited to DSB in S/G2 phase, BRCA1 promotes DSB repair pathway choice towards HR by antagonizing 53BP1 complex’s block on DNA end resection (Densham & Morris, 2019). This is achieved in part by repelling 53BP1 complex from the center of DSB. Although 53BP1 foci can still be observed, it relocates from the center to the periphery of DSB in S/G2 phase (Chapman et al., 2012). More strikingly, the foci of RIF1 (and likely its downstream effector Shieldin complex/CST complex) can barely be observed in S/G2 phase (Escribano-Diaz et al., 2013). Since PTIP is downstream of 53BP1, it is likely that PTIP foci are also repelled by BRCA1 in S/G2 phase. Supporting the idea that BRCA1 actively repels 53BP1 complex from DSB, BRCA1 depletion significantly increases RIF1’s foci and restores 53BP1’s foci at the center of DSB in S/G2 phase (Escribano-Diaz et al., 2013).
It remains elusive how BRCA1 antagonizes 53BP1 complex and promotes DNA end resection. Nearly half of BRCA1 protein is encoded by exon 11 in both human and mice. Loss of this region in Brca1Δ11/Δ11 mice results in defective DNA end resection, compromised HR repair, and embryonic lethality (Bunting et al., 2010; Shen et al., 1998; Xu et al., 1999). A recent study reports that part of this region is required for BRCA1’s function in repelling 53BP1 complex from DSB and promoting DNA end resection (Nacson et al., 2020). However, no specific motifs have been identified in this region and the mechanism requires further investigation.
As a coordinator for DNA end resection, CtIP promotes both MRN-dependent short-range resection and DNA2-mediated long-range resection (Ceppi et al., 2020; Daley et al., 2017). BRCA1 binds phosphorylated CtIP at S327 and regulates its recruitment to DSB (Yu et al., 2006). An early study reports that CtIP is also required for repelling RIF1 foci in S/G2 phase, but CtIP S327A mutant that is unable to bind BRCA1 fails to do so (Escribano-Diaz et al., 2013). These observations lead to an appealing hypothesis that CtIP cooperates with BRCA1 in counteracting 53BP1 complex’s block on DNA end resection. However, several lines of evidence argue against it. In chicken DT40 cells, CtIP S332A mutant with disrupted BRCA1 binding has no defect in HR repair (Nakamura et al., 2010). In mice, 53bp1 KO can rescue the embryonic lethality of Brca1 KO but not Ctip KO (Polato et al., 2014). Importantly, CtipS326A/S326A mice carrying corresponding human S327A mutation are viable and have no developmental defect, and CtipS326A/S326A mouse embryonic fibroblasts (MEFs) are also proficient in HR (Reczek et al., 2013). These studies suggest that despite the importance of BRCA1 and CTIP per se, their interaction is dispensable for DNA end resection and HR repair. It remains to be clarified how CtIP counteracts 53BP1 complex’s block on DNA end resection.
The RING domains of BRCA1 and BARD1 dimerize to form an E3 ubiquitin ligase (Brzovic et al., 2003; Ruffner et al., 2001; Wu et al., 1996), which has been shown to ubiquitinate several substrates, especially histone H2A on lysine 125, 127, and 129 (Kalb et al., 2014; Stewart et al., 2018; Witus et al., 2021). However, its activity in promoting DNA end resection and HR repair is still controversial. Using BRCA1 I26A mutant and BARD1 R99E mutant that disrupt the E3 ubiquitin ligase activity without affecting dimerization, one study reports that BRCA1–BARD1’s E3 ubiquitin ligase activity is required for promoting DNA end resection, HR pair, and PARPi resistance (Densham et al., 2016). This group further demonstrates that chromatin remodeler SMARCAD1 binds ubiquitinated H2A and repels 53BP1 complex, offering a possible link between BRCA1–BARD1’s E3 ubiquitin ligase activity and 53BP1 counteraction (Densham et al., 2016). However, opposite results are obtained in several other studies. An early study shows that BRCA1 I26A mutant can still inhibit RIF1 foci in S/G2 phase (Escribano-Diaz et al., 2013), which is consistent with the finding that I26A mutation in mice has no impact on HR repair, genomic stability, mouse viability, or cancer susceptibility (Shakya et al., 2011). One study also shows that BARD1 R99E can fully restore PARPi resistance in BARD1-depeletd cells (Nakamura et al., 2019). A more recent study reveals that Brca1Δ2/Δ2 MEFs, which express a RING-less BRCA1 that cannot dimerize with BARD1, still exhibit robust DNA end resection activity (Callen et al., 2020). The discrepancy between these studies prompts further investigation into the contribution of BRCA1–BARD1’s E3 ubiquitin ligase activity in DNA end resection and HR repair.
Taken together, BRCA1 promotes DSB repair pathway choice towards HR in S/G2 phase by actively repelling 53BP1 complex’s from DSB and lifting the block on DNA end resection (Fig. 2). How BRCA1 executes its function in this process is still not clear despite that several domains are potentially involved. Further studies are needed to reveal the underlying mechanisms. It is reported recently that H2AK15Ub, which is recognized by both 53BP1 and BRCA1–BARD1 complex, can be phosphorylated at T12 of ubiquitin (Walser et al., 2020). H2AK15pUbT12 is inhibitory to 53BP1 binding but is permissive for BRCA1–BARD1 complex binding (Walser et al., 2020). It remains to be studied if this modification regulates BRCA1’s function in repelling 53BP1 complex’s from DSB.
BRCA1 facilitates the recruitment of RAD51 to DSB
DNA end resection produces 3’ ssDNA that is immediately bound and protected by RPA complex, which is subsequently replaced by RAD51 recombinase. The loading of RAD51 to DSB and RPA complex replacement is critical for the formation of RAD51-ssDNA presynaptic filaments that mediate homologous DNA pairing and strand invasion (Wright et al., 2018). BRCA2, together with DSS1, directly binds RAD51 and promotes the formation of RAD51-ssDNA presynaptic filaments (Daley et al., 2014; Holloman, 2011). As a major partner of BRCA2, PALB2 is required for the localization of BRCA2/RAD51 at DSB (Xia et al., 2006).
It has been known for a long time that BRCA1 interacts with BRCA2 and is required for the localization of RAD51 at DSB (Bhattacharyya et al., 2000; Chen et al., 1998). However, the underlying mechanism remains elusive until the identification of BRCA1–PALB2 interaction, which is mediated by the coiled-coil (CC) domains on BRCA1 and PALB2, respectively (Sy et al., 2009; Zhang, Fan, et al., 2009; Zhang, Ma, et al., 2009). Loss of PALB2 does not affect the localization of BRCA1 at DSB. On the contrary, loss of BRCA1 or disrupting its interaction with PALB2 by cancer-associated CC domain mutations compromises PALB2, BRCA2, and RAD51 foci at DSB (Sy et al., 2009; Zhang, Fan, et al., 2009; Zhang, Ma, et al., 2009). Therefore, BRCA1 functions upstream of PALB2 in loading BRCA2/RAD51 to DSB. Interestingly, BRCA1–PALB2 interaction occurs only in S/G2 phase but is abolished in G1 (Orthwein et al., 2015). This is because CUL3/RBX1/KEAP1 E3 ubiquitin ligase ubiquitinates PALB2 at its CC domain and prevents the recognition by BRCA1 in G1 phase, which is counteracted by USP11, a deubiquitylase that is more stable and specifically interacts with PALB2 in S/G2 phase (Orthwein et al., 2015). Therefore, in addition to the block on DNA end resection, disassembly of BRCA1–PALB2 complex and inhibition of BRCA2/RAD51 loading to DSB provide another layer of control to suppress HR in G1. Recently, it is reported that human BRCA1Δ11q protein, an equivalent to mouse BRCA1Δ11 protein with intact CC domain, interacts with PALB2 with reduced efficiency, suggesting that there is additional regulation of BRCA1–PALB2 interaction yet to be revealed (Zong et al., 2019).
To investigate the physiological significance of BRCA1–PALB2 interaction, mice carrying mutations on their CC domains have been generated. Palb2CC6/CC6 mice have three amino acids on CC domains of PALB2 mutated to alanine so that PALB2–BRCA1 interaction is disrupted. Unlike Palb2 KO mice that are embryonic lethal, Palb2CC6/CC6 mice are viable (Simhadri et al., 2014). No obvious developmental defects are identified in these mice except for reduced male fertility. DNA damage-induced RAD51 foci are decreased in Palb2CC6/CC6 B cells but are not abolished as in Palb2 KO cells. Consistently, Palb2CC6/CC6 B cells have moderate elevation of chromosome aberrations and cell death after mitomycin treatment (Simhadri et al., 2014). These observations suggest that disrupting BRCA1-interacting motifs on PALB2 mildly compromises RAD51 loading and HR repair.
Two mice carrying mutations on BRCA1 CC domains that disrupts PALB2 binding have been generated recently. One carries three amino acids deletions (Brca1CC/CC) and the other one carries L1363P mutation that mimics the cancer-associated L1407P mutation in human (Brca1LP/LP) (Nacson et al., 2020; Park et al., 2020). Interestingly, both Brca1CC/CC and Brca1LP/LP mice have similar phenotypes that are much stronger than Palb2CC6/CC6 mice. Both of them are viable but have Fanconi anemia (FA)-like developmental defects, including decreased body size and weight, reduction of blood cellularity, bone marrow failure, reduced hematopoietic stem cells, and infertility. In addition, both mice succumb to death due to acute T-lymphoblastic leukemia within 4 months. Mechanistically, mutations on BRCA1 CC domain seem to have a stronger impact on RAD51 loading and HR repair than the CC6 mutation on PALB2 CC domain (Nacson et al., 2020; Park et al., 2020; Simhadri et al., 2014). IR-induced RAD51 foci are undetectable in Brca1CC/CC cells and is severely impaired in Brca1LP/LP cells. Further studies are needed to clarify why mutations on CC domains of BRCA1 and PALB2 have such dramatic difference at cellular and organismal levels.
BRCA1 promotes RAD51-mediated homologous DNA pairing
After RAD51-ssDNA presynaptic filaments are formed, RAD51 mediates template DNA engagement, homolog searching, and limited homologous DNA pairing to assemble the synaptic complex. RAD51 further promotes strand invasion and extensive homologous DNA pairing to generate post-synaptic complex that contains a DNA structure known as the displacement loop (D-loop). The D-loop is then extended by DNA synthesis and subsequently resolved by different mechanisms to complete repair (Daley et al., 2014; Holloman, 2011). The regulation of RAD51’s activity is poorly understood.
Interestingly, BRCA1–BARD1 complex promotes this process. Both BRCA1 and BARD1 can bind DNA, especially the D-loop structure. Both of them interact with RAD51 directly as well (Zhao et al., 2017). In vitro assay reveals that BRCA1–BARD1 complex enhances RAD51’s activity in strand invasion to form D-loop, which is likely achieved by promoting the assembly of synaptic complex (Zhao et al., 2017). Mutating key residues for RAD51 binding on BRCA1–BARD1 complex not only disrupts its ability to enhance RAD51’s activity in synaptic complex assembly and D-loop formation in vitro, but also compromises HR repair without affecting RAD51 loading at DSB in vivo (Zhao et al., 2017). Therefore, BRCA1 regulates HR repair after RAD51-ssDNA presynaptic filament formation. Interestingly, the interaction between BRCA1–BARD1 complex and RAD51 is enhanced by BRCA1 S114 phosphorylation and its isomerization by phosphorylation-directed prolyl isomerase PIN1 (Daza-Martin et al., 2019). It seems that the enhanced interaction specifically contributes to stalled replication fork protection, but its potential role in synaptic complex assembly and D-loop formation is worth further investigation.
Rescue of HR repair defects caused by BRCA1 deficiency
As discussed above, BRCA1 functions at multiple stages of HR in DSB repair. Consistently, cancer cells carrying disruptive mutations on BRCA1 gene have severe HR deficiency. These cells can be effectively killed by PARPi initially, but resistance always develops eventually. Multiple mechanisms contribute to PARPi resistance, and some of them are due to the restoration of HR without rescuing the production of functional BRCA1 protein (Noordermeer & Attikum, 2019). Clarifying how HR can be restored in BRCA1 mutant cancer cells will reveal clues for targeting PARPi resistance. Therefore, great efforts have been invested to study the mechanisms that restores HR in BRCA1 mutant cancer cells.
A major breakthrough in this aspect originates from the study of embryonic development of Brca1 mutant mice. Similar to knockout mice of other key HR proteins, Brca1 knockout and most Brca1 mutant mice are embryonic lethal (Liu & Lu, 2020). Early studies show that loss of p53 signaling by p53, Chk2, or Atm KO fully rescues the lethality of Brca1Δ11/Δ11 mice, but HR defect is not rescued (Cao et al., 2006; Xu et al., 1999). Interestingly, loss of another DNA damage response protein 53BP1 fully rescues embryonic lethality and HR defects of Brca1Δ11/Δ11 mice (Bunting et al., 2010; Cao et al., 2009). Soon after these seminal discoveries, it is found that 53bp1 KO also fully rescues embryonic lethality and HR defects of Brca1Δ2/Δ2 mice, which were thought to be Brca1 null mice at that time (Bunting et al., 2012).
Accompanying the studies in mice, several groups have demonstrated that 53BP1 depletion rescues the HR defects in BRCA1 mutant ES cells, cancer cell lines, or mouse tumors (Bouwman et al., 2010; Grotsky et al., 2013; Jaspers et al., 2013). Extensive studies have been carried out to examine factors that function together with 53BP1. As mentioned above, many factors downstream of 53BP1 have been identified, including PTIP/Artemis, RIF1/Shieldin complex/CST complex, DYNLL1, and ATMIN. Indeed, loss of these factors rescues the HR deficiency and leads to PARPi resistance in BRCA1 mutant cells (Becker et al., 2018; Boersma et al., 2015; Chapman et al., 2013; Dev et al., 2018; Escribano-Diaz et al., 2013; Findlay et al., 2018; Gao et al., 2018; Ghezraoui et al., 2018; Gupta et al., 2018; He et al., 2018; Mirman et al., 2018; Noordermeer et al., 2018; Tomida et al., 2018; Virgilio et al., 2013; West et al., 2019; Xu et al., 2015; Zimmermann et al., 2013). Based on these studies, it is well received that loss of 53BP1 or its associated members rescues HR defects caused by BRCA1 deficiency (Mirman & Lange, 2020; Panier & Boulton, 2014; Zimmermann & Lange, 2014), and the underlying mechanism is the antagonism between BRCA1 and 53BP1 in DSB repair pathway choice (Aparicio et al., 2014; Ceccaldi et al., 2016; Ronato et al., 2020). In S/G2 phase, BRCA1 counteracts 53BP1’s block on DNA end resection and directs DSB repair pathway choice towards HR. As a consequence of defective DNA end resection, BRCA1 mutant cells have HR deficiency and are sensitive to PARPi. 53BP1 depletion removes the barrier on DNA end resection, circumventing the requirement for BRCA1 in this process, so that DSB repair pathway choice is directed to HR in the absence of BRCA1. In line with this mechanism, it is generally believed that 53BP1 depletion restores PARPi resistance in BRCA1-deficient cells (Mirman & Lange, 2020; Panier & Boulton, 2014; Zimmermann & Lange, 2014).
It is worth noting that 53BP1 loss does not always restore HR in BRCA1-deficient cells. This is likely due to the fact that BRCA1 functions at multiple steps of HR, which is often overlooked recently. As discussed above, BRCA1 not only counteracts 53BP1’s block on DNA end resection, but also functions downstream of DNA end resection to promote the assembly of RAD51-ssDNA presynaptic filament and synaptic complex, both of which are important steps of HR. Theoretically, 53BP1 loss can only restore HR in cells expressing a BRCA1 mutant that retains the functions downstream of DNA end resection. Early studies show that 53bp1 KO fully rescues embryonic lethality and HR defects of Brca1Δ11/Δ11 mice (Bunting et al., 2010; Cao et al., 2009). BRCA1Δ11 protein still contains all known functional domains and can dimerize with BARD1 through its RING domain, interact with PALB2 through its CC domain, and localize to DSB through its BRCT domains. Therefore, BRCA1Δ11 is a hypomorphic protein that is defective for DNA end resection but retains most functions of BRCA1. 53bp1 KO also fully rescues embryonic lethality and HR defects of Brca1Δ2/Δ2 mice (Bunting et al., 2012). Originally thought to be a null allele, Brca1Δ2 allele actually produces a RING-less BRCA1 protein, which fails to dimerize with BARD1 but retains CC domain to bind PALB2 and BRCT domains to localize to DSB (Li et al., 2016). On the contrary, recent studies report that 53bp1 KO rescues embryonic lethality but not HR defects of Brca1Δ5-13/Δ5-13 mice and Brca1ΔC/ΔC mice, both of which are total Brca1 KO or close to Brca1 KO (Chen et al., 2020; Nacson et al., 2018). Therefore, a mutant BRCA1 that retains its ability to localize to DSB and to interact with PALB2 is a prerequisite for 53BP1 KO to restore HR in mice. Similarly, 53BP1 loss can only restore HR and PARPi resistance in cancer cells expressing hypomorphic BRCA1 proteins that interact and recruit PALB2 (Nacson et al., 2018).
The recruitment of 53BP1 to DSB requires histone ubiquitination, which is established by γH2AX–MDC1–RNF8–RNF168 signaling pathway (Mattiroli & Penengo, 2021; Uckelmann & Sixma, 2017). Interestingly, although H2ax KO, Rnf8 KO, or Rnf168 KO abolishes the recruitment of 53BP1 to DSB, none of them rescues the embryonic lethality of Brca1Δ11/Δ11 mice (Zong et al., 2019). Further characterization reveals that BRCA1Δ11 protein actually has reduced ability to interact with PALB2 (Zong et al., 2019), and the normal HR repair in Brca1Δ11/Δ11;53bp1−/− cells partially relies on RNF168, which binds PALB2’s WD40 domain and functions in parallel with BRCA1 in loading PALB2 to DSB (Luijsterburg et al., 2017). Although DNA end resection is rescued in Brca1Δ11/Δ11;H2ax−/−, Brca1Δ11/Δ11;Rnf8−/−, or Brca1Δ11/Δ11;Rnf168−/− cells, RNF168 is either absent or unable to localize to DSB to mediate PALB2 loading so that BRCA1Δ11 protein alone is not sufficient for PALB2 loading. Similarly, in addition to inhibiting EXO1 in DNA end resection, RIF1/Shieldin complex also blocks RNF168-mediated PALB2 loading (Callen et al., 2020; Dev et al., 2018). 53BP1 S25A mutation abolishes the recruitment of PTIP, releases the block on DNA2, and restores DNA end resection (Callen et al., 2020). However, 53BP1 S25A protein can still recruit RIF1/Shieldin complex and block RNF168-mediated PALB2 loading. As a consequence, unlike 53bp1 KO, 53bp1S25A/S25A rescues embryonic lethality but fails to rescue HR of Brca1Δ11/Δ11 mice due to defective PALB2 loading, and Brca1Δ11/Δ11;53bp1S25A/S25A mice have premature aging phenotype (Callen et al., 2020). On the contrary, inactivating Shieldin complex by Shld1 KO or Shld2 KO lifts the block on both DNA end resection and RNF168-mediated PALB2 loading. Therefore, Shld1 KO or Shld2 KO rescues embryonic lethality of Brca1Δ11/Δ11 mice and no obvious abnormalities are reported for Brca1Δ11/Δ11;Shld1−/− or Brca1Δ11/Δ11;Shld2−/− mice, suggesting that HR is likely restored (Ling et al., 2020; Shinoda et al., 2021). In line with these observations, Rnf168 KO rescues both embryonic lethality and HR of Brca1Δ2/Δ2 mice, which produce BRCA1ΔRING protein that efficiently interacts with PALB2 (Zong et al., 2019). Collectively, a BRCA1 mutant with full capacity of PALB2 loading is required for Rnf168 KO to rescue embryonic lethality and HR. It will be interesting to see if 53bp1S25A/S25A can rescue embryonic lethality and HR of Brca1Δ2/Δ2 mice. It should be noted that RNF168 alone is not sufficient for PALB2 loading, since HR defects are present in Brca1Δ5−13/Δ5−13;53bp1−/− and Brca1ΔC/ΔC;53bp1−/− cells, in which BRCA1-mediated PALB2 loading is completely abolished while RNF168-mediated PALB2 loading is retained (Chen et al., 2020; Nacson et al., 2018). Taken together, the capability of PALB2 loading in BRCA1-deficient cells determines if HR can be rescued by the loss of 53BP1 complex or its upstream regulators.
Perspectives
As a key player of HR in DSB repair, BRCA1 functions at different stages of HR (Fig. 3). Once localized to DSB in S/G2 phase, BRCA1 directs DSB repair pathway choice to HR, facilitates the recruitment of RAD51 to DSB, and promotes RAD51-mediated homologous DNA pairing. Although significant advances have been made in revealing BRCA1’s role in DSB repair pathway choice in the past decade, all functions of BRCA1 mentioned above are equally important so that loss of either of them compromises HR. Therefore, the functions of BRCA1 downstream of DSB repair pathway choice should not be overlooked. Mechanistic studies are still needed to reveal how BRCA1 executes it functions in all stages of HR.
Fig. 3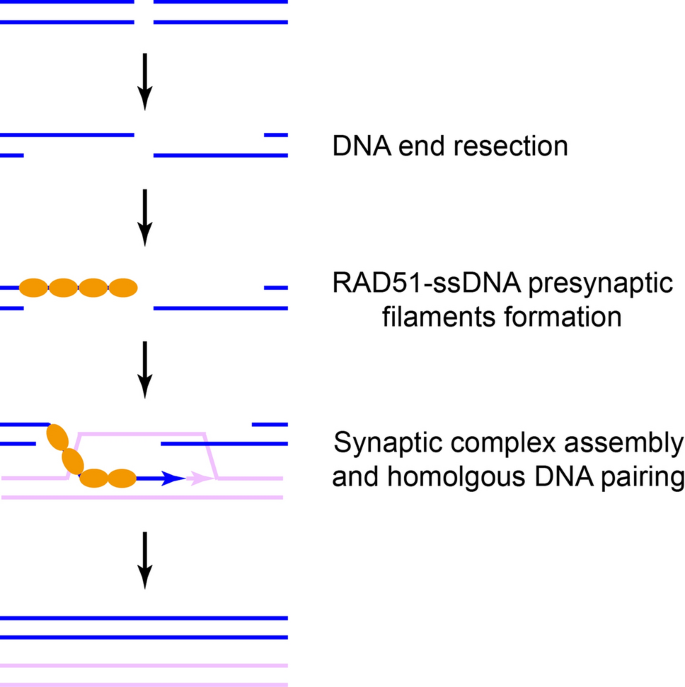
BRCA1 functions at different stages of HR. In S/G2 phase of the cell cycle, BRCA1 promotes DNA end resection and directs DSB repair pathway choice to HR. BRCA1 facilitates the recruitment of RAD51 to DSB to form RAD51-ssDNA presynaptic filaments. BRCA1 also promotes RAD51-mediated homologous DNA pairing by enhancing RAD51’s activity in synaptic complex assembly
Full size imageDespite being a great threat to genomic stability, programmed DSB are generated in meiotic prophase, which are repaired by HR exclusively to promote the obligate crossover between homologous chromosomes (Gray & Cohen, 2016). There are some unique features that distinguish DSB repair in meiotic prophase from that in somatic cells (Phadnis et al., 2011). Unlike in Brca1Δ11/Δ11 somatic cells, the recruitment of RAD51 and its meiosis-specific ortholog DMC1 to meiotic DSB is largely unaffected in Brca1Δ11/Δ11 mice, which leads to the hypothesis that BRCA1 is dispensable for DSB repair in meiotic prophase (Broering et al., 2014; Xu et al., 2003). However, since Brca1Δ11/Δ11 mice express hypomorphic BRCA1 that still retains the majority of its functions, further studies are need to clarify the role of BRCA1 in programmed DSB in meiotic prophase.
References
Aparicio, T., Baer, R., & Gautier, J. (2014). DNA double-strand break repair pathway choice and cancer. DNA Repair, 19, 169–175.
Becker, J. R., Bonnet, C., Clifford, G., Groth, A., Wilson, M. D., & Chapman, J. R. (2020). BARD1 links histone H2A Lysine-15 ubiquitination to initiation of BRCA1-dependent homologous recombination. bioRxiv. https://doi.org/10.1101/2020.06.01.127951
Becker, J. R., Cuella-Martin, R., Barazas, M., Liu, R., Oliveira, C., Oliver, A. W., Bilham, K., Holt, A. B., Blackford, A. N., Heierhorst, J., et al. (2018). The ASCIZ-DYNLL1 axis promotes 53BP1-dependent non-homologous end joining and PARP inhibitor sensitivity. Nature Communications, 9, 5406.
Bhattacharyya, A., Ear, U. S., Koller, B. H., Weichselbaum, R. R., & Bishop, D. K. (2000). The breast cancer susceptibility gene BRCA1 is required for subnuclear assembly of Rad51 and survival following treatment with the DNA cross-linking agent cisplatin. Journal of Biological Chemistry, 275, 23899–23903.
Boersma, V., Moatti, N., Segura-Bayona, S., Peuscher, M. H., van der Torre, J., Wevers, B. A., Orthwein, A., Durocher, D., & Jacobs, J. J. L. (2015). MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5’ end resection. Nature, 521, 537–540.
Bothmer, A., Robbiani, D. F., Di Virgilio, M., Bunting, S. F., Klein, I. A., Feldhahn, N., Barlow, J., Chen, H. T., Bosque, D., Callen, E., et al. (2011). Regulation of DNA end joining, resection, and immunoglobulin class switch recombination by 53BP1. Molecular Cell, 42, 319–329.
Bouwman, P., Aly, A., Escandell, J. M., Pieterse, M., Bartkova, J., van der Gulden, H., Hiddingh, S., Thanasoula, M., Kulkarni, A., Yang, Q., et al. (2010). 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nature Structural and Molecular Biology, 17, 688–695.
Broering, T. J., Alavattam, K. G., Sadreyev, R. I., Ichijima, Y., Kato, Y., Hasegawa, K., Camerini-Otero, R. D., Lee, J. T., Andreassen, P. R., & Namekawa, S. H. (2014). BRCA1 establishes DNA damage signaling and pericentric heterochromatin of the X chromosome in male meiosis. Journal of Cell Biology, 205, 663–675.
Brzovic, P. S., Keeffe, J. R., Nishikawa, H., Miyamoto, K., Fox, D., 3rd., Fukuda, M., Ohta, T., & Klevit, R. (2003). Binding and recognition in the assembly of an active BRCA1/BARD1 ubiquitin-ligase complex. Proceedings of the National Academy of Sciences of the United States of America, 100, 5646–5651.
Bunting, S. F., Callen, E., Kozak, M. L., Kim, J. M., Wong, N., Lopez-Contreras, A. J., Ludwig, T., Baer, R., Faryabi, R. B., Malhowski, A., et al. (2012). BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Molecular Cell, 46, 125–135.
Bunting, S. F., Callen, E., Wong, N., Chen, H. T., Polato, F., Gunn, A., Bothmer, A., Feldhahn, N., Fernandez-Capetillo, O., Cao, L., et al. (2010). 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell, 141, 243–254.
Callen, E., Di Virgilio, M., Kruhlak, M. J., Nieto-Soler, M., Wong, N., Chen, H. T., Faryabi, R. B., Polato, F., Santos, M., Starnes, L. M., et al. (2013). 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell, 153, 1266–1280.
Callen, E., Zong, D., Wu, W., Wong, N., Stanlie, A., Ishikawa, M., Pavani, R., Dumitrache, L. C., Byrum, A. K., Mendez-Dorantes, C., et al. (2020). 53BP1 enforces distinct pre- and post-resection blocks on homologous recombination. Molecular Cell, 77, 26–38.
Cao, L., Kim, S., Xiao, C., Wang, R. H., Coumoul, X., Wang, X., Li, W. M., Xu, X. L., De Soto, J. A., Takai, H., et al. (2006). ATM-Chk2-p53 activation prevents tumorigenesis at an expense of organ homeostasis upon Brca1 deficiency. EMBO Journal, 25, 2167–2177.
Cao, L., Xu, X., Bunting, S. F., Liu, J., Wang, R. H., Cao, L. L., Wu, J. J., Peng, T. N., Chen, J., Nussenzweig, A., et al. (2009). A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Molecular Cell, 35, 534–541.
Ceccaldi, R., Rondinelli, B., & D’Andrea, A. D. (2016). Repair pathway choices and consequences at the double-strand break. Trends in Cell Biology, 26, 52–64.
Ceppi, I., Howard, S. M., Kasaciunaite, K., Pinto, C., Anand, R., Seidel, R., & Cejka, P. (2020). CtIP promotes the motor activity of DNA2 to accelerate long-range DNA end resection. Proceedings of the National Academy of Sciences of the United States of America, 117, 8859–8869.
Chapman, J. R., Barral, P., Vannier, J. B., Borel, V., Steger, M., Tomas-Loba, A., Sartori, A. A., Adams, I. R., Batista, F. D., & Boulton, S. J. (2013). RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Molecular Cell, 49, 858–871.
Chapman, J. R., Sossick, A. J., Boulton, S. J., & Jackson, S. P. (2012). BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. Journal of Cell Science, 125, 3529–3534.
Chen, C. C., Feng, W., Lim, P. X., Kass, E. M., & Jasin, M. (2018). Homology-directed repair and the role of BRCA1, BRCA2, and related proteins in genome integrity and cancer. Annual Review of Cancer Biology, 2, 313–336.
Chen, J., Li, P., Song, L., Bai, L., Huen, M. S. Y., Liu, Y., & Lu, L. Y. (2020). 53BP1 loss rescues embryonic lethality but not genomic instability of BRCA1 total knockout mice. Cell Death and Differentiation, 27, 2552–2567.
Chen, J., Silver, D. P., Walpita, D., Cantor, S. B., Gazdar, A. F., Tomlinson, G., Couch, F. J., Weber, B. L., Ashley, T., Livingston, D. M., et al. (1998). Stable interaction between the products of the BRCA1 and BRCA2 tumor suppressor genes in mitotic and meiotic cells. Molecular Cell, 2, 317–328.
Curtin, N. J., & Szabo, C. (2020). Poly(ADP-ribose) polymerase inhibition: past, present and future. Nature Reviews. Drug Discovery, 19, 711–736.
Daley, J. M., Gaines, W. A., Kwon, Y., & Sung, P. (2014). Regulation of DNA pairing in homologous recombination. Cold Spring Harbor Perspectives Biology, 6, 017954.
Daley, J. M., Jimenez-Sainz, J., Wang, W., Miller, A. S., Xue, X., Nguyen, K. A., Jensen, R. B., & Sung, P. (2017). Enhancement of BLM-DNA2-mediated long-range DNA end resection by CtIP. Cell Reports, 21, 324–332.
Daza-Martin, M., Starowicz, K., Jamshad, M., Tye, S., Ronson, G. E., MacKay, H. L., Chauhan, A. S., Walker, A. K., Stone, H. R., Beesley, J. F. J., et al. (2019). Isomerization of BRCA1-BARD1 promotes replication fork protection. Nature, 571, 521–527.
Densham, R. M., Garvin, A. J., Stone, H. R., Strachan, J., Baldock, R. A., Daza-Martin, M., Fletcher, A., Blair-Reid, S., Beesley, J., Johal, B., et al. (2016). Human BRCA1-BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nature Structural and Molecular Biology, 23, 647–655.
Densham, R. M., & Morris, J. R. (2019). Moving mountains: The BRCA1 promotion of DNA resection. Frontiers in Molecular Biosciences, 6, 79.
Dev, H., Chiang, T. W., Lescale, C., de Krijger, I., Martin, A. G., Pilger, D., Coates, J., Sczaniecka-Clift, M., Wei, W., Ostermaier, M., et al. (2018). Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nature Cell Biology, 20, 954–965.
Di Virgilio, M., Callen, E., Yamane, A., Zhang, W., Jankovic, M., Gitlin, A. D., Feldhahn, N., Resch, W., Oliveira, T. Y., Chait, B. T., et al. (2013). Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science, 339, 711–715.
Doil, C., Mailand, N., Bekker-Jensen, S., Menard, P., Larsen, D. H., Pepperkok, R., Ellenberg, J., Panier, S., Durocher, D., Bartek, J., et al. (2009). RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell, 136, 435–446.
Escribano-Diaz, C., Orthwein, A., Fradet-Turcotte, A., Xing, M., Young, J. T., Tkac, J., Cook, M. A., Rosebrock, A. P., Munro, M., Canny, M. D., et al. (2013). A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Molecular Cell, 49, 872–883.
Findlay, S., Heath, J., Luo, V. M., Malina, A., Morin, T., Coulombe, Y., Djerir, B., Li, Z., Samiei, A., Simo-Cheyou, E., et al. (2018). SHLD2/FAM35A co-operates with REV7 to coordinate DNA double-strand break repair pathway choice. EMBO Journal, 37, e100158.
Gao, S., Feng, S., Ning, S., Liu, J., Zhao, H., Xu, Y., Shang, J., Li, K., Li, Q., Guo, R., et al. (2018). An OB-fold complex controls the repair pathways for DNA double-strand breaks. Nature Communications, 9, 3925.
Ghezraoui, H., Oliveira, C., Becker, J. R., Bilham, K., Moralli, D., Anzilotti, C., Fischer, R., Deobagkar-Lele, M., Sanchiz-Calvo, M., Fueyo-Marcos, E., et al. (2018). 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature, 560, 122–127.
Gray, S., & Cohen, P. E. (2016). Control of meiotic crossovers: From double-strand break formation to designation. Annual Review of Genetics, 50, 175–210.
Grotsky, D. A., Gonzalez-Suarez, I., Novell, A., Neumann, M. A., Yaddanapudi, S. C., Croke, M., Martinez-Alonso, M., Redwood, A. B., Ortega-Martinez, S., Feng, Z., et al. (2013). BRCA1 loss activates cathepsin L-mediated degradation of 53BP1 in breast cancer cells. Journal of Cell Biology, 200, 187–202.
Gupta, R., Somyajit, K., Narita, T., Maskey, E., Stanlie, A., Kremer, M., Typas, D., Lammers, M., Mailand, N., Nussenzweig, A., et al. (2018). DNA repair network analysis reveals shieldin as a key regulator of NHEJ and PARP inhibitor sensitivity. Cell, 173, 972–988.
He, Y. J., Meghani, K., Caron, M. C., Yang, C., Ronato, D. A., Bian, J., Sharma, A., Moore, J., Niraj, J., Detappe, A., et al. (2018). DYNLL1 binds to MRE11 to limit DNA end resection in BRCA1-deficient cells. Nature, 563, 522–526.
Holloman, W. K. (2011). Unraveling the mechanism of BRCA2 in homologous recombination. Nature Structural and Molecular Biology, 18, 748–754.
Horn, V., Uckelmann, M., Zhang, H., Eerland, J., Aarsman, I., le Paige, U. B., Davidovich, C., Sixma, T. K., & van Ingen, H. (2019). Structural basis of specific H2A K13/K15 ubiquitination by RNF168. Nature Communications, 10, 1751.
Huen, M. S., Grant, R., Manke, I., Minn, K., Yu, X., Yaffe, M. B., & Chen, J. (2007). RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell, 131, 901–914.
Huen, M. S., Sy, S. M., & Chen, J. (2010). BRCA1 and its toolbox for the maintenance of genome integrity. Nature Reviews Molecular Cell Biology, 11, 138–148.
Hustedt, N., & Durocher, D. (2016). The control of DNA repair by the cell cycle. Nature Cell Biology, 19, 1–9.
Jaspers, J. E., Kersbergen, A., Boon, U., Sol, W., van Deemter, L., Zander, S. A., Drost, R., Wientjens, E., Ji, J., Aly, A., et al. (2013). Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discovery, 3, 68–81.
Johnson, N., Johnson, S. F., Yao, W., Li, Y. C., Choi, Y. E., Bernhardy, A. J., Wang, Y., Capelletti, M., Sarosiek, K. A., Moreau, L. A., et al. (2013). Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proceedings of the National Academy of Sciences of the United States of America, 110, 17041–17046.
Kalb, R., Mallery, D. L., Larkin, C., Huang, J. T., & Hiom, K. (2014). BRCA1 is a histone-H2A-specific ubiquitin ligase. Cell Reports, 8, 999–1005.
Kass, E. M., Moynahan, M. E., & Jasin, M. (2016). When Genome Maintenance Goes Badly Awry. Molecular Cell, 62, 777–787.
Kim, H., Chen, J., & Yu, X. (2007). Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science, 316, 1202–1205.
Kolas, N. K., Chapman, J. R., Nakada, S., Ylanko, J., Chahwan, R., Sweeney, F. D., Panier, S., Mendez, M., Wildenhain, J., Thomson, T. M., et al. (2007). Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science, 318, 1637–1640.
Li, M., Cole, F., Patel, D. S., Misenko, S. M., Her, J., Malhowski, A., Alhamza, A., Zheng, H., Baer, R., Ludwig, T., et al. (2016). 53BP1 ablation rescues genomic instability in mice expressing “RING-less” BRCA1. EMBO Reports, 17, 1532–1541.
Li, M. L., & Greenberg, R. A. (2012). Links between genome integrity and BRCA1 tumor suppression. Trends in Biochemical Sciences, 37, 418–424.
Ling, A. K., Munro, M., Chaudhary, N., Li, C., Berru, M., Wu, B., Durocher, D., & Martin, A. (2020). SHLD2 promotes class switch recombination by preventing inactivating deletions within the Igh locus. EMBO Reports, 21, e49823.
Liu, Y., & Lu, L. Y. (2020). BRCA1 and homologous recombination: implications from mouse embryonic development. Cell and Bioscience, 10, 49.
Lord, C. J., & Ashworth, A. (2017). PARP inhibitors: Synthetic lethality in the clinic. Science, 355, 1152–1158.
Luijsterburg, M. S., Typas, D., Caron, M. C., Wiegant, W. W., van den Heuvel, D., Boonen, R. A., Couturier, A. M., Mullenders, L. H., Masson, J. Y., & van Attikum, H. (2017). A PALB2-interacting domain in RNF168 couples homologous recombination to DNA break-induced chromatin ubiquitylation. eLife. https://doi.org/10.7554/eLife.20922
Mailand, N., Bekker-Jensen, S., Faustrup, H., Melander, F., Bartek, J., Lukas, C., & Lukas, J. (2007). RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell, 131, 887–900.
Manke, I. A., Lowery, D. M., Nguyen, A., & Yaffe, M. B. (2003). BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science, 302, 636–639.
Mattiroli, F., & Penengo, L. (2021). Histone ubiquitination: An integrative signaling platform in genome stability. Trends in Genetics. https://doi.org/10.1016/j.tig.2020.12.005
Mattiroli, F., Vissers, J. H., van Dijk, W. J., Ikpa, P., Citterio, E., Vermeulen, W., Marteijn, J. A., & Sixma, T. K. (2012). RNF168 ubiquitinates K13–15 on H2A/H2AX to drive DNA damage signaling. Cell, 150, 1182–1195.
Michl, J., Zimmer, J., & Tarsounas, M. (2016). Interplay between Fanconi anemia and homologous recombination pathways in genome integrity. EMBO Journal, 35, 909–923.
Mirman, Z., & de Lange, T. (2020). 53BP1: a DSB escort. Genes and Development, 34, 7–23.
Mirman, Z., Lottersberger, F., Takai, H., Kibe, T., Gong, Y., Takai, K., Bianchi, A., Zimmermann, M., Durocher, D., & de Lange, T. (2018). 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polalpha-dependent fill-in. Nature, 560, 112–116.
Munoz, I. M., Jowsey, P. A., Toth, R., & Rouse, J. (2007). Phospho-epitope binding by the BRCT domains of hPTIP controls multiple aspects of the cellular response to DNA damage. Nucleic Acids Research, 35, 5312–5322.
Nacson, J., Di Marcantonio, D., Wang, Y., Bernhardy, A. J., Clausen, E., Hua, X., Cai, K. Q., Martinez, E., Feng, W., Callen, E., et al. (2020). BRCA1 mutational complementation induces synthetic viability. Molecular Cell, 78, 951–959.
Nacson, J., Krais, J. J., Bernhardy, A. J., Clausen, E., Feng, W., Wang, Y., Nicolas, E., Cai, K. Q., Tricarico, R., Hua, X., et al. (2018). BRCA1 mutation-specific responses to 53BP1 loss-induced homologous recombination and PARP inhibitor resistance. Cell Reports, 24, 3513–3527.
Nakamura, K., Kogame, T., Oshiumi, H., Shinohara, A., Sumitomo, Y., Agama, K., Pommier, Y., Tsutsui, K. M., Tsutsui, K., Hartsuiker, E., et al. (2010). Collaborative action of Brca1 and CtIP in elimination of covalent modifications from double-strand breaks to facilitate subsequent break repair. PLoS Genetics, 6, 1000828.
Nakamura, K., Saredi, G., Becker, J. R., Foster, B. M., Nguyen, N. V., Beyer, T. E., Cesa, L. C., Faull, P. A., Lukauskas, S., Frimurer, T., et al. (2019). H4K20me0 recognition by BRCA1-BARD1 directs homologous recombination to sister chromatids. Nature Cell Biology, 21, 311–318.
Noordermeer, S. M., Adam, S., Setiaputra, D., Barazas, M., Pettitt, S. J., Ling, A. K., Olivieri, M., Alvarez-Quilon, A., Moatti, N., Zimmermann, M., et al. (2018). The shieldin complex mediates 53BP1-dependent DNA repair. Nature, 560, 117–121.
Noordermeer, S. M., & van Attikum, H. (2019). PARP inhibitor resistance: A tug-of-war in BRCA-mutated cells. Trends in Cell Biology, 29, 820–834.
Nowsheen, S., Aziz, K., Aziz, A., Deng, M., Qin, B., Luo, K., Jeganathan, K. B., Zhang, H., Liu, T., Yu, J., et al. (2018). L3MBTL2 orchestrates ubiquitin signalling by dictating the sequential recruitment of RNF8 and RNF168 after DNA damage. Nature Cell Biology, 20, 455–464.
Orthwein, A., Noordermeer, S. M., Wilson, M. D., Landry, S., Enchev, R. I., Sherker, A., Munro, M., Pinder, J., Salsman, J., Dellaire, G., et al. (2015). A mechanism for the suppression of homologous recombination in G1 cells. Nature, 528, 422–426.
Pandey, N., & Black, B. E. (2021). Rapid detection and signaling of DNA damage by PARP-1. Trends in Biochemical Sciences. https://doi.org/10.1016/j.tibs.2021.01.014
Panier, S., & Boulton, S. J. (2014). Double-strand break repair: 53BP1 comes into focus. Nature Reviews Molecular Cell Biology, 15, 7–18.
Park, D., Bergin, S. M., Jones, D., Ru, P., Koivisto, C. S., Jeon, Y. J., Sizemore, G. M., Kladney, R. D., Hadjis, A., Shakya, R., et al. (2020). Ablation of the Brca1-Palb2 interaction phenocopies fanconi anemia in mice. Cancer Research, 80, 4172–4184.
Paull, T. T. (2019). RNA-DNA hybrids and the convergence with DNA repair. Critical Reviews in Biochemistry and Molecular Biology, 54, 371–384.
Phadnis, N., Hyppa, R. W., & Smith, G. R. (2011). New and old ways to control meiotic recombination. Trends in Genetics, 27, 411–421.
Polato, F., Callen, E., Wong, N., Faryabi, R., Bunting, S., Chen, H. T., Kozak, M., Kruhlak, M. J., Reczek, C. R., Lee, W. H., et al. (2014). CtIP-mediated resection is essential for viability and can operate independently of BRCA1. Journal of Experimental Medicine, 211, 1027–1036.
Reczek, C. R., Szabolcs, M., Stark, J. M., Ludwig, T., & Baer, R. (2013). The interaction between CtIP and BRCA1 is not essential for resection-mediated DNA repair or tumor suppression. Journal of Cell Biology, 201, 693–707.
Ronato, D. A., Mersaoui, S. Y., Busatto, F. F., Affar, E. B., Richard, S., & Masson, J. Y. (2020). Limiting the DNA double-strand break resectosome for genome protection. Trends in Biochemical Sciences, 45, 779–793.
Ruffner, H., Joazeiro, C. A., Hemmati, D., Hunter, T., & Verma, I. M. (2001). Cancer-predisposing mutations within the RING domain of BRCA1: Loss of ubiquitin protein ligase activity and protection from radiation hypersensitivity. Proceedings of the National Academy of Sciences of the United States of America, 98, 5134–5139.
Saredi, G., Huang, H., Hammond, C. M., Alabert, C., Bekker-Jensen, S., Forne, I., Reveron-Gomez, N., Foster, B. M., Mlejnkova, L., Bartke, T., et al. (2016). H4K20me0 marks post-replicative chromatin and recruits the TONSL-MMS22L DNA repair complex. Nature, 534, 714–718.
Scully, R., Panday, A., Elango, R., & Willis, N. A. (2019). DNA double-strand break repair-pathway choice in somatic mammalian cells. Nature Reviews Molecular Cell Biology, 20, 698–714.
Shakya, R., Reid, L. J., Reczek, C. R., Cole, F., Egli, D., Lin, C. S., deRooij, D. G., Hirsch, S., Ravi, K., Hicks, J. B., et al. (2011). BRCA1 tumor suppression depends on BRCT phosphoprotein binding, but not its E3 ligase activity. Science, 334, 525–528.
Shen, S. X., Weaver, Z., Xu, X., Li, C., Weinstein, M., Chen, L., Guan, X. Y., Ried, T., & Deng, C. X. (1998). A targeted disruption of the murine Brca1 gene causes gamma-irradiation hypersensitivity and genetic instability. Oncogene, 17, 3115–3124.
Shibata, A., & Jeggo, P. A. (2020). Roles for 53BP1 in the repair of radiation-induced DNA double strand breaks. DNA Repair, 93, 102915.
Shinoda, K., Zong, D., Callen, E., Wu, W., Dumitrache, L. C., Belinky, F., Chari, R., Wong, N., Ishikawa, M., Stanlie, A., et al. (2021). The dystonia gene THAP1 controls DNA double-strand break repair choice. Molecular Cell. https://doi.org/10.1016/j.molcel.2021.03.034
Simhadri, S., Peterson, S., Patel, D. S., Huo, Y., Cai, H., Bowman-Colin, C., Miller, S., Ludwig, T., Ganesan, S., Bhaumik, M., et al. (2014). Male fertility defect associated with disrupted BRCA1-PALB2 interaction in mice. Journal of Biological Chemistry, 289, 24617–24629.
Sobhian, B., Shao, G., Lilli, D. R., Culhane, A. C., Moreau, L. A., Xia, B., Livingston, D. M., & Greenberg, R. A. (2007). RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science, 316, 1198–1202.
Stewart, G. S., Panier, S., Townsend, K., Al-Hakim, A. K., Kolas, N. K., Miller, E. S., Nakada, S., Ylanko, J., Olivarius, S., Mendez, M., et al. (2009). The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell, 136, 420–434.
Stewart, M. D., Zelin, E., Dhall, A., Walsh, T., Upadhyay, E., Corn, J. E., Chatterjee, C., King, M. C., & Klevit, R. E. (2018). BARD1 is necessary for ubiquitylation of nucleosomal histone H2A and for transcriptional regulation of estrogen metabolism genes. Proceedings of the National Academy of Sciences of the United States of America, 115, 1316–1321.
Sy, S. M., Huen, M. S., & Chen, J. (2009). PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proceedings of the National Academy of Sciences of the United States of America, 106, 7155–7160.
Tarsounas, M., & Sung, P. (2020). The antitumorigenic roles of BRCA1-BARD1 in DNA repair and replication. Nature Reviews Molecular Cell Biology, 21, 284–299.
Thorslund, T., Ripplinger, A., Hoffmann, S., Wild, T., Uckelmann, M., Villumsen, B., Narita, T., Sixma, T. K., Choudhary, C., Bekker-Jensen, S., et al. (2015). Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature, 527, 389–393.
Tomida, J., Takata, K. I., Bhetawal, S., Person, M. D., Chao, H. P., Tang, D. G., & Wood, R. D. (2018). FAM35A associates with REV7 and modulates DNA damage responses of normal and BRCA1-defective cells. EMBO Journal, 37, e99543.
Uckelmann, M., & Sixma, T. K. (2017). Histone ubiquitination in the DNA damage response. DNA Repair, 56, 92–101.
Walser, F., Mulder, M. P. C., Bragantini, B., Burger, S., Gubser, T., Gatti, M., Botuyan, M. V., Villa, A., Altmeyer, M., Neri, D., et al. (2020). Ubiquitin phosphorylation at Thr12 modulates the DNA damage response. Molecular Cell, 80, 423–436.
Wang, B., & Elledge, S. J. (2007). Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proceedings of the National Academy of Sciences of the United States of America, 104, 20759–20763.
Wang, B., Matsuoka, S., Ballif, B. A., Zhang, D., Smogorzewska, A., Gygi, S. P., & Elledge, S. J. (2007). Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science, 316, 1194–1198.
Wang, J., Aroumougame, A., Lobrich, M., Li, Y., Chen, D., Chen, J., & Gong, Z. (2014). PTIP associates with Artemis to dictate DNA repair pathway choice. Genes and Development, 28, 2693–2698.
West, K. L., Kelliher, J. L., Xu, Z., An, L., Reed, M. R., Eoff, R. L., Wang, J., Huen, M. S. Y., & Leung, J. W. C. (2019). LC8/DYNLL1 is a 53BP1 effector and regulates checkpoint activation. Nucleic Acids Research, 47, 6236–6249.
Witus, S. R., Burrell, A. L., Farrell, D. P., Kang, J., Wang, M., Hansen, J. M., Pravat, A., Tuttle, L. M., Stewart, M. D., Brzovic, P. S., et al. (2021). BRCA1/BARD1 site-specific ubiquitylation of nucleosomal H2A is directed by BARD1. Nature Structural and Molecular Biology, 28, 268–277.
Wright, W. D., Shah, S. S., & Heyer, W. D. (2018). Homologous recombination and the repair of DNA double-strand breaks. Journal of Biological Chemistry, 293, 10524–10535.
Wu, L. C., Wang, Z. W., Tsan, J. T., Spillman, M. A., Phung, A., Xu, X. L., Yang, M. C., Hwang, L. Y., Bowcock, A. M., & Baer, R. (1996). Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nature Genetics, 14, 430–440.
Xia, B., Sheng, Q., Nakanishi, K., Ohashi, A., Wu, J., Christ, N., Liu, X., Jasin, M., Couch, F. J., & Livingston, D. M. (2006). Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Molecular Cell, 22, 719–729.
Xu, G., Chapman, J. R., Brandsma, I., Yuan, J., Mistrik, M., Bouwman, P., Bartkova, J., Gogola, E., Warmerdam, D., Barazas, M., et al. (2015). REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature, 521, 541–544.
Xu, X., Aprelikova, O., Moens, P., Deng, C. X., & Furth, P. A. (2003). Impaired meiotic DNA-damage repair and lack of crossing-over during spermatogenesis in BRCA1 full-length isoform deficient mice. Development, 130, 2001–2012.
Xu, X., Wagner, K. U., Larson, D., Weaver, Z., Li, C., Ried, T., Hennighausen, L., Wynshaw-Boris, A., & Deng, C. X. (1999). Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nature Genetics, 22, 37–43.
Yu, X., Chini, C. C., He, M., Mer, G., & Chen, J. (2003). The BRCT domain is a phospho-protein binding domain. Science, 302, 639–642.
Yu, X., Fu, S., Lai, M., Baer, R., & Chen, J. (2006). BRCA1 ubiquitinates its phosphorylation-dependent binding partner CtIP. Genes and Development, 20, 1721–1726.
Zhang, F., Fan, Q., Ren, K., & Andreassen, P. R. (2009). PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Molecular Cancer Research, 7, 1110–1118.
Zhang, F., Ma, J., Wu, J., Ye, L., Cai, H., Xia, B., & Yu, X. (2009). PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Current Biology, 19, 524–529.
Zhao, W., Steinfeld, J. B., Liang, F., Chen, X., Maranon, D. G., Jian Ma, C., Kwon, Y., Rao, T., Wang, W., Sheng, C., et al. (2017). BRCA1-BARD1 promotes RAD51-mediated homologous DNA pairing. Nature, 550, 360–365.
Zhao, W., Wiese, C., Kwon, Y., Hromas, R., & Sung, P. (2019). The BRCA tumor suppressor network in chromosome damage repair by homologous recombination. Annual Review of Biochemistry, 88, 221–245.
Zimmermann, M., & de Lange, T. (2014). 53BP1: pro choice in DNA repair. Trends in Cell Biology, 24, 108–117.
Zimmermann, M., Lottersberger, F., Buonomo, S. B., Sfeir, A., & de Lange, T. (2013). 53BP1 regulates DSB repair using Rif1 to control 5’ end resection. Science, 339, 700–704.
Zong, D., Adam, S., Wang, Y., Sasanuma, H., Callen, E., Murga, M., Day, A., Kruhlak, M. J., Wong, N., Munro, M., et al. (2019). BRCA1 haploinsufficiency is masked by RNF168-mediated chromatin ubiquitylation. Molecular Cell, 73, 1267–1281.
Acknowledgements
This work is funded by National Key R&D Program of China (2018YFC1004900 and 2016YFC1000600), National Natural Science Foundation of China (32070829, 81602263, and 81471494), and Zhejiang Natural Science Foundation (LZ21H040001 and LY19C050002).
Author information
Affiliations
Key Laboratory of Reproductive Genetics (Ministry of Education) and Women’s Reproductive Health Laboratory of Zhejiang Province, Women’s Hospital, Zhejiang University School of Medicine, Hangzhou, China
Yidan Liu & Lin-Yu Lu
Institute of Translational Medicine, Zhejiang University School of Medicine, Hangzhou, China
Lin-Yu Lu
Zhejiang University Cancer Center, Hangzhou, China
Lin-Yu Lu
Corresponding authors
Rights and permissions
About this article
Cite this article
Liu, Y., Lu, LY. BRCA1: a key player at multiple stages of homologous recombination in DNA double-strand break repair. GENOME INSTAB. DIS. 2, 164–174 (2021). https://doi.org/10.1007/s42764-021-00042-1
Received03 April 2021
Revised07 May 2021
Accepted20 May 2021
Published27 May 2021
Issue DateJune 2021
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
BRCA1
Homologous recombination
DNA double-strand break repair
DNA end resection
用户登录
还没有账号?
立即注册