






Regulation of Rothmund–Thomson syndrome protein RecQL4 functions in DNA replication by SIRT1-mediated deacetylation
Original Research Paper
Yuxia Yang, Wei Fan, Rong Wang, Rui Wang, Wei Gu & Jianyuan Luo
Genome Instability & Disease 2, 240–252 (2021)
Abstract
NAD dependent histone deacetylase SIRT1 has demonstrated involvement in the regulation of stress responses, cellular metabolism, and cell survival. SIRT1 overexpression has been demonstrated to induce G1 arrest, but its function in the cell cycle remains unclear. Here, we identified RecQL4 as a SIRT1 interacting protein through complex purification. RecQL4 is a member of the RecQ DNA helicase family involved in DNA replication, recombination, and repair. Mutations in the RECQL4 gene are responsible for Rothmund–Thomson syndrome (RTS), a severe autosomal recessive disorder causing premature aging and predisposition to cancers. RecQL4 can be acetylated by CBP at lysine 88. Transfection of wild-type RecQL4 into cells derived from an RTS patient can rescue cell proliferation, while a RecQL4 acetylation mutant severely impairs this function. We demonstrated that the acetylation of RecQL4 can regulate both DNA replication activity and the timing of replication firing by dynamically regulating its nuclear localization during the S phase. SIRT1 deacetylates RecQL4 both in vitro and vivo. The acetylation status of RecQL4 affects its loading to the chromatin during the S phase of the cell cycle, consequently affecting DNA replication initiation. Our findings provided new insights on the role of protein acetylation in regulating DNA replication initiation.
Introduction
Mutations in the RECQL4 gene are responsible for Rothmund–Thomson syndrome (RTS), a severe autosomal recessive genetic disorder causing premature aging and predisposition to specific cancers. RecQL4 is a member of a RecQ DNA helicase family involved in DNA replication, recombination, and repair (Bachrati & Hickson, 2003; Hickson, 2003; Kitao et al., 1998; Larizza et al., 2006; Vennos & James, 1995). Other members of the RecQ family include BLM and WRN, whose mutations also cause Bloom syndrome and Werner syndrome, respectively. RTS patients show diverse premature aging phenotypes and are very frequently associated with one characteristic type of cancer: osteosarcoma. Analysis of RTS cells has revealed the genomic instability, including an unusually high frequency of chromosomal rearrangements, trisomies, translocations, and deletions. These aberrations can even be of a different nature in different cells isolated from the same patient (Der Kaloustian et al., 1990; Orstavik et al., 1994; Ying et al., 1990).
Recent studies indicate that RecQL4 functions in different DNA metabolic processes (Croteau et al., 2012, 2014). RecQL4 has been found to function in the initiation of DNA replication with its N-Terminus, which shares homology with yeast DNA replication initiation factor Sld2, which is required for the recruitment of DNA polymerase α (Matsuno et al., 2006; Sangrithi et al., 2005). It is required for the association of MCM10 and CTF4 with replication origins in human cells (Im et al., 2015). Petkovic and colleagues report that after etoposide treatment RecQL4 nuclear foci coincide with the foci formed by Rad51, a crucial protein functioning in homologous recombination of the double strand break (DSB) repair pathway (Petkovic et al., 2005). It also participates in the non-homologous end joining repair pathway by interacting with the Ku complex (Shamanna et al., 2014). In addition, fibroblasts from RTS patients (RTS cells) are sensitive to ionizing radiation (Vennos & James, 1995) and Kumata and colleagues recently provide the evidence that RecQL4 participates in DSB repair in Xenopus egg extracts (Kumata et al., 2007). Woo and Werner report that RecQL4 plays a role in oxidative stress (Werner et al., 2006; Woo et al., 2006). Cells defective in RecQL4 escaped from the S-phase arrest following UV or HU treatment (Park et al., 2006). Duan et al. (2020) show that RECQL4 promotes repair of oxidative base lesion 8-oxoG through interaction with OGG1. Our previous studies also show that RecQL4 interacts with XPA and can facilitate UV-induced DNA repair (Fan & Luo, 2008). These data altogether suggest that RecQL4 is involved in normal DNA replication, distinct DNA repair processes, and cell cycle arrest.
Class III histone deacetylase SIRT1 plays an important role in regulating DNA metabolism. SIRT1 knockout mice display altered histone modification, impaired DNA damage response, and reduced ability to repair DNA damage (Wang et al., 2008). Yeast Sir2 translocates to the site of DNA double-strand breaks (DSBs) and functions in the non-homologous end joining (NHEJ) pathway (McAinsh et al., 1999). SIRT1 deacetylates Ku70 and enhances DNA repair capacity upon exposure to ionizing radiation (Jeong et al., 2007). SIRT1 facilitates DNA repair in cells by deacetylating WRN protein to regulate its enzymatic activities and cellular localization (Li et al., 2008). It also plays a positive role in repairing double-strand DNA breaks through maintaining NBS1 in a hypoacetylated state by deacetylation (Yuan et al., 2007). In response to oxidative stress and ionizing irradiation, SIRT1 dissociates from repetitive DNA foci and relocalizes to DNA breaks to promote DNA repair (Oberdoerffer et al., 2008). SIRT1 null MEF cells are found to be more sensitive than control MEF cells in response to UV irradiation, suggesting that SIRT1 may be involved in UV-induced DNA repair (Wang et al., 2008). Further studies found that SIRT1 facilitates NER pathway for UV-induced DNA repair by deacetylating XPA (Fan & Luo, 2010). SIRT1 also participates in regulating DNA replication by targeting TopBP1 (Liu et al., 2014; Wang et al., 2014) and indirectly modulating BER of 8-oxoG by deacetylating RECQL4 (Duan et al., 2020).
In this study, we isolated a naturally forming SIRT1 containing complex from human cells. Through mass spectrometry, we identified RecQL4 as a SIRT1 interacting protein. Our data showed that SIRT1 interacts with RecQL4 both in vitro and in cells. RecQL4 can be acetylated by CBP in cells. We further found that the acetylation of RecQL4 regulates its dynamic nuclear translocation during S phase. Transfection of wild-type RecQL4 into cells derived from an RTS patient can rescue cell proliferation, while a RecQL4 acetylation mutant severely impairs this function. SIRT1 deacetylates RecQL4 and the acetylation status of RecQL4 affects its loading to the chromatin during the S phase of the cell cycle, therefore, affecting the DNA replication initiation. Our study provides new insights into the regulation of the RecQL4 protein during DNA replication and helps to understand the precise role of both RecQL4 and SIRT1 proteins in maintaining genomic integrity.
Materials and methods
Cell culture, plasmids, and treatments
HeLa, H1299, and 293T cells were obtained from American Type Culture Collection and maintained at 37 °C and 5% CO2 in Dubelcco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). The AG05013 fibroblasts from an RTS patient were purchased from Coriell Cell Repositories. Cells were maintained in the DMEM medium supplemented with essential and non-essential amino acids, vitamins, and 10% FBS.
RecQL4 RNAi and antibodies
RecQL4 siRNA duplex was designed and synthesized by Invitrogen stealth RNAi system. The sequence for the RNAi is: UUGUGGUGAAGGAACCAGUGGCUCA. The RNA was transfected by Lipofectamin 2000 (Invitrogen). The rabbit polyclonal antibody of RecQL4 was raised against GST-RecQL4 (239–357). Antisera from immunized rabbits were affinity purified by use of GST-RecQL4 (239–357) protein coupled with agarose beads. Anti-acetylated lysine antibody was purchased from Cell Signaling Inc (9441). Anti-acetylated RecQL4 specific antibody was produced by immunizing rabbits with a synthetic acetylated peptide (KLH-coupled) corresponding to residues surrounding Lys88 of human RecQL4 and affinity purified with acetylated peptide after depletion of antibodies to unacetylated RecQL4 by passing through the same peptide without acetylation of the lysine.
Co-immunoprecipitation assay
Whole cell lysates from H1299 were incubated with 2 μg of mouse anti-SIRT1 (santa cruz) or control IgG for 10–14 h at 4 °C with end-over-end rotation. Protein A/G-agarose beads were added and the reaction mixtures were further mixed for 1 h at 4 °C. After being washed with PBST, proteins were extracted from the agarose beads by boiling in 1 × SDS gel loading buffer and resolved on 8% SDS–polyacrylamide gels.
Endogenous RecQL4 acetylation assay
H1299 cells were treated with HU or HU, TSA, and nicotinamide combine for 12 h or without treatment as controls. Whole cell lysates were incubated with anti-RecQL4 antibody for O/N at 4 °C with end-over-end rotation. Protein A/G-agarose beads were added and the reaction mixtures were further mixed for 1 h at 4 °C. After being washed with PBST 4 times, bound proteins were eluted with 0.1 M glycine (PH 2.5) and then neutralized with saturated Tris buffer. The eluted proteins were then analyzed by western blot with anti-acetylated lysine and anti-RecQL4 antibodies.
In vitro acetylation assay
In vitro acetylation assays were performed essentially as described (Wang et al., 2005) with some modifications. 20 μl reactions contain 50 mM HEPES (pH 8.0), 10% glycerol, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 10 mM sodium butyrate, 1 μl of [14C]acetyl-CoA (55 mci/mmol, Amersham Biosciences), about 1 μg of GST fusion proteins, and 100 ng of p300 (1195–1810) HAT domain were incubated at 30 °C for 2 h. The reaction mixtures were subjected to SDS-PAGE and autoradiography.
In vitro deacetylation assay
The 14C-labeled acetylated RecQL4 (2.5 μg) was incubated with purified SIRT1 (10 ng) in the presence of 250 μM NAD at 30 °C for 1 h. The purified SIRT1 was prepared essentially as previously described (Luo et al., 2001). The reactions were performed in a buffer containing 50 mM Tris–HCl (pH 9.0), 50 mM NaCl, 4 mM MgCl2, 0.5 mM DTT, 0.2 mM PMSF, 0.02% NP-40, and 5% glycerol. The reactions were resolved on SDS-PAGE and analyzed by autoradiography and Coomassie Blue staining.
RTS cell rescue assay
The AG05013 fibroblasts from an RTS patient were grown in MEM medium supplemented with non-essential amino acids and 20% FBS. The RTS cells were infected by either a pBabe retrovirus empty vector or a pBabe retrovirus containing wild-type RecQL4 or RecQL4K88R mutant. Cells were selected in medium containing 1.0 μg/ml puromycine for 3 days before the assays. Viable cell counting: cell numbers were enumerated every 3 days by trypan-blue exclusion. Cell suspension was mixed with same amounts of Trypan blue solution. Unstained cells were counted on the hemacytometer under a microscope. MTT assay: 1000 infected RTS cells were seeded into 96-well plate. After incubating in 5% CO2 at 37 °C for 72 h, 0.5 mg/ml MTT was added to cells. Cells were incubated in 5% CO2 at 37 °C for another 3–4 h. Supernatants were carefully removed and 100 µl DMSO was added to each well. Absorbance was measured at 570 nm on spectrophotometer. BrdU incorporation assay: 3000 infected RTS cells were plated on coverslips in 24-well plate. Cells were treated with 2 mM HU for 16 h. After being released from HU arrest for 3 h, 10 uM BrdU were added to cells and incubated at 37˚C for 1 h. Cells were fixed with 70% ethanol and incubated with 2 N HCl/Triton X-100 at room temperature. Cells were incubated with Anti-BrdU FITC for 1 h at room temperature and the nuclei was stained with 0.2 µg/ml DAPI. The fluorescence was examined under a microscope. [3H]-thymidine incorporation assay: Infected RTS cells were arrested by 2 mM HU for 16 h followed by 10 uCi/ml [3H]-thymidine incubation for 6 h. Incorporated [3H] thymidine was measured after 5% TCA precipitation using a Beckman scintillation counter. For statistical analysis of significance between the groups, a t Test was performed. A value of p < 0.05 was considered to be significant.
Results
Identification of RecQL4 as SIRT1 target
To understand the functions of SIRT1 in vivo, we performed affinity purification of the naturally forming SIRT1 containing complexes from human lung cancer cell line H1299, which stably expresses a Flag-tagged full-length SIRT1. Through mass spectrometry, we identified a SIRT1 interacting protein RecQL4 (Fig. 1A). To verify the interaction between RecQL4 and SIRT1, Flag-tagged RecQL4 was transfected into 293T cells and cell extracts were immunoprecipitated by anti-Flag M2 beads. As expected, endogenous SIRT1 protein was clearly co-immunoprecipitated with Flag-RecQL4 (Fig. 1B, lane 2, upper panel). We immunoprecipitated endogenous SIRT1 from cell extracts and endogenous RecQL4 was also clearly co-immunoprecipitated (Fig. 1C, lane 3, upper panel). The interaction between RecQL4 and SIRT1 was confirmed through in vitro assay. GST pulldown assay was performed using GST-RecQL4NT, GST-RecQL4M, GST-RecQL4CT, and [35S] labeled in vitro translated SIRT1 proteins. As shown in Fig. 1D, SIRT1 can strongly interact with the N-terminus of RecQL4 (lane 2) but not other parts of RecQL4, which is consistent with previous studies showing the deacetylation of RecQL4 mediated by SIRT1 Duan et al. (2020).
Fig. 1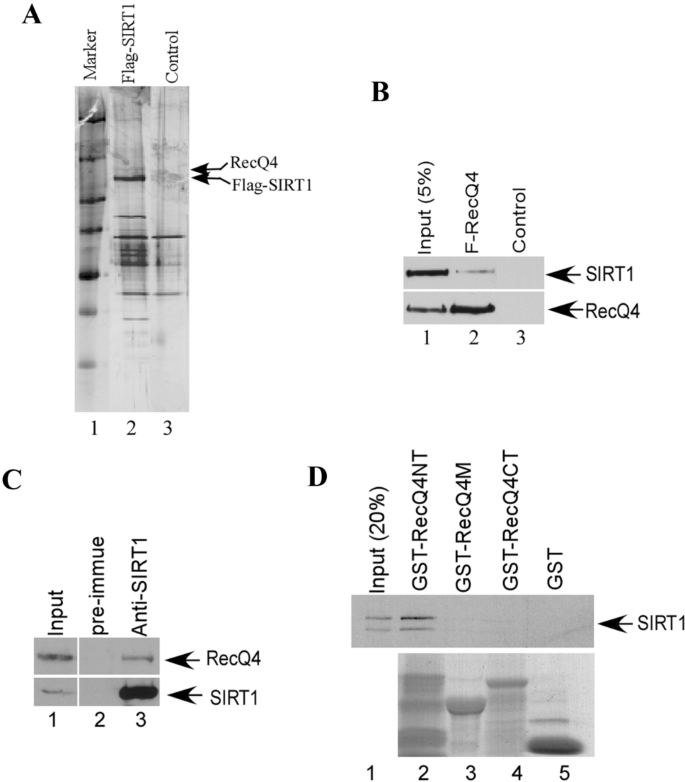
RecQL4 is a target for SIRT1. A Affinity purification of SIRT1 associated protein complexes. Silver staining of an SDS-PAGE gel from the anti-Flag affinity column eluates either from the FLAG-SIRT1 expressing H1299 cells (lane 2) or the control H1299 cells (lane 3), arrows indicate the Flag-SIRT1 and RecQL4 protein by Mass-spectrometry analysis. B Co-IP for the interaction between RecQL4 and SIRT1. Flag-tagged RecQL4 was transfected into 293 cells (lane 2). After immunoprecipitation with M2 beads, the IP products were subjected to SDS-PAGE and a western blot analysis with anti-RecQL4 antibody or anti-SIRT1 antibody. C Endogenous interaction between RecQL4 and SIRT1. The whole cell lysates from H1299 were immunoprecipitated with control IgG, anti-SIRT1 antibody, and precipitated proteins were detected with anti-RecQL4 and anti-SIRT1 antibodies. 2% of the whole cell lysates were also included as input. D GST pulldown assay. GST-RecQL4NT, M, CT, and GST protein beads were incubated with [35S] labeled in vitro translated SIRT1 protein at 4° C for 1 h with rotation. After washing, the GST beads were eluted with SDS-sample buffer and subjected to SDS-PAGE and autoradiography. The input was 10% of the labeled SIRT1 in each reaction
Full size imageRecQL4 is acetylated by CBP in cells
The interaction between RecQL4 and SIRT1 prompts us to examine if RecQL4 is a target for SIRT1. We first tested if RecQL4 could be modified by acetylation. Flag-tagged RecQL4 and CBP expression vectors were co-transfected into 293T cells and the acetylation of RecQL4 protein was detected (Fig. 2A, lane 2). CBP was also clearly detected in RecQL4 immunoprecipitates in cells that had been co-transfected with CBP and RecQL4 expression vectors (Fig. 2B, lane 3), indicating that RecQL4 can interact with CBP in cells, which is consistent with previous reports Duan et al. (2020). We further examined the acetylation of RecQL4 using an in vitro acetylation assay with the three GST-RecQL4 proteins generated above. RecQL4 protein can be acetylated in vitro and the acetylation sites are located at the N-terminal domain (Fig. 2C, lane 1, upper panel). The acetylation status of endogenous RecQL4 protein was also examined. We clearly detected the acetylation of endogenous RecQL4 in cells that had been treated with the DNA damaging agent HU overnight (Fig. 2D, lane 2, upper panel). The elevated acetylation of RecQL4 after HU treatment is mediated by CBP. The RecQL4-CBP interaction becomes stronger post HU treatment (Supplementary Fig. 4a). As expected, when cells were simultaneously treated with HU, the histone deacetylase inhibitors TSA, and nicotinamide together, the amount of acetylated RecQL4 protein was significantly increased (Fig. 2D, lane 3, upper panel).
Fig. 2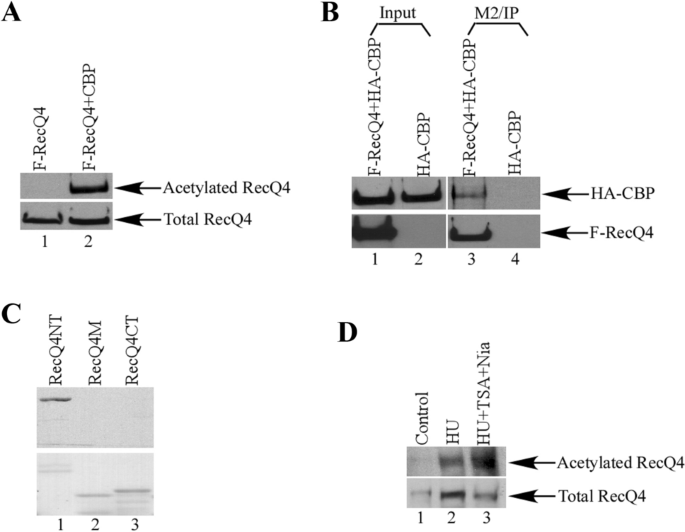
RecQL4 is acetylated by CBP. A Western blot analysis of the purified RecQL4 proteins from transient transfection of Flag-RecQL4 alone or with CBP in 293T cells with anti-acetylated lysine (upper panel) or anti-RecQL4 (lower panel) antibodies. B RecQL4 interacts with CBP in cells. 293T cells were transfected with HA-CBP alone or with Flag-RecQL4. After anti-Flag M2 beads immunoprecipitation, proteins were analyzed by western blot with anti-HA antibody (upper panel) for CBP or anti-RecQL4 antibody (lower panel). C RecQL4NT protein can be acetylated in vitro. GST-RecQL4 proteins were incubated with [14C]-acetyl-CoA and HAT proteins for in vitro acetylation assay. The Coomassie staining of the GST-RecQL4 proteins in SDS-PAGE gel were shown in the lower panel. D RecQL4 protein can be acetylated in vivo. Endogenous RecQL4 proteins were immunoprecipitated after H1299 cells were treated with HU or HU, TSA, and nicotinamide for 12 h. The acetylated RecQL4 with anti-acetylated lysine (upper panel) or anti-RecQL4 (lower panel) antibodies was analyzed by western blot
Full size imageRecQL4 is acetylated at lysine 88
To further map the acetylation sites of RecQL4 in N-terminus, we divided the GST-RecQL4NT protein into four distinct fragments and prepared the GST fusion proteins: GST-RecQL4 (1–119), GST-RecQL4 (120–238), GST-RecQL4 (239–357), and GST-RecQL4 (358–478). The purified proteins were examined for acetylation by the in vitro acetylation assay. GST-RecQL4 (1–119) exhibited a strong acetylation signal in the assay (Fig. 3A, lane 1, upper panel). Examination of the sequence of this protein discovered that there are four lysines located within this region (Fig. 3B). We further divided this region into two smaller fragments and prepared the GST fusion proteins: GST-RecQL4 (1–89) and GST-RecQL4 (89–119) and found that GST-RecQL4 (1–89) was clearly acetylated (Fig. 3B, upper panel, lane 2), suggesting that the major acetylation sites of RecQL4 are located within residues 1–89. We mutated the two lysines to arginines individually in the GST-RecQL4 (1–89) protein and performed the same acetylation assay. The acetylation level of GST-RecQL4 (1-89K88R) protein was significantly reduced, while no detectable change on the GST-RecQL4 (1-89K49R) protein was observed compared to wild-type protein (Fig. 3C, upper panel, lane 3 versus lane 2). Hence, the major acetylation site was mapped at K88 by the in vitro acetylation assay. To verify this site in cells, we made the K to R mutation in full-length RecQL4 protein and transfected the constructs with CBP into 293T cells. The acetylation of RecQL4K88R protein was dramatically decreased, while the acetylation level of RecQL4K49R protein was similar to that of wild-type RecQL4 protein (Fig. 3D, upper panel, lane 5 versus lane 3). This result was also confirmed by mass spectrometry (Fig. 3E). Taken together, these results demonstrated that the acetylation of RecQL4 protein occurs predominately at lysine 88.
Fig. 3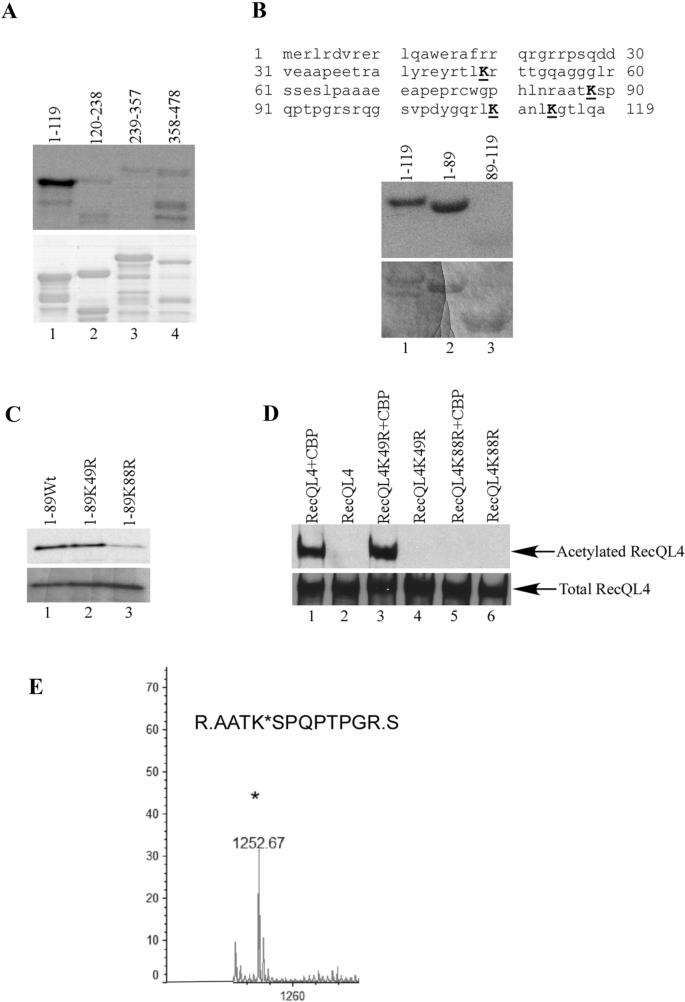
Identification of the acetylation sites of RecQL4. A In vitro acetylation assay for GST N-terminal proteins: GST-RecQL4 (1–119), GST-RecQL4 (120–238), GST-RecQL4 (239–357) and GST-RecQL4 (358–478). The acetylation of GST-RecQ4 proteins was shown by autoradiography (upper panel). The GST-RecQL4 proteins were shown in Coomassie stained SDS-PAGE gel (lower panel). B In vitro acetylation assay mapping the major acetylation site of RecQL4. RecQL4 protein sequence 1–119 contains four lysines (upper panel). GST-RecQL4 (1–89) was acetylated (lane 2, middle panel). The GST-RecQL4 proteins were shown in Coomassie stained SDS-PAGE gel (lower panel). C GST-RecQL4 (1-89wt), GST-RecQL4 (1-89K49R), and GST-RecQL4 (1-89K88R) were in vitro acetylated. The acetylation of GST-RecQL4 proteins was shown by autoradiography (upper panel). The GST-RecQL4 proteins were shown in Coomassie stained SDS-PAGE gel (lower panel). D Confirming the acetylation of lysine 88 by full-length RecQL4 mutant in cells. Western blot analysis of purified RecQL4 proteins from transient transfection of wild-type Flag-RecQL4, K49R and, K88R mutants alone or with CBP in 293T cells with anti-acetylated lysine (upper panel) and anti-RecQL4 (lower panel) antibodies. E Identifying the acetylation site of RecQL4 K88 by mass spectrometry
Full size imageRecQL4 is deacetylated by SIRT1
To investigate whether SIRT1 is able to deacetylate RecQL4 in cells, we co-transfected increasing amounts of SIRT1 together with FLAG-RecQL4 and CBP for the acetylation assay. SIRT1 effectively deacetylated RecQL4 in a dose-dependent manner (Fig. 4A, lanes 2–4). To test whether RecQL4 could be specifically deacetylated by SIRT1 in vitro, the SIRT1 protein was expressed with the N-terminal Flag epitope in cells and purified to near homogeneity on the M2-agarose affinity column. As shown in Fig. 4B, 14C-labeled acetylated RecQL4 was efficiently deacetylated by purified SIRT1 (lane 2), but not by a control eluate (lane 1). Importantly, NAD is required for the SIRT1-mediated deacetylation of RecQL4 (lane 2 versus lane 3), which is consistent with previous reports Duan et al. (2020). However, the deacetylation activity was completely inhibited in the presence of the SIRT1 inhibitor nicotinamide (lane 4 versus lane 2). We further examined the acetylation status of endogenous RecQL4 after the down-regulation of SIRT1 in cells. As expected, we could clearly detect the acetylation of RecQL4 after the transfection of siRNA targeting SIRT1 (Fig. 4C, lane 2, upper panel). We also tested the possibility that Class I and II HDACs may deacetylate RecQL4 by adding the inhibitor TSA into SIRT1 down-regulated cells. The acetylation level of RecQL4 displayed little change (Fig. 4C, lane 3 versus lane 2, upper panel), indicating that SIRT1 is the major deacetylase for RecQL4.
Fig. 4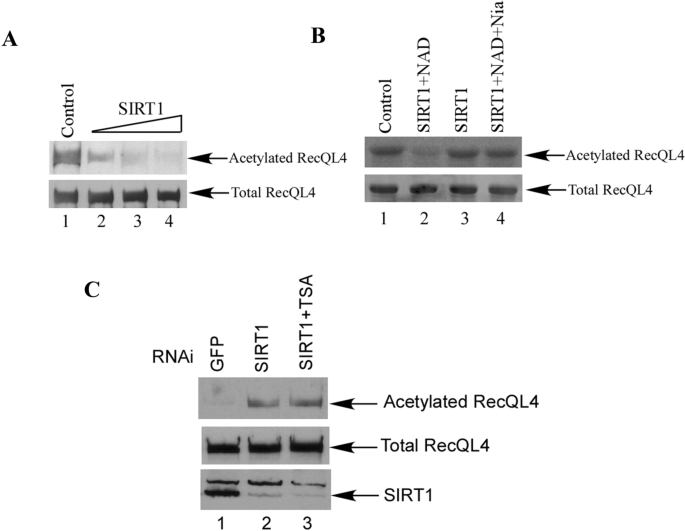
RecQL4 can be deacetylated by SIRT1. A RecQL4 deacetylation assay in the cells. 293 cells were co-transfected with Flag-RecQL4 and CBP with different amount of SIRT1 as indicated. After anti-Flag M2 beads immunoprecipitation, the IP products were subjected to SDS-PAGE and a western blot assay with anti-acetylated lysine and anti-RecQL4 antibodies. B RecQL4 deacetylation assay in vitro. 1.0 μg of 14C-labeled acetylated RecQL4NT was incubated with either the control eluate (lane 1), the purified 10 ng of SIRT1 (lanes 2 and 3), or the same amount of SIRT1 in the presence of nicotinamide (lane 4) for 60 min at 30 °C. NAD (50 μM) was also added in each reaction except lane 3. The proteins were analyzed by resolution on SDS-PAGE and autoradiography (upper) or Coomassie blue staining (lower). C Down-regulation of SIRT1 increased the acetylation of RecQL4. HeLa cells were transfected with siRNA for GFP (lane 1), or SIRT1 (lanes 2, 3). Cells were also treated with TSA for 6 h before harvest (lane 3)
Full size imageAcetylation of RecQL4 regulates the timing of DNA replication initiation
To study the regulation of RecQL4 protein in DNA replication initiation in human cells, we first knocked down the expression of RecQL4 protein by siRNA in H1299 cells. Using these cells, we performed the BrdU incorporation assay for DNA replication during the S phase. After the knockdown of RecQL4 protein, cells could not initiate DNA replication in both early and late S phases (Fig. 5A, upper panel). However, the cells transfected with siRNA targeting GFP displayed normal DNA replication (Fig. 5A, lower panel). We then examined whether the RecQL4 protein is located in the DNA replication foci by immunostaining. As shown in Supplementary Fig. 1, RecQL4 was clearly detected co-localizing with BrdU in the DNA replication foci, which suggests that RecQL4 protein is indeed required for DNA replication initiation in human cells.
Fig. 5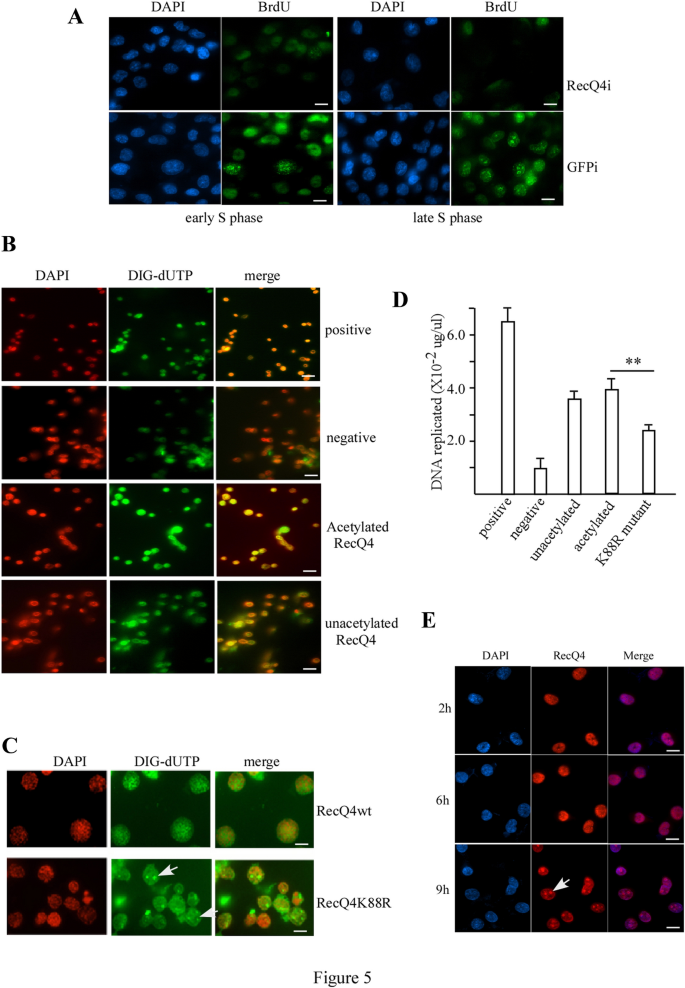
Acetylation of RecQL4 regulates DNA replication initiation. A BrdU incorporation assay for H1299 cells transfected with SiRNA for RecQL4 (upper panel) or GFP (lower panel) in early S phase (left panel) or late S phase (right panel). Scale bar = 10 µm. B In vitro DNA replication assay upon positive, negative, acetylated RecQL4, or unacetylated RecQL4 proteins added into assay. Scale bar = 20 µm. C In vitro DNA replication assay with wild-type RecQL4 or RecQL4K88R mutant proteins. Scale bar = 5 µm. D Quantitative DNA replication assay. The same in vitro DNA replication assay was done by substituting DIG-dUTP with [α-32P]-dATP and dTTP. **p < 0.01 (t test). E Dynamic re-localization of RecQL4 protein in S phase. Immunostaining with anti-RecQL4 antibody of H1299 cells that were arrested by HU for 24 h before being released on indicated time. Scale bar = 10 µm
Full size imageTo further explore the mechanism of RecQL4 acetylation in DNA replication regulation, we adopted an in vitro DNA replication assay system (Krude et al., 1997; Stoeber et al., 1998). A schematic representation of the replication experiment was shown (Supplementary Fig. 2). Briefly, the nuclei from HeLa cells of G1 phase can initiate DNA replication after the addition of the cytosolic and nuclear factors from the cell extracts of S phase (Krude et al., 1997). We first transfected HeLa cells with siRNA targeting RecQL4 or GFP as controls to down regulate the expression of RecQL4 protein. Twenty-four hours after siRNA transfection, cells were treated with mimosine for 24 h to synchronize them at late G1 phase before being harvested to isolate the nuclei. In parallel, the whole cell extracts of HeLa were prepared by immune-depletion of the RecQL4 protein by RecQL4 antibody or pre-immune serum as controls. With supplemented exogenous purified RecQL4, acetylated RecQL4, or RecQL4K88R protein, the five in vitro DNA replication reactions were assembled (Supplementary Fig. 2). After these reactions, the nuclei were immunostained for incorporated Digoxigenin-11-dUTP (DIG-dUTP) with anti-DIG antibody and DAPI. As expected, most of the nuclei in the positive group initiated the DNA replication reaction, while most nuclei in the negative group did not initiate the DNA replication (Fig. 5B, upper two panels). Both the acetylated and unacetylated RecQL4 proteins showed the initiation of DNA replication (Fig. 5B, lower two panels). However, the acetylated RecQL4 protein showed a stronger DNA replication reaction than the unacetylated RecQL4 protein (Fig. 5B, lower two panels). To quantify DNA synthesis and confirm this result, we substituted DIG-dUTP with [α-32P]-dATP in the DNA replication assay. Indeed, both the acetylated and unacetylated RecQL4 synthesized new DNA in the reaction. However, the acetylated RecQL4 initiated the replication more efficiently (3.9 × 10–2 μg/μl) than did the unacetylated RecQL4 (3.6 × 10–2 μg/μl, Fig. 5D). To further confirm that the acetylation of RecQL4 increases the efficiency of DNA replication initiation, we tested the RecQL4 acetylation mutant K88R in the same assay. Although the K88R mutant could still initiate the DNA replication, the efficiency was much lower (2.5 × 10–2 μg/μl) (Fig. 5C, D), indicating that the acetylation of RecQL4 indeed increases its DNA replication efficiency.
Although both wild-type RecQL4 and RecQL4K88R mutant proteins can initiate DNA replication, the replication pattern was different (Fig. 5C). The wild-type RecQL4 protein initiated DNA replication homogeneously distributed in nuclei and mimicked the DNA replication of early S phase in H1299 cells (Fig. 5C, upper panel versus Fig. 5A, lower left panel). In contrast, the RecQL4K88R mutant protein initiated DNA replication with a different pattern and mimicked the DNA replication of late S phase in H1299 cells (Fig. 5C, lower panel versus Fig. 5A, lower right panel). These results suggested that the acetylation of RecQL4 also participated in the regulation of DNA replication timing with the acetylated form initiating the DNA replication of early S phase or the unacetylated form initiating the DNA replication of late S phase.
DNA replication follows a reproducible temporal pattern (Donaldson, 2005; Machida et al., 2005) and the replication-related proteins are dynamically re-located in nuclei during S phase (Stoeber et al., 1998). We further examined whether the cellular localization of RecQL4 protein during S phase progression corroborated this replication pattern. H1299 cells were arrested at late G1 by mimosine treatment for 24 h before being released to S phase. Cells were immunostained by anti-RecQL4 antibody at 2 h (early S phase), 6 h and 9 h (late S phase) after release. As shown in Fig. 5E, RecQL4 proteins were homogeneously distributed in nuclei except for nucleoli in early S phase (2 h, upper panel). In contrast, RecQL4 proteins shifted to nucleoli in late S phase (9 h, lower panel).
Acetylation of RecQL4 participates in the regulation of DNA replication and cell proliferation
To elucidate the physiological significance of RecQL4 acetylation regulation, we examined its function in cells derived from an RTS patient (AG05013) that had no detectable RecQL4 protein (Yin et al., 2004). The RTS cells were infected with either a pBabe retrovirus empty vector or a pBabe retrovirus containing wild-type RecQL4 or mutant RecQL4K88R. Three days after puromycin selection, 2.5 × 104 cells from each population were plated for growth curve analysis. The expression of the wild-type RecQL4 significantly increased the cell growth compared to that of the mutant RecQL4K88R (Fig. 6A). Although both wild type and K88R mutant RecQL4 increased the cell numbers compared to vector controls at day 3, cells that expressed the K88R mutant and vector were significantly reduced in numbers compared to cells that expressed wide type after 6 and 9 days (Fig. 6A, B). Cell proliferation was also confirmed by the MTT assay. The same set of cells described above was scored 72 h after stable selection. The cell viability was significantly increased in wild-type RecQL4 expressed cells, compared with the cells that expressed the RecQL4K88R mutant and vector control (Fig. 6C). Moreover, the increase in cell proliferation was accompanied with DNA replication change. [3H]-thymidine incorporation assays revealed that the DNA replication was significantly increased in the wild-type RecQL4 transfected RTS cells. In contrast, there was slightly increased DNA replication in K88R mutant transfected RTS cells compared to RTS cells transfected with vectors alone (Fig. 6D). We further confirmed the DNA replication status of these cells by BrdU incorporation assay. There was an increase in the percentage of positive cells for BrdU incorporation in wild-type RecQL4 transfected RTS cells than that in the K88R mutant or vector control transfected cells (Fig. 6E, F and Supplementary Fig. 3).
Fig. 6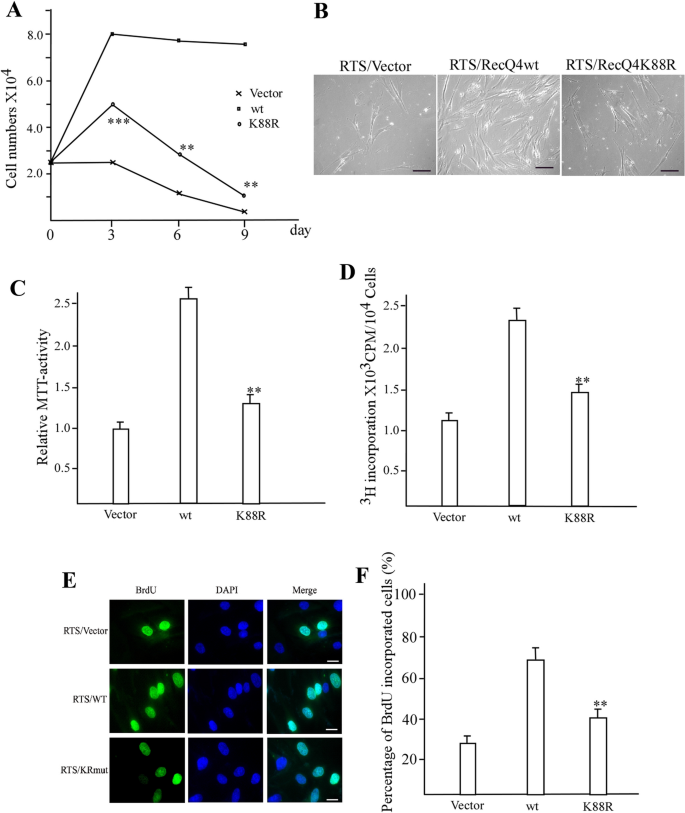
RecQL4 acetylation mutant fails to rescue the phenotype of cells from an RTS patient. A Growth curve of RTS cells transfected with pBabe vector alone, wild-type RecQL4, or K88R mutant. The result is average from three independent experiments. **p < 0.01 and ***p < 0.001 (t test). Comparison between pBabe vector alone group and K88R mutant group. B Representation of stably transfected RTS cells after 9 days of growth. Scale bar = 20 µm. C Cell proliferation analysis by MTT assay for stably transfected RTS cells. **p < 0.01 (t test). Comparison between pBabe vector alone group and K88R mutant group. D [3H]-thymidine incorporation assay for the stably transfected RTS cells. **p < 0.01 (t test). Comparison between pBabe vector alone group and K88R mutant group. E BrdU incorporation assay for the stably transfected RTS cells. F Quantification of BrdU incorporated RTS cells shown in E. **p < 0.01 (t test). Comparison between pBabe vector alone group and K88R mutant group. Scale bar = 10 µm
Full size imageAcetylation of RecQL4 regulates its chromatin binding in S phase
To further define the function of the acetylation of RecQL4 during DNA replication, we used RecQL4 acetylation mutant to examine its effect on RecQL4 association with chromatin. RTS cells stably transfected with wild-type RecQL4 or K88R mutant were arrested at late G1 by mimosine treatment for 24 h before being released to the S phase. In agreement with the in vitro and in RTS cell replication assay results (Figs. 5 and 6), wild-type RecQL4 showed a stronger association with chromatin than the acetylation mutant K88R both in the early and late S phase (Fig. 7A, lanes 1, 3 versus lanes 2, 4), indicating that the acetylation of RecQL4 indeed plays an important role in the regulation of DNA replication. Down-regulation of SIRT1 achieved similar results. RecQL4 associates with chromatin stronger both in the early or late S phase cells when SIRT1 was down regulated by siRNA (Fig. 7B, lanes 6, 8 versus lanes 5, 7), suggesting that RecQL4 is the physiological target for SIRT1 at the chromatin. The acetylation level of RecQL4 in S phase was further examined. As expected, we detected the strong acetylation signal of RecQL4 in the early S phase (Fig. 7C, lane 1), while in the late S phase, the acetylation level of RecQL4 was reduced (Fig. 7C, lane 2). Coincidentally, the expression level of SIRT1 was low in the early S phase and increased in the late S phase (Fig. 7D), indicating that SIRT1 regulates the acetylation level of RecQL4 in the S phase through its differential protein expression.
Fig. 7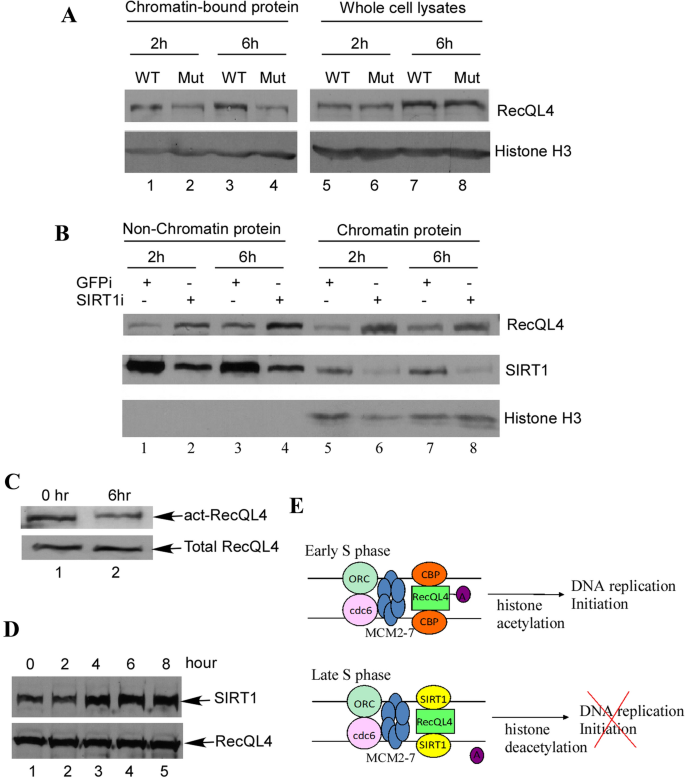
SIRT1 regulates the acetylation status of RecQL4 in S phase. A Acetylation of RecQL4 increases its chromatin binding. Wild-type RecQL4 and acetylated mutant stably transfected RTS cells were arrested by HU for 24 h before being released. Cells were harvested at indicated time for the isolation of chromatin binding proteins. B SIRT1 regulates RecQL4 chromatin binding. HeLa cells were transfected with siRNA for SIRT1 or GFP for controls. 24 h after transfection, cells were treated with HU for 24 h before being released. Cells were harvested at indicated time for the isolation of chromatin binding proteins. C, D HeLa cells were arrested by HU for 24 h before being released. Cells were harvested at indicated time followed by western blotting with anti-acetylated RecQL4 (upper panel) or anti-RecQL4 (lower panel) antibodies. E SIRT1 is up-regulated in late S phase. HeLa cells were arrested by HU for 24 h before being released. Cells were harvested at indicated time followed by western blotting with anti-SIRT1 (upper panel) or anti-RecQL4 (lower panel) antibodies. F Hypothetical model for the acetylation of RecQL4 and SIRT1 regulation during DNA replication
Full size imageDiscussion
The acetylation of proteins has been demonstrated to regulate the pattern of DNA replication in human cells (Kemp et al., 2005) and RecQL4 plays an important role in DNA replication initiation (Sangrithi et al., 2005). However, the regulating mechanism of the function of RecQL4 on DNA replication is unknown. Here, we present the evidence that the function of RecQL4 on DNA replication is regulated by the acetylation/deacetylation mediated by SIRT1. RecQL4 can be acetylated by CBP and the major acetylation site was identified at its N-terminal lysine 88. Transfection of wild-type RecQL4 into cells derived from an RTS patient can rescue the cell proliferation, while a RecQL4 acetylation mutant severely impairs this function. We demonstrated that the acetylation of RecQL4 protein can regulate both DNA replication activity and the timing of replication firing by dynamically regulating its nuclear localization during the S phase. SIRT1 deacetylates RecQL4 in vitro, which is consistent with previous studies showing the deacetylation of RecQL4 mediated by SIRT1 Duan et al. (2020). We further confirmed the function of SIRT1 on the deacetylation of RecQL4 in vivo. The acetylation status of RecQL4 affects its loading to the chromatin during the S phase of the cell cycle, therefore, affecting the DNA replication initiation.
The N-terminal region of the RecQL4 protein can stimulate chromosomal DNA replication in Xenopus egg extracts depleted of xRTS (Sangrithi et al., 2005), underlining the importance of its role in the regulation of the function of RecQL4 on DNA replication initiation. This region contains homology to the yeast replication factors Sld2/DRC1, which interact with Dpb11 after being phosphorylated by cyclin dependent kinases. This is an essential step for the formation of the active replicative helicase complex on replication origins and is crucial for the initiation of DNA replication (Tanaka et al., 2013; Wang & Elledge, 1999). Our discovery on the acetylation site of RecQL4 located in this region further demonstrated that the N-terminal region of the RecQL4 is the important regulatory domain for its function on DNA replication initiation.
Cells replicate their DNA according to a precise and reproducible temporal pattern (Cvetic & Walter, 2005; Donaldson, 2005; Forsburg, 2004; Lin et al., 2003; Machida et al., 2005; Takeda & Dutta, 2005; Zhou et al., 2005). Histone acetylation state has been suggested as a candidate for a molecular determinant of replication timing (Aparicio et al., 2004; Ouspenski et al., 2003; Vogelauer et al., 2002). Treating cells with histone deacetylase inhibitor TSA can alter the pattern of DNA replication in human cells (Kemp et al., 2005). The role of RecQL4 in the initiation of DNA replication has been extensively studied from Drosophila, Xenopus, and mammalians (Im et al., 2009, 2015; Matsuno et al., 2006; Sangrithi et al., 2005; Thangavel et al., 2010; Wu et al., 2008; Xu et al., 2009a, 2009b; Xu et al., 2009a, 2009b). However, how RecQL4 protein is regulated remains unclear. Our results demonstrated that RecQL4 can be regulated by the acetylation/deacetylation pathway during DNA replication initiation. Acetylation increased RecQL4 binding to chromatin during the S phase (Fig. 7A, B). The reduced acetylation level of RecQL4 during the S phase accompanied with the gradually increasing expression of SIRT1 (Fig. 7C, D), indicating that the acetylation of RecQL4 is dynamically regulated in the S phase.
SIRT1 plays an important role in DNA metabolism. SIRT1 knockout mice showed a severe phenotype in genomic instability, including altered histone modification, impaired DNA damage response, and reduced ability to repair DNA damage (Wang et al., 2008). SIRT1 deacetylates Ku70 and enhances DNA repair capacity upon exposure to ionizing radiation (Jeong et al., 2007). SIRT1 facilitates DNA repair in cells by deacetylating the WRN protein to regulate its enzymatic activities and cellular localization (Li et al., 2008). It also interacts with MRN and plays a positive role in repairing double-strand DNA breaks through maintaining NBS1 in the hypoacetylated state by deacetylation (Yuan et al., 2007). SIRT1 also facilitates the NER pathway for UV-induced DNA repair by deacetylating XPA (Fan & Luo, 2010). Recently, SIRT1 has been found participating in the regulation of DNA replication by targeting TopBP1 (Liu et al., 2014; Wang et al., 2014). The activation of SIRT1 deacetylated TopBP1 resulted in TopBP1–Treslin disassociation and DNA replication inhibition (Liu et al., 2014). SIRT1 null MEF cells showed the increased BrdU incorporation and increased DNA replication origin firing (Wang et al., 2014). These results align with our observation that the acetylation level of RecQL4 was increased in the early S phase (Figs. 5, 6). Hence, the lower expression level of SIRT1 is necessary for the efficient replication initiation. Indeed, the expression level of SIRT1 is low in the early S phase and increased throughout the S phase progression (Fig. 7D). It is possible that SIRT1 regulates both TopBP1 and RecQL4 to coordinate the DNA replication initiation.
RecQL4 has been reported to be acetylated by p300 at the sites of lysine 376, 380, 382, 385 and 386. This acetylation is important for the translocation of RecQL4 from nucleus to cytoplasm (Dietschy et al., 2009). However, we only detected a very weak acetylation signal in this region in our in vitro acetylation assay (Fig. 3A, lane 4) and were unable to detect the acetylation signal in mass spectrometry. The different results may be caused by different methods to identify the acetylation sites. We believe our results that K88, identified by in vitro assay (Figs. 2C, 3A–C) and confirmed by in vivo assay (Fig. 3D) and mass spectrometry (Fig. 3E), is the major acetylation site for RecQL4. The acetylation of RecQL4 may facilitate RecQL4 interaction with DNA initiation complex, and the loading to DNA replication initiation site of the chromatin.
RecQL4 is the physiological target for SIRT1 that regulates the acetylation status of RecQL4 in the S phase. The nuclear localization of RecQL4 in the S phase collaborated with the DNA replication foci, and the acetylation mutant initiated the replication of late S phase, suggesting that the acetylation of RecQL4 protein is an important molecular determinant of DNA replication timing. Many studies have suggested that the acetylation of histone can regulate the timing of replication origin firing (Aparicio et al., 2004; Ouspenski et al., 2003; Vogelauer et al., 2002). Our results indicate that the acetylation of RecQL4 protein also plays an important role in the regulation of DNA replication initiation timing. It is possible that the RecQL4 protein not only interacts with CBP for acetylation but also recruits CBP to replication origins for the acetylation of histone in the replication of early S phase. While in the late S phase, the RecQL4 protein is deacetylated by SIRT1 and localizes to late replication origin for replication. The observation that the treatment of cells with histone deacetylase inhibitor TSA alters the replication pattern with earlier initiation from a later origin (Kemp et al., 2005) supports this notion. The model in Fig. 7E summarizes the functional regulation of the acetylation of RecQL4 during DNA replication initiation. Our findings added the complexity of the functions of SIRT1 in stress responses, cellular metabolism, and aging.
References
Aparicio, J. G., Viggiani, C. J., Gibson, D. G., & Aparicio, O. M. (2004). The Rpd3-Sin3 histone deacetylase regulates replication timing and enables intra-S origin control in Saccharomyces cerevisiae. Molecular and Cellular Biology, 24, 4769–4780.
Bachrati, C. Z., & Hickson, I. D. (2003). RecQ helicases: Suppressors of tumorigenesis and premature aging. The Biochemical Journal, 374, 577–606.
Croteau, D. L., Singh, D. K., Hoh, F. L., Lu, H., & Bohr, V. A. (2012). RECQL4 in genomic instability and aging. Trends in Genetics, 28, 624–631.
Croteau, D. L., Popuri, V., Opresko, P. L., & Bohr, V. A. (2014). Human RecQ helicases in DNA repair, recombination, and replication. Annual Review of Biochemistry, 83, 519–552.
Cvetic, C., & Walter, J. C. (2005). Eukaryotic origins of DNA replication: Could you please be more specific? Seminars in Cell & Developmental Biology, 16, 343–353.
Der Kaloustian, V. M., McGill, J. J., Vekemans, M., & Kopelman, H. R. (1990). Clonal lines of aneuploid cells in Rothmund Thomson syndrome. American Journal of Medical Genetics, 37, 336–339.
Dietschy, T., Shevelev, I., Pena-Diaz, J., Hühn, D., Kuenzle, S., Mak, R., et al. (2009). p300-mediated acetylation of the Rothmund-Thomson-syndrome gene product RECQL4 regulates its subcellular localization. Journal of Cell Science, 122, 1258–1267.
Donaldson, A. D. (2005). Shaping time: Chromatin structure and the DNA replication programme. Trends in Genetics, 21, 444–449.
Duan, S. L., Han, X. R., Akbari, M., Croteau, D. L., Rasmussen, L. J., & Bohr, V. A. (2020). Interaction between RECQL4 and OGG1 promotes repair of oxidative base lesion 8-oxoG and is regulated by SIRT1 deacetylase. Nucleic Acids Research, 9, 6530–6546.
Fan, W., & Luo, J. (2008). RecQ4 facilitates UV light-induced DNA damage repair through interaction with nucleotide excision repair factor xeroderma pigmentosum group A (XPA). Journal of Biological Chemistry, 283, 29037–29044.
Fan, W., & Luo, J. (2010). SIRT1 regulates UV-induced DNA repair through deacetylating XPA. Molecular Cell, 39, 247–258.
Forsburg, S. L. (2004). Eukaryotic MCM proteins: Beyond replication initiation. Microbiology and Molecular Biology Reviews, 68, 109–131.
Hickson, I. D. (2003). RecQ helicases: Caretakers of the genome. Nature Reviews Cancer, 3, 169–178.
Im, J. S., Ki, S. H., Farina, A., Jung, D. S., Hurwitz, J., & Lee, J. K. (2009). Assembly of the Cdc45-Mcm2-7-GINS complex in human cells requires the Ctf4/And-1, RecQL4, and Mcm10 proteins. Proceedings of the National Academy of Sciences of the United States of America, 106, 15628–15632.
Im, J. S., Park, S. Y., Cho, W. H., Bae, S. H., Hurwitz, J., & Lee, J. K. (2015). RecQL4 is required for the association of Mcm10 and Ctf4 with replication origins in human cells. Cell Cycle, 14, 1001–1009.
Jeong, J., Juhn, K., Lee, H., Kim, S. H., Min, B. H., Lee, K. M., et al. (2007). SIRT1 promotes DNA repair activity and deacetylation of Ku70. Experimental & Molecular Medicine, 39, 8–13.
Kemp, M. G., Ghosh, M., Liu, G., & Leffak, M. (2005). The histone deacetylase inhibitor trichostatin A alters the pattern of DNA replication origin activity in human cells. Nucleic Acids Research, 33, 325–336.
Kitao, S., Ohsugi, I., Ichikawa, K., Goto, M., Furuichi, Y., & Shimamoto, A. (1998). Cloning of two new human helicase genes of the RecQ family: Biological significance of multiple species in higher eukaryotes. Genomics, 54, 443–452.
Krude, T., Jackman, M., Pines, J., & Laskey, R. A. (1997). Cyclin/Cdk-dependent initiation of DNA replication in a human cell-free system. Cell, 88, 109–119.
Kumata, Y., Tada, S., Yamanada, Y., Tsuyama, T., Kobayashi, T., Dong, Y. P., et al. (2007). Possible involvement of RecQL4 in the repair of double-strand DNA breaks in Xenopus egg extracts. Biochimica Et Biophysica Acta, 1773, 556–564.
Larizza, L., Magnani, I., & Roversi, G. (2006). Rothmund-Thomson syndrome and RECQL4 defect: Splitting and lumping. Cancer Letters, 232, 107–120.
Li, K., Casta, A., Wang, R., Lozada, E., Fan, W., Kane, S., et al. (2008). Regulation of WRN protein cellular localization and enzymatic activities by SIRT1-mediated deacetylation. Journal of Biological Chemistry, 283, 7590–7598.
Lin, C. M., Fu, H., Martinovsky, M., Bouhassira, E., & Aladjem, M. I. (2003). Dynamic alterations of replication timing in mammalian cells. Current Biology, 13, 1019–1028.
Liu, T., Lin, Y. H., Leng, W., Jung, S. Y., Zhang, H., Deng, M., et al. (2014). A divergent role of the SIRT1-TopBP1 axis in regulating metabolic checkpoint and DNA damage checkpoint. Molecular Cell, 56, 681–695.
Luo, J., Nikolaev, A. Y., Imai, S., Chen, D., Su, F., Shiloh, A., Guarente, L., & Gu, W. (2001). Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell, 107, 137–148.
Machida, Y. J., Hamlin, J. L., & Dutta, A. (2005). Right place, right time, and only once: Replication initiation in metazoans. Cell, 123, 13–24.
Matsuno, K., Kumano, M., Kubota, Y., Hashimoto, Y., & Takisawa, H. (2006). The N-terminal noncatalytic region of Xenopus RecQ4 is required for chromatin binding of DNA polymerase alpha in the initiation of DNA replication. Molecular and Cellular Biology, 26, 4843–4852.
McAinsh, A. D., Scott-Drew, S., Murray, J. A., & Jackson, S. P. (1999). DNA damage triggers disruption of telomeric silencing and Mec1p-dependent relocation of Sir3p. Current Biology, 9, 963–966.
Oberdoerffer, P., Michan, S., McVay, M., Mostoslavsky, R., Vann, J., Park, S. K., et al. (2008). SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell, 135, 907–918.
Orstavik, K. H., McFadden, N., Hagelsteen, J., Ormerod, E., & van der Hagen, C. B. (1994). Instability of lymphocyte chromosomes in a girl with Rothmund-Thomson syndrome. Journal of Medical Genetics, 31, 570–572.
Ouspenski, I. I., Van Hooser, A. A., & Brinkley, B. R. (2003). Relevance of histone acetylation and replication timing for deposition of centromeric histone CENP-A. Experimental Cell Research, 285, 175–188.
Park, S. J., Lee, Y. J., Beck, B. D., & Lee, S. H. (2006). A positive involvement of RecQL4 in UV-induced S-phase arrest. DNA and Cell Biology, 25, 696–703.
Petkovic, M., Dietschy, T., Freire, R., Jiao, R., & Stagljar, I. (2005). The human Rothmund-Thomson syndrome gene product, RECQL4, localizes to distinct nuclear foci that coincide with proteins involved in the maintenance of genome stability. Journal of Cell Science, 118, 4261–4269.
Sangrithi, M. N., Bernal, J. A., Madine, M., Philpott, A., Lee, J., Dunphy, W. G., et al. (2005). Initiation of DNA replication requires the RECQL4 protein mutated in Rothmund-Thomson syndrome. Cell, 121, 887–898.
Shamanna, R. A., Singh, D. K., Lu, H., Mirey, G., Keijzers, G., Salles, B., et al. (2014). RECQ helicase RECQL4 participates in non-homologous end joining and interacts with the Ku complex. Carcinogenesis, 35, 2415–2424.
Stoeber, K., Mills, A. D., Kubota, Y., Krude, T., Romanowski, P., Marheineke, K., et al. (1998). Cdc6 protein causes premature entry into S phase in a mammalian cell-free system. EMBO Journal, 17, 7219–7229.
Takeda, D. Y., & Dutta, A. (2005). DNA replication and progression through S phase. Oncogene, 24, 2827–2843.
Tanaka, S., Komeda, Y., Umemori, T., Kubota, Y., Takisawa, H., & Araki, H. (2013). Effi cient initiation of DNA replication in eukaryotes requires Dpb11/TopBP1-GINS interaction. Molecular and Cellular Biology, 33, 2614–2622.
Thangavel, S., Mendoza-Maldonado, R., Tissino, E., Sidorova, J. M., Yin, J., Wang, W., et al. (2010). Human RECQ1 and RECQ4 helicases play distinct roles in DNA replication initiation. Molecular and Cellular Biology, 30, 1382–1396.
Vennos, E. M., & James, W. D. (1995). Rothmund-Thomson syndrome. Dermatologic Clinics, 13, 143–150.
Vogelauer, M., Rubbi, L., Lucas, I., Brewer, B. J., & Grunstein, M. (2002). Histone acetylation regulates the time of replication origin firing. Molecular Cell, 10, 1223–1233.
Wang, H., & Elledge, S. J. (1999). DRC1, DNA replication and checkpoint protein 1, functions with DPB11 to control DNA replication and the S-phase checkpoint in Saccharomyces cerevisiae. Proceedings of the National Academy of Sciences of the United States of America, 96, 3824–3829.
Wang, R., Cherukuri, P., & Luo, J. (2005). Activation of Stat3 sequence-specific DNA binding and transcription by p300/CREB-binding protein-mediated acetylation. Journal of Biological Chemistry, 280, 11528–11534.
Wang, R. H., Sengupta, K., Li, C., Kim, H. S., Cao, L., Xiao, C., et al. (2008). Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell, 14, 312–323.
Wang, R. H., Lahusen, T. J., Chen, Q., Xu, X., Jenkins, L. M., Leo, E., et al. (2014). SIRT1 deacetylates TopBP1 and modulates intra-S-phase checkpoint and DNA replication origin firing. International Journal of Biological Sciences, 10, 1193–1202.
Werner, S. R., Prahalad, A. K., Yang, J., & Hock, J. M. (2006). RECQL4-deficient cells are hypersensitive to oxidative stress/damage: Insights for osteosarcoma prevalence and heterogeneity in Rothmund-Thomson syndrome. Biochemical and Biophysical Research Communications, 345, 403–409.
Woo, L. L., Futami, K., Shimamoto, A., Furuichi, Y., & Frank, K. M. (2006). The Rothmund-Thomson gene product RECQL4 localizes to the nucleolus in response to oxidative stress. Experimental Cell Research, 312, 3443–3457.
Wu, J., Capp, C., Feng, L., & Hsieh, T. S. (2008). Drosophila homologue of the Rothmund-Thomson syndrome gene: Essential function in DNA replication during development. Developmental Biology, 323, 130–142.
Xu, X., Rochette, P. J., Feyissa, E. A., Su, T. V., & Liu, Y. (2009a). MCM10 mediates RECQ4 association with MCM2-7 helicase complex during DNA replication. EMBO Journal, 28, 3005–3014.
Xu, Y., Lei, Z., Huang, H., Dui, W., Liang, X., Ma, J., et al. (2009b). dRecQ4 is required for DNA synthesis and essential for cell proliferation in Drosophila. PLoS ONE, 4, 6107.
Yin, J., Kwon, Y. T., Varshavsky, A., & Wang, W. (2004). RECQL4, mutated in the Rothmund-Thomson and RAPADILINO syndromes, interacts with ubiquitin ligases UBR1 and UBR2 of the N-end rule pathway. Human Molecular Genetics, 13, 2421–2430.
Ying, K. L., Oizumi, J., & Curry, C. J. R. (1990). Rothmund-Thomson syndrome associated with trisomy-8 mosaicism. Journal of Medical Genetics, 27, 258–260.
Yuan, Z., Zhang, X., Sengupta, N., Lane, W. S., & Seto, E. (2007). SIRT1 regulates the function of the Nijmegen breakage syndrome protein. Molecular Cell, 27, 149–162.
Zhou, J., Chau, C. M., Deng, Z., Shiekhattar, R., Spindler, M. P., Schepers, A., et al. (2005). Cell cycle regulation of chromatin at an origin of DNA replication. EMBO Journal, 24, 1406–1417.
Acknowledgements
We specially thank Drs. D. Altieri, J. Chen, and S. Cantor for critical discussion on the manuscript, and other members of J. Luo’s lab for comments. We thank the Nucleic Acid facility of UMass Medical School for sequencing the plasmids.
Funding
This work was supported by National Natural Science Foundation of China (No. 81270427 to J. Luo, No. 81471405 to J. Luo), and Major State Basic Research Development Program of China (973 Program, No. 2013CB530801 to J. Luo).
Author information
Yuxia Yang, Wei Fan and Rong Wang contributed equally to this work.
Affiliations
Department of Medical Genetics, Center for Medical Genetics, Peking University Health Science Center, 38 Xueyuan Road, Beijing, 100191, China
Yuxia Yang & Jianyuan Luo
Department of Cancer Biology and the Cancer Center, University of Massachusetts Medical School, 364 Plantation Street, Worcester, MA, 01605, USA
Wei Fan, Rong Wang, Rui Wang & Jianyuan Luo
Institute for Cancer Genetics, Columbia University, 1130 St. Nicholas Ave., New York, NY, 10032, USA
Wei Gu
Corresponding author
Correspondence to Jianyuan Luo.
Ethics declarations
Conflict of interest
The authors declare no completing financial interests.
Ethics approval
Not applicable.
Consent for publication
All authors have approved the final submission and consented to publication.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file1 (TIF 1047 kb)
Supplementary file2 (TIF 4989 kb)
Supplementary file3 (TIF 4328 kb)
42764_2021_48_MOESM4_ESM.tif
S4. The elevated acetylation of RecQL4 after HU treatment is mediated by CBP. The RecQL4-CBP interaction becomes stronger post HU treatment (a). b, The early or late S phase by the time post releasing from G1 arrest was confirmed using Cyclin E1 (TIF 4100 kb)
Rights and permissions
About this article
Cite this article
Yang, Y., Fan, W., Wang, R. et al. Regulation of Rothmund–Thomson syndrome protein RecQL4 functions in DNA replication by SIRT1-mediated deacetylation. GENOME INSTAB. DIS. 2, 240–252 (2021). https://doi.org/10.1007/s42764-021-00048-9
Received17 March 2021
Revised14 July 2021
Accepted27 July 2021
Published30 July 2021
Issue DateAugust 2021
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
RecQL4
DNA replication
SIRT1
Deacetylation
用户登录
还没有账号?
立即注册