






The activation mechanisms of master kinases in the DNA damage response
Review Article
Genome Instability & Disease 2, 211–224 (2021)
Abstract
DNA damage can be introduced by intrinsic or extrinsic stimuli and threaten the transmission of genetic information and ultimately, survival. Eukaryotes have evolved a sophisticated mechanism, DNA damage response (DDR), to counteract this threat posed by DNA damage. The DDR requires a series of proteins involved in multiple DNA repair pathways, of which ATM, ATR and DNA–PK serve as the three most critical kinases. The signaling mechanisms underlying these pathways have been extensively studied and these three master kinases structures determined by cryo-electron microscopy (cryo-EM) have led to tremendous progress in understanding the molecular mechanism of the DDR. In this review, we discuss the recent advances made in understanding the structures of ATM, ATR, and DNA–PK. We highlight the implications of these structures in terms of their function and regulation, and speculate on the critical role of PIKK regulatory domain (PRD) in their activation.
Introduction
Genome integrity in eukaryotic cells is continually under threat from various DNA lesions. DNA damage can be induced by either environmental stimuli [such as ionizing radiation (IR), ultraviolet light (UV), or chemical agents], or physiological cellular processes [such as meiotic recombination and RAG-induced DNA cleavage in V(D)J recombination] (Ciccia & Elledge, 2010; Hailemariam et al., 2019; Jackson & Bartek, 2009; Kumar & Alt, 2016; Schatz & Swanson, 2011). Estimates suggest that every cell could receive tens of thousands of DNA damage lesions per day (Hoeijmakers, 2009).
To counteract DNA damage, cells have evolved a highly organized mechanism known as the DNA damage response (DDR), which serves to sense DNA breaks, arrest the cell cycle, and then activate appropriate DNA repair pathways and various other cellular processes (Blackford & Jackson, 2017; Marechal & Zou, 2013). DNA damage signaling is transduced by protein phosphorylation that is regulated by three master kinases: ataxia telangiectasia-mutated (ATM), ataxia–telangiectasia mutated- and Rad3-related (ATR), and DNA-dependent protein kinase catalytic subunit (DNA–PKcs). ATM and DNA–PKcs are key initiators of two crucial repair pathways of DNA double-stranded breaks (DSBs): homologous recombination (HR) and non-homologous end-joining (NHEJ), respectively (Aparicio et al., 2014). ATR is recruited to replication protein A (RPA)-coated single-stranded DNA (ssDNA) and stalled DNA replication forks by its stable binding partner ATRIP (Cimprich & Cortez, 2008).
ATM, ATR, and DNA–PKcs belong to the evolutionarily conserved phosphoinositide 3-kinase-related kinase (PIKK) family. Besides ATM, ATR, and DNA–PK, the PIKK family also includes: mechanistic target of rapamycin (mTOR) that is involved in metabolism regulation; suppressor of morphogenesis in genitalia 1 (SMG-1), which controls mRNA decay; and transformation/transcription domain-associated protein (TRRAP), which regulates transcription (Baretic & Williams, 2014; Lempiainen & Halazonetis, 2009; Lovejoy & Cortez, 2009). All PIKKs are capable of phosphorylating serine and threonine residues in their substrates, except for TRRAP, which is a kinase-dead adaptor protein (Murr et al., 2007). Moreover, all PIKK family members share a similar domain organization that consists of a highly conserved kinase domain (KD), surrounded by a FRAP–ATM–TRRAP (FAT) domain that is N-terminal to the catalytic domain, and a FATC domain at very C-terminal, all of which regulate the kinase activity. They also share a large N-terminal α-solenoid domain that is made up of Huntington, Elongation Factor 3, Protein phosphatase 2A and TOR kinase (HEAT) repeats that serve as a platform for binding partners (Fig. 1A) (Baretic et al., 2019; Lovejoy & Cortez, 2009; Paull, 2015; Rivera-Calzada et al., 2015).
Fig. 1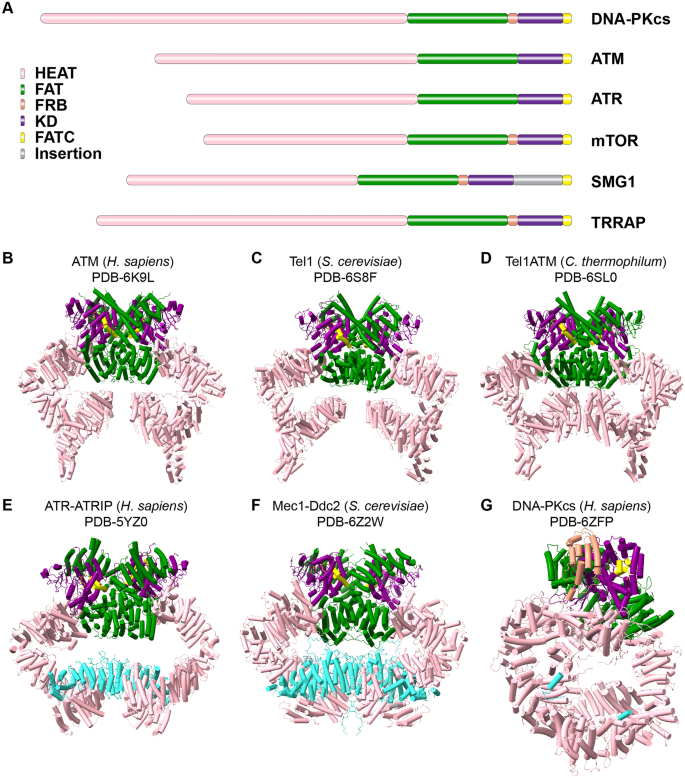
The domain organization of PIKK family members. A A comparison of the domain organization across PIKK family members. B–G The representative structures of H. sapiens ATM (B), S. cerevisiae Tel1 (C), C. thermophilum Tel1/ATM (D), H. sapiens ATR (E), S. cerevisiae Mec1 (F), and H. sapiens DNA-PKcs (G). Cyan colors ATRIP (E), Ddc2 (F), and Ku80 (G), and the other colors are coded as in (A)
Full size imageDue to the central role of the three kinases in genome integrity and human disease, their functions and mode of signaling have been extensively studied (Bhatti et al., 2011; Harper & Elledge, 2007; Jansma et al., 2020; Marechal & Zou, 2013; McKinnon, 2012). However, many details concerning the mechanisms of activation remain elusive. Recent advances in cryo-electron microscopy (cryo-EM) have, however, enabled researchers to determine the structures of these large kinases (Fig. 1B–G and Table 1), which in turn has helped to shed light on the molecular basis of kinase activity regulation.
Table 1 A summary of the cryo-EM structures of ATM, ATR, and DNAPKFull size tableIn this review, we provide a summary of the structures of ATM, ATR, and DNA–PK. We also outline how recent structural and biochemical advances have helped clarify the mechanisms by which kinase activity is regulated following the detection of DNA damage.
ATM
ATM was originally identified in 1995 (Savitsky et al., 1995). Loss of ATM activity in human cells results in the pleiotropic neurodegenerative disorder known as ataxia-telangiectasia (A-T); this disorder is characterized by immunodeficiency, cancer predisposition, premature aging, and insulin-resistant diabetes (Blackford & Jackson, 2017; Lavin, 2008; McKinnon, 2012). ATM is most homologous to Tel1—a budding yeast protein involved in telomere length maintenance, and that is currently considered to be the yeast ATM ortholog (Greenwell et al., 1995; Lustig & Petes, 1986).
ATM exists in a dimeric configuration that is inactive in resting state. DNA damage-evoked monomerization has been reported to accompany autophosphorylation at Ser1981 (Bakkenist & Kastan, 2003). The MRN (Mre11–Rad50–Nbs1) complex senses the DSBs and stimulates ATM kinase activity (Berkovich et al., 2007; Hartlerode et al., 2015; Lee & Paull, 2004, 2005). Damaged DNA is also involved in ATM transformation and activation (Dupre et al., 2006; Hailemariam et al., 2019; Lee & Paull, 2005; Shiotani & Zou, 2009; Xiao et al., 2019). Interestingly, ATM activity is also involved in the oxidative stress response, which is independent of its role in DNA damage signaling (Guo et al., 2010b; Lee et al., 2018).
Reconstructions of ATM have been produced using cryo-EM (Fig. 1B–D) (Baretic et al., 2017; Jansma et al., 2020; Llorca et al., 2003; Wang et al., 2016; Xin et al., 2019; Yates et al., 2020). In these studies, C. thermophilum (up to 3.7 Å) (Jansma et al., 2020), S. cerevisiae (up to 3.9 Å) (Xin et al., 2019; Yates et al., 2020), and human (up to 4.3 Å) (Baretić and Williams, 2017; Xiao et al., 2019) dimeric ATM structures were solved. They consistently revealed a butterfly architecture with two protomers in a head-to-head arrangement. The large, spiral-shaped, α-solenoid domain is followed by a FAT domain-containing three tetratricopeptide repeat domains (TRD) and a short HEAT-repeats domain (HRD). The FAT domain adopts a C-shaped conformation and wraps around the KD with FATC. In addition to dimeric ATM, data from negative-stain electron microscopy studies have suggested the existence of an ATM monomer (Lau et al., 2016; Llorca et al., 2003). In particular, one recent study reported that this monomeric form of ATM exhibits enhanced enzymatic activity compared to the dimeric form (Xiao et al., 2019). While monomeric ATM (at 7.8 Å resolution) has a similar overall fold to the one protomer of dimeric ATM, it has a more open catalytic pocket (Xiao et al., 2019).
The ATM N terminus consists of a 1.3-turn, left-handed α-solenoid followed by an extended HEAT repeat and a disconnected helix-bundle. This region is thought to provide a hub for protein interactions, as demonstrated by performing HEAT repeats studies (Rubinson et al., 2010; Takai et al., 2007). In the case of fission yeast, Nbs1 interacts with repeats 17–18 and repeats 21–22 in the Tel1 N terminus (You et al., 2005). It seems that ATMIN and Nbs1 competitively interact with this region in human cells, suggesting a role for this region in regulating protein activity and stabilization (Kanu & Behrens, 2007, 2008). In addition, biochemical studies have indicated that the N terminus is also involved in targeting substrates, including BRCA1, p53, and LKB1 (Fernandes et al., 2005). All ATM/Tel1 structures derived thus far have identified a flexible N-terminal domain. A structural model of the N-terminal of C. thermophilum Tel1 (ctTel1) was built, which showed both an open and a closed form (Jansma et al., 2020). The researchers of this study observed that the N terminus of the protomers can move independently, leading to either a symmetric open/closed conformation, or an asymmetric configuration. The ctTel1 N terminal (residues 1–1864), a region with a number of positively charged residues, can bind DNA and has a higher binding affinity to longer DNA in vitro (Jansma et al., 2020), which is consistent with the studies that activity of ATM can be more accumulated by longer DNA oligomers (Dupre et al., 2006; Lee & Paull, 2005; Xiao et al., 2019).
The ATM kinase domain resembles a canonical protein kinase that can be separated into two parts: an N-terminal lobe (N-lobe) and a C-terminal lobe (C-lobe). The ATM kinase domain consists of specific structural motifs including an activation loop, a catalytic loop that contributes to Mg2+ and substrate binding, a PIKK regulatory domain (PRD), and an LST8 binding element (LBE) that might be involved in kinase regulation. Two structures of ATM bounded with ATPγS and AMP–PNP were recently determined by two different groups (Jansma et al., 2020; Yates et al., 2020). As previously thought, the PRD in apo form could block the substrate binding cleft (Baretić and Williams, 2017; Xiao et al., 2019; Xin et al., 2019). ATPγS/AMP-PNP and Mg2+ are bound in the cleft between two kinase lobes: they can produce a conformation necessary for catalysis by rearranging the PRD but lift active site access (Fig. 2A).
Fig. 2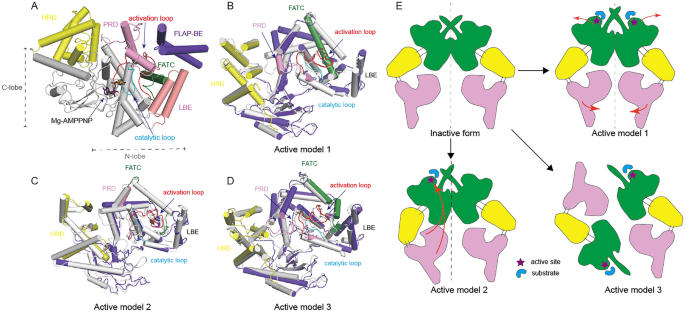
The mechanisms of ATM/Tel1 activation. A The structure of AMP-PNP bound to the yeast Tel1(PDB:6S8F) kinase domain. B–D A comparison of the active state (colored) and the inactive state (gray) according to three current models. The models used are as follows: B model 1 (PDB:5NP0, 5NP1); C model 2 (PDB: 6JXA, 6JXC); D model 3 (PDB: 6K9K, 6K9L). E Three proposed models that illustrate the mechanism for ATM/Tel1 activation in response to DNA damage
Full size imageATM dimerization is mediated by two contact interfaces via a process that is highly conserved (Baretić and Williams, 2017; Jansma et al., 2020; Xiao et al., 2019; Xin et al., 2019; Yates et al., 2020). The lower interface is mainly introduced by an interaction between TRD2-TRD2 with a bulk of hydrophobic residues. In the upper interface, a four-helix bundle (fα19 to fα22) is found that comprises the TRD3 named FLAP binding element (FLAP-BE), FATC, PRD, and LBE and that includes many charged and polar residues that have extensive contact. In the inactive ATM dimer, FATC, LBE and PRD wrap around the activation loop to form a compact arrangement. The FLAP-BE from the adjacent protomer packs against most of the kinase regulatory elements, including FATC, PRD, and the activation loop. For this reason, FLAP-BE is thought to be a critical regulator of ATM kinase activity and a fulcrum of the upper interface.
Three hypotheses of ATM/Tel1 activation have been raised as a result of data from recent structural studies (Fig. 2). The first is the symmetric motion model. Here, the open dimer has two protomers with only a limited contact interface, and a tightly packed closed dimer with a larger interface. The open dimer lacks the intermolecular interactions that block the peptide-binding site in the closed dimer and reveals an active form (Baretić and Williams, 2017). The second is the symmetric motion model. Here, motion of the N-lobe generally leads to a compact and active conformation in which PRD and FATC disordered, allowing ATP and other substrates to bind to the active site inside the central cleft (Xin et al., 2019). The final hypothesis is the dimer-to-monomer transition model. Here, ATM monomerization can be induced by DNA damage. The monomer is catalytically more active due to a lack of restriction by FLAP-BE, as well as a more open catalytic pocket (Xiao et al., 2019).
Compared to the inactive ATM dimer, the three potential active forms all reveal a more open catalytic pocket, mainly due to the PRD disorder (Fig. 2B–D), lack of FLAP-BE restriction and a less compact of LBE (Fig. 2B, C), and FATC disorder (Fig. 2C, D). Notably, ATM monomer shows a more open catalytic pocket relative to open dimer, suggesting that the open dimer is an intermediate state between closed dimer and monomer (Xiao et al., 2019).
Nonetheless, how these conformation changes are generated and stabilized is unclear. According to data from a biochemical study, the MRN complex can bind both the ATM N and C termini and stimulate kinase activity (Dupre et al., 2006; Lee & Paull, 2004; You et al., 2005). It also seems that the NuA4/Tip60 acetyltransferase complex binds to the FATC and acetylates Lys3016 to activate ATM (Adamowicz et al., 2016; Sun et al., 2005, 2007). Furthermore, damaged DNA can bind the ATM protein to mediate ATM activation (Dupre et al., 2006; Jansma et al., 2020; Lee & Paull, 2005; Llorca et al., 2003; Xiao et al., 2019). These data suggest that post-translational modifications (PTMs) and various protein regulators might be able to disrupt interactions made with the ATM dimer or induce allosteric regulation; how these changes are induced remains to be studied.
ATR
Compared to ATM and DNA–PKcs, which primarily respond to DSBs, ATR is required to respond to a wide range of genotoxic stresses, among which the most characterized are DNA damage and DNA replication stress (Harper & Elledge, 2007; Zeman & Cimprich, 2014). ATR might also be involved in other processes, including DSB repair, inter-strand crosslink repair, meiosis, and the response to mechanical and osmotic stresses (Saldivar et al., 2017). To fulfill its functions, ATR needs to interact with its associated protein, ATRIP, to assemble into a stable complex that is critical for its localization, stability, and kinase activation (Cortez et al., 2001; Zou & Elledge, 2003). Silencing or deleting ATRIP results in a phenotype that echoes ATR loss—namely, lethality and DNA damage sensitivity (Brown & Baltimore, 2000; de Klein et al., 2000).
ATR kinase activation can be triggered by various types of damage and stress, and seems to involve a common DNA structure—ssDNA coated with RPA (Cimprich & Cortez, 2008; Zou & Elledge, 2003). Such RPA-coated ssDNA serves as a platform for the recruitment of many proteins, including ATRIP, which facilitates the recruitment of the ATR-ATRIP complex (Mec1-Ddc2 in yeast) to DNA damage sites or stressed replication forks (Namiki & Zou, 2006). However, this action is not sufficient for optimal activation: several activator proteins are also needed. The best-characterized ATR activators are topoisomerase II binding protein 1 (TopBP1) (Kumagai et al., 2006) and Ewing tumor-associated antigen 1 (ETAA1) (Bass et al., 2016; Haahr et al., 2016; Lee et al., 2016) in human cells, while the Mec1 activators in budding yeast are DNA replication regulator Dpb11(human ortholog of TopBP1) (Mordes et al., 2008b; Navadgi-Patil & Burgers, 2008), DNA damage checkpoint 1 (Ddc1) (Navadgi-Patil & Burgers, 2009), and DNA replication ATP-dependent helicase/nuclease 2 (Dna2) (Kumar & Burgers, 2013). The observed common domain among these activators is the ATR-activation domain (AAD), which stimulates ATR kinase activity via contacts with both ATRIP and the C-terminal PRD in ATR, implying a common mechanism of ATR activation by activators. Meanwhile, the number of various ATR activators highlights the essential involvement of ATR in diverse processes.
ATR activator proteins are thought to be recruited in a manner that is distinct from ATR–ATRIP complex recruitment. TopBP1 recruitment is mediated by its interaction with the C-terminal tail of the RAD9 subunit of the checkpoint clamp RAD9-RAD1-HUS1 (9–1–1) complex (Delacroix et al., 2007; Lee et al., 2007; Navadgi-Patil & Burgers, 2009), which is loaded onto the ss/ds-DNA junction by the RAD17-RFC complex (Zou et al., 2002). This recruitment process is also partially dependent on the MRN complex and RAD9–RAD1–HUS1-interacting nuclear orphan (RHINO) (Cotta-Ramusino et al., 2011; Duursma et al., 2013). Unlike TopBP1, ETAA1 is recruited to RPA-coated ssDNA via a direct interaction with RPA (Bass et al., 2016; Haahr et al., 2016; Lee et al., 2016). It is thus tempting to speculate that TopBP1 and ETAA1 might be recruited to different types of aberrant DNA structures to stimulate ATR toward discrete sets of substrates. TopBP1 might be the superior activator, as a point mutation (W1147R) in its AAD is lethal in mice (Zhou et al., 2013), whereas mutations in ETAA1 AAD do not significantly alter the growth of U2OS or HEK293T cells (Bass et al., 2016).
The recently determined cryo-EM structures of Mec1-Ddc2 and data generated from biochemical studies have helped us gain a more comprehensive understanding of Mec1 kinase activation in yeast, and how structural conformational changes allow a functional transformation from an inactive to an active state. The Mec1-Ddc2 structure forms a similar butterfly shape as seen in ATM/Tel1, forming a dimer of a heterodimer with two-fold rotational symmetry in a head-to-head manner (Fig. 1F) (Tannous et al., 2021; Wang et al., 2017). The cryo-EM reconstructions of Mec1–Ddc2 have revealed that the activation loop attaches to the PRD insert (PRD-I), which seems to be key to inhibiting substrate access (Tannous et al., 2021; Wang et al., 2017). This conformation pushes the activation loop far away from the ATP gamma-phosphate for optimal hydrolysis (Fig. 3A). Both the inhibitory effect of PRD-I and the inappropriate binding site of ATP together attribute to low kinase activity in absence of the activators.
Fig. 3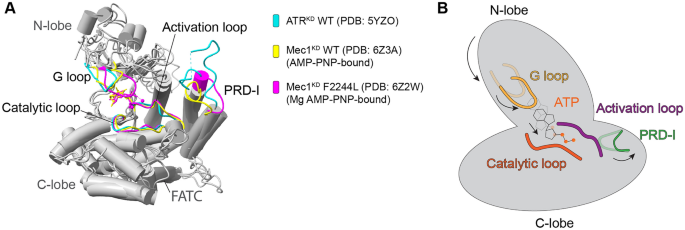
The mechanisms of ATR/Mec1 activation. A A structural comparison of the kinase domains in apo state ATR (human), AMP-PNP-bound Mec1 (yeast), and Mg2+-AMP-PNP-bound Mec1 F2244L (yeast). B One possible model of ATR/Mec1 activation. The arrows indicate the conformational motions of each subdomain or element from an inactive state (transparent) to an active state (solid) that allow for ATR/Mec1 activation
Full size imageThe structure of an Mg2+–AMP–PNP-bound Mec1 (F2244L)–Ddc2 mutant mimics Mec1 in its pre-catalytic state, indicating that PRD-I must move away from the nucleotide-binding pocket to facilitate substrate access and Mec1 activation (Tannous et al., 2021). The disruption between PRD-I and the activation loop allows for the C-terminal of FAT (C-FAT) to move in a clockwise direction relative to the kinase C-lobe and N-lobe. This effect induces an enclosed nucleotide-binding pocket that enhances ATP binding and hydrolysis (Fig. 3) (Tannous et al., 2021). This activation model is similar to another previous model, in which the potential charge–charge interaction between the AAD of Dpb11 and PRD is capable of destabilizing the interaction between PRD-I and the activation loop to eliminate the PRD-I block (Wang et al., 2017).
Substantially differing from the structure of the yeast homolog, human ATR-ATRIP presents as an asymmetric dimer of a heterodimer with a twist in the HEAT domain of one ATR molecule (Fig. 1E) (Rao et al., 2018). The mechanism underlying ATR auto-inhibition in human cells is poorly understood. The PRD-I in human ATR–ATRIP is 25-residues longer than that in yeast. Although its structure has not been accurately solved because of its flexibility, it unlikely contacts with the activation loop in the apo state that is reminiscent of an active state. Such structural conformation, however, adopts low kinase activity (Fig. 3A) (Rao et al., 2018). Based on findings from Mec1, we think the mechanism of ATR auto-inhibition might be based on instability of the nucleotide-binding pocket or low accessibility of substrate. This hypothesis needs further investigation.
While ATR activation might be achieved through increased substrate access or structural conformation changes induced by the binding of TopBP1 to both PRD and phosphorylated Thr1989 (Rao et al., 2018). Even though there is no structure show how ATR/Mec1 be stimulated by activators, biochemical and indirect structural evidence suggest that an interaction between the AAD and the PRD-I might introduce the necessary conformational changes for kinase activation (Mordes et al., 2008a; Wang et al., 2017). Organism-specific differences in the structures of Mec1–Ddc2 and ATR–ATRIP now require exploration.
DNA–PKcs
DNA–PK activity—which was discovered by three independent groups (Carter et al., 1988; Jackson et al., 1990; Lees-Miller et al., 1990; Yates et al., 2020)—has an essential role in NHEJ (Blackford & Jackson, 2017; Gottlieb & Jackson, 1993; Jeggo et al., 1995; Suwa et al., 1994). NHEJ repairs DSBs throughout interphase but predominates in G1 phase when HR is unavailable. NHEJ is also an essential step of V(D)J recombination, as it serves to establish diverse antigen receptors during lymphocyte development (Chi et al., 2020; Little et al., 2015; Schatz & Swanson, 2011).
When DSBs are present, NHEJ is initiated through the recognition of DSBs by the Ku70/80 heterodimer, which assembles around broken dsDNA ends and subsequently recruits DNA–PKcs, the kinase catalytic subunit, to the damage sites (Lieber, 2010; Scully et al., 2019). The complex consisting of DNA, Ku70/80, and DNA–PKcs is referred to as the DNA–PK holoenzyme (Gottlieb & Jackson, 1993; Hartley et al., 1995; Lees-Miller et al., 1990; Suwa et al., 1994; Walker et al., 1985). DNA–PK might prompt DNA-end synapsis (DeFazio et al., 2002; Hammel et al., 2010; Spagnolo et al., 2006), creating a stage for further recruitment of other NHEJ regulatory factors, including DNA ligase IV, X-ray cross complementing protein 4 (XRCC4), XRCC4-like factor (XLF), endonucleases (such as Artemis), and other factors (Lieber, 2010; Ochi et al., 2014).
A number of proteins are DNA–PK substrates, including the transcription factor p53 (Lees-Miller et al., 1990), heat shock protein 90 (HSP90) (Lees-Miller & Anderson, 1989), MYC (Iijima et al., 1992), and most NHEJ factors (Jette & Lees-Miller, 2015). The kinase activity of DNA–PKcs is vital for NHEJ (Callen et al., 2009; Kurimasa et al., 1999); it is regulated by extensive phosphorylation, auto-phosphorylation, and the activity of various binding partners (Chan & Lees-Miller, 1996; Davis et al., 2014; Dobbs et al., 2010; Jette & Lees-Miller, 2015).
To better understand the architecture and activation of DNA–PKcs, structural studies on DNA–PK in isolation or different complexes have been performed over the past two decades using various techniques including atomic force microscopy (AFM), crystallography, and electron microscopy. The results of these studies have provided an insight into the overall structure and regulation of DNA–PK at a low resolution (Boskovic et al., 2003; Chiu et al., 1998; Davis et al., 2014; Rivera-Calzada et al., 2005; Sibanda et al., 2010; Yaneva et al., 1997). Since 2017, members of the Blundell research group reported crystals of selenomethionine (Se-Met)-labeled DNA–PKcs complexed with native C-terminal of Ku80 (residues 539–732) diffracting to 4.3 Å resolution (Sibanda et al., 2017). Furthermore, several cryo-EM structures have been reported of the apo DNA–PKcs at 4.8 Å (Sharif et al., 2017), DNA–PK holoenzyme at 6.6 Å (Yin et al., 2017), apo DNA–PKcs at 2.8 Å and dimeric DNA–PK at 7.2 Å (Chaplin et al., 2021), and DNA–PK in a series of different states up to 3.7 Å (Chen et al., 2021b). The data from these studies have revealed how the DNA–PK holoenzyme might be assembled and activated.
The structural units of DNA–PKcs comprise the N-terminal region (residues 1–892), the circular cradle (residues 893–2801), and the C-terminal head (residues 2802–4128, FAT, FRB, kinase, and FATC) (Fig. 1G) (Chaplin et al., 2021; Chen et al., 2021b; Sharif et al., 2017; Sibanda et al., 2017; Yin et al., 2017). DNA–PKcs alone have weak basal activity (Gottlieb & Jackson, 1993; Hartley et al., 1995; Lees-Miller et al., 1990; Yin et al., 2017). In the KD, the FKBP12-rapamycin-binding (FRB) and LBE form two sides of the substrate entry groove. The PRD domain lies adjacent to FRB and blocks the active site entrance, suggesting an inactive conformation (Fig. 4A) (Chen et al., 2021b; Sibanda et al., 2017).
Fig. 4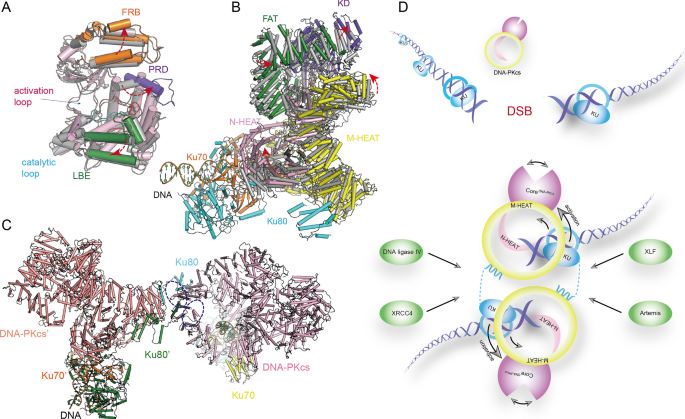
DNA-PK and DNA-PKcs structures. A, B Superimposition of apo and holo form DNA-PKcs. The overall structure (PDB: 7K17,7K0Y) (A) and structure details of the kinase domain (PDB: 7K11,7K10) (B) indicate the conformational changes of DNA-PKcs induced by KU70/80-DNA. The arrows indicate the directions of movement of the structural units. C The overall structure of dimeric DNA-PK. The purple circle indicates Domains-swapped Ku80 C-terminal bound to the cradle regions of DNA-PKcs. D A model for NHEJ initiation: Ku70/80 senses the DSBs, followed by recruitment DNA-PKcs, subsequently Ku70/80 and DNA together active the DNA-PKcs via an allosteric activation mechanism. DNA-PKcs can assembly a dimer and recruit the NHEJ factors allowing completion of NHEJ
Full size imageIn the DNA–PK holoenzyme, Ku70/80 heterodimer binds the DNA–PKcs N-terminal at four main interaction regions. The DNA duplex inserts into a DNA-binding tunnel within the DNA–PKcs N-terminal, which is rich in positively charged residues. The blocked or Y-shaped end is found outside Ku70/80 and the free DNA end is located within the open cradle (Fig. 4B) where it is protected by DNA–PKcs from unnecessary DNA end processing (Chen et al., 2021b; Yin et al., 2017). The structure of apo DNA–PKcs with a DNA oligomer has been determined: the data suggest that DNA–PKcs alone has weak DNA-end binding affinity (Chen et al., 2021b). Ku70/80 is situated outside DNA–PKcs, where it bridges the two sides of the DNA-binding groove in DNA–PKcs and stabilizes the DNA–PKcs–DNA interaction (Chaplin et al., 2021; Chen et al., 2021b; Yin et al., 2017).
The binding of Ku70/80 and DNA results in a drastic conformational change to DNA–PKcs, from the N-terminal to the kinase domain. The FRB and LBE wrapping around the substrate entry groove move leftward, and the PRD-I pops open, leading to more favorable substrate recognition or efficient phosphorylation (Fig. 4A) (Yin et al., 2017). The allosteric activation mechanism nicely explains how the DNA and Ku70/80 heterodimer stimulate the kinase activity of DNA–PKcs demonstrated in previous studies (Gottlieb & Jackson, 1993; Lees-Miller et al., 1990; Walker et al., 1985).
Negative-stain electron microscopy and small-angle X-ray scattering (SAXS) studies have identified a dimeric DNA–PK form that might bridge two damaged dsDNA ends together (Baretic et al., 2019; Graham et al., 2016; Hammel et al., 2010; Spagnolo et al., 2006). Data from a recent cryo-EM structural study suggested a dimeric DNA–PK form at 7.2 Å and identified two new binding sites between DNA–PKcs and the Ku70/80 complex (Fig. 4C). The dimeric DNA–PK complex exhibits an asymmetric configuration; moreover, the Ku80 C terminus can interact with circular cradle regions in the opposing DNA–PKcs protomer, creating a flexible arm that is capable of connecting two DNA–PK subunits (Chaplin et al., 2021). The structures of DNA–PK complexed with NHEJ factors, including Ku70/80, ligase IV, XRCC4, and XLF, have also been solved: it seems that a single XLF homodimer bridges the two halves and form a long-range synaptic complex that indicates a critical state in NHEJ (Chen et al., 2021b).
All the findings from these aforementioned studies draw us a picture of NHEJ initiation. When DSB occurs, the Ku70/80 heterodimer senses the damaged DNA ends and binds to the DNA. DNA–PKcs is subsequently recruited to the DNA ends and binds to Ku70/80. Ku70/80-DNA boosts the kinase activity of DNA–PKcs through an allosteric activation mechanism. Finally, DNA–PK assembles a dimeric form bringing two damaged DNA ends together, and recruits downstream factors creating a stage for NHEJ (Fig. 4E).
Generality in activation mechanisms
As the structures of ATM, ATR, and DNA–PK have been solved in their own different states, the mechanism underlying how kinase activity is regulated is gradually becoming clear. All share a highly conserved KD. LBE, FATC (ATM, ATR) or FRB (DNA–PK), and PRD together assemble a gate through which substrates enter, and extend into the active cleft. The same configuration has been observed within the mTOR complex (Yang et al., 2017) and the SMG1-8–9 complex (Langer et al., 2020; Zhu et al., 2019). For this reason, these enzymes exhibit auto-inhibition forms in the absence of activators.
In the presence of genotoxic stress, ATM, ATR, and DNA–PKcs can be recruited to damaged DNA sites. The activation of ATM and DNA–PKcs is accompanied by a dramatic conformation change. The binding of Ku70/80-DNA forces the N-terminal of DNA–PKcs to twist and stretch, leading to a squeezing motion of the extension of the kinase C-lobe that ultimately reveal a more open catalytic pocket (Fig. 4B) (Chen et al., 2021b; Yin et al., 2017). Furthermore, neither an N-terminal compaction hypothesis nor the other two activation models of ATM activation suggest the active transition is introduced by the conformation change from N-terminal (Baretić and Williams, 2017; Xiao et al., 2019; Xin et al., 2019). The binding of Nbs1 or other factors might generate an activation switch and serve to stabilize the active form. A similar regulation has also been observed in other PIKK members: RHEB binds to mTORC1 and activates it by allosterically realigning the active site residues for catalysis (Yang et al., 2017). However, N-terminal-mediated ATR activation has been rarely reported.
PRD has an important role in regulating PIKK. Comparing the structures of the inactive and active forms, the motion of PRD has been observed in the activation of ATM, ATR and, DNA–PKcs. In ATM, PRD interacts with the FLAP-BE from the opposing protomer to assemble a part of the dimeric interface. Deletion of PRD-I or partial deletion of the PRD seems to boost the kinase activity and accumulate the dimer to monomer transition (Xiao et al., 2019). In addition, a point mutation K2751E within the PRD of Tel1 affects the interaction with Xrs2 (Nbs1 ortholog in yeast) in vitro (Xin et al., 2019). Cys2991 within the PRD-I binds Cys2991 in the opposing protomer via a disulfide bond to form an active dimer in response to oxidative stress (Guo et al., 2010a; Xiao et al., 2019). Several studies have indicated that PRD-I blocks the substrate entrance as a pseudo-substrate in Tel1/ctTel1 and the SMG complex (Jansma et al., 2020; Langer et al., 2020; Yates et al., 2020). In ATR, TopBP1 might directly interact with the PRD to facilitate kinase activation (Mordes et al., 2008a). These findings suggest a common activation mechanism through PRD regulation.
Previous studies have reported that autophosphorylation is critical for the activation of ATM, ATR, and DNA–PK (Bakkenist & Kastan, 2003; Meek et al., 2008; So et al., 2009). In ATM, early findings suggested that Ser1981 autophosphorylation was induced by DNA damage (Bakkenist & Kastan, 2003), leading to dimer dissociation and activation. Subsequent studies showed that activation of ATM by DNA and the MRN complex does not require autophosphorylation in vitro (Dupre et al., 2006). Ser1981 in TRD1 is distal to the dimeric interface, suggesting that autophosphorylation is unlikely to control the intrinsic activation state (Baretić and Williams, 2017). However, in cells, Ser1981 phosphorylation enables ATM to remain associated with DSB sites by binding to MDC1 (Adamowicz, 2018). In ATR, Thr1989 autophosphorylation acts as a molecular switch for ATR activation (Liu et al., 2011; Nam et al., 2011). In trans-mode, ATR-ATRIP is recruited by RPA-coated ssDNA and directly interacts with the BRCT 7–8 domain of TopBP1 for its further activation (Liu et al., 2011). In DNA–PKcs, the ABCDE cluster that is found in the circular cradle proximal to the FAT domain is autophosphorylated and is crucial for NHEJ (Chan et al., 2002; Ding et al., 2003). Indeed, inhibiting ABCDE autophosphorylation blocks DNA-end processing and repair by NHEJ (Nagasawa et al., 2011; Reddy et al., 2004; Zhang et al., 2011). A recent structural study reported two DNA–PK structures with different ABCDE cluster positions. This finding suggests that ABCDE autophosphorylation exposes the DNA ends, allowing for NHEJ factor recognition and/or efficient repair processing (Chen et al., 2021b). DNA–PKcs autophosphorylation might trigger their simultaneous dissociation from DSBs, allowing for a transition from the long-range to short-range synaptic complex (Chan et al., 2002; Ding et al., 2003). This process could serve as a checkpoint to ensure that there are two free DNA ends close together (Chen et al., 2021b).
Damaged DNA is not only a substrate of the DDR, but also a crucial ATM, ATR and DNA–PKcs activator. Similar to the effect of DNA ends on DNA–PK activation, it has been extensively reported dsDNA can stimulate ATM and ATR kinase activity. It was originally considered that DNA ends induced ATM monomerization and activation together with the MRN complex (Dupre et al., 2006; Lee & Paull, 2005). We now know that dsDNA alone can stimulate the kinase activity of both dimeric and monomeric ATM, indicating a combinatorial mechanism for activation of ATM via a conformational change and DNA (Xiao et al., 2019). Moreover, data from several in vitro biochemical assays have revealed that ATR can be activated by different DNA structures, such as the ssDNA-dsDNA overhang (MacDougall et al., 2007). The details underlying this mechanism now need to be dissected.
References
Adamowicz, M. (2018). Breaking up with ATM. Journal of Immunology Science, 2, 26–31.
Adamowicz, M., Vermezovic, J., & di Fagagna, F. D. (2016). NOTCH1 inhibits activation of ATM by impairing the formation of an ATM-FOXO3a-KAT5/Tip60 complex. Cell Reports, 16, 2068–2076.
Aparicio, T., Baer, R., & Gautier, J. (2014). DNA double-strand break repair pathway choice and cancer. DNA Repair (amst), 19, 169–175.
Bakkenist, C. J., & Kastan, M. B. (2003). DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature, 421, 499–506.
Baretic, D., Maia de Oliveira, T., Niess, M., Wan, P., Pollard, H., Johnson, C. M., Truman, C., McCall, E., Fisher, D., Williams, R., et al. (2019). Structural insights into the critical DNA damage sensors DNA-PKcs, ATM and ATR. Progress in Biophysics and Molecular Biology, 147, 4–16.
Baretic, D., Pollard, H.K., Fisher, D.I., Johnson, C.M., Santhanam, B., Truman, C.M., Kouba, T., Fersht, A.R., Phillips, C., and Williams, R.L. (2017). Structures of closed and open conformations of dimeric human ATM. Science Advances, 3, e1700933
Baretic, D., & Williams, R. L. (2014). PIKKs–the solenoid nest where partners and kinases meet. Current Opinion in Structural Biology, 29, 134–142.
Bass, T.E., Luzwick, J.W., Kavanaugh, G., Carroll, C., Dungrawala, H., Glick, G.G., Feldkamp, M.D., Putney, R., Chazin, W.J., and Cortez, D. (2016). ETAA1 acts at stalled replication forks to maintain genome integrity. Nature Cell Biology, 18, 1185–1195.
Berkovich, E., Monnat, R. J., Jr., & Kastan, M. B. (2007). Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nature Cell Biology, 9, 683–690.
Bhatti, S., Kozlov, S., Farooqi, A. A., Naqi, A., Lavin, M., & Khanna, K. K. (2011). ATM protein kinase: The linchpin of cellular defenses to stress. Cellular and Molecular Life Sciences, 68, 2977–3006.
Blackford, A. N., & Jackson, S. P. (2017). ATM, ATR, and DNA-PK: The trinity at the heart of the DNA damage response. Molecular Cell, 66, 801–817.
Boskovic, J., Rivera-Calzada, A., Maman, J. D., Chacon, P., Willison, K. R., Pearl, L. H., & Llorca, O. (2003). Visualization of DNA-induced conformational changes in the DNA repair kinase DNA-PKcs. EMBO Journal, 22, 5875–5882.
Brown, E. J., & Baltimore, D. (2000). ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Gene Development, 14, 397–402.
Callen, E., Jankovic, M., Wong, N., Zha, S., Chen, H. T., Difilippantonio, S., Di Virgilio, M., Heidkamp, G., Alt, F. W., Nussenzweig, A., et al. (2009). Essential role for DNA-PKcs in DNA double-strand break repair and apoptosis in ATM-deficient lymphocytes. Molecular Cell, 34, 285–297.
Carter, T. H., Kopman, C. R., & James, C. B. (1988). DNA-stimulated protein phosphorylation in HeLa whole cell and nuclear extracts. Biochemical and Biophysical Research Communications, 157, 535–540.
Chan, D. W., Chen, B. P., Prithivirajsingh, S., Kurimasa, A., Story, M. D., Qin, J., & Chen, D. J. (2002). Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes & Development, 16, 2333–2338.
Chan, D. W., & Lees-Miller, S. P. (1996). The DNA-dependent protein kinase is inactivated by autophosphorylation of the catalytic subunit. Journal of Biological Chemistry, 271, 8936–8941.
Chaplin, A. K., Hardwick, S. W., Liang, S., Kefala Stavridi, A., Hnizda, A., Cooper, L. R., De Oliveira, T. M., Chirgadze, D. Y., & Blundell, T. L. (2021). Dimers of DNA-PK create a stage for DNA double-strand break repair. Nature Structural & Molecular Biology, 28, 13–19.
Chen, S., Lee, L., Naila, T., Fishbain, S., Wang, A., Tomkinson, A. E., Lees-Miller, S. P., & He, Y. (2021a). Structural basis of long-range to short-range synaptic transition in NHEJ. Nature, 593, 294–298.
Chen, X., Xu, X., Chen, Y., Cheung, J.C., Wang, H., Jiang, J., de Val, N., Fox, T., Gellert, M., and Yang, W. (2021b). Structure of an activated DNA-PK and its implications for NHEJ. Molecular Cell, 81, 801–810.
Chi, X., Li, Y., & Qiu, X. (2020). V(D)J recombination, somatic hypermutation and class switch recombination of immunoglobulins: Mechanism and regulation. Immunology, 160, 233–247.
Chiu, C. Y., Cary, R. B., Chen, D. J., Peterson, S. R., & Stewart, P. L. (1998). Cryo-EM imaging of the catalytic subunit of the DNA-dependent protein kinase. Journal of Molecular Biology, 284, 1075–1081.
Ciccia, A., & Elledge, S. J. (2010). The DNA damage response: making it safe to play with knives. Molecular Cell, 40, 179–204.
Cimprich, K. A., & Cortez, D. (2008). ATR: an essential regulator of genome integrity. Nature Reviews Molecular Cell Biology, 9, 616–627.
Cortez, D., Guntuku, S., Qin, J., & Elledge, S. J. (2001). ATR and ATRIP: partners in checkpoint signaling. Science, 294, 1713–1716.
Cotta-Ramusino, C., McDonald, E. R., 3rd., Hurov, K., Sowa, M. E., Harper, J. W., & Elledge, S. J. (2011). A DNA damage response screen identifies RHINO, a 9-1-1 and TopBP1 interacting protein required for ATR signaling. Science, 332, 1313–1317.
Davis, A. J., Chen, B. P., & Chen, D. J. (2014). DNA-PK: a dynamic enzyme in a versatile DSB repair pathway. DNA Repair (amst), 17, 21–29.
de Klein, A., Muijtjens, M., van Os, R., Verhoeven, Y., Smit, B., Carr, A. M., Lehmann, A. R., & Hoeijmakers, J. H. J. (2000). Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Current Biology, 10, 479–482.
DeFazio, L. G., Stansel, R. M., Griffith, J. D., & Chu, G. (2002). Synapsis of DNA ends by DNA-dependent protein kinase. EMBO Journal, 21, 3192–3200.
Delacroix, S., Wagner, J. M., Kobayashi, M., Yamamoto, K., & Karnitz, L. M. (2007). The Rad9-Hus1-Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes & Development, 21, 1472–1477.
Ding, Q., Reddy, Y. V., Wang, W., Woods, T., Douglas, P., Ramsden, D. A., Lees-Miller, S. P., & Meek, K. (2003). Autophosphorylation of the catalytic subunit of the DNA-dependent protein kinase is required for efficient end processing during DNA double-strand break repair. Molecular and Cellular Biology, 23, 5836–5848.
Dobbs, T. A., Tainer, J. A., & Lees-Miller, S. P. (2010). A structural model for regulation of NHEJ by DNA-PKcs autophosphorylation. DNA Repair (amst), 9, 1307–1314.
Dupre, A., Boyer-Chatenet, L., & Gautier, J. (2006). Two-step activation of ATM by DNA and the Mre11-Rad50-Nbs1 complex. Nature Structural & Molecular Biology, 13, 451–457.
Duursma, A. M., Driscoll, R., Elias, J. E., & Cimprich, K. A. (2013). A role for the MRN complex in ATR activation via TOPBP1 recruitment. Molecular Cell, 50, 116–122.
Fernandes, N., Sun, Y., Chen, S., Paul, P., Shaw, R. J., Cantley, L. C., & Price, B. D. (2005). DNA damage-induced association of ATM with its target proteins requires a protein interaction domain in the N terminus of ATM. Journal of Biological Chemistry, 280, 15158–15164.
Gottlieb, T. M., & Jackson, S. P. (1993). The DNA-dependent protein kinase: Requirement for DNA ends and association with Ku antigen. Cell, 72, 131–142.
Graham, T. G., Walter, J. C., & Loparo, J. J. (2016). Two-stage synapsis of DNA ends during non-homologous end joining. Molecular Cell, 61, 850–858.
Greenwell, P. W., Kronmal, S. L., Porter, S. E., Gassenhuber, J., Obermaier, B., & Petes, T. D. (1995). TEL1, a gene involved in controlling telomere length in S. cerevisiae, is homologous to the human ataxia telangiectasia gene. Cell, 82, 823–829.
Guo, Z., Deshpande, R., & Paull, T. T. (2010a). ATM activation in the presence of oxidative stress. Cell Cycle, 9, 4805–4811.
Guo, Z., Kozlov, S., Lavin, M. F., Person, M. D., & Paull, T. T. (2010b). ATM activation by oxidative stress. Science, 330, 517–521.
Haahr, P., Hoffmann, S., Tollenaere, M. A., Ho, T., Toledo, L. I., Mann, M., Bekker-Jensen, S., Raschle, M., & Mailand, N. (2016). Activation of the ATR kinase by the RPA-binding protein ETAA1. Nature Cell Biology, 18, 1196–1207.
Hailemariam, S., Kumar, S., & Burgers, P. M. (2019). Activation of Tel1(ATM) kinase requires Rad50 ATPase and long nucleosome-free DNA but no DNA ends. Journal of Biological Chemistry, 294, 10120–10130.
Hammel, M., Yu, Y., Mahaney, B. L., Cai, B., Ye, R., Phipps, B. M., Rambo, R. P., Hura, G. L., Pelikan, M., So, S., et al. (2010). Ku and DNA-dependent protein kinase dynamic conformations and assembly regulate DNA binding and the initial non-homologous end joining complex. Journal of Biological Chemistry, 285, 1414–1423.
Harper, J. W., & Elledge, S. J. (2007). The DNA damage response: ten years after. Molecular Cell, 28, 739–745.
Hartlerode, A. J., Morgan, M. J., Wu, Y., Buis, J., & Ferguson, D. O. (2015). Recruitment and activation of the ATM kinase in the absence of DNA-damage sensors. Nature Structural & Molecular Biology, 22, 736–743.
Hartley, K. O., Gell, D., Smith, G. C., Zhang, H., Divecha, N., Connelly, M. A., Admon, A., Lees-Miller, S. P., Anderson, C. W., & Jackson, S. P. (1995). DNA-dependent protein kinase catalytic subunit: a relative of phosphatidylinositol 3-kinase and the ataxia telangiectasia gene product. Cell, 82, 849–856.
Hoeijmakers, J. H. (2009). DNA damage, aging, and cancer. New England Journal of Medicine, 361, 1475–1485.
Iijima, S., Teraoka, H., Date, T., & Tsukada, K. (1992). DNA-activated protein kinase in Raji Burkitt’s lymphoma cells. Phosphorylation of c-Myc oncoprotein. European Journal of Biochemistry, 206, 595–603.
Jackson, S. P., & Bartek, J. (2009). The DNA-damage response in human biology and disease. Nature, 461, 1071–1078.
Jackson, S. P., MacDonald, J. J., Lees-Miller, S., & Tjian, R. (1990). GC box binding induces phosphorylation of Sp1 by a DNA-dependent protein kinase. Cell, 63, 155–165.
Jansma, M., Linke-Winnebeck, C., Eustermann, S., Lammens, K., Kostrewa, D., Stakyte, K., Litz, C., Kessler, B., and Hopfner, K.P. (2020). Near-Complete Structure and Model of Tel1ATM from Chaetomium thermophilum Reveals a Robust Autoinhibited ATP State. Structure, 28, 83–95
Jeggo, P. A., Taccioli, G. E., & Jackson, S. P. (1995). Menage a trois: double strand break repair, V(D)J recombination and DNA-PK. BioEssays, 17, 949–957.
Jette, N., & Lees-Miller, S. P. (2015). The DNA-dependent protein kinase: a multifunctional protein kinase with roles in DNA double strand break repair and mitosis. Progress in Biophysics and Molecular Biology, 117, 194–205.
Kanu, N., & Behrens, A. (2007). ATMIN defines an NBS1-independent pathway of ATM signalling. EMBO Journal, 26, 2933–2941.
Kanu, N., & Behrens, A. (2008). ATMINistrating ATM signalling: regulation of ATM by ATMIN. Cell Cycle, 7, 3483–3486.
Kumagai, A., Lee, J., Yoo, H. Y., & Dunphy, W. G. (2006). TopBP1 activates the ATR-ATRIP complex. Cell, 124, 943–955.
Kumar, S., & Burgers, P. M. (2013). Lagging strand maturation factor Dna2 is a component of the replication checkpoint initiation machinery. Gene Development, 27, 313–321.
Kumar, V., Alt, F.W. (2016). NHEJ and Other Repair Factors in V(D)J Recombination. In Encyclopedia of Immunobiology, pp. 107–114
Kurimasa, A., Kumano, S., Boubnov, N. V., Story, M. D., Tung, C. S., Peterson, S. R., & Chen, D. J. (1999). Requirement for the kinase activity of human DNA-dependent protein kinase catalytic subunit in DNA strand break rejoining. Molecular and Cellular Biology, 19, 3877–3884.
Langer, L.M., Gat, Y., Bonneau, F., Conti, E. (2020). Structure of substrate-bound SMG1–8–9 kinase complex reveals molecular basis for phosphorylation specificity. Elife, 57127
Lau, W. C., Li, Y., Liu, Z., Gao, Y., Zhang, Q., & Huen, M. S. (2016). Structure of the human dimeric ATM kinase. Cell Cycle, 15, 1117–1124.
Lavin, M. F. (2008). Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nature Reviews Molecular Cell Biology, 9, 759–769.
Lee, J., Kumagai, A., & Dunphy, W. G. (2007). The Rad9-Hus1-Rad1 checkpoint clamp regulates interaction of TopBP1 with ATR. Journal of Biological Chemistry, 282, 28036–28044.
Lee, J.H., Mand, M.R., Kao, C.H., Zhou, Y., Ryu, S.W., Richards, A.L., Coon, J.J., and Paull, T.T. (2018). ATM directs DNA damage responses and proteostasis via genetically separable pathways. Science Signal, 11, eaan5598.
Lee, J. H., & Paull, T. T. (2004). Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science, 304, 93–96.
Lee, J. H., & Paull, T. T. (2005). ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science, 308, 551–554.
Lee, Y. C., Zhou, Q., Chen, J. J., & Yuan, J. S. (2016). RPA-binding protein ETAA1 Is an ATR activator involved in DNA replication stress response. Current Biology, 26, 3257–3268.
Lees-Miller, S. P., & Anderson, C. W. (1989). The human double-stranded DNA-activated protein kinase phosphorylates the 90-kDa heat-shock protein, hsp90 alpha at two NH2-terminal threonine residues. Journal of Biological Chemistry, 264, 17275–17280.
Lees-Miller, S. P., Chen, Y. R., & Anderson, C. W. (1990). Human cells contain a DNA-activated protein kinase that phosphorylates simian virus 40 T antigen, mouse p53, and the human Ku autoantigen. Molecular and Cellular Biology, 10, 6472–6481.
Lempiainen, H., & Halazonetis, T. D. (2009). Emerging common themes in regulation of PIKKs and PI3Ks. EMBO Journal, 28, 3067–3073.
Lieber, M. R. (2010). The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annual Review of Biochemistry, 79, 181–211.
Little, A.J., Matthews, A., Oettinger, M., Roth, D.B., Schatz, D.G. (2015). The Mechanism of V(D)J Recombination. In Molecular Biology of B Cells, pp. 13–34.
Liu, S. Z., Shiotani, B., Lahiri, M., Marechal, A., Tse, A., Leung, C. C. Y., Glover, J. N. M., Yang, X. H. H., & Zou, L. (2011). ATR autophosphorylation as a molecular switch for checkpoint activation. Molecular Cell, 43, 192–202.
Llorca, O., Rivera-Calzada, A., Grantham, J., & Willison, K. R. (2003). Electron microscopy and 3D reconstructions reveal that human ATM kinase uses an arm-like domain to clamp around double-stranded DNA. Oncogene, 22, 3867–3874.
Lovejoy, C. A., & Cortez, D. (2009). Common mechanisms of PIKK regulation. DNA Repair (amst), 8, 1004–1008.
Lustig, A. J., & Petes, T. D. (1986). Identification of yeast mutants with altered telomere structure. Proceedings of National Academy Science of United States of America, 83, 1398–1402.
MacDougall, C. A., Byun, T. S., Van, C., Yee, M. C., & Cimprich, K. A. (2007). The structural determinants of checkpoint activation. Genes & Development, 21, 898–903.
Marechal, A., & Zou, L. (2013). DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol, 5.
McKinnon, P. J. (2012). ATM and the molecular pathogenesis of ataxia telangiectasia. Annual Review of Pathology: Mechanisms of Disease, 7, 303–321.
Meek, K., Dang, V., & Lees-Miller, S. P. (2008). DNA-PK: the means to justify the ends? Advances in Immunology, 99, 33–58.
Mordes, D. A., Glick, G. G., Zhao, R., & Cortez, D. (2008a). TopBP1 activates ATR through ATRIP and a PIKK regulatory domain. Genes & Development, 22, 1478–1489.
Mordes, D. A., Nam, E. A., & Cortez, D. (2008b). Dpb11 activates the Mec1-Ddc2 complex. Proceedings of National Academy Science of United States of America, 105, 18730–18734.
Murr, R., Vaissiere, T., Sawan, C., Shukla, V., & Herceg, Z. (2007). Orchestration of chromatin-based processes: mind the TRRAP. Oncogene, 26, 5358–5372.
Nagasawa, H., Little, J. B., Lin, Y. F., So, S., Kurimasa, A., Peng, Y., Brogan, J. R., Chen, D. J., Bedford, J. S., & Chen, B. P. (2011). Differential role of DNA-PKcs phosphorylations and kinase activity in radiosensitivity and chromosomal instability. Radiation Research, 175, 83–89.
Nam, E. A., Zhao, R. X., Glick, G. G., Bansbach, C. E., Friedman, D. B., & Cortez, D. (2011). Thr-1989 phosphorylation is a marker of active ataxia telangiectasia-mutated and Rad3-related (ATR) kinase. Journal of Biological Chemistry, 286, 28707–28714.
Namiki, Y., & Zou, L. (2006). ATRIP associates with replication protein A-coated ssDNA through multiple interactions. Proceedings of National Academy Science of United States of America, 103, 580–585.
Navadgi-Patil, V. M., & Burgers, P. M. (2008). Yeast DNA replication protein Dpb11 activates the Mec1/ATR checkpoint kinase. Journal of Biological Chemistry, 283, 35853–35859.
Navadgi-Patil, V. M., & Burgers, P. M. (2009). The unstructured C-terminal tail of the 9-1-1 clamp subunit Ddc1 activates Mec1/ATR via two distinct mechanisms. Molecular Cell, 36, 743–753.
Ochi, T., Wu, Q., & Blundell, T. L. (2014). The spatial organization of non-homologous end joining: from bridging to end joining. DNA Repair (amst), 17, 98–109.
Paull, T. T. (2015). Mechanisms of ATM activation. Annual Review of Biochemistry, 84, 711–738.
Rao, Q., Liu, M., Tian, Y., Wu, Z., Hao, Y., Song, L., Qin, Z., Ding, C., Wang, H. W., Wang, J., et al. (2018). Cryo-EM structure of human ATR-ATRIP complex. Cell Research, 28, 143–156.
Reddy, Y. V., Ding, Q., Lees-Miller, S. P., Meek, K., & Ramsden, D. A. (2004). Non-homologous end joining requires that the DNA-PK complex undergo an autophosphorylation-dependent rearrangement at DNA ends. Journal of Biological Chemistry, 279, 39408–39413.
Rivera-Calzada, A., López-Perrote, A., Melero, R., Boskovic, J., Muñoz-Hernández, H., Martino, F., & Llorca, O. (2015). Structure and assembly of the PI3K-like protein kinases (PIKKs) revealed by electron microscopy. AIMS Biophysics, 2, 36–57.
Rivera-Calzada, A., Maman, J. D., Spagnolo, L., Pearl, L. H., & Llorca, O. (2005). Three-dimensional structure and regulation of the DNA-dependent protein kinase catalytic subunit (DNA-PKcs). Structure, 13, 243–255.
Rubinson, E. H., Gowda, A. S., Spratt, T. E., Gold, B., & Eichman, B. F. (2010). An unprecedented nucleic acid capture mechanism for excision of DNA damage. Nature, 468, 406–411.
Saldivar, J. C., Cortez, D., & Cimprich, K. A. (2017). The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nature Reviews Molecular Cell Biology, 18, 622–636.
Savitsky, K., Bar-Shira, A., Gilad, S., Rotman, G., Ziv, Y., Vanagaite, L., Tagle, D. A., Smith, S., Uziel, T., Sfez, S., et al. (1995). A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science, 268, 1749–1753.
Schatz, D. G., & Swanson, P. C. (2011). V(D)J recombination: mechanisms of initiation. Annual Review of Genetics, 45, 167–202.
Scully, R., Panday, A., Elango, R., & Willis, N. A. (2019). DNA double-strand break repair-pathway choice in somatic mammalian cells. Nature Reviews Molecular Cell Biology, 20, 698–714.
Sharif, H., Li, Y., Dong, Y., Dong, L., Wang, W. L., Mao, Y., & Wu, H. (2017). Cryo-EM structure of the DNA-PK holoenzyme. Proceedings of National Academy Science of United States of America, 114, 7367–7372.
Shiotani, B., & Zou, L. (2009). Single-Stranded DNA Orchestrates an ATM-to-ATR Switch at DNA Breaks. Molecular Cell, 33, 547–558.
Sibanda, B. L., Chirgadze, D. Y., Ascher, D. B., & Blundell, T. L. (2017). DNA-PKcs structure suggests an allosteric mechanism modulating DNA double-strand break repair. Science, 355, 520–524.
Sibanda, B. L., Chirgadze, D. Y., & Blundell, T. L. (2010). Crystal structure of DNA-PKcs reveals a large open-ring cradle comprised of HEAT repeats. Nature, 463, 118–121.
So, S., Davis, A. J., & Chen, D. J. (2009). Autophosphorylation at serine 1981 stabilizes ATM at DNA damage sites. Journal of Cell Biology, 187, 977–990.
Spagnolo, L., Rivera-Calzada, A., Pearl, L. H., & Llorca, O. (2006). Three-dimensional structure of the human DNA-PKcs/Ku70/Ku80 complex assembled on DNA and its implications for DNA DSB repair. Molecular Cell, 22, 511–519.
Sun, Y., Jiang, X., Chen, S., Fernandes, N., & Price, B. D. (2005). A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proceedings of National Academy Science of United States of America, 102, 13182–13187.
Sun, Y., Xu, Y., Roy, K., & Price, B. D. (2007). DNA damage-induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Molecular and Cellular Biology, 27, 8502–8509.
Suwa, A., Hirakata, M., Takeda, Y., Jesch, S. A., Mimori, T., & Hardin, J. A. (1994). DNA-dependent protein kinase (Ku protein-p350 complex) assembles on double-stranded DNA. Proceedings of National Academy Science of United States of America, 91, 6904–6908.
Takai, H., Wang, R. C., Takai, K. K., Yang, H., & de Lange, T. (2007). Tel2 regulates the stability of PI3K-related protein kinases. Cell, 131, 1248–1259.
Tannous, E. A., Yates, L. A., Zhang, X., & Burgers, P. M. (2021). Mechanism of auto-inhibition and activation of Mec1(ATR) checkpoint kinase. Nature Structural & Molecular Biology, 28, 50–61.
Walker, A. I., Hunt, T., Jackson, R. J., & Anderson, C. W. (1985). Double-stranded DNA induces the phosphorylation of several proteins including the 90 000 mol. wt. heat-shock protein in animal cell extracts. EMBO Journal, 4, 139–145.
Wang, X., Chu, H., Lv, M., Zhang, Z., Qiu, S., Liu, H., Shen, X., Wang, W., & Cai, G. (2016). Structure of the intact ATM/Tel1 kinase. Nature Communications, 7, 11655.
Wang, X., Ran, T., Zhang, X., Xin, J., Zhang, Z., Wu, T., Wang, W., & Cai, G. (2017). 3.9 A structure of the yeast Mec1-Ddc2 complex, a homolog of human ATR-ATRIP. Science, 358, 1206–1209.
Xiao, J., Liu, M., Qi, Y., Chaban, Y., Gao, C., Pan, B., Tian, Y., Yu, Z., Li, J., Zhang, P., et al. (2019). Structural insights into the activation of ATM kinase. Cell Research, 29, 683–685.
Xin, J., Xu, Z., Wang, X., Tian, Y., Zhang, Z., & Cai, G. (2019). Structural basis of allosteric regulation of Tel1/ATM kinase. Cell Research, 29, 655–665.
Yaneva, M., Kowalewski, T., & Lieber, M. R. (1997). Interaction of DNA-dependent protein kinase with DNA and with Ku: biochemical and atomic-force microscopy studies. EMBO Journal, 16, 5098–5112.
Yang, H., Jiang, X., Li, B., Yang, H. J., Miller, M., Yang, A., Dhar, A., & Pavletich, N. P. (2017). Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature, 552, 368–373.
Yates, L.A., Williams, R.M., Hailemariam, S., Ayala, R., Burgers, P., and Zhang, X. (2020). Cryo-EM Structure of Nucleotide-Bound Tel1(ATM) Unravels the Molecular Basis of Inhibition and Structural Rationale for Disease-Associated Mutations. Structure, 28, 96–104. e103
Yin, X., Liu, M., Tian, Y., Wang, J., & Xu, Y. (2017). Cryo-EM structure of human DNA-PK holoenzyme. Cell Research, 27, 1341–1350.
You, Z., Chahwan, C., Bailis, J., Hunter, T., & Russell, P. (2005). ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Molecular and Cellular Biology, 25, 5363–5379.
Zeman, M. K., & Cimprich, K. A. (2014). Causes and consequences of replication stress. Nature Cell Biology, 16, 2–9.
Zhang, S., Yajima, H., Huynh, H., Zheng, J., Callen, E., Chen, H. T., Wong, N., Bunting, S., Lin, Y. F., Li, M., et al. (2011). Congenital bone marrow failure in DNA-PKcs mutant mice associated with deficiencies in DNA repair. Journal of Cell Biology, 193, 295–305.
Zhou, Z.W., Liu, C., Li, T.L., Bruhn, C., Krueger, A., Min, W., Wang, Z.Q., & Carr, A.M. (2013). An essential function for the ATR-activation-domain (AAD) of TopBP1 in mouse development and cellular senescence. PLoS Genet, 9, e1003702.
Zhu, L., Li, L., Qi, Y., Yu, Z., & Xu, Y. (2019). Cryo-EM structure of SMG1-SMG8-SMG9 complex. Cell Research, 29, 1027–1034.
Zou, L., Cortez, D., & Elledge, S. J. (2002). Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin. Genes & Development, 16, 198–208.
Zou, L., & Elledge, S. J. (2003). Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science, 300, 1542–1548.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (31830107 and 31821002). We thank Dr Jessica Tamanini at Shenzhen University Health Science Center for editing the manuscript prior to submission. We would like to acknowledge the vast number of other highly valuable papers that could not be cited in this review due to space limitations.
Author information
Affiliations
Fudan University Shanghai Cancer Center, Institutes of Biomedical Sciences, State Key Laboratory of Genetic Engineering and Key Laboratory of Medical Epigenetics and Metabolism, Shanghai Medical College of Fudan University, Shanghai, 200032, China
Jianxiong Xiao, Qinhui Rao & Yanhui Xu
Department of Immunobiology, Yale School of Medicine, Yale University, New Haven, CT, 06515, USA
Jianxiong Xiao
Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, CT, 06529, USA
Qinhui Rao
Corresponding authors
Correspondence to Jianxiong Xiao or Qinhui Rao.
Rights and permissions
About this article
Cite this article
Xiao, J., Rao, Q. & Xu, Y. The activation mechanisms of master kinases in the DNA damage response. GENOME INSTAB. DIS. 2, 211–224 (2021). https://doi.org/10.1007/s42764-021-00045-y
Received07 April 2021
Revised14 June 2021
Accepted15 June 2021
Published30 June 2021
Issue DateAugust 2021
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
ATM
ATR
DNA–PK
Activation
PIKK kinases
Structure
用户登录
还没有账号?
立即注册