






The emergence of a unified mechanism in the Fanconi anemia pathway
Review Article
Genome Instability & Disease 2, 281–291 (2021)
Abstract
Fanconi anemia (FA) is a rare genetic disorder characterized by bone marrow failure, developmental abnormalities and a predisposition to cancer, particularly acute myeloid leukemia. So far, 22 FA genes (FANCA-W) have been identified. Germline inactivation of any one of the 22 currently known FA genes causes FA. Proteins encoded by FA genes are involved in the Fanconi anemia pathway, which repairs DNA interstrand crosslinks (ICLs). The main function of the FA pathway in repairing ICLs has been extensively studied. In addition, several lines of evidence indicate that the FA pathway has crucial roles in genome maintenance upon replication stress, and is involved in common fragile sites, R-loops, and mitotic DNA synthesis. Recently, we found that all the functions of the FA pathway are possibly derived from one unified mechanism, namely that FA proteins play a role in the cleavage-coupled break-induced replication pathway to restart stalled replication forks. In the review, we summarize our current understanding of the functions of the FA pathway, and review our knowledge of the potential endogenous pathogenic factors that contribute to FA.
Introduction
Fanconi anemia (FA) is a rare chromosome instability syndrome that affects one in every 100,000 births. In 1927 the Swiss pediatrician Guido Fanconi first described a family containing three boys with aplastic anemia associated with developmental defects (Lobitz & Velleuer, 2006). FA is a genetic disease mainly seen in children and is characterized by progressive bone marrow failure, developmental abnormalities, and an increased predisposition to malignancies early in life. Congenital anomalies vary from patient to patient; approximately 25–40% of FA patients are physically normal (D'Andrea, 2010), but half of children with FA have skeletal anomalies, frequently of the thumb and forearm. The thumbs are usually smaller (known as hypoplastic thumbs), duplicated, or absent. The radius of the forearm is usually smaller or absent (Giampietro et al., 1993). Moreover, many FA patients show subtle endocrine defects, including insufficient production of growth hormone production and hyperthyroidism, leading to short stature. In addition to developmental abnormalities, FA individuals exhibit other clinical symptoms, such as ear anomalies and hearing loss as well as reduced fertility and café-au-lait spots on the skin (Kee & D'Andrea, 2012; Tsui & Crismani, 2019). FA patients are predisposed to a variety of cancers, particularly acute myeloid leukemia and squamous cell carcinoma of the head and neck (2003a), (2003b), (2016).
So far, 22 FA genes (FANCA–FANCW) have been identified. Autosomal biallelic germline inactivation of one of the multiple FA genes causes FA except for FANCB and FANCR. Mutations in FANCB, which is X-linked, and mutations in FANCR in which the mode of inheritance is autosomal dominant. Proteins encoded by the FA genes are involved in a specific DNA repair pathway—the Fanconi anemia pathway. Cells derived from FA patients are hypersensitive to agents that induce DNA interstrand crosslinks (ICLs), which covalently link the two strands of DNA, thereby inhibiting the unwinding of the DNA helix that is required for DNA replication and transcription. Based on these findings, it was predicted that FA was caused by defective repair of endogenously produced ICLs, and repair of ICLs was thought to be the main function of the FA pathway. Indeed, the role of FA proteins in the repair of ICLs has been extensively studied over the past two decades. Several lines of evidence indicate that the FA pathway has additional crucial roles in genome maintenance by protecting replication fork stability upon fork stalling and controlling chromosome segregation during mitosis (Chaudhury et al., 2013; Naim & Rosselli, 2009). The FA pathway is also involved in the resolution of DNA–RNA hybrids, which arise from impaired transcription or replication–transcription collisions (García-Rubio et al., 2015; Okamoto et al., 2018; Schwab et al., 2015). Recently, it was discovered that the FA pathway promotes the restart of stalled replication forks (Xu et al., 2021). Here, we summarize the functions of the FA pathway in ICL repair and replication fork restart, and propose a unified model for those processes. In addition, we analyze and summarize the endogenous DNA lesions which cause FA, and provide a new idea for preventing and treating FA.
FA genes and proteins
There are at least 22 known genes associated with FA. However, if stringent criteria are followed, which state that the mutated gene must be found in at least two patients with bone marrow failure and result in a positive chromosome fragility test, some FA genes should be classified as FA-like genes, because they don’t result in the traditional bone marrow failure phenotype. Such genes include BRCA1 (also known as FANCS), RAD51 and the RAD51 paralog RAD51C (also known as FANCO). Although FANCM associates with FA proteins, its mutations were only found in one FA patient, who also carries biallelic FANCA mutations (Meetei et al., 2005). Two Finnish individuals with loss-of-function mutations in FANCM do not show FA phenotype (Lim et al., 2014). Proteins encoded by FA genes are involved in the FA pathway, which coordinates multiple layers of signaling in response to DNA damage and enzymatic DNA repair processes. The FA core complex, which comprises eight FA proteins (FANCA, B, C, E, F, G, L, M) and associated subunits (FAAP100, FAAP24 and FAAP20) functions as a large ubiquitin ligase for the FANCD2–FANCI (hereafter referred to as FANCD2-I) heterodimer during the DNA damage response (Walden & Deans, 2014). The remaining FA proteins catalyze nucleolytic, synthetic and recombination reactions downstream of FANCD2-I in the FA pathway.
The function of the FA pathway
The FA pathway for repairing ICLs
ICLs are extremely toxic DNA lesions that create an impassable roadblock to DNA replication. The function of the FA pathway in removing the roadblock induced by ICLs has been extensively studied. Several proteins are required at different steps in the process, including lesion recognition and initiation of repair, FANCD2-I mono-ubiquitination by the FA core complex, nucleases recruitment and incision, lesion bypass by translesion synthesis and homologous recombination.
Lesion recognition and initiation
This pathway mainly occurs in the S phase of the cell cycle, when DNA replication forks stall at the ICLs. FANCM was an early candidate for lesion sensing. It is a DNA-binding protein containing a helicase motif that recognizes the stalled fork structure together with Fanconi anemia-associated protein 24 (FAAP24) and the histone fold proteins MHF1 (also known as FAAP16 or CENPS) and MHF2 (also known as FAAP10 or CENPX). The FANCM–FAAP24 complex recruits replication protein A or Telo2/HCLK2, thereby promoting ATR–CHK1 checkpoint activation (Collis et al., 2008; Huang et al., 2010). MHF1 and MHF2 assemble into a heterodimer that binds DNA and enhances the DNA branch migration activity of FANCM to stimulate its association with chromatin (Singh et al., 2010). Once bound to chromatin, FA signaling is activated and FANCM functions as a landing platform for the FA core complex. Moreover, FANCM interacts with proliferation cell nuclear antigen (PCNA) to aid replisome to traverse ICLs via its translocase and DNA binding activities (Rohleder et al., 2016). FANCM also links the FA core complex and the BTRR (BLM-TOP3a-RMI1-RMI2) complex, forming a supercomplex BRAFT, which facilitates both repair and traverse of ICLs (Ling et al., 2016; Meetei et al., 2003).
In Xenopus egg extracts, the recognition of ICLs requires converging replication forks (Zhang et al., 2015). In this stalled replication fork, the CMG replicative helicase complex (comprised of Cdc45, MCM2-7, and GINS), which is a 3' to 5' ssDNA translocase, needs to be unloaded for the leading strand to approach the ICLs (Fu et al., 2011). The RING-type ubiquitin E3 ligase TRAIP is required for polyubiquitination of MCM2-7 and to promote unloading of CMG by the p97 ATPase, which increases the accessibility of the stalled fork for FANCD2-I (Wu et al., 2019). BRCA1 was also shown to promote CMG unloading, but a later study from the same group indicated that this result may be due to inadvertent depletion of TRAIP with BRCA1 antiserum (Wu et al., 2019). However, several lines of in vivo evidence shows that BRCA1 is required for the efficient recruitment of FANCD2-I to DNA damage sites (Garcia-Higuera et al., 2001; Vandenberg et al., 2003; Xu et al., 2021). Interestingly, 53BP1 negatively regulates this recruitment by antagonizing BRCA1, possibly by controlling the accessibility of the stalled forks for FANCD2-I and subsequent nucleases (Xu et al., 2021).
FANCD2/I mono-ubiquitination by the FA core complex
The FANCM–FAAP24–MHF1/2 complex acts as a landing platform to recruit the FA core complex to ICLs (Ciccia et al., 2007; Meetei et al., 2005; Yan et al., 2010). The FA core complex, which is composed of at least eight FA proteins, namely FANCA, B, C, E, F, G, L, M, together with the FA-associated proteins FAAP100 and FAAP20 function as a large ubiquitin ligase for two other FA proteins, FANCD2 and FANCI (Ling et al., 2007; Meetei et al., 2003; Walden & Deans, 2014; Yan et al., 2012). Structure analysis reveals an asymmetric assembly of the core complex with two copies of all subunits except for FANCC, FANCE and FANCF (Wang et al., 2021). FANCL, a member of FA core complex that has the ubiquitin ligase activity, together with the corresponding E2 ubiquitin-conjugating enzyme UBE2T (also known as FANCT) or UBE2W is responsible for the mono-ubiquitination of FANCD2 and FANCI at lysine 561 and lysine 523, respectively (Alpi et al., 2008; Hira et al., 2015; Meetei et al., 2003; Rickman et al., 2015). The accumulation of ubiquitinated FANCD2-I at the ICL lesion is the key step in the FA pathway; after this step downstream nucleases and other repair proteins are recruited to complete the ICL repair. The cryo-EM structure of FANCD2-I revealed that the ubiquitination of FANCD2-I occurs after it is recruited to the ICLs (Liang et al., 2016). Another cryo-EM structural analysis found that mono-ubiquitination triggers a complete rearrangement of FANCD2-I from an open structure to a closed ring structure that encircles the DNA, and this ring structure can act as a clamp that can slide on DNA to coordinate downstream repair factors (Alcon et al., 2020; Wang et al., 2020). The appropriate functions of ubiquitylation and deubiquitylation are crucial for the normal functioning of the FA pathway, but the timings and locations of these steps are incompletely understood. It is known that ubiquitin carboxy-terminal hydrolase 1 (USP1) and USP1-associated factor (UAF1) are critical for FANCD2-I de-ubiquitination (Cohn et al., 2007).
DNA incision
Ubiquitinated FANCD2/I at ICLs can recruit various DNA repair proteins, including structure-specific nucleases and translesion synthesis polymerases and other repair factors (Knipscheer et al., 2009; Yang et al., 2015). Structure-selective nucleases have critical roles in unhooking the stalled forks. In a cell-free system, it was shown that ubiquitinated FANCD2 recruits the nuclease scaffold protein SLX4 (also known as FANCP) and its associated 3’-flap structure-specific endonuclease XPF (also known as ERCC4 or FANCQ)–ERCC1 heterodimer to ICLs (Klein Douwel et al., 2014). In addition, SLX4 interacts and activates MUS81–EME1 endonucleases and SLX1 (Fekairi et al., 2009). The N-terminal region of SLX4 contains a ubiquitin-binding zinc finger domain, which is required for interaction of SLX4 with ubiquitylated FANCD2 and its recruitment to ICL sites in DT40 cells (Yamamoto et al., 2011). However, this result could not be verified in other systems (Tan et al., 2020; Xu et al., 2021). FAN1 (Fanconi anemia-associated nuclease 1), another structure-specific nuclease that can be recruited to ICLs by mono-ubiquitinated FANCD2-I, also has a crucial role in replication fork unhooking. FAN1 interacts directly with mono-ubiquitinated FANCD2/I in a manner dependent on its ubiquitin-binding domain, and possesses intrinsic 5'-3' exonuclease activity and endonuclease activity that cleaves nicked and branched DNA structures (Liu et al., 2010; Smogorzewska et al., 2010). Recent structural and biochemical studies showed that mono-ubiquitination is required for the conformational change of FANCD2-I into a clamp structure for double-stranded DNA binding, but not for the interaction of FANCD2-I with SLX4 and FAN1 (Alcon et al., 2020; Tan et al., 2020; Wang et al., 2020; Xu et al., 2021). Moreover, BRCA1 promotes the recruitment of SLX4 to stalled replication forks through an interaction of their N-terminal regions (Xu et al., 2021).
Cells from individuals with FAN1 mutations do not exhibit chromosome breakage or cell-cycle arrest after treatment with ICL-inducing agents, unlike other FA-mutated cells. Rather, mutations in FAN1 cause the rare kidney disease karyomegalic interstitial nephritis (Zhou et al., 2012). Moreover, one study showed that the recruitment of Fan1 by ubiquitinated Fancd2 is dispensable for ICL repair and the FAN1 ubiquitin-binding zinc finger domain is not required for efficient ICL repair (Lachaud et al., 2016). These results indicate that FAN1 may be involved in other aspects of ICL repair independently of the FA pathway. Deletion of nucleases such as ERCC1-XPF, MUS81, FAN1, SNM1A and SLX1 renders cells sensitive to ICL-inducing agents, indicating each of these nucleases are involved in ICL repair. However, there is no experimental system available to determine where the specific location of where these nucleases unhook from the ICLs at parental DNA strands, or to determine the length of the duplex surrounding an unhooked ICL in the product of this reaction.
Lesion bypass and repair
The incision step generates a double-strand DNA break at the stalled replication fork, and the ICLs remain at opposite parental DNA strands. The nucleotide containing the damaged base needs to be bypassed for DNA replication. The nascent leading strand is restored by the translesion DNA synthesis, which involves multiple polymerases. The bypass step is executed by REV1 and the extension step is executed by REV3 and REV7 (subunits of Pol ζ) (Roy & Schärer, 2016). The translesion synthesis polymerases recognize mono-ubiquitylated PCNA through its ubiquitin-binding domains and can be recruited to ICL repair intermediates (Bomar et al., 2007; Kannouche et al., 2004). RFWD3 (also known as FANCW) promotes ubiquitylation of proteins on ssDNA to drive recruitment of effectors of PCNA ubiquitylation and DNA damage bypass (Gallina et al., 2021). In addition, lesion bypass by REV1 requires its interaction with FAAP20 and is promoted by FA core complex (Budzowska et al., 2015; Kim et al., 2012). Translesion DNA synthesis bypasses the ICLs and the repaired DNA strand can function as a template for subsequent homologous recombination.
The double-stranded breaks at the stalled fork induced by the incision step need to be repaired by homologous recombination for the repair of ICL to be completed. The initiation of homologous recombination repair is an end-resection step finished by CTIP (also known as RBBP8), which is recruited by FANCD2/I (Murina et al., 2014; Unno et al., 2014). The MRN complex (MRE11–RAD50–NBS1) and DNA2 also have a role in the resection step. Strand invasion is promoted by RAD51 (also known as FANCR). PALB2 (also known as FANCN) binds directly to BRCA2 (also known as FANCD1) and BRCA1 to promote the formation of RAD51 nucleofilaments; this step also needs the help of DNA helicase BRIP1 (also known as FANCJ), the RAD51 paralogs RAD51C and XRCC2 (also known as FANCU) (Clauson et al., 2013; Kottemann & Smogorzewska, 2013; Michl et al., 2016).
The FA/break-induced replication (BIR) pathway in the replication stress response
FA proteins in replication stress
In addition to repairing ICLs, it is well-established that the FA pathway is activated by replication stress. Replication forks are vulnerable to stalling upon encountering various obstacles, including damaged template DNA, protein–DNA crosslinks, RNA–DNA hybrids (R-loops), collisions with transcription complexes, difficult-to-replicate DNA sequences, and replication inhibitors (Cortez, 2015). Hydroxyurea and aphidicolin, which induce replication fork stalling but do not cause ICLs, strongly activate the FA pathway (Howlett et al., 2005). Common fragile sites (CFSs) are specific chromosome loci that contain an increased frequency of gaps and breaks on metaphase chromosomes following partial inhibition of DNA synthesis. FANCD2 and FANCA maintain the stability of CFS under mild replication stress (Howlett et al., 2005), and FANCD2 promotes efficient DNA replication across CFSs (Madireddy et al., 2016). Moreover, FANCD2 and FANCI localize to under-replicated regions of CFS sites during mitotic DNA synthesis, failure in which induces ultra-fine DNA bridges and causes abnormal chromosome separation (Minocherhomji et al., 2015). FANCD2-I also forms sister foci at the root of ultra-fine bridges, promoting the resolution of ultra-fine bridges and suppressing aneuploidy (Chan et al., 2009; Naim & Rosselli, 2009). Consistent with this observation, chromatin immunoprecipitation sequence analysis revealed that upon mild replication stress, FANCD2 mostly accumulates to the middle portion of large transcribed genes, which often correspond to known CFSs (Okamoto et al., 2018). Transcription bubbles often collide with replication forks in the S phase at CFS sites, causing the formation of R-loops. Consistent with this finding, FA proteins protect genome integrity by resolving R-loops (García-Rubio et al., 2015; Okamoto et al., 2018; Schwab et al., 2015). All those studies suggest that FA proteins are important in the cellular response to replication stress. However, the molecular mechanisms underlying this response remain elusive, and it was unclear whether the replication stress response mechanisms are different from those that function in ICL repair (Ceccaldi et al., 2016; Duxin & Walter, 2015; Michl et al., 2016).
The FA/BIR pathway in the restart of stalled replication forks
Previously, we found that there are two major pathways for restarting stalled replication forks (Fig. 1) (Xu et al., 2017). One is the 53BP1-dependent cleavage-free pathway, which acts at the early stage of replication stress. The other is the BRCA1-dependent BIR-like pathway, which is a late-acting pathway. BIR is a unique recombination mechanism that repairs one-ended DNA breaks. The canonical concept of BIR is that it repairs one-ended breaks resulting from a passive breakage of stalled replication forks, fork collapse, or telomere erosion (Kramara et al., 2018). The BRCA1-dependent BIR pathway is activated differently: it is initiated by active and programmed fork cleavage (Xu et al., 2017). BRCA1 antagonizes 53BP1 to promote the cleavage of stalled replication forks and initiation of the BIR pathway. This occurs similarly to the role of BRCA1 in homologous recombination-mediated double-strand break repair by promoting DNA end resection (Xu et al., 2017). The switch between the 53BP1-dependent cleavage-free pathway and the BRCA1-dependent BIR-like pathway is regulated by PLK1/CDK1 activity over time of replication stress.
Fig. 1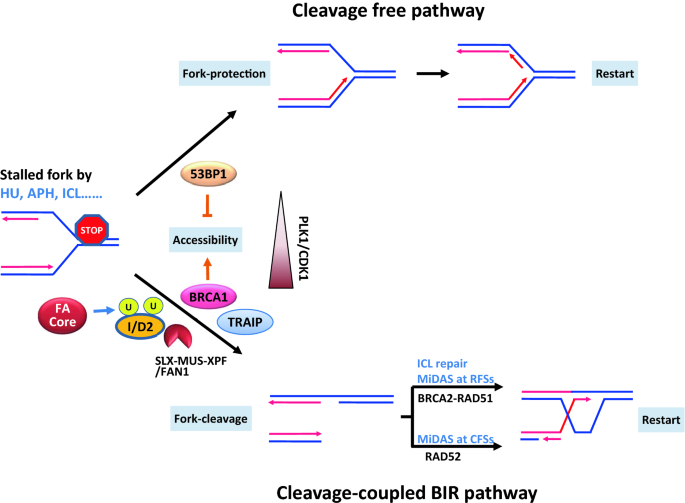
A unified model of FA proteins in fork restart. There are two major restart pathways for stalled replication forks: the cleavage free pathway and cleavage- coupled BIR pathway. 53BP1 and BRCA1 antagonistically regulate the two pathway by controlling the accessibility of the stalled forks for downstream proteins FANCD2-I and endonucleases. The BIR pathway is promoted by PLK1/CDK1 activity when stress time is prolonged. TRAIP also contributes the accessibility by unloading MCM complex. The recombination in the BIR pathway is dependent on BRCA1-RAD51 or RAD52 possibly based on the structure of the stalled forks at different situations and chromosome sites
Full size imageRecently, we showed that the functions of the FA pathway in response to replication stress and ICL repair derive from one unified molecular mechanism (Xu et al., 2021). The FA pathway and the BIR pathway share almost identical mechanisms in DNA damage response signaling and subsequent fork cleavage/unhooking. Both pathways are antagonistically controlled by BRCA1 and 53BP1, require TRAIP-mediated disassembly of the replisome and mono-ubiquitination of the FANCD2-I complex, and are executed by the SLX4 complex or/and FAN1 for fork cleavage (Sonneville et al., 2019; Wu et al., 2019; Xu et al., 2021). Moreover, both pathways require POLD3 for DNA synthesis (Xu et al., 2021). However, the pathways have different recombination reactions: while ICL repair and the FA pathway depend on the activity of BRCA2-RAD51 (Ceccaldi et al., 2016; Deans & West, 2011; Wang, 2007), the BIR pathway contains RAD52- or RAD51-dependent mechanisms based on the structures of the stalled replication forks (Garribba et al., 2020). Therefore, the FA pathway may be one of the BIR pathways (Fig. 1).
Research over the past two decades has established that FA-deficient cells (except for FANCJ- or FANCD2-deficient cells) are not sensitive to aphidicolin- or hydroxyurea-induced replication stress, although they are sensitive to ICL-induced replication stress (Chen et al., 2016; Schlacher et al., 2012; Tian et al., 2017). We discovered that FA-mutated cells are hypersensitive to persistent replication stress induced by a low concentration of hydroxyurea or aphidicolin, which also triggers bone marrow failure in FA-deficient mice (Xu et al., 2021). Moreover, acute myeloid leukemia patients with FA have an elevated frequency of monosomy 7 (loss of one chromosome 7) compared with de novo acute myeloid leukemia cases (Rochowski et al., 2012). Monosomy 7 is associated with an increased risk of developing myelodysplastic syndrome/acute myeloid leukemia and a poor prognosis in FA patients (Mehta et al., 2010). However, the elevated frequency of monosomy 7 in FA is unlikely to arise from defects in ICL repair. We found that replication stress induced by aphidicolin, but not ICLs, causes monosomy 7 in FA-deficient cells due to dense CFSs on chromosome 7, resembling that seen in FA patients (Xu et al., 2021). Therefore, physiological replication stress, which resembles that replication stress induced by aphidicolin but not by ICLs, may induce at least some symptoms in FA patients.
FA proteins in fork protection
Upstream FA proteins, including the FA core complex and FANCD2-I, and a subset of homologous recombination proteins, including BRCA1, BRCA2 and RAD51, protect the nascent DNA strands of stalled replication forks from nucleolytic degradation (Schlacher et al., 2011, 2012). Although degradation of nascent DNA has been reported to correlate with cellular sensitivity to genotoxic drugs in mouse embryonic stem cells and some cancer cells (Ray Chaudhuri et al., 2016), it does not significantly increase cell death or even chromosomal breakage (Feng & Jasin, 2017; Rickman et al., 2020). Therefore, nascent DNA degradation is unlikely responsible for the exhaustion of hematopoietic stem cells in FA patients. Moreover, the structure-specific endonucleases SLX4/FANCP and XPF/FANCQ, which execute fork cleavage/unhooking in the FA pathway, are not required to protect replication forks from nascent DNA degradation, and conversely, they promote nascent DNA degradation in WRNIP-deficient cells (Porebski et al., 2019). Therefore, nascent DNA protection only requires a subset of FA proteins, and protecting nascent DNA is not the major function of the entire FA pathway.
Independent functions of FA proteins in response to replication stress
Both FANCD2 and FANCI have been reported to fulfil several of their functions independently of each other and independently of mono-ubiquitination by the FA core complex. In response to low levels of replication stress, FANCI and FANCD2 have opposite functions in dormant origin firing (Chen et al., 2015). FANCI, but not FANCD2, is required for efficient foci formation of FA core complex (Castella et al., 2015). FANCD2 interacts with the MCM2-MCM7 replicative helicase and is required for replisome surveillance independently of FANCD2 mono-ubiquitination (Lossaint et al., 2013). Moreover, FANCD2 mono-ubiquitination-independent functions are essential for cellular resistance to pulsed replication stress (Tian et al., 2017).
Causes of FA
Most FA patients develop bone marrow failure with age. At birth, blood counts are normal, but by a median age of 7 years, the hematopoietic system becomes dysfunctional. The fact that all blood lineages eventually become deficient strongly implies hematopoietic stem cell dysfunction. The bone marrow CD34( +) cell compartment is significantly depleted in FA patients even before severe pancytopenia is present (Kelly et al., 2007). Indeed the FA pathway likely has a crucial role in hematopoiesis in humans, as knockdown of FANCA and FANCD2 in human embryonic stem cells reduces hematopoietic fates and progenitor numbers (Tulpule et al., 2010). These findings highlight two questions: Is unresolved DNA damage the initiator of bone marrow failure? And what is the nature of DNA damage in hematopoietic stem cells?
Based on the notion that FA patient-derived cells are hypersensitive to ICL-inducing agents, it is widely believed that FA is caused by defective repair of endogenously produced ICLs, although the endogenous mutagen that generates these lesions remains elusive. We revealed that FA-deficient cells are hypersensitive to persistently low levels of replication stress (Xu et al., 2021), implying that physiological replication stress may be responsible for FA symptoms. Replication stress is possibly a more general cause of FA symptoms, since ICLs also mediate replication stress to replication forks. Replication stress is a better candidate for the cause of some FA symptoms, such as monosomy 7, than ICLs. Indeed, replication stress is very common in stem cells, particularly in hematopoietic stem cells (Flach & Milyavsky, 2018). Rapid cell-cycle progression and a short G1 phase impose constitutive replication stress and fork remodeling in mouse embryonic stem cells (Ahuja et al., 2016). In addition, replication stress limits fetal erythropoiesis and of hematopoietic stem cells (Alvarez et al., 2015), and is also a potent driver of functional decline in ageing haematopoietic stem cells (Flach et al., 2014). Moreover, additional causes of FA are gradually emerging, including reactive aldehydes, oxidative stress and other factors.
Reactive aldehydes
Increased reactive aldehyde levels are the best candidates for the pathogenesis of FA, since the elevated levels of reactive aldehydes can form a wide variety of DNA adducts including ICLs (Voulgaridou et al., 2011). A series of mouse genetic studies from the Patel laboratory connected the FA pathway with aldehyde metabolism. Initial research showed that chicken cells (DT40) deficient in the BRCA/FANC pathway are hypersensitive to formaldehyde (Ridpath et al., 2007). Patel and colleagues then established double mutant deficient in the acetaldehyde detoxifying enzyme aldehyde dehydrogenase 2 (Aldh2) and the FA gene FANCD2 to explore the interplay between aldehyde metabolism and the FA pathway in mice. Their data showed that Aldh2 is essential for the development of Fancd2 mutant embryos. Aldh2−/− Fancd2−/− mice spontaneously develop acute leukemia, and more rapidly develop bone marrow failure after ethanol consumption than single mutant mice (Langevin et al., 2011). This finding suggests that acetaldehyde might be able to cause the offending lesion that causes FA. Consistent with this finding, the severity of FA patients correlates with the presence of a dominant-negative polymorphism (E487K), which causes a dramatic decrease in ALDH2 activity and is present in 36% of the population in East Asia (Hira et al., 2013). In subsequent research, Patel and colleagues also examined mice deficient in alcohol dehydrogenase 5 (Adh5) and Fancd2 genes. Double mutant mice were viable, but the average life expectancy was shorter than Aldh2−/− Fancd2−/− mice, and depletion of hematopoietic stem cells happened much earlier than in Aldh2−/− Fancd2−/− mice (Pontel et al., 2015). These findings suggest that formaldehyde might be more cytotoxic than acetaldehyde or that endogenous formaldehyde might be produced at higher levels than endogenous acetaldehyde. More recently, the Patel lab established mice lacking two aldehyde detoxifying enzymes: Aldh2 and Adh5. Double mutant mice have greatly shortened lifespans compared with single knock-out mice and develop leukemia. They also found that formaldehyde is a common substrate of ALDH2 and ADH5 (Dingler et al., 2020), which indicates formaldehyde might be the primary culprit underlying the pathogenesis of FA. Certain metabolic processes such as alcohol metabolism, histone demethylation, lipid peroxidation can produce formaldehyde and acetaldehyde (Stone et al., 2008). Lei Li’s group showed that during the differentiation of hematopoietic stem cells, rapid and extensive transcription reprogramming poses a major threat to genome stability and cell viability in the absence of the FA pathway, since oxidative protein demethylation that occurs during the regulation of transcription produces formaldehyde (Shen et al., 2020). Although the relationship between aldehydes and FA is well-studied, aldehydes not only cause ICLs, but also DNA–protein crosslinks (DPCs) and base adducts (Cohen Hubal et al., 1997; Duxin & Walter, 2015). A key question remains unanswered, namely what is the nature form of DNA damage of aldehydes in causing FA? It has been suggested that aldehyde-induced DNA damage may not include ICLs, because cells mutant for translesion synthesis or homologous recombination, which play critical roles in ICL repair, are not sensitive to aldehydes (Langevin et al., 2011; Oberbeck et al., 2014; Rosado et al., 2011).
DPCs are ubiquitous in cells. The formation of DPCs requires the proteins to be close to DNA, then the two molecules covalently and irreversibly bond together through a chemical reaction that engages an endogenous or exogenous crosslinker (Vaz et al., 2017). They are cytotoxic as they create physical obstacles to the progression of DNA replication and transcription. If DPCs are not repaired, then replication forks stall, which finally leads to DNA double-strand breaks. Reactive aldehydes, particularly formaldehyde, induce DPCs as well as ICLs (Klages-Mundt & Li, 2017). In addition, ICLs-inducing drugs also induce DPCs. These findings indicate that the FA pathway may be strongly activated by DPCs as well as ICLs (Chválová et al., 2007). Despite these initial findings, the connection between the FA pathway and DPC repair remains unclear. Some studies have shown that the FA pathway plays a dispensable role in DPC repair, as knockdown of FANCD2 doesn’t sensitize cells to DPCs (Vaz et al., 2016). And knockout of FANCD2 in mouse embryonic fibroblasts did not impact DPC repair. (Stingele et al., 2016). However, several lines of evidence indicate that FA proteins are important in DPC repair. FANCG-deficient cells are hypersensitive to the cytotoxic effects of 5-aza-2'-deoxycytidine, which is a DNA methyltransferase inhibitor that is incorporated into DNA and traps DNA methyltransferase. FANCG-knockout cells fail to trigger homologous recombination-mediated repair after 5-aza-2'-deoxycytidine treatment, which leads to accumulation of chromatid breaks and inter-chromosomal radial fusions (Orta et al., 2013). The different conclusions drawn from the studies described above may be due to the use of different drugs to induce DPCs; some drugs not only induce DPCs but also lead to other lesions. Further research in this area will help elaborate our understanding of the connection between the FA pathway and DPC repair.
Mono-adducts, which can stall replication forks, are the major DNA lesions induced by aldehydes. Although the FA pathway is not required for the repair of DNA mono-adducts, further research is needed to determine if the FA pathway is required for the restart of monoadduct-stalled forks.
Although increasing evidence shows that elevated levels of reactive aldehydes underlie the pathology of FA, almost all results were acquired in an ALDH2- or ADH5-deficient background. Therefore it is unclear if aldehydes are the major cause of pathology in FA patients, who have normal ALDH2 and ADH5 to mediate aldehyde metabolism.
Oxidative stress and inflammatory cytokines
Oxidative stress is defined as an imbalance between the production of reactive oxygen species (ROS) and antioxidant defense systems. Oxidative stress is common to many human diseases, and ROS are the most pervasive types of reactive molecules in human cells. They are generated as a by-product of lipid peroxidation and the electron transport chain, and can impact not only DNA, but also RNA and cellular proteins. Many studies have indicated the crucial connection between FA and oxidative stress, which is an important factor in the pathogenesis of FA (Bogliolo et al., 2002; Cohen-Haguenauer et al., 2006; Cumming et al., 1996; Futaki et al., 2002; Hadjur et al., 2001; Kruyt et al., 1998; Pagano et al., 2005; Park et al., 2004; Saadatzadeh et al., 2004). FANCC knockout in primary hematopoietic progenitors and murine embryonic fibroblasts makes the cells hypersensitive to oxidative stress induced by hydrogen peroxide. Furthermore, pretreatment with N-acetylcysteine or the antioxidant selenomethionine preferentially enhanced the survival of Fancc−/− cells (Saadatzadeh et al., 2004). Knocking out superoxide dismutase 1, which is a key enzyme that protecting against ROS-mediated damage, in Fancc−/− mice causes bone marrow hypocellularity, where erythroid, myeloid, and early-B lymphoid colonies show decreased proliferation and survival capabilities (Hadjur et al., 2001). Together, these findings provide direct and indirect evidence that oxidative stress is another potential candidate that underlies the pathogenesis of FA, although the type of DNA damage induced by oxidative stress to activate the FA pathway remains unclear.
FA cells display hypersensitivity to certain cytokines, such as tumor necrosis factor α and interferon γ (Haneline et al., 1998; Rathbun et al., 1997; Wang et al., 1998). These cytokines also induce bone marrow dysfunction in FA-deficient mice (Li et al., 2004; Si et al., 2006). One hypothesis is that cytokines stimulate the production of ROS, which induces certain types of DNA damage and leads to bone marrow dysfunction (Walter et al., 2015).
Future perspectives
A unified molecular mechanism of FA proteins in ICL repair and stalled fork restart has been established. This unified mechanism centres on the function of FA proteins in the cleavage-coupled BIR pathway, which plays a more general role in the restart of stalled replication fork restarts. The differences between the FA pathway and other BIR pathways remain elusive, particularly with regards to the recombination step that involves RAD51–BRCA1 and RAD52. Moreover, the relationship between BRCA1 and TRAIP to promote accessibility of stalled forks for FANCD2-I is unclear. More importantly, in light of the discovery of a new role for FA proteins in BIR, it remains to be investigated whether all DNA lesions that are induced by aldehydes, oxidative stress or other factors capable of causing replication stress jointly contribute to the symptoms of FA.
References
Ahuja, A. K., et al. (2016). A short G1 phase imposes constitutive replication stress and fork remodelling in mouse embryonic stem cells. Nature Communications, 7, 10660. https://doi.org/10.1038/ncomms10660
Alcon, P., et al. (2020). FANCD2-FANCI is a clamp stabilized on DNA by monoubiquitination of FANCD2 during DNA repair. Nature Structural & Molecular Biology. https://doi.org/10.1038/s41594-020-0380-1
Alpi, A. F., Pace, P. E., Babu, M. M., & Patel, K. J. (2008). Mechanistic insight into site-restricted monoubiquitination of FANCD2 by Ube2t, FANCL, and FANCI. Molecular Cell, 32, 767–777. https://doi.org/10.1016/j.molcel.2008.12.003
Alvarez, S., et al. (2015). Replication stress caused by low MCM expression limits fetal erythropoiesis and hematopoietic stem cell functionality. Nature Communications, 6, 8548. https://doi.org/10.1038/ncomms9548
Bogliolo, M., et al. (2002). The Fanconi anaemia genome stability and tumour suppressor network. Mutagenesis, 17, 529–538. https://doi.org/10.1093/mutage/17.6.529
Bomar, M. G., Pai, M. T., Tzeng, S. R., Li, S. S., & Zhou, P. (2007). Structure of the ubiquitin-binding zinc finger domain of human DNA Y-polymerase eta. EMBO Reports, 8, 247–251. https://doi.org/10.1038/sj.embor.7400901
Budzowska, M., Graham, T. G., Sobeck, A., Waga, S., & Walter, J. C. (2015). Regulation of the Rev1-pol ζ complex during bypass of a DNA interstrand cross-link. The EMBO Journal, 34, 1971–1985. https://doi.org/10.15252/embj.201490878
Castella, M., et al. (2015). FANCI regulates recruitment of the FA core complex at sites of DNA damage independently of FANCD2. PLoS Genetics, 11, e1005563. https://doi.org/10.1371/journal.pgen.1005563
Ceccaldi, R., Sarangi, P., & D’Andrea, A. D. (2016). The Fanconi anaemia pathway: new players and new functions. Nature Reviews. Molecular Cell Biology, 17, 337–349. https://doi.org/10.1038/nrm.2016.48
Chan, K. L., Palmai-Pallag, T., Ying, S., & Hickson, I. D. (2009). Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nature Cell Biology, 11, 753–760. https://doi.org/10.1038/ncb1882
Chaudhury, I., Sareen, A., Raghunandan, M., & Sobeck, A. (2013). FANCD2 regulates BLM complex functions independently of FANCI to promote replication fork recovery. Nucleic Acids Research, 41, 6444–6459. https://doi.org/10.1093/nar/gkt348
Chen, Y. H., et al. (2015). ATR-mediated phosphorylation of FANCI regulates dormant origin firing in response to replication stress. Molecular Cell, 58, 323–338. https://doi.org/10.1016/j.molcel.2015.02.031
Chen, X., Bosques, L., Sung, P., & Kupfer, G. M. (2016). A novel role for non-ubiquitinated FANCD2 in response to hydroxyurea-induced DNA damage. Oncogene, 35, 22–34. https://doi.org/10.1038/onc.2015.68
Chválová, K., Brabec, V., & Kaspárková, J. (2007). Mechanism of the formation of DNA-protein cross-links by antitumor cisplatin. Nucleic Acids Research, 35, 1812–1821. https://doi.org/10.1093/nar/gkm032
Ciccia, A., et al. (2007). Identification of FAAP24, a Fanconi anemia core complex protein that interacts with FANCM. Molecular Cell, 25, 331–343. https://doi.org/10.1016/j.molcel.2007.01.003
Clauson, C., Schärer, O. D., & Niedernhofer, L. (2013). Advances in understanding the complex mechanisms of DNA interstrand cross-link repair. Cold Spring Harbor Perspectives in Biology, 5, a012732. https://doi.org/10.1101/cshperspect.a012732
Cohen Hubal, E. A., Schlosser, P. M., Conolly, R. B., & Kimbell, J. S. (1997). Comparison of inhaled formaldehyde dosimetry predictions with DNA–protein cross-link measurements in the rat nasal passages. Toxicology and Applied Pharmacology, 143, 47–55. https://doi.org/10.1006/taap.1996.8076
Cohen-Haguenauer, O., et al. (2006). In vivo repopulation ability of genetically corrected bone marrow cells from Fanconi anemia patients. Proceedings of the National Academy of Sciences of the United States of America, 103, 2340–2345. https://doi.org/10.1073/pnas.0510613103
Cohn, M. A., et al. (2007). A UAF1-containing multisubunit protein complex regulates the Fanconi anemia pathway. Molecular Cell, 28, 786–797. https://doi.org/10.1016/j.molcel.2007.09.031
Collis, S. J., et al. (2008). FANCM and FAAP24 function in ATR-mediated checkpoint signaling independently of the Fanconi anemia core complex. Molecular Cell, 32, 313–324. https://doi.org/10.1016/j.molcel.2008.10.014
Cortez, D. (2015). Preventing replication fork collapse to maintain genome integrity. DNA Repair, 32, 149–157. https://doi.org/10.1016/j.dnarep.2015.04.026
Cumming, R. C., Liu, J. M., Youssoufian, H., & Buchwald, M. (1996). Suppression of apoptosis in hematopoietic factor-dependent progenitor cell lines by expression of the FAC gene. Blood, 88, 4558–4567.
D’Andrea, A. D. (2010). Susceptibility pathways in Fanconi’s anemia and breast cancer. The New England Journal of Medicine, 362, 1909–1919. https://doi.org/10.1056/NEJMra0809889
Deans, A. J., & West, S. C. (2011). DNA interstrand crosslink repair and cancer. Nature Reviews Cancer, 11, 467–480. https://doi.org/10.1038/nrc3088
Dingler, F. A., et al. (2020). Two aldehyde clearance systems are essential to prevent lethal formaldehyde accumulation in mice and humans. Molecular Cell, 80, 996-1012.e1019. https://doi.org/10.1016/j.molcel.2020.10.012
Duxin, J. P., & Walter, J. C. (2015). What is the DNA repair defect underlying Fanconi anemia? Current Opinion in Cell Biology, 37, 49–60. https://doi.org/10.1016/j.ceb.2015.09.002
Fekairi, S., et al. (2009). Human SLX4 is a Holliday junction resolvase subunit that binds multiple DNA repair/recombination endonucleases. Cell, 138, 78–89. https://doi.org/10.1016/j.cell.2009.06.029
Feng, W., & Jasin, M. (2017). BRCA2 suppresses replication stress-induced mitotic and G1 abnormalities through homologous recombination. Nature Communications, 8, 525. https://doi.org/10.1038/s41467-017-00634-0
Flach, J., & Milyavsky, M. (2018). Replication stress in hematopoietic stem cells in mouse and man. Mutation Research, 808, 74–82. https://doi.org/10.1016/j.mrfmmm.2017.10.001
Flach, J., et al. (2014). Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature, 512, 198–202. https://doi.org/10.1038/nature13619
Fu, Y. V., et al. (2011). Selective bypass of a lagging strand roadblock by the eukaryotic replicative DNA helicase. Cell, 146, 931–941. https://doi.org/10.1016/j.cell.2011.07.045
Futaki, M., et al. (2002). The FANCG Fanconi anemia protein interacts with CYP2E1: possible role in protection against oxidative DNA damage. Carcinogenesis, 23, 67–72. https://doi.org/10.1093/carcin/23.1.67
Gallina, I., et al. (2021). The ubiquitin ligase RFWD3 is required for translesion DNA synthesis. Molecular Cell, 81, 442-458.e449. https://doi.org/10.1016/j.molcel.2020.11.029
Garcia-Higuera, I., et al. (2001). Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Molecular Cell, 7, 249–262. https://doi.org/10.1016/s1097-2765(01)00173-3
García-Rubio, M. L., et al. (2015). The Fanconi Anemia Pathway Protects Genome Integrity from R-loops. PLoS Genetics, 11, e1005674. https://doi.org/10.1371/journal.pgen.1005674
Garribba, L., et al. (2020). Folate stress induces SLX1- and RAD51-dependent mitotic DNA synthesis at the fragile X locus in human cells. Proceedings of the National Academy of Sciences of the United States of America. https://doi.org/10.1073/pnas.1921219117
Giampietro, P. F., et al. (1993). The need for more accurate and timely diagnosis in Fanconi anemia: a report from the International Fanconi Anemia Registry. Pediatrics, 91, 1116–1120.
Hadjur, S., et al. (2001). Defective hematopoiesis and hepatic steatosis in mice with combined deficiencies of the genes encoding Fancc and Cu/Zn superoxide dismutase. Blood, 98, 1003–1011. https://doi.org/10.1182/blood.v98.4.1003
Haneline, L. S., et al. (1998). Multiple inhibitory cytokines induce deregulated progenitor growth and apoptosis in hematopoietic cells from Fac-/- mice. Blood, 91, 4092–4098.
Hira, A., et al. (2013). Variant ALDH2 is associated with accelerated progression of bone marrow failure in Japanese Fanconi anemia patients. Blood, 122, 3206–3209. https://doi.org/10.1182/blood-2013-06-507962
Hira, A., et al. (2015). Mutations in the gene encoding the E2 conjugating enzyme UBE2T cause Fanconi anemia. American Journal of Human Genetics, 96, 1001–1007. https://doi.org/10.1016/j.ajhg.2015.04.022
Howlett, N. G., Taniguchi, T., Durkin, S. G., D’Andrea, A. D., & Glover, T. W. (2005). The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Human Molecular Genetics, 14, 693–701. https://doi.org/10.1093/hmg/ddi065
Huang, M., et al. (2010). The FANCM/FAAP24 complex is required for the DNA interstrand crosslink-induced checkpoint response. Molecular Cell, 39, 259–268. https://doi.org/10.1016/j.molcel.2010.07.005
Kannouche, P. L., Wing, J., & Lehmann, A. R. (2004). Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Molecular Cell, 14, 491–500. https://doi.org/10.1016/s1097-2765(04)00259-x
Kee, Y., & D’Andrea, A. D. (2012). Molecular pathogenesis and clinical management of Fanconi anemia. The Journal of Clinical Investigation, 122, 3799–3806. https://doi.org/10.1172/jci58321
Kelly, P. F., et al. (2007). Stem cell collection and gene transfer in Fanconi anemia. Molecular Therapy: THe Journal of the American Society of Gene Therapy, 15, 211–219. https://doi.org/10.1038/sj.mt.6300033
Kim, H., Yang, K., Dejsuphong, D., & D’Andrea, A. D. (2012). Regulation of Rev1 by the Fanconi anemia core complex. Nature Structural & Molecular Biology, 19, 164–170. https://doi.org/10.1038/nsmb.2222
Klages-Mundt, N. L., & Li, L. (2017). Formation and repair of DNA-protein crosslink damage. Science China. Life Sciences, 60, 1065–1076. https://doi.org/10.1007/s11427-017-9183-4
Klein Douwel, D., et al. (2014). XPF-ERCC1 acts in Unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Molecular Cell, 54, 460–471. https://doi.org/10.1016/j.molcel.2014.03.015
Knipscheer, P., et al. (2009). The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science (new York, N.y.), 326, 1698–1701. https://doi.org/10.1126/science.1182372
Kottemann, M. C., & Smogorzewska, A. (2013). Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature, 493, 356–363. https://doi.org/10.1038/nature11863
Kramara, J., Osia, B., & Malkova, A. (2018). Break-induced replication: the where, the why, and the how. Trends in Genetics, 34, 518–531. https://doi.org/10.1016/j.tig.2018.04.002
Kruyt, F. A., et al. (1998). Abnormal microsomal detoxification implicated in Fanconi anemia group C by interaction of the FAC protein with NADPH cytochrome P450 reductase. Blood, 92, 3050–3056.
Kutler, D. I., et al. (2003a). High incidence of head and neck squamous cell carcinoma in patients with Fanconi anemia. Archives of Otolaryngology Head & Neck Surgery, 129, 106–112. https://doi.org/10.1001/archotol.129.1.106
Kutler, D. I., et al. (2003b). A 20-year perspective on the International Fanconi Anemia Registry (IFAR). Blood, 101, 1249–1256. https://doi.org/10.1182/blood-2002-07-2170
Kutler, D. I., et al. (2016). Natural history and management of Fanconi anemia patients with head and neck cancer: A 10-year follow-up. The Laryngoscope, 126, 870–879. https://doi.org/10.1002/lary.25726
Lachaud, C., et al. (2016). Ubiquitinated Fancd2 recruits Fan1 to stalled replication forks to prevent genome instability. Science (new York, N.y.), 351, 846–849. https://doi.org/10.1126/science.aad5634
Langevin, F., Crossan, G. P., Rosado, I. V., Arends, M. J., & Patel, K. J. (2011). Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature, 475, 53–58. https://doi.org/10.1038/nature10192
Li, X., et al. (2004). Continuous in vivo infusion of interferon-gamma (IFN-gamma) preferentially reduces myeloid progenitor numbers and enhances engraftment of syngeneic wild-type cells in Fancc-/- mice. Blood, 104, 1204–1209. https://doi.org/10.1182/blood-2004-03-1094
Liang, C. C., et al. (2016). The FANCD2-FANCI complex is recruited to DNA interstrand crosslinks before monoubiquitination of FANCD2. Nature Communications, 7, 12124. https://doi.org/10.1038/ncomms12124
Lim, E. T., et al. (2014). Distribution and medical impact of loss-of-function variants in the Finnish founder population. PLoS Genetics, 10, e1004494. https://doi.org/10.1371/journal.pgen.1004494
Ling, C., et al. (2007). FAAP100 is essential for activation of the Fanconi anemia-associated DNA damage response pathway. The EMBO Journal, 26, 2104–2114. https://doi.org/10.1038/sj.emboj.7601666
Ling, C., et al. (2016). Bloom syndrome complex promotes FANCM recruitment to stalled replication forks and facilitates both repair and traverse of DNA interstrand crosslinks. Cell Discovery, 2, 16047. https://doi.org/10.1038/celldisc.2016.47
Liu, T., Ghosal, G., Yuan, J., Chen, J., & Huang, J. (2010). FAN1 acts with FANCI-FANCD2 to promote DNA interstrand cross-link repair. Science (new York, NY), 329, 693–696. https://doi.org/10.1126/science.1192656
Lobitz, S., & Velleuer, E. (2006). Guido Fanconi (1892–1979): a jack of all trades. Nature Reviews Cancer, 6, 893–898. https://doi.org/10.1038/nrc2009
Lossaint, G., et al. (2013). FANCD2 binds MCM proteins and controls replisome function upon activation of s phase checkpoint signaling. Molecular Cell, 51, 678–690. https://doi.org/10.1016/j.molcel.2013.07.023
Madireddy, A., et al. (2016). FANCD2 facilitates replication through common fragile sites. Molecular Cell, 64, 388–404. https://doi.org/10.1016/j.molcel.2016.09.017
Meetei, A. R., et al. (2003). A novel ubiquitin ligase is deficient in Fanconi anemia. Nature Genetics, 35, 165–170. https://doi.org/10.1038/ng1241
Meetei, A. R., et al. (2003). A multiprotein nuclear complex connects Fanconi anemia and Bloom syndrome. Molecular and Cellular Biology, 23, 3417–3426. https://doi.org/10.1128/mcb.23.10.3417-3426.2003
Meetei, A. R., et al. (2005). A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nature Genetics, 37, 958–963. https://doi.org/10.1038/ng1626
Mehta, P. A., et al. (2010). Numerical chromosomal changes and risk of development of myelodysplastic syndrome–acute myeloid leukemia in patients with Fanconi anemia. Cancer Genetics and Cytogenetics, 203, 180–186. https://doi.org/10.1016/j.cancergencyto.2010.07.127
Michl, J., Zimmer, J., & Tarsounas, M. (2016). Interplay between Fanconi anemia and homologous recombination pathways in genome integrity. The EMBO Journal, 35, 909–923. https://doi.org/10.15252/embj.201693860
Minocherhomji, S., et al. (2015). Replication stress activates DNA repair synthesis in mitosis. Nature, 528, 286–290. https://doi.org/10.1038/nature16139
Murina, O., et al. (2014). FANCD2 and CtIP cooperate to repair DNA interstrand crosslinks. Cell Reports, 7, 1030–1038. https://doi.org/10.1016/j.celrep.2014.03.069
Naim, V., & Rosselli, F. (2009). The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nature Cell Biology, 11, 761–768. https://doi.org/10.1038/ncb1883
Oberbeck, N., et al. (2014). Maternal aldehyde elimination during pregnancy preserves the fetal genome. Molecular Cell, 55, 807–817. https://doi.org/10.1016/j.molcel.2014.07.010
Okamoto, Y., et al. (2018). Replication stress induces accumulation of FANCD2 at central region of large fragile genes. Nucleic Acids Research, 46, 2932–2944. https://doi.org/10.1093/nar/gky058
Orta, M. L., et al. (2013). 5-Aza-2’-deoxycytidine causes replication lesions that require Fanconi anemia-dependent homologous recombination for repair. Nucleic Acids Research, 41, 5827–5836. https://doi.org/10.1093/nar/gkt270
Pagano, G., et al. (2005). Oxidative stress as a multiple effector in Fanconi anaemia clinical phenotype. European Journal of Haematology, 75, 93–100. https://doi.org/10.1111/j.1600-0609.2005.00507.x
Park, S. J., et al. (2004). Oxidative stress/damage induces multimerization and interaction of Fanconi anemia proteins. The Journal of Biological Chemistry, 279, 30053–30059. https://doi.org/10.1074/jbc.M403527200
Pontel, L. B., et al. (2015). Endogenous formaldehyde is a hematopoietic stem cell genotoxin and metabolic carcinogen. Molecular Cell, 60, 177–188. https://doi.org/10.1016/j.molcel.2015.08.020
Porebski, B., et al. (2019). WRNIP1 protects reversed DNA replication forks from SLX4-dependent nucleolytic cleavage. iScience, 21, 31–41. https://doi.org/10.1016/j.isci.2019.10.010
Rathbun, R. K., et al. (1997). Inactivation of the Fanconi anemia group C gene augments interferon-gamma-induced apoptotic responses in hematopoietic cells. Blood, 90, 974–985.
Ray Chaudhuri, A., et al. (2016). Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature, 535, 382–387. https://doi.org/10.1038/nature18325
Rickman, K. A., et al. (2015). Deficiency of UBE2T, the E2 ubiquitin ligase necessary for FANCD2 and FANCI Ubiquitination, causes FA-T subtype of Fanconi anemia. Cell Reports, 12, 35–41. https://doi.org/10.1016/j.celrep.2015.06.014
Rickman, K. A., et al. (2020). Distinct roles of BRCA2 in replication fork protection in response to hydroxyurea and DNA interstrand cross-links. Genes & Development, 34, 832–846. https://doi.org/10.1101/gad.336446.120
Ridpath, J. R., et al. (2007). Cells deficient in the FANC/BRCA pathway are hypersensitive to plasma levels of formaldehyde. Cancer Research, 67, 11117–11122. https://doi.org/10.1158/0008-5472.Can-07-3028
Rochowski, A., et al. (2012). Patients with Fanconi anemia and AML have different cytogenetic clones than de novo cases of AML. Pediatric Blood & Cancer, 59, 922–924. https://doi.org/10.1002/pbc.24168
Rohleder, F., et al. (2016). FANCM interacts with PCNA to promote replication traverse of DNA interstrand crosslinks. Nucleic Acids Research, 44, 3219–3232. https://doi.org/10.1093/nar/gkw037
Rosado, I. V., Langevin, F., Crossan, G. P., Takata, M., & Patel, K. J. (2011). Formaldehyde catabolism is essential in cells deficient for the Fanconi anemia DNA-repair pathway. Nature Structural & Molecular Biology, 18, 1432–1434. https://doi.org/10.1038/nsmb.2173
Roy, U., & Schärer, O. D. (2016). Involvement of translesion synthesis DNA polymerases in DNA interstrand crosslink repair. DNA Repair, 44, 33–41. https://doi.org/10.1016/j.dnarep.2016.05.004
Saadatzadeh, M. R., Bijangi-Vishehsaraei, K., Hong, P., Bergmann, H., & Haneline, L. S. (2004). Oxidant hypersensitivity of Fanconi anemia type C-deficient cells is dependent on a redox-regulated apoptotic pathway. The Journal of Biological Chemistry, 279, 16805–16812. https://doi.org/10.1074/jbc.M313721200
Schlacher, K., et al. (2011). Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell, 145, 529–542. https://doi.org/10.1016/j.cell.2011.03.041
Schlacher, K., Wu, H., & Jasin, M. (2012). A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell, 22, 106–116. https://doi.org/10.1016/j.ccr.2012.05.015
Schwab, R. A., et al. (2015). The Fanconi anemia pathway maintains genome stability by coordinating replication and transcription. Molecular Cell, 60, 351–361. https://doi.org/10.1016/j.molcel.2015.09.012
Shen, X., et al. (2020). A surge of DNA damage links transcriptional reprogramming and hematopoietic deficit in Fanconi anemia. Molecular Cell, 80, 1013-1024.e1016. https://doi.org/10.1016/j.molcel.2020.11.040
Si, Y., et al. (2006). Continuous in vivo infusion of interferon-gamma (IFN-gamma) enhances engraftment of syngeneic wild-type cells in Fanca-/- and Fancg-/- mice. Blood, 108, 4283–4287. https://doi.org/10.1182/blood-2006-03-007997
Singh, T. R., et al. (2010). MHF1-MHF2, a histone-fold-containing protein complex, participates in the Fanconi anemia pathway via FANCM. Molecular Cell, 37, 879–886. https://doi.org/10.1016/j.molcel.2010.01.036
Smogorzewska, A., et al. (2010). A genetic screen identifies FAN1, a Fanconi anemia-associated nuclease necessary for DNA interstrand crosslink repair. Molecular Cell, 39, 36–47. https://doi.org/10.1016/j.molcel.2010.06.023
Sonneville, R., et al. (2019). TRAIP drives replisome disassembly and mitotic DNA repair synthesis at sites of incomplete DNA replication. eLife. https://doi.org/10.7554/eLife.48686
Stingele, J., et al. (2016). Mechanism and regulation of DNA-protein crosslink repair by the DNA-dependent metalloprotease SPRTN. Molecular Cell, 64, 688–703. https://doi.org/10.1016/j.molcel.2016.09.031
Stone, M. P., et al. (2008). Interstrand DNA cross-links induced by alpha, beta-unsaturated aldehydes derived from lipid peroxidation and environmental sources. Accounts of Chemical Research, 41, 793–804. https://doi.org/10.1021/ar700246x
Tan, W., et al. (2020). Monoubiquitination by the human Fanconi anemia core complex clamps FANCI:FANCD2 on DNA in filamentous arrays. eLife. https://doi.org/10.7554/eLife.54128
Tian, Y., et al. (2017). Constitutive role of the Fanconi anemia D2 gene in the replication stress response. The Journal of Biological Chemistry, 292, 20184–20195. https://doi.org/10.1074/jbc.M117.814780
Tsui, V., & Crismani, W. (2019). The Fanconi anemia pathway and fertility. Trends in Genetics, 35, 199–214. https://doi.org/10.1016/j.tig.2018.12.007
Tulpule, A., et al. (2010). Knockdown of Fanconi anemia genes in human embryonic stem cells reveals early developmental defects in the hematopoietic lineage. Blood, 115, 3453–3462. https://doi.org/10.1182/blood-2009-10-246694
Unno, J., et al. (2014). FANCD2 binds CtIP and regulates DNA-end resection during DNA interstrand crosslink repair. Cell Reports, 7, 1039–1047. https://doi.org/10.1016/j.celrep.2014.04.005
Vandenberg, C. J., et al. (2003). BRCA1-independent ubiquitination of FANCD2. Molecular Cell, 12, 247–254. https://doi.org/10.1016/s1097-2765(03)00281-8
Vaz, B., et al. (2016). Metalloprotease SPRTN/DVC1 orchestrates replication-coupled DNA-protein crosslink repair. Molecular Cell, 64, 704–719. https://doi.org/10.1016/j.molcel.2016.09.032
Vaz, B., Popovic, M., & Ramadan, K. (2017). DNA-protein crosslink proteolysis repair. Trends in Biochemical Sciences, 42, 483–495. https://doi.org/10.1016/j.tibs.2017.03.005
Voulgaridou, G. P., Anestopoulos, I., Franco, R., Panayiotidis, M. I., & Pappa, A. (2011). DNA damage induced by endogenous aldehydes: current state of knowledge. Mutation Research, 711, 13–27. https://doi.org/10.1016/j.mrfmmm.2011.03.006
Walden, H., & Deans, A. J. (2014). The Fanconi anemia DNA repair pathway: structural and functional insights into a complex disorder. Annual Review of Biophysics, 43, 257–278. https://doi.org/10.1146/annurev-biophys-051013-022737
Walter, D., et al. (2015). Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature, 520, 549–552. https://doi.org/10.1038/nature14131
Wang, W. (2007). Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nature Reviews Genetics, 8, 735–748. https://doi.org/10.1038/nrg2159
Wang, J., et al. (1998). Overexpression of the fanconi anemia group C gene (FAC) protects hematopoietic progenitors from death induced by Fas-mediated apoptosis. Cancer Research, 58, 3538–3541.
Wang, R., Wang, S., Dhar, A., Peralta, C., & Pavletich, N. P. (2020). DNA clamp function of the monoubiquitinated Fanconi anaemia ID complex. Nature, 580, 278–282. https://doi.org/10.1038/s41586-020-2110-6
Wang, S., Wang, R., Peralta, C., Yaseen, A., & Pavletich, N. P. (2021). Structure of the FA core ubiquitin ligase closing the ID clamp on DNA. Nature Structural & Molecular Biology, 28, 300–309. https://doi.org/10.1038/s41594-021-00568-8
Wu, R. A., et al. (2019). TRAIP is a master regulator of DNA interstrand crosslink repair. Nature, 567, 267–272. https://doi.org/10.1038/s41586-019-1002-0
Xu, Y., et al. (2017). 53BP1 and BRCA1 control pathway choice for stalled replication restart. eLife. https://doi.org/10.7554/eLife.30523
Xu, X., et al. (2021). Fanconi anemia proteins participate in a break-induced-replication-like pathway to counter replication stress. Nature Structural & Molecular Biology, 28, 487–500. https://doi.org/10.1038/s41594-021-00602-9
Yamamoto, K. N., et al. (2011). Involvement of SLX4 in interstrand cross-link repair is regulated by the Fanconi anemia pathway. Proceedings of the National Academy of Sciences of the United States of America, 108, 6492–6496. https://doi.org/10.1073/pnas.1018487108
Yan, Z., et al. (2010). A histone-fold complex and FANCM form a conserved DNA-remodeling complex to maintain genome stability. Molecular Cell, 37, 865–878. https://doi.org/10.1016/j.molcel.2010.01.039
Yan, Z., et al. (2012). A ubiquitin-binding protein, FAAP20, links RNF8-mediated ubiquitination to the Fanconi anemia DNA repair network. Molecular Cell, 47, 61–75. https://doi.org/10.1016/j.molcel.2012.05.026
Yang, Y., et al. (2015). FANCD2 and REV1 cooperate in the protection of nascent DNA strands in response to replication stress. Nucleic Acids Research, 43, 8325–8339. https://doi.org/10.1093/nar/gkv737
Zhang, J., et al. (2015). DNA interstrand cross-link repair requires replication-fork convergence. Nature Structural & Molecular Biology, 22, 242–247. https://doi.org/10.1038/nsmb.2956
Zhou, W., et al. (2012). FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nature Genetics, 44, 910–915. https://doi.org/10.1038/ng.2347
Acknowledgements
This work was supported in part by the National Natural Science Foundation of China (32071285 and 31870807) to R.G.
Author information
Affiliations
State Key Laboratory of Protein and Plant Gene Research, School of Life Sciences, Peking University, Beijing, 100871, China
Xinlin Xu, Rong Guo & Dongyi Xu
Corresponding authors
Correspondence to Rong Guo or Dongyi Xu.
Rights and permissions
About this article
Cite this article
Xu, X., Guo, R. & Xu, D. The emergence of a unified mechanism in the Fanconi anemia pathway. GENOME INSTAB. DIS. 2, 281–291 (2021). https://doi.org/10.1007/s42764-021-00053-y
Received11 September 2021
Revised05 October 2021
Accepted07 October 2021
Published16 October 2021
Issue DateOctober 2021
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
Fanconi anemia
Replication stress
DNA interstrand crosslinks
BIR
BRCA1
用户登录
还没有账号?
立即注册