






Chromosome instability induced by mutations in TAD anchors leads to tumors
Review Article
Genome Instability & Disease (2021)
Abstract
In the light of recent data, a way is described between mutations in TAD anchors and stimulation of tumor growth driven by chromosome instability. At the end of this way every tumor sacrifices by a portion of its cells, dying necrotically, to stimulate remaining cells to proliferate. Necrosis (cell puncture and leakage of chemokine CXCL12) launches the mechanism of wound healing that stimulates cells to divide. Necrosis in cancer cells is induced by chromosome aberrations hindering anaphase cleavage furrow ingression and leading to membrane rupture. Inherited chromosome instability arises, because some of DNA loops (gene blocks, also called TADs) are torn off chromatid axes at their attachment points (anchors) because of mutations at some of these points. Namely, CTCF target sites (CCXXXAGGGGG) at the basements of TAD loops acquire mutations in central Adenine and in 3–5 nucleotides just beyond the borders of this 11 bp sequence. The sequence itself is degenerate between different TADs mainly at the X nucleotides, rarely at some others. Interstitial satellite DNA (SAT) adjacent to TADs anchors begins to behave such as telomeric satellite DNA. This launches transcription of such erupted satellites not leaned against TADs and production of their DNA copies due to the action of TERT reverse transcriptase just like in telomeres and leads to insertion of telomere sequences into such SATs. Some sticky ends in satellites also add to chromosome instability.
Introduction
Numerous hallmarks of cancer are widely discussed during last 20 years. However, almost neither of them embrace all types of cancer. Various types of tumors have their own characteristic hallmarks. Only two hallmarks are intrinsic to all types of cancer.
(1)
Chromosome instability is revealed in all types of cancer as was concluded in 1998 by Lengauer et al. (1998). Later this conclusion was confirmed in thousands of publications.
(2)
Tumors in vivo never consist of cancer cell mass alone, their aggregation, bulge or lump. They are always embedded into special “organ” or “pocket” filled with specific collection of non-cancer (stromal) cells. This “society” of specialized cells is often called “tumor stroma”, “tumor niche” or “tumor microenvironment” (TME) or even “homing” for cancer cells.
All cells of tumor stroma originate from bone marrow and are recruited into stroma through blood and lymph vessels (Bianchi & Mezzapelle, 2020; Guo et al., 2016; Lopez-Gil et al., 2021; Mortezaee, 2020). There are two types of bone marrow cells: Hematopoietic stem cells and Bone Marrow Stem (Stromal) Cells (BMSC). The later consist of pluripotent Mesenchymal Stem Cells (MSC) capable to differentiate into stromal fibroblasts, chondrocytes, adipocytes, osteoblasts and vascular cells (endothelial ones and pericytes) and others (Bianchi & Mezzapelle, 2020; Guo et al., 2016; Lopez-Gil et al., 2021; Mortezaee, 2020).
All these cells are capable for proteolysis and synthesis of extracellular matrix (ECM) that embraces almost every cell in our body. Stromal fibroblasts are capable to move within stroma due to proteolysis and synthesis of ECM and so to expand the volume of stroma, thus allowing other components of stroma to grow and expand (e.g., blood vessels). This gives opportunity for tumor cells (homed into stroma) to expand also. Tumor cells are engaged in “chemical conversations” with stroma, using chemokine CXCL12 (originating from tumor) and cytokine CXCR4 (originating from BMSC in stroma). Such conversations give signals to stroma for its expansion and to tumor cells for to expand too.
Malignant cancer cells inhabiting stroma exhibit their chromosome instability in every cell cycle. This instability drives the growth of tumor cells via their chemical feedback connection with stromal cells. Necrosis (membrane rupture) in a portion of cancer cells happens every cell cycle due to inherent chromosome instability in cancer cells, that leads in every cell cycle to stochastic output of about 1–40% (no more than critical 50%) of cells harboring novel chromosome aberrations and this leads to necrotic death of those cells due to a failure of anaphase furrow to split those cells. This rips membrane and induces leakage of cytoplasm containing chemokine CXCL12. Definition of various types of necrosis may be found in recent review (Nirmala & Lopus, 2020). All types of necrosis are united by one common feature—cytoplasmic membrane rupture, sometimes even membrane abscission. Apoptosis and related types of cell death may sometimes detach pieces of eliminated cell but without membrane rupture, via digestion of engulfed cells or in the case of autophagy (Nirmala & Lopus, 2020).
Every cell in our organism either normal or malignant contains cytoplasmic factors that launch a mechanism of wound healing—a well-defined evolutionary mechanism of survival in warm-blooded amniotic organisms including humans. These factors (chemokines) get out of cytoplasm only after damage to cytoplasmic membrane inflicted by occasional wound from kitchen knife or got in a battle or from wild beast or from inflammatory infection and launch the process of wound healing. These chemokines attract MSC from bone marrow. Cells recruited from bone marrow may differentiate into fibroblasts, osteoblasts, adipocytes etc. and also into vascular cells within a wound, thus creating wound stroma. Remarkably, the type of cells composing stroma does not have any relation to the type of cells in surrounding tissue, so the scars are clearly visible either in external or internal wounds. The only exception in the type of wound healing is liver, where its own dormant hepatocytes participate in restoration of damaged sites.
The constituents of healing wound stroma seem to have no difference with constituents of cancer stroma. The only difference is the absence of cancer cells in real wounds. The similarity between wound and cancer consists in cytoplasmic membrane rupture leading to formation of stroma. In one case occasional knife cutting or bacterial infection inflict damage to membrane, in another case—occasional necrosis of some cancer cells leads to cytoplasm leakage and stimulation of stroma growth. However, even “chronic wounds” or “chronic inflammation” in humans which may last for years very rarely enhance risk of cancer.
Main cytoplasmic factor (chemokine) responsible for stimulation of cell growth either within a wound or within a tumor stroma was former known under the name Stromal cell-derived factor-1 (SDF-1), but now is named “CXCL12” (“L” for “ligand”). It is only 89 amino acids long. After getting out of a cell it forms a complex with chemokine receptor located on membrane surface of BMSC. This receptor is called CXCR4 (“R” for “receptor”) and is 352 amino acids long. It is embedded into cytoplasmic membrane of BMSC and is folded in such a way that forms 3 extra-cellular loops and 3 intra-cellular loops connected with 7 transmembrane helices. Its N-end sticks out of membrane and its C-end is on the other side of membrane and looks into cytoplasm of BMSC. This receptor may form dimers with itself or with another receptor on the same membrane, called ACKR3 (formerly known as CXCR7).
CXCL12 ligand may also form heterodimers with another ligand released from necrotic cells. It is called High mobility group box 1 (HMGB1).
To stimulate the growth of wound or cancer stroma the formation of CXCL12/CXCR4 complex is absolutely required. The participation of ACKR3 or HMGB1 in this complex is facultative and depends on many other factors.
Thus, so-called “CXCL12/CXCR4 axis” is a prerequisite, i.e., is absolutely required, necessary and sometimes even sufficient for the formation of stroma in wounds and malignant tumors. For more detail see recent reviews and experimental papers (Bianchi & Mezzapelle, 2020; Guo et al., 2016; Lopez-Gil et al., 2021; Mortezaee, 2020; Xu et al., 2013).
Unlike real wounds, tumors in addition contain a clone of cancer stem cells (CSC) exhibiting chromosome instability and thus inducing necrosis every cell cycle in a portion of CSC stimulating further growth of both cancer cells and stroma (fibroblasts, blood vessels and novel extracellular matrix instead of the old one, thus allowing the expansion of tumor volume).
A clone produced from one cancer cell may be drawn as a fir tree (or pyramid) put upside-down and giving new generations of cells (fir tree tiers, layers or stories of elongating branches (1–2–4–8–16–32 etc.). Some cells (“branches”) in this “fir tree” in some layers (levels) die necrotically due to chromosome aberrations. Therefore, the cone of such upset “fir tree” becomes narrower but still expanding and widening its basement up to the sky endlessly, due to a fuel (chemokine CXCL12) emitted by dead branches (necrotic cells) thus stimulating stroma expansion (see Fig. 2 in Luchnik (2003)).
Why broken chromosomes induce necrosis?
So as chromosome aberrations block anaphase furrow, let us consider types of chromosome aberrations. These are classified here in Fig. 1. This figure represents simple organism containing only two chromosomes: one acrocentric and one metacentric, and their diploid set. Such representation allows to consider all types of chromosome aberrations, avoiding color FISH staining of individual chromosomes at the background of surrounding other chromosomes in experiment. The classification presented in Fig. 1 uses terminology of “paired” and “simple” chromosome breaks and exchanges instead of “chromosome”, “isochromatid” and “chromatid” ones. Indeed, what we call chromatid in metaphase becomes chromosome in anaphase during few minutes.
Fig. 1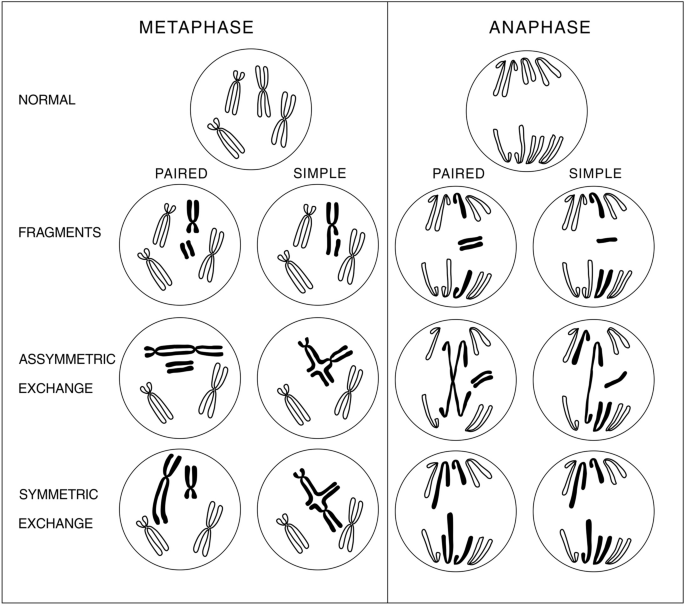
Classification of chromosome aberrations. This simple organism has only two chromosomes: one acrocentric and one metacentric and diploid set of chromosomes. This allows to look at chromosome aberrations excluding background of some more chromosomes in a set. Terms “paired” and “simple” are used instead of “chromosome” and “chromatid”, because metaphase chromatid becomes chromosome in a few minutes (in anaphase). Isochromatid aberrations are also termed “paired”. Consequences of particular aberrations for necrotic cell death are considered in the main text
Full size imageFrom Fig. 1, it is seen that asymmetric exchanges form bridges in anaphase that block anaphase furrow (contractile ring made of actin and type II myosin) and lead to blowing up of a cell and leaking out of its contents. Symmetric exchanges lead to lagging (too long) chromosomes that may also block anaphase furrow ingression and block final occlusion of tightening lasso segregating daughter cells, and so this also leads to leakage of cytoplasm, thus these aberrations induce necrotic death and induce “wound healing” that stimulates proliferation of remaining cancer cells and building wound stroma (angiogenesis, fibrosis and extracellular matrix). Acentric chromosome fragments, either simple or paired, distribute to only one of the daughter cells thus leading to genetic imbalance and necrotic death either immediate or delayed in several subsequent cell divisions. In addition, they may lead to micronuclei and so to genetic imbalance leading to their necrotic death.
Here we encounter with serious difference between wound healing after kitchen knife injury and “healing” in cancer. Both in tumor and in real wound bone marrow cells infiltrating wounds or tumors build stroma made from cells of bone marrow. However, unlike real wounds tumors have another proliferating pool of cells—those usually called cancer stem cells (CSC) which are not only cycling but are also genetically unstable. Therefore, it is these cells which permanently produce proliferative (healing) signal due to their stochastic death. These cycling cells often arise in differentiated tissues composed of non-dividing cells. Cells in an adult organism are mostly non-cycling except bone marrow and cells lining intestine and also skin derma cells. In fact, tumor cells in vivo belong to only a few classes—epithelial, mesenchymal, endodermal and several others—much less than a number of types of differentiated cells in various tissues. Therefore, where from tumor stem cells are coming? This question will be clarified later in this article after consideration of the nature of lesions leading to instability.
Constitutive and inducible repair
Chromosome instability in cancer cells arises due to inability to repair spontaneous DNA DSB by constitutive repair machinery acting in every cell in any organism. Cancer cells contain “potentially lethal lesions” in their chromosomes as argued by many authors. In addition, these lesions are sometimes referred to as “fragile sites”.
Constitutive repair acts in all cycling cells to prevent spontaneous mutation accumulation during prenatal and postnatal development and during all-lifelong in all actively proliferating cells. It acts also in actively proliferating cancer cells.
Up to several ten percent (no more than 50%) of cancer cells harbor novel chromosome aberrations in every cell cycle. Normal cells in culture (stimulated ex vivo lymphocytes) produce less than 0.5% of cells containing at most one aberration per cell in every cell cycle (0.5 aberrations per 100 cells). This figure for cells in vivo could be much lower but difficult to measure. Obviously, constitutive repair of spontaneous DNA lesions is impaired in cancer cells.
Chromosome aberrations are known to arise in normal or cancer cells as a result of unrepaired DNA double-strand breaks (DSB). It is difficult to quantify the number of DSB in isolated high molecular weight DNA; moreover, there is no “control” for spontaneous breaks in DNA from cells untreated with mutagens.
Nevertheless, such estimates were made using other methods. First estimates were made by Billen (1990). According to his calculations, every normal cell produces 4000 DNA DSB per hour plus thousands of DNA lesions of other types. Ionizing radiation produces 400 DSB/Gy according to this author. Analogous estimates made by Lindall and Barnes (2000) gave 25 DSB per cell per day. This means 25,000 DSB per 100 cells. All other estimates were made using immunostaining of cells with antibodies to DNA repair markers, such as gamma-H2AX, pATM, 53BP1, RAD59, RAD 51, BRCA1, and others and counting repair “foci” under microscope. Critical review on “foci” measurements by Barnard et al. (2013) disproves this method as an unsuitable for quantitative estimates of DSB. In addition, these authors analyze numerous artifacts produced by these methods and advise to use double or triple different antibody staining to diminish artifacts, nevertheless they doubt on reliability of this method anyway. Later it was shown that every “focus” hides many DSB gathered into “repair centers” (Neumaier et al., 2012).
Notwithstanding of this we mention here the results that more or less deserve consideration. Vilenchik and Knudson (2003) calculated 50 triple-stained foci in every normal not irradiated cell using doubling dose extrapolations to zero doze and foci counting. This means 5000 DSB per 100 cells per cell cycle, i.e., much higher than less than 0.5% of chromosome aberrations (not foci) per 100 cells visible under microscope in normal cells. Pustovalova et al. (2016) found only two gH2AX stained foci per cell in early passage primary human mesenchymal stromal cells and 5 foci at 18–22 passages. This means 200–500 DSB per 100 cells. In any way it is clear that constitutive repair performs error-free repair of hundreds of spontaneous DSB at every cell cycle in all normal cells.
Unlike constitutive repair, inducible one (induced by experimentalists who use mutagens), is often called “DNA damage response” (DDR). It is always local, i.e., it does not act on all genomic DNA, instead it deals with separate lesions. Constitutive repair always acts all over genome and is homology directed (HDR). For example, repair of all lesions and replication errors in satellite DNA without any “frameshift” or unequal crossing-over is possible only through HDR, promoted by SMC5/6 complex, see (Amaral et al., 2017; Caridi et al., 2017; Diaz & Pecinka, 2018; Ryu et al., 2016; Verver et al., 2016) for reviews. Gene-rich areas (TADs) are repaired with participation of SMC1/3 (cohesin) complex with the help of some accessory protein complexes (see below).
The evidence for regular constitutive repair of hundreds of spontaneous DSB in every cell cycle comes also from studies of rare clonal conditional mutations (found in cell cultures) in protein subunits of Structural Maintenance of Chromosomes (SMC) complexes performing constitutive repair every cell cycle. In vivo all these mutations are lethal for organism if they are not clonal. In case when some of these protein subunits can be knocked out in cell cultures, up to hundreds of chromosome aberrations may be observed in nearest mitosis. Some other protein complexes also participate in constitutive repair as evidenced from their conditional knock out. All these will be considered below, after a glance at what is known about DNA repair systems.
DNA repair systems
How multicellular organisms manage to repair so many spontaneous DNA DSB beginning from zygote to an adult organism after passing millions of cell divisions during development? The matter is that in every cell cycle lesion in DNA accumulate due to errors in replication in S-stage of a cell cycle or due to errors happening in mitosis and also in G0 stage sometimes lasting for years. Two critical stages of a cell cycle (“S” and “M”) are so complex machineries that errors are inevitable. Both after S and M stages cells need to check their DNA for integrity and absence of errors.
DNA DSB repair systems considered in literature are mostly referred to as DNA Damage Response (DDR) that means repair systems induced in response to mutagen treatment of cell cultures made by experimentalists. Spontaneous damage is difficult to measure and quantify as considered in the above section. Moreover, constitutive repair systems may also intersect and participate in inducible ones.
Modern views and reviews divide all DNA DSB repair systems into two pathways—non-homologous end joining (NHEJ) and homologous recombination (HR) repair. NHEJ in turn is subdivided into several sub-pathways (branches). According to current views, NHEJ repairs 100% of DNA DSB in G1 stage of a cell cycle and about 80% in G2 stage (Sasanuma et al., 2020). HR repair is supposed to act only upon 20% of DNA DSB in G2. In addition, NHEJ usually is considered as “error-prone”, and HR-repair as error-free, even if it occurs inside the same cell, repairing 80% of damage through “error-prone” NHEJ. Such state of affairs does not allow to pass millions of cell divisions in an error-free way from zygote to an adult organism.
What is wrong with the term “NHEJ”? Irradiating cells in G1 gives much less chromosome aberrations than irradiating in G2. Therefore, how could NHEJ be considered as such an error-prone system? (Key word here is “non-homologous”, i.e., “erroneous”). To understand it better let us consider what is meant by the term “NHEJ”. The general and prerequisite trait of this system is immediate protection of bare DNA DSB ends from endogenous attack by exonucleases and from going apart from one another due to diffusion. For this purpose, every cell nucleus in every eukaryote has a pair of proteins (Ku70 and Ku80) overwhelming in their abundance any other protein in the nucleus.
After occurrence of DNA DSB in any place in genome, Ku70/80 dimer caps two ends of a break in less than a second to protect them from nuclease attack. Each Ku dimer covers about 20 bp of DNA at the end of a break and usually is called Ku-ring, embracing the DNA. However, it is not a ring in a true sense of a word. Rather, it is cradle embracing DNA duplex from three sides. A thin protein lid of a cradle is allosterically movable, allowing access for many proteins and even for homologous or non-homologous DNA molecules to damaged DNA embraced by Ku70/80 dimer. Surprisingly, Ku knock out mice are born alive, yet radiosensitive (Li et al., 2007). This means that some other proteins may partially substitute Ku protein.
Next, also extremely rapidly, two Ku protected ends are joined together by proteins creating continuity between two ends disjoined by DSB. This process may use several types of proteins depending on a cell type or a site of damage within genome. One protein often bridging two Ku-capped ends is DNA protein kinase catalytic subunit (DNA-PKcs) sometimes termed simply PKcs. It is one of the only three DNA-dependent protein kinases existing in a cell (ATM, ATR, PKcs) (Menolfi & Zha, 2020). However, PKcs is the largest one (4128 aa) and like Ku70/80 dimer has a channel inside, accommodating DNA duplex diameter and lined with positively charged amino acids. Complexed with Ku dimers, PKcs is named simply as DNA-PK. It retains its kinase catalytic subunit (at its C-end) and may exhibit its kinase activity upon binding to Ku dimer. Genetic manipulations inactivating kinase activity but retaining whole protein intact make cells unviable. Strikingly, genetic inactivation of the whole giant protein (knock out) does not have any impact on cellular viability in spite of concomitant disappearance of catalytic subunit domain of the kinase. Moreover, mice with knock out of the whole PKcs protein are viable and fertile. However, they suffer from certain immunodeficiency.
Here we came to very important aspect of NHEJ. This system aimed to repair the DNA DSB is very ancient in evolution. However, only in vertebrates it acquired a novel function—not only to repair DNA properly but also to repair it improperly for to create a diversity of “wrong” DNA joints used by immune system to create the diversity of antibodies. For this reason, vertebrates acquired a novel protein—DNA-PKcs and some other proteins to create antibody diversity. That is why knock out of DNA-PKcs does not spoil DNA repair but only impairs immunologic response. PKcs is not induced by DNA damage unlike many other repair proteins. Indeed, it was found that without DNA-PKcs other participants of NHEJ may successfully bridge two DNA ends capped with Ku70/80 dimer. These are: XRCC4 (X-ray cross-complementing protein4), XLF (XRCC4-like factor), PAXX (paralogue of XRCC4 and XLF) and Lig4 (DNA ligase), the latter is needed as structural protein for DNA bridging, because its ligase activity is used much later in NHEJ process or used in immunologic response. Lig4 knock outs are alive in cell lines but are embryonically lethal.
To cope with this diversity of proteins we should take into account that knock out of anyone of them does not lead to cell death in cell lines but enhance their radiosensitivity. However, only XRCC4 and Lig4 mutants are embryonic lethal in mice, all other single mutants are not. Conversely, all double knock out pairwise combinations are lethal in cell lines. This is more surprising, taking into account that XRCC4, XLF and PAXX all resemble each other in overall construction. All three represent protein homodimers made of two identical subunits. All have doubled head domain, coiled-coil helical domain and two identical disordered tails. XRCC4 is 336 aa long (one subunit), XLF is 299 aa long and PAXX is 204 aa long. Only XRCC4 has the ability to bind Lig4 (912 aa long) to its disordered tails. One tail binds one Lig4 using BRCT1 domain of Lig4, another tail binds another Lig4 through its BRCT2 domain, also containing in Lig4 protein. XLF and PAXX do not bind Lig4. Lig4 has cradle structure capable to embrace one DNA duplex, thus performing its structural role.
The XRCC4–Lig4 complex may hold together two DNA molecules in parallel arrangement using two Lig4 proteins embracing two DNA molecules. Now it is clear why sole XRCC4 or Lig4 knockouts from the above set are embryonic lethal even in the presence of non-mutant companion. They are lethal only in non-dividing cells, such as neurons appearing in embryos at early stages. In newborns almost all cells are non-dividing. (In proliferating cell lines double knockouts of XRCC4 and XLF are not viable).
In this connection it is clear why Ku knock out causes early aging in mice (Li et al., 2007). This is because NHEJ is fully competent only in the dividing cells, comprising minority of cells in adults. This observation is important for further consideration in “Proofreading” section.
Returning to lethal pairwise knock out combinations of XRCC4, XLF, PAXX and Lig4 let us turn to their interactions in vitro in presence of bare DNA. These studies revealed that some of the mentioned pairwise combinations of proteins added to bare DNA in solution demonstrate that these pairwise combinations promote not only bridging of DNA ends but also are capable to tether two DNA molecules to align them side by side in parallel arrangement so that two DNA duplexes become capable to slide along each other or move together through protein complex forth and back.
Brower et al. (2016) demonstrated that only a pair of proteins XRCC4 and XLF are capable not only to held together two DNA ends (as they indeed do) but also form sleeves embracing two DNA molecules in parallel arrangement allowing DNA movement forth and back within a sleeve, either for both DNA’s simultaneously or one along the other. This observation is important for our further consideration in the “Proofreading” section.
Mentioned another pair of proteins (XRCC4 and Lig4) was tested with two bare DNA fragments one of which was immobilized at one end on the surface (Zhao et al., 2019). These authors also added Ku protein which is known to promote weak and transient interaction between DNA ends with rather short dwell time (Ramsden & Gellert, 1998). A pair of proteins XRCC4 and Lig4 are capable to form complex between two XRCC4 disordered tails and two Lig4 molecules attached to two tails of XRCC4 dimer (Liang et al., 2020; Wu, 2019). Each one of the two Lig4 molecules is capable to embrace tightly only one DNA duplex. Zhao et al. (2019) found that XRCC4–Lig4 complex is capable to bridge two DNA molecules in “flexible complex” about 2 orders of magnitude stronger than does Ku protein alone. They also found that at least 64% of “flexible complex” bridging was between two DNA molecules in parallel arrangement observed somewhere in the middle of DNA length, the remaining bridging was closer to DNA ends. Addition of XLF protein shifted some percent of XRCC4–Lig4 complex to the ends of bridged DNA. Addition of PAXX protein had minor effect on bridging.
These results demonstrating parallel arrangement of two DNA molecules by XRCC4–Lig4 complex and by XRCC4–XLF complex imply the possibility of tethering of the two DNA’s in parallel by capturing them by two Lig4 molecules attached to XRCC4 protein dimer or by placing them into XRCC–XLF sleeve.
Thus lethal combinations of double knockouts of proteins described in previous paragraphs imply that parallel alignment of two DNA duplexes is vitally necessary for cell survival. Later in this paper we consider parallel arrangement of homologous DNA molecules promoted by topoisomerase 2B-cohesin (SMC1/3) complex for to repair DNA without errors.
Returning to “homologous recombination” (HR) repair it should be noted that this type of repair was always studied after extensive mutagen treatment of cells. The main features of so-called “HR-repair” described in literature are the following. HR acts only in G2 stage of a cell cycle, because it presumably uses only sister chromatids. Main protein complex involved in this repair is MRE11–RAD50–NBS1, called for brevity MRN complex. This complex recognizes DNA DSB and so gives signals for further processing of the ends by other proteins participating in this type of repair. These are CtIP exonuclease (C-terminal binding interacting protein), BLM (DNA-helicase), DNA2 and Exo1 exonucleases used for DNA end resection making DNA ends single-stranded to the length of about 300–1000 nucleotides, ending at 3’ ends, because 5’ ends were removed by resection. Then, single-stranded ends are covered by Rad 51, BRCA2, BRCA1 proteins to promote search for homologous DNA sequence in sister chromatid.
This description in much more detail is given in almost all reviews on HR-repair. Recall that all this information was obtained after considerable mutagenic treatment of cell cultures. Recently a number of papers revised some of the details in this scheme. All these revisions were made after application of enormous doses of various mutagens usually exceeding lethal dose several-fold. What happens in the absence of irradiation or exposure to some other mutagen?
Three central proteins of HR-repair (MRN complex) completely block NHEJ after extensive mutagen treatment of cells if either one of them is knocked out (Chen et al., 2001; Huang & Dynan, 2002; Moore & Haber, 1996; Xie et al., 2009). Moreover, it was established that MRN complex may tether two DNA ends to each other which is usually considered as characteristic of NHEJ repair. In addition, it was found that MRN complex promotes joining of DNA ends by XRCC4/Lig4 complex (Chen et al., 2001; Huang & Dynan, 2002). At last it was found that proper and successful ligation of DNA ends by XRCC4/Lig4 in a cell-free assay is possible only after addition of highly purified MRN complex (Chen et al., 2001; Huang & Dynan, 2002) from either mammalian or yeast cells. DNA end resection is equally induced in G1 stage after mutagen treatments and uses the same enzymes as in G2 stage, also questioning end-resection as unique characteristic of HR-repair restricted to G2 stage (Biehs et al., 2017; Rass et al., 2009; Xie et al., 2009).
Where from second copy of DNA is taken?
Here we come to important point. HDR requires second copy of DNA. If second copy were absent then all DNA lesions should be repaired in an error-prone way through non-homologous end joining (NHEJ) at least in G1 stage. However, it would lead to numerous mutations accumulating during not only development but also in germ-line cells. It is impossible to imagine active error-prone repair in every cell cycle during embryo development without affecting the fidelity of genome.
To cope with this situation, eukaryotes harbored second copy of DNA in a form of binemic structure (depicted in Fig. 2). Understanding evolutionary obligation of a second copy for maintenance, reliability and fidelity of eukaryotic genome, in 1982 a binemic model of eukaryotic chromosome was substantiated relying on experimental data existing at that time (Luchnik, 1982).
Fig. 2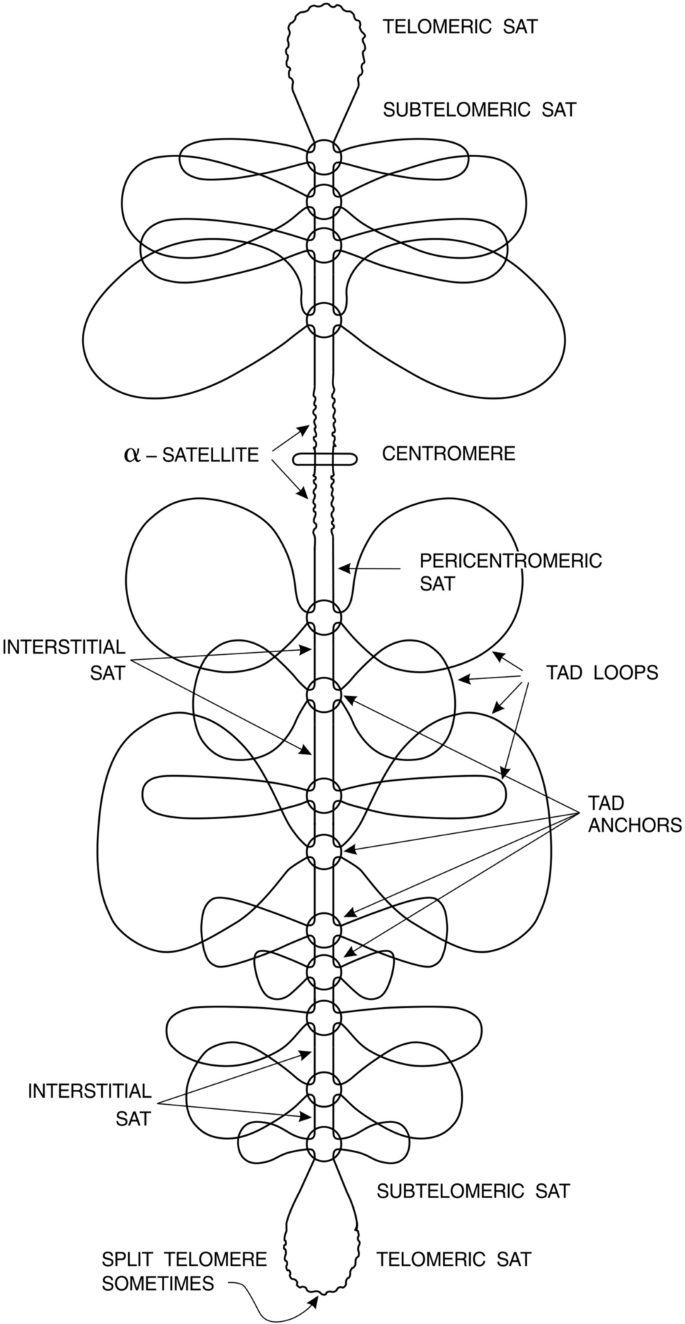
Presentation of chromosome in a framework of inlaid genome structure. TADs alternate with SATs (satellite interstitial DNA and some other reiterated DNA’s). Such a structure allows checking and repair of both TADs and SATs beginning from TAD anchors acting as zipper locks for to begin and to end the repair of compartments. Continuous line represents DNA double helix
Full size imageLater on, experimental evidence for it was provided using probes for unique genes to find their number in the genome (Luchnik et al., 1995). For this purpose, DNA from individual human sperm cells was divided into 4 portions and the number of some definitely unique genes was tested in every tube using PCR. In addition, serial dilutions of human DNA to 1/2 of the haploid genome per tube were used and a number of positive signals were counted in many series of 10 tubes (3.7 score wait for uninemy, 6.3 score wait for binemy according to Poisson distribution). All possible controls and precautions were done. Both approaches gave certain and unambiguous result favoring binemic structure of human chromosomes. Those experiments may be reproduced by anyone using PCR primers for any of the unique genes, taking care of contamination with human DNA present in every dust particle in the air and in every drop of buffers.
Binemic structure enhances reliability of genome against environmental mutagens or spontaneous double-strand lesions arising during replication of huge length of DNA in human genome at least 6.2 × 109 fold. In other words, probability of arising of two DSB in one and the same site in two identical DNA duplexes is estimated as 6.2 × 10–9. (Estimated as probability of arising of two independent coincidental DSB (separated by no more than 10 base pairs either upstream or downstream from primary break in a first (reference) molecule) in two homologous DNA duplexes if to admit that haploid DNA length in humans is about 3200 Mbp = 3.2 × 109).
Really
Pc=nN=20 bp3.2×109bp=6.25×10−9where Pc is probability of coincidence of two DNA DSB at the same site (within 10 bp upstream or downstream from first break site in a reference molecule) in two homologous DNA duplexes.
In numerator—number of base pairs in “second” duplex around the same site (primary DSB) in a “first” (reference) duplex, upstream or downstream from this point. In denominator—haploid DNA length in humans (in bp).
Three or more copies are unable to enhance reliability, because the probability of coincidence of damage in the third or fourth copy is astronomically negligible.
The main objection against binemic structure is that chromosome behaves as one DNA molecule in recombination and replication. However, the main evolutionary task for binemic structure is to make two DNA duplexes within chromosome absolutely identical in every cell cycle to repair errors and to enhance reliability of genome at least 6.2 × 109 fold. Scheme of proofreading leading to strand exchange in the whole genome, considered below in this article, leads to the expected result of semiconservative replication of chromosomes seen under microscope in first and second metaphases after tritium labeling of DNA at the beginning of the first cycle (Fig. 4).
Evolutionary border between uninemy and binemy is found in budding yeast S. cerevisiae, where some mutants show meiosis with fertile “diploid” ascospores (Scild et al., 1981). Higher multicellular fungi (and possibly unicellular fission ones) seem to have binemic chromosomes expected from their low radio-sensitivity in G1 stage. Usual strains of diploid S. cerevisiae exhibit very high radio-sensitivity in G1 stage unlike in G2 stage. However, if to block G1 stage for 1–2 days then viability restores as well as repair of DNA double-strand breaks via contact of homologous chromosomes in a synapsis-like manner (Luchnik et al., 1977). This repair is rather error-prone as seen in sectored colonies demonstrating genetic instability. Probably it is explained by heterozygous nature of homologous chromosomes.
For all that, uninemic chromosomes in budding yeast seem to be temporal evolutionary sideway to achieve cell division rate similar to that in bacteria, where deleterious mutations are rapidly sorted out from population. This way is impossible in multicellular organisms. However, even bacteria use second copy of DNA when it is possible (Vickridge et al., 2017).
Inlaid genome
Eukaryotic chromosomes are subdivided into alternating regions of active chromatin (“euchromatin”) and inactive heterochromatin first discovered in giant chromosomes in salivary glands in Drosophila. Alternating regions were also termed “bands”. Euchromatin bands, when become active, unpack and unravel giant chromatin loops for transcription. Similar loop unpacking was observed in lamp-brush chromosomes in Amphibians when they became transcriptionally active. Now these loops of chromatin are termed “topological domains” or “topologically associated domains” (Nora et al., 2012; Dixon et al., 2012; Luchnik et al., 1977; Scild et al., 1981), after cytological observations were confirmed by biochemical data. Alternating potentially transcriptionally active and constitutively inactive compartments were earlier termed as “A” and “B” compartments, respectively (Lieberman-Aiden et al., 2009; Vickridge et al., 2017).
New method introduced in 2009 (Lieberman-Aiden et al., 2009; Vickridge et al., 2017) used formaldehyde crosslinks within chromatin in isolated nuclei with subsequent shearing or restriction endonuclease cutting, then ligating fragments again “by chance” and, afterwards, massive sequencing DNA fragments for to determine proximity of genome fragments to each other within genome. This method in many modifications reveals numerous contacts between proximal and distal sites within chromosomes and even between chromosomes. The majority of contacts were found within 0.1–3 Mb zones and were later termed “topologically associated domains” (TADs) by other authors (Nora et al., 2012; Dixon et al., 2012; Luchnik et al., 1977; Scild et al., 1981). Within large megabase-long zones, shorter zones of about 100 kb were later revealed by many authors who used considerably enhanced resolution of the technique. Borders between both large and small zones as well as borders of TADs themselves were enriched in CTCF binding sites. Those authors (Nora et al., 2012; Dixon et al., 2012; Lieberman-Aiden et al., 2009; Luchnik et al., 1977; Scild et al., 1981; Vickridge et al., 2017) provided no evidence of topological closeness of their TADs. It was made later by other authors (Naughton et al., 2013).
Mentioned authors (Naughton et al., 2013) compared their results with results of formaldehyde technique, revealing TADs. They found that about 30% of TAD boundaries revealed by formaldehyde technique overlapped with supercoiling domains boundaries. However, only about 10% of supercoiling domains (SCD) overlapped with TAD domains. This is consistent with supercoiling domains being a subdomains of larger TADs. However, TADs themselves are also topologically closed. In addition, it was found that transcriptionally active supercoiling domains are negatively supercoiled and transcriptionally inactive ones were positively supercoiled (Naughton et al., 2013). Negatively supercoiled domains became positively supercoiled after block in their transcription, and positively supercoiled ones became negatively supercoiled upon activation of transcription. Satellite DNA between TADs was found to be non-supercoiled at all. Thus, TADs are subdivided into independently supercoiling domains (SCD) of about 100–300 kb, (Naughton et al., 2013). Both SCD and TADs have sequence-specific borders, but these sequences differ in TAD and SCD borders.
Taken together the data on topologically closed (either active or inactive) domains allow to assemble them together into non-controversial model of genome organization. According to this model, genome is divided into alternating regions of gene-containing TADs and transcriptionally inert often lamina-associated constitutive heterochromatin composed mostly of highly reiterated satellite DNA (gene deserts).
These domains which we call SATs comprise 30–40% of genome (including centromere and telomere satellites) and are 30–1000 kb in length (or much longer at centromeres and telomeres). Interstitial satellites (SATs) are unable to supercoil into elastic spring. In addition, they can be properly repaired only through homology directed repair promoted by SMC5/6 repair complexes (Reviews (Amaral et al., 2017; Caridi et al., 2017; Diaz & Pecinka, 2018; Ryu et al., 2016; Verver et al., 2016)). SATs belong to so-called constitutive heterochromatin abruptly restricting TAD borders (Dixon et al., 2012; Luchnik et al., 1977).
The model of Inlaid genome (or “chopped genome”) is drawn here in Fig. 2 with inscriptions corresponding to modern terminology (taken from Luchnik (1982) with modern inscriptions).
Here we should say about what is satellite DNA according to contemporary views. We should mention that almost all of satellite DNA evolved from 5 to 6 bp long original units arrayed head to tail, but some nucleotides interchanged their places within original units, some were replaced by novel ones (In humans, ancestral unit is 5 bp long, except telomere DNA having 6 bp basic unit). Therefore, satellite arrays evolved to be degenerate, for not to allow neighboring 5 bp units to be often identical, usually no more than 2–3 neighboring identical units are rarely allowed (Altemose et al., 2014). This is mostly considered in publication (Altemose et al., 2014) in relation to human Hsat2 and Hsat3, comprising 90% of human satellite DNA. In other organisms the story is the same, but less studied in the detail. Degenerate 5 bp units are arrayed in 23–26 bp units (never 25) differing in sequence, thus forming subfamilies, which in turn are combined in longer arrays of several kilobases or megabases long. These longer interstitial arrays are never entirely homologous to each other within one genome. Moreover, they differ between individual humans at one and the same chromosome site. Of course they also differ in different SAT sites in one and the same individual but never change their individuality during individual development.
It is worth mentioning that strict recognition of homology between two DNA pieces is achieved when their length is about 25 ± 5 bp, not less, sometimes more in a wide range of temperature and counter-ion conditions. To prove that, it is sufficient to look at various PCR methodologies used by thousands of experimentalists. They assured by trial and error that to obtain reliable results they should use primers 25 ± 5 bp long, not less, sometimes more.
So the unit of recognition found in human satellite DNA is determined by chemical and physical nature of DNA molecule itself. It is important for further consideration. (Telomere and centromere satellites will be considered separately, because they do not have such an enormous level of degeneracy between repeating units).
Structure of TAD anchors
The compendium of TAD anchor structure in vertebrates in a framework of current literature is depicted in Fig. 3. This figure shows only CTCF target sites and topoisomerase 2 subunits. All other important elements of TAD anchor are described below. It is convenient to describe them referring to coordinates of CTCF target sites in Fig. 3 with their indicated polarities.
Fig. 3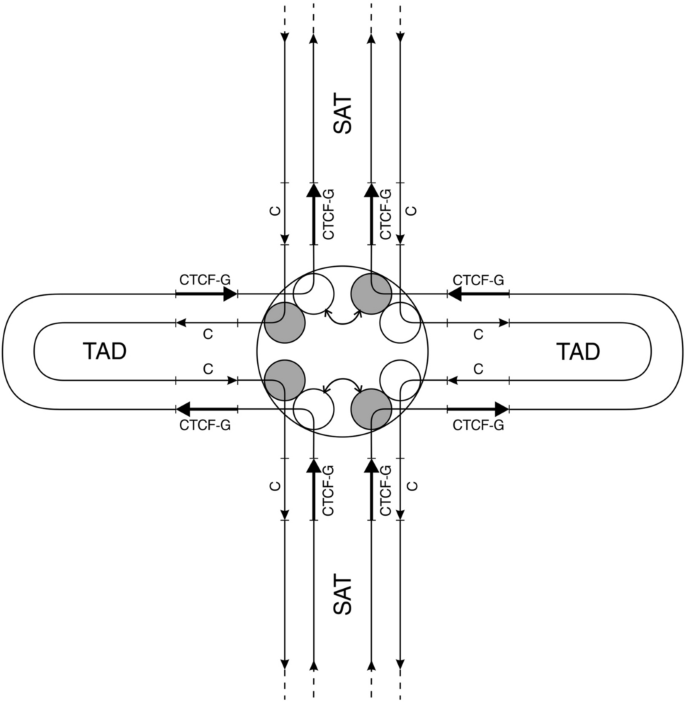
Presentation of TAD anchor according to recent achievements. CTCF-G denotes G-rich CTCF target sites, “C” denotes C-rich complementary sequence (CTCF-C). Grey balls within TAD anchor represent one subunit of topoisomerase 2B. White balls represent one subunit of topoisomerase 2A. Bent arrows show exchange between topoisomerase subunits leading to DNA strand exchange all over genome (in all anchors) for to perform proofreading of DNA. Straight arrows indicate polarity of DNA strands. SAT denotes interstitial satellite DNA between TADs
Full size imageTAD anchors contain doubled tandem CTCF target sites (CTS) (Lobanenkov & Zentner, 2018; Pugacheva et al., 2015) shown in Fig. 3. Often CTCF target sites are called CBS (CTCF binding sites). Cohesin (SMC1/3 complex) binding sites are also often called CBS. Therefore, to avoid confusion we will call CTCF target sites as CTS according to original publications (Lobanenkov & Zentner, 2018; Pugacheva et al., 2015). Doubled tandem CTS are separated by 50–60 bp of DNA between two CTS centers. Each CTS may be bound by either CTCF or CTCF-like protein, also called BORIS (Pugacheva et al., 2015). Majority of researchers used methods, such as immunoprecipitation, which do not allow to distinguish doubled CTS and single CTS.
The matter is that commercially available antibodies do not embrace the majority of isoforms of CTCF or BORIS. These isoforms are following. CTCF gene has 13 exons with alternative splicing and produces at least 17 protein variants (Voutsadakis, 2018) with different immunogenic properties. Experimentalists usually use only one or few of the same commercially available antibodies. CTCFL (BORIS) gene has 16 exons and 22–23 splice variants (Loukinov, 2018; Voutsadakis, 2018). In addition, posttranslational modifications add to diversity of variants of both proteins. This problem disvalues many of the results obtained using immunoprecipitations which could precipitate some BORIS variants together with some CTCF variants. In a review by Lukinov (2018) it is said that among all “commercially available anti CTCF/BORIS monoclonal antibodies” they “did not find one reliable candidate for immunochemistry”. Therefore, this group generated their own set of nine monoclonal antibodies. None of them gave reasonable immunostaining of CTCFL (BORIS) protein in spermatogonia definitely expressing this protein. On the other hand, these authors used next generation RNA sequencing to determine expression of CTCF-like (BORIS) protein in cancer cells (Loukinov, 2018).
More than 90% expression of BORIS in various types of cancer was found (Loukinov, 2018). In addition, these authors determined 34–45 thousand doubled binding sites for CTCF and/or CTCF-like (BORIS) per haploid genome. In accordance with this result, later it was found that CTCFL (BORIS) protein determines cell stemness of cancer stem cells (see last section). Many single CTCF target sites also occupy promoters of c-MYC, hTERT, p53 and many other cancer relevant promoters.
All CTCF target sites (CTS) always have polarity and this polarity is usually indicated according G-rich strand in 5’- > 3’ direction. Polarity is usually determined using central 11 bp, having predominantly adenine in the center and five guanines to the right of “A”. Five nucleotides to the left of “A” have rather degenerate sequence, most often having “CC” − 4 and − 5 nucleotides from “number zero” adenine at the center of conditional degenerate palindrome (CCXXXAGGGGG). Central adenine is very often mutated in cancer, as well as several nucleotides right at the two borders of 11 bp core sequence (see last section for the detail). Some variants at central 11 bp sequence alleviate BORIS binding to CTCF target sites.
The paper by Uuskula-Reimand et al. (2016) established binding of Top2B and cohesin subunits (SMC1/3 based complex) around CTCF target sites (CTS) at TAD anchors. Top2B subunits were found to bind mainly to both separate strands (forward and reverse) upstream of CTS “shade” (two superimposed CTS due to the method used) when analyzed together with G-rich 5’- > 3’ oriented CTS by ChIP-exonuclease method. Rad21 (kleisin, cohesin subunit) was found to bind both strands downstream of CTS “shade” and only one (forward) strand upstream of CTS “shade”.
CTCF protein was found to bind to forward and reverse strands both upstream and downstream of CTS “shade” which is explained by “superimposing” of two CTCF sites one over another in such type of experiment.
Relative strength of CTCF protein binding to either Top2B or Rad21 (cohesin subunit, kleisin) is very weak in cycling cells (Uuskula-Reimand et al., 2016). Nevertheless, zinc fingers 9–11 of CTCF coincide with Top2B binding region (so-called “upstream region” located -13–22 nucleotides upstream from central adenine of the first CTCF-G target site, just between two CTS target sites). Therefore, it looks like CTS sequence itself plays major role in determining binding strength and positions of Top2B and cohesin, not solely CTCF protein binding to its target sequence. Indeed, all three proteins (CTCF, Topo2B, Rad21) are successfully immuno-precipitated with antibodies to CTCF (in dormant liver cells) (Uuskula-Reimand et al., 2016). This may be explained by the finding of Vian et al. (2018) that strength of binding of CTCF is strongly dependent on cohesin (SMC1/3) binding. Many other proteins were found to bind to Top2B protein in the paper by Uuksula-Reimand et al. (2016). Important are STAG1 and 2 (cohesin subunits) and cohesin loaders to DNA (NIPBL and PDS5A).
Important development of the ideas on TADs anchors structure was provided in the paper by Canela et al. (2017) giving mutual cumulative clarification of the question of anchors in the two papers. First of all, Canela et al. (2017) clarify some discrepancy about Topo2B asymmetrical binding mainly to only one of the DNA strands downstream of CTS “shade”, provided topoisomerase 2 is a two subunit protein binding to both DNA strands. They found that Topo2A and Topo2B location exactly coincides at one and the same point at TAD anchors just beside CTCF target site. Taken together with data by Uuksula-Reimand et al. (2016) these data may be understood as Top2A–Top2B heterodimer binding in between doubled CTCF target sites (CTS).
Also, both groups of authors independently determine that the total number of Rad21/CTS sites is about 40,000 per genome (40,308 in publication (Uuskula-Reimand et al., 2016) versus 40,034 in publication by Vian et al. (2018) [the same group as Canela et al.]), and that exactly half of this figure (20,251) contains also topoisomerase 2B, another half (20,057) lacks topo2B (Vian et al., 2018). This implies that all Rad21/CTS sites contain Topo2B/Topo2A dimers; exactly half of sites contain Topo2A/B in reverse/forward strands location, another half contains this dimer in forward/reverse strands, respectively (see Fig. 3). The same figure for CTCF consensus target sites per genome at TAD anchors (39,838) was also found by Nakahashi et al. (2013). Close estimate (34,000–45,000 sites) was made by Lukinov (2018) for doubled CTCF/BORIS sites.
TAD anchor structure shown in Fig. 3 has important consequences for promotion of strand exchanges between two homologous DNA duplexes. Indeed, two halves of binemic chromosome do not differ in anything but asymmetric position of Top2A and Top2B subunits in relation to strand polarity. In addition, CTCF-G strand polarity in right and left halves of binemic chromosome belongs to single strands of same polarity! This makes the two halves of binemic chromosome fundamentally distinct all over the length of every chromosome and the entire genome. This allows to begin genome proofreading (strand exchange) from the exchange between one Topo2B subunit tightly bound to DNA with one Topo2A subunit from DNA strand of the same polarity, as depicted in Fig. 3 by bent arrow. In this figure, TAD origin is depicted in a form of a circle with equal nearness of all topoisomerase subunits. DNA strands polarity and polarities of CTS’s are also indicated. From the figure, it is clear how strand exchange all over genome is feasible and attainable. This exchange begins and ends at separate “zipper locks” (TAD anchors) and proceeds in two directions: into TADs (directed by cohesin-STAG2 bound to topo2B homodimers) and into SATs (directed by SMC5/6 bound to topo2A homodimers). (Details are in below sections).
From Fig. 3, it also follows that topoisomerase 2 strongly bends DNA. Indeed, it was found by many authors that it bends DNA at least by 60° in vitro. In vivo it could be even greater in response to cohesin and other factors. CTCF itself also bends DNA.
All this makes sense if we consider one-way movement of cohesin-STAG2 along TAD loop (Canela et al., 2017) during proofreading. After proofreading cohesin stops at the opposite side of a TAD loop and then induces proofreading of SAT domain by another complex (SMS5/6) in a relay race mechanism. However, it should be taken into account that 20% of cohesin (SMC complex) does not move; it forms cohesin dimers that keep sister chromatids together (Shi et al., 2020) (“cohesin” in a true sense).
Proofreading will be considered in the detail in following section. Of notice is that cohesin found between CTCF target sites contains stromal antigen 1 (STAG1) which helps to bind cohesin to DNA and stay at that place. Cohesin found just out of CTCF second (of a pair) hind target site contains stromal antigen STAG2 which allows this cohesin to travel along all the TAD length (Countryman et al., 2018). Interesting, that unlike STAG1 cohesin, STAG2 cohesin recognizes repair and replication intermediates; ssDNA gaps, double-strand ends, single-strand overhangs, flaps, forks etc. (Countryman et al., 2018). Indeed, it is important for repair of DNA during movement of such STAG2-cohesin (SMC1/3-based complex) along TAD’s.
SMC complexes
Constitutive repair machinery reconverts hundreds of spontaneous DNA lesions every cell cycle in an error-free way and thus guarantees the fidelity of genome through thousands of cellular generations. Only recently it is becoming clear that these are Structural Maintenance of Chromosomes (SMC) complexes that perform constitutive repair during G1 and G2 stages. (They also participate in inducible repair, usually created by experimentalists, so to distinguish one from another sometimes is rather difficult). In addition, they participate in DNA repair in replication forks during S stage of a cell cycle.
SMC complexes belong to the most ancient enzymatic complexes coeval to DNA and RNA polymerases. They are found among all bacterial species as well as among all eukaryotes. They always consist of two proteins’ ‘rods’ connected at one end through so-called ‘hinge’; so they form a structure resembling letter V. Two apices (“heads”) of letter V are connected by so-called kleisin protein—a flexible molecule allowing V-letter sometimes to flap into double rod usually called “I-conformation”. Kleisin remains associated with two heads of a double rod. “I-conformation” may open back to V-structure closed with kleisin lid. This conformation is often called “O-conformation”—this means that two rods may flex into two arcs forming a kind of O-letter in their shape. Many other conformations may be assumed by SMC complexes because of unique flexible secondary structure of the rods.
Each of the two rods represents one protein molecule folded back at its center so that its N and C ends meet together and form ‘head’ possessing ATPase activity. Folded back protein forms antiparallel coiled coil resembling DNA double-helical molecule. This resemblance is important for our further consideration. All coiled coil protein molecules such as muscle myosin may retract and relax reversibly many times.
In bacteria SMC complexes are formed by two identical coiled coils (rods). Eukaryotic SMC complexes differ from bacterial ones, because they are formed by two different coiled coil proteins (the left one and the right one). However, the length of these two coiled coils is identical. Eukaryotes have three types of SMC complexes. These are: cohesin (SMC1/SMC3), condensin (SMC2/SMC4) and the most ancient safeguardase SMC5/SMC6. Names of the two former safeguardases do not reflect their main functions, because they were discovered as DNA repair complexes; their metaphase functions were discovered later. In addition, they may substitute each other in mitosis, but not in DNA repair.
Besides three classical eukaryotic SMC complexes the SMC-like complex called MRN (Mre11-Rad50-Nbs1) also fulfills repair functions. It has two identical coiled-coil “rods” (Rad50), like bacterial SMC complexes. However, in prokaryotes it is represented by only two subunits (Mre11 and Rad50). Third subunit (Nbs1) is found only in eukaryotes. Instead of “hinge” MRN complex has “zinc hook” connection between two Rad50 coiled-coils rods. This connection may be replaced by hinge connection from another SMC without loss of function (Tatebe et al., 2020). In addition, MRN does not have kleisin. Instead, two Rad50 heads may connect to each other through two Mre11 proteins. In addition, zinc hooks may sometimes detach one from the other and the complex reversibly forms W-shaped structure with two free zinc hooks and two Mre11 connecting two Rad50 heads. (For the details see recent reviews (Reginato & Cejka, 2020; Zabolotnaya et al., 2020)). Unlike SMC complexes, MRN complex performs functions mostly in inducible repair mentioned in the above section “DNA repair systems”.
Recently it was found that SMC complexes travel along DNA molecules both in vitro and in vivo (Countryman et al., 2018; Kanke et al., 2016; Stigler et al., 2016). They recognize lesions in DNA such as DNA DSB, double strand ends, flaps, forks, ssDNA gaps and overhangs and others (Countryman et al., 2018) for to perform constitutive repair. Inducible alarm (SOS) repair is induced by experimentalists and is called DDR (DNA Damage Responce) and it resolves repair task using error-prone systems. Constitutive SMC complexes repair DNA in an error-free way.
Cohesin (SMC1/3 protein complex) clonal mutations are often found in many types of cancer clones in ex vivo cultures (Hill et al., 2016; Leylek et al., 2020; Solomon et al., 2014). Most frequently mutated is STAG2 subunit. MRN complex (SMC-like complex) also was found frequently mutated in cancer clones (Brandt et al., 2017).
Most interesting was the finding that cancer cell lines with STAG2 mutations display synthetic lethality with knockouts of genes participating in either NHEJ or “Homologous recombination” or both systems of repair of DNA DSB (Mondal et al., 2019). This result shows that STAG2 protein participates in ALL (or almost all) of repair of double-stranded lesions in DNA (Mondal et al., 2019), because synthetic lethality was absent with mutations in BER or NER repair systems, responsible for repair of single-stranded DNA lesions (Mondal et al., 2019).
These results indicate that SMC1/3 complex containing STAG2 subunits represents universal constitutive repair system responsible for the repair of DNA DSB at least in gene-rich areas (TADs). More detailed consideration of this system is given in “Proofreading” section.
Recently SMC complexes were found to retract and relax using their capability to twist and writhe within their linking number (coupling writhe and twist just like DNA linking number couples its relaxation and tension) (Krepel et al., 2020). The authors established that each coil-coiled rod may twist and writhe on its own way. In the same time, it was found that both coiled-coils may unite to form braided coil containing four protein strands and this braided coil may also twist and wreath on its own way. These authors make inference that DNA is entwined with protein braided coils and this property is required for SMC movement along DNA.
The holoenzyme SMC complex contains many more subunits than simple SMC heterodimer. These will be considered later on.
Some recent works investigated SMC holoenzyme binding to DNA in vivo (Li et al., 2018; Lobanenkov & Zentner, 2018) (In yeast STAG is called Scc3). SMC1/3 complex begins to bind DNA through two identical stromal antigen (STAG) molecules bound to kleisin (Rad21). Then cohesin sticks to DNA with all its long body (50 nm) including hinge at its hind end. There are two types of STAG molecules considered later on. In addition, SMC1/3 complex in vivo strongly binds two topoisomerase 2B subunits which in turn strongly bind to DNA. SMC5/6 complex holoenzyme binds to DNA through NSE3/NSE1/NSE4 (kleisin) subunits complex (Zabrady et al., 2016). In addition, SMC5/6 complex in vivo strongly binds two topoisomerase 2A subunits instead of topo 2B (Deiss et al., 2019; Kanno et al., 2015).
After loading onto DNA the long coiled-coils are entwined into DNA molecules (Krepel et al., 2020). These are 50 nm long for cohesin (SMC1/3), covering 150 bp of DNA, and 37 nm long for SMC5/6 safeguardase, covering 110 bp of DNA.
(It does not escape from our attention that the length of Okazaki fragments in eukaryotes is the same as the length of cohesin (SMC1/3) that covers also 150 bp of DNA. SMC5/6 coverage in eukaryotes is somewhat shorter, because it covers only reiterated DNA sequences. In prokaryotes Okazaki fragments are about 1200 nucleotides long in spite of their very small genomes).
Proofreading
Recently it was found that homologous DNA duplexes (double helices) may recognize each other in water buffers devoid of divalent cations and proteins and form quadruplexes (four-stranded DNA (Danilowicz et al., 2009; Lee et al., 2016)). Non-homologous duplexes are unable to do so. Physical nature of such strong recognition and bindings is not fully understood. Thus, homologous DNA duplexes may form tight intertwined (braided) structure only if they are homologous to each other and it is connected with nature of DNA itself. DNA synapsis and formation of quadruplexes in vivo of course are regulated by cellular enzymology.
Recently it was also found by Mayaji et al. (2020) that topoisomerase 2B is capable to find homology between remote stretches of DNA in vivo and align them in parallel side by side promoting attachment (linkage) between two duplexes thus forming quadruplexes (four-stranded DNA) (Miyaji et al., 2020).
Also it was found recently that Topo2A or Topo2B homodimers preferentially bind DNA quadruplexes (Oliveri et al., 2020) after exposure of cells to some (not all) Topo2 inhibitors (pyridostatin, etoposide, CX5461) (Oliveri et al., 2020). This means that Topo2 homodimers have strong propensity to bind quadruplexes.
If we fold a paper sheet having a drawing on it (Fig. 3) at central vertical axis we get chromosome made of DNA quadruplexes instead of binemic chromosome made of two homologous identical duplexes. This imaginary procedure taken together with findings by Mayaji et al. (2020) about formation of quadruplexes from homologous duplexes performed by topo2B enzyme and taken together with experimental discovery of formation of quadruplexes from long homologous DNA molecules proceeding with considerable cooperativity, i.e., the strong force of interaction without any enzymatic help (Danilowicz et al., 2009; Lee et al., 2016), and taking into account strong propensity of Topo2 homodimers to bind DNA quadruplexes (Oliveri et al., 2020), we get the following provisional scheme of constitutive DNA proofreading in every cell cycle.
First stage of DNA synapsis begins from formation of quadruplexes from CTCF target sites belonging to opposite (symmetrical) TADs, after topo2A–topo2B subunits exchange. These quadruplexes are needed to position two homologous TAD sequences in proper register without shift for even one nucleotide for proper proofreading even if possible errors aroused in separate nucleotides outside CTCF site. This is why doubled CTCF target sites are needed for proper proofreading.
Then cohesin–STAG2–Topo2B complex begins to move starting from CTS quadruplexes formed by topo2B/Topo2A exchange. Long quadruplexes form and propagate cooperatively beginning from CTS and allowing strand exchange inside these quadruplexes, promoted by Topo2B homodimers moving forward, being a part of cohesin–STAG2–Topo2B complex, thus forming Holliday structure and repair, leaving two repaired heteroduplexes (made of two DNA single-strands originating from different halves of binemic chromosome) behind it.
Return to proofreading. The matter is that formation of quadruplexes from remote homologous GC-rich “unique” DNA stretches needs the existence of two homologous DNA duplexes within one genome. In the same time, 95% of Topo2B reactions were not found to be connected with DNA homology (Miyaji et al., 2020). They simply performed usual reactions between non-homologous DNA sites close to each other (less than 1 kb) and they are usually concentrated around TSS sites (Miyaji et al., 2020). Rare reactions between remote homologous DNA sometimes (not always) was found at the borders between AT-rich quadruplexes made of partially non-homologous satellite DNA and GC-rich quadruplexes made of “unique” DNA sequences homologous to each other. Therefore, these rare topo-sites obviously belong to TAD/SAT borders (Miyaji et al., 2020).
Each quadruplex is proofread using cohesin–STAG2 complex moving ahead and also propagating quadruplex ahead together with bound top2B-STAG2 and other subunits of cohesin. In case of DSB’s (in one of the two symmetric TADs) they become single-strand breaks after strand exchange and are easily repaired. In case of inter-crosslinks or DNA gaps (single or double-stranded) strand exchange stops. Such errors (and other complex errors) are recognized by STAG2 moving behind Topo2B and ahead of other parts of cohesin (Sun et al., 2013; Xu et al., 2018). The doubled head of cohesin (equipped with two STAG2 subunits) skips over such complex lesions at a distance of 50 nm (15 full turns of DNA or about 150 nucleotides) (Brandt et al., 2017). This is a length of cohesin between its head and hinge. (The doubled head of cohesin sticks to yet non-disjoined quadruplex). Braided coiled-coil of cohesin (SMC1/3) also contains four strands (Krepel et al., 2020) and binds DNA over all its length (50 nm). However, this braided protein coil is very flexible in its nature (Krepel et al., 2020) and may either retract or relax or even adopt conformations, where two SMC rods detach from each other in some places or even detach over the whole length of cohesin, forming a kind of “O” conformation and even skip over DNA damage. Such plasticity and elasticity resembles a muscle (also containing coiled-coil protein fibers).
SMC1/3 complex reminds caterpillars (butterfly larvae) travelling along twigs. Some caterpillars (not all of the species) have their foothold only at their head and at their tail, not in between. They move along twig by sticking their head and tail to one point at a twig, their body forming convex arc between head and tail, not touching a twig (Xu et al., 2018). Then caterpillar detaches its head (doubled head of SMC1/3) from this twig and throws it ahead to the whole length of its body and sticks it again to the twig at a considerable distance from its tail. Then it drags its tail (hinge of SMC1/3) always remaining touched to the twig (DNA quadruplex) on its way to the head until meets a damage between head and hinge. Then it stops and waits for damage removal, then moves further until meets undamaged DNA at the SMC head. For this purpose, it has 18 nm hole (within hinge) which allows to pass only for healthy DNA, 18 nm in diameter. Then head makes another jump ahead and so on.
Now return to cancer. It arises from spontaneous DNA double-strand breaks if these are left unrepaired in an error-free way, as we described in first section. Mostly it happens from the absence of second copy of DNA (uninemic sites in binemic chromosomes arising from mutations in some of the TAD’s loop anchors—see last section). SMC complexes are unable to work on only one DNA molecule in uninemic sites for to perform error-free repair in such regions in cancer chromosomes. This leads to arising of chromosome aberrations in every cell cycle leading to necrotic cell death (in a portion of cancer cells, less than 50%) and launching “wound healing” as was described in “Introduction” section. This stimulates further cell divisions in remaining portion (exceeding 50%) of cancer cells (see “Introduction” section for citations).
In any way, strand exchange is possible, while topo2B or topo2A move along a pair of homologous DNA duplexes (Holliday structure or “quadruplex”) with a help of other participants (SMC complexes). Details of overall proofreading are considered in the next section.
Thus topo2A dimers (within SATs) or topo2B dimers (within TADs) move along four-stranded structure (two homologous DNA duplexes side by side) and make strand exchange between two DNA duplexes. Homologous DNA duplexes themselves may form tight intertwined structure (Danilowicz et al., 2009; Lee et al., 2016). Therefore, conditions for strand exchange between two homologous DNA are quite appropriate. Strand exchange within quadruplex is often called “Holliday structure strand exchange” in a more familiar terminology.
Genome-wide strand exchange as a consequence of proofreading
For to allow to check the whole genome for its fidelity evolution created long chain of alternating TADs and SATs (A and B compartments) thus splitting genome into individual checking domains in every TAD and SAT (see “Inlaid genome” section). Fastening “zippers” (strand exchange between two homologous DNA duplexes) allows to compare DNA duplexes in binemic chromosomes for their identity, fidelity and absence of errors, thus scoping and embracing the whole genome split into many compartments. “Zipper locks” for to begin and to end fastening, as we assured from previous section, are located at Topo2A/B heterodimers sandwiched (locked) between two CTCF target sites (CTS) within TAD anchors. Strand exchange begins from exchange between Top2A and Top2B subunits belonging to DNA strands of the same polarity in otherwise identical DNA double helices within binemic chromosomes. It is these doubled CTSs that determine the position of Top2A/Top2B heterodimers on DNA, just at the region between them.
The concept of two proofreading points leading to genome-wide DNA strand exchange in every cell cycle was formulated by my late father; Prof. Luchnik (1971). This concept was not bound to any cellular structure and was based only on peculiarities of chromosome aberration production either spontaneous or after irradiation at different stages of a cell cycle. Now the former concept becomes much simpler and easier to comprehend when it is bound to cellular structures. In addition, I get rid of concept of “multinemy” discussed by my father. It is clear that binemic structure is necessary and sufficient to maintain genome fidelity as is discussed above in the “Where from second DNA copy is taken?” section.
General scheme of genome-wide strand exchanges during two stages of verification of DNA fidelity is depicted in Fig. 4. These stages are designated as P1 and P2 and they are within G1 and G2 stages of the cell cycle, respectively.
Fig. 4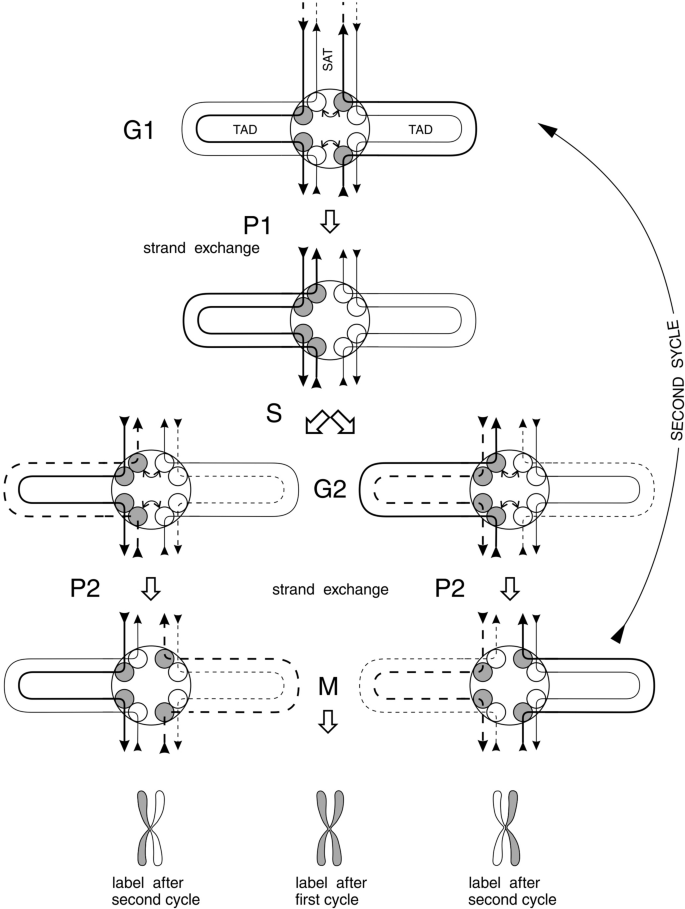
Strand exchange and proofreading (P1 and P2) during the cell cycle. Bent arrow denotes exchange between topo 2A (white ball) and topo 2B (grey ball) subunits leading to DNA strand exchange. Straight arrows indicate DNA strand polarity. Bold lines represent DNA strands bound to topo 2B. Light lines represent DNA strands bound to topo 2A. Dashed lines (bold and light) represent freshly replicated DNA. Grey and white metaphase chromatids below the figure represent distribution of tritium or BrdU label in first and second cell cycles after pulse labeling of DNA at the beginning of the first cycle
Full size imageThe difference between “checkpoints” and “proof points” is in that the former are inducible and the later are constitutive. “Checkpoints” are defined as delays in a cell cycle induced by mutagens and other experimental interference used by experimentalist, so they are scattered in all stages of a cell cycle, depending on mutagen and experimental conditions. On the other hand, “proof points” are constitutional, strictly bound to two timepoints in the cell cycle, and usually are not dependent on experimentalist interference.
The cell cycle depicted in Fig. 4 is repeated for millions of times during prenatal and postnatal development. In addition, it repeats discontinuously in everlasting cell cultures in vitro, except cultures from healthy donors, where cells have a limit for further dividing as well as cells in adult organisms—this leads to in vitro and in vivo aging (senescence).
In Fig. 4, TAD anchors are depicted as circular round plaques with DNA duplexes sticking out in 8 directions; four of them into TADs and four others into SATs. Chromosome axis is formed by SATs connecting TAD anchors along whole chromosome. Therefore, we do not need to depict whole chromosome understanding that situation along this axis repeats many times, as shown in Fig. 2.
TAD anchor is in fact bipartite, i.e., having right and left halves as follows from the behavior of DNA strands during the cell cycle. Other proteins, beside Topo 2A and Topo 2B, participate in the formation of this anchor, such as cohesin protein complex and CTCF proteins (CTCF and BORIS) and some others.
For the simplicity of the consideration we depicted in Fig. 4, only Topo 2A and Topo 2B subunits as main actors within anchors, and omitted machinery performing further strand exchange deeper into TADs and SATs and proofreading of heteroduplexes after primary exchange of topo 2A and topo 2B subunits described in previous section.
It should be explained that SAT DNA forming chromosome axis guarantees strand exchange simultaneously along axis, because this exchange is initiated by Topo 2A–Topo 2B subunits exchange only on SAT sides, where Topo2A and Topo2B subunits are sandwiched between CTCF-G doubled sites (see Fig. 3).
First proofreading point (P1) leads to overall strand exchange as depicted in the upper part of the figure (see Fig. 4). Then, it’s time for DNA replication. Nascent strands are depicted as dashed lines (third raw from above in Fig. 4).
Just after replication second proofreading (P2) is required to repair all errors accumulated during replication. This event is depicted between third and fourth rows (counting from above) in Fig. 4. It results in the second overall strand exchange. Now two daughter genomes are ready for mitosis.
After mitosis everything repeats once again in daughter cells and so on until cells stop cycling and become dormant (senescent) or differentiated.
Thus, two stages of genome-wide proofreading (P1 and P2) begin with local DNA proofreadings’ initiated by exchange between subunits of Topo2B and Topo2A at the borders between TADs and SATs (TAD anchors). Corresponding DNA strands also begin to exchange following to topoisomerases bound to DNA. Topo 2B subunit binds to DNA with a strength comparable to covalent bond. One-directional movement of strand exchange through TAD or through SAT is performed by SMC complexes as described above. Such complexes were identified as SMC1/3 (cohesin) complex and SMC5/6 complex. The first one binds and moves along TAD’s DNA, as established by many authors, the second one moves along SAT DNA (Amaral et al., 2017; Caridi et al., 2017; Diaz & Pecinka, 2018; Ryu et al., 2016; Verver et al., 2016).
The existence of two genome-wide strand exchanges during one cell cycle answers the long standing question. Why we see under microscope classic semiconservative labeling of sister chromatids just as if they were simple DNA duplexes, not binemic structures? (see labeled chromatids in below part of Fig. 4 for the answer).
Structure of cancer chromosomes
Above we described powerful and error-free mechanism of regular surveillance and repair of the genome. Consider what happens with this machinery in cancer cells and why all of them exhibit chromosome instability in spite of proper proofreading.
We already mentioned that cancer cells lose one of the two symmetric (bilateral) halves of some TADs (mutations in TAD anchor DNA at some occasional CTCF target sites) in one or the other site in chromosomes. This loss is heritable trait in every given cell lineage. One of the two symmetric bilateral halves of TAD within binemic chromosome always remains in place, so chromosome continuity is not broken at such sites in cancer cells. However, the chance of accidental breakage increases millions of times as discussed in the “Where from second copy of DNA is taken?” section.
To be more concise let us look at a chromosome of a cancer cell (Fig. 5). For simplicity consider only a short section of a chromosome devoid of one of the two symmetric and identical TAD halves at one of the genomic anchors. It looks like you tear off one of the two identical wings of a butterfly. However, not only one of the two symmetric TAD loops disappears; half of the anchor plaque is also distorted due to mutations at CTCF target sites.
Fig. 5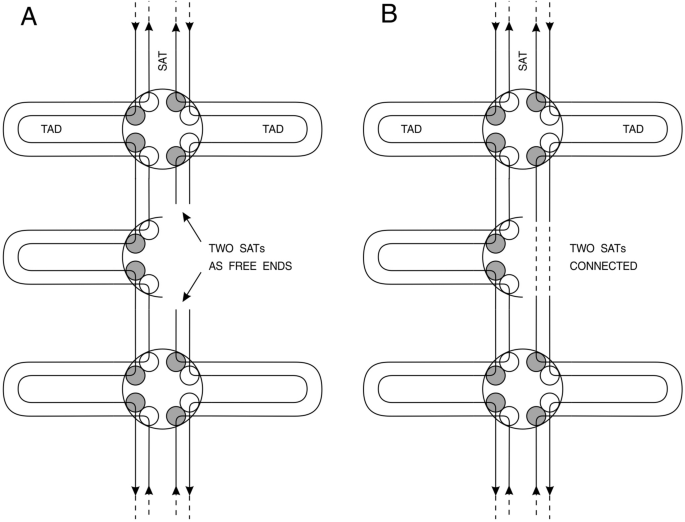
Presentation of a piece of cancer chromosome containing potentially lethal damage (PLD) leading to chromosome instability. A The absence of one half of TAD anchor (due to microdeletion in CTCF cassette DNA) leads to free ends in interstitial satellite DNA (SAT). B connection of two SATs with each other due to mutations in CTCF target sites preventing CTCF and topoisomerase B proteins binding to DNA. Both situations lead to chromosome instability as discussed in the text
Full size imageThis crippled halve of the anchor does not have four of the eight topoisomerase subunits (two of Top 2A and two of Top 2B are absent) because of mutation in CTCF target sites, necessary for topoisomerase 2B to find its place properly (between two tandem CTCF target sites).
Mutations in CTCF target sites in cancer cells were found to be either nucleotide substitutions (Guo et al., 2018; Hnisz et al., 2016; Kaiser et al., 2016; Katainen et al., 2015; Pinoli et al., 2020) or microdeletions (Hnisz et al., 2016). The former ones are now well documented and detailed for many cancers. The later are more difficult to reveal, nevertheless those are also documented in cancers (Hnisz et al., 2016). Spectra of nucleotide substitutions at 11 bp consensus core sequence of CTCF target sites are almost the same in all publications; this is central adenine replaced by another nucleotide and several (5–6) nucleotides just at the borders of 11 bp consensus, beginning from − 6 and further upstream and beginning from + 6 and further downstream by 5–6 nucleotides, either one of them can be replaced by improper nucleotide.
A publication by Pinoli et al. (2020) embraced thousands of various cancer cases from 26 different cancer types thus producing a generalization putting a conclusive statement affirming primary reports of separate cancer cases (Guo et al., 2018; Hnisz et al., 2016; Kaiser et al., 2016; Katainen et al., 2015). This publication (Pinoli et al., 2020) may be considered as a TABLE for various cancers for to classify all of them as being marked by the same nucleotide mutations at CCXXXAGGGGG site. (In fact several tables and table-like figures are presented). Some bias is observed in some pictures around position of central “A” because of “shade” formed due to superposition of two neighboring CTCF target sites, as we already discussed above.
We see two different consequences of microdeletions and substitutions at CTCF target sites. In the first case two SAT’s ends become unprotected so they become dangerous for the cell (Fig. 5A). In the second case two satellite DNA ends connect to each other directly through crippled CTCF sites because of their vicinity to each other (Fig. 5B). It makes no difference for the further fate of cancer cell. In case of unprotected SAT ends one may await the fate of unprotected telomere ends and subsequent trials of TERT reverse transcriptase to keep them long. In case of two connected SATs no DNA breaks are awaited. However, proper proofreading of lonely (without a pair) TAD also becomes impossible; this leads to chromosome aberrations.
Here we should recall what are SATs briefly characterized in the “Inlaid genome” section and what is their role in proofreading described in previous section.
Only recently it is becoming clear that not only telomeres’ satellite transcription participates in cancer. It is found recently that other satellites (mainly interstitial Hsat II and Hsat III) become transcribed in cancer and those transcripts are reverse transcribed into DNA and this DNA integrates into existing SAT regions (Bersani et al., 2015; Ting et al., 2011; Zhu et al., 2018). Moreover, telomeres’ DNA is inserted into different SAT regions using TERT action on telomeres’ transcripts (reviewed in Aksenova and Mirkin (2019)).
Thus some SAT regions become unstable because of their distorted structure in cancer cells. This is not natural diversity of SATs between different loci or between individuals. Such unstable SATs belong to those around deleted ½ TAD anchors depicted in Fig. 5. The failure to participate properly in P1 and P2 strand exchange and repair to unify both copies of particular SAT or TAD (because of the absence of starting point at deleted or mutated CTS sites) leads to instability in this SAT locus leading to chromosome deletions and translocations of pieces of chromosomes into other SAT loci including telomeres and centromeres. In addition, ‘lonely TADs’ become highly susceptible both to induced mutagenesis and spontaneous double-strand breaks due to their inability to undergo proofreading; this leads to chromosome instability (enhanced spontaneous aberration level in every cell cycle).
Also, some types of cancer may arise without distortion of binemic structure, i.e., they are not connected with loss of one of the two halves of symmetric (binemic) TADs. It is reminiscent of the two types of cancer: 1. Related to telomere maintenance and elongation by TERT (85%) and 2. Those maintaining telomeres through ALT mechanism by homologous recombination. In connection with said above about participation of TERT in violation of SAT structure we suppose that 85% of cancers are connected with violation of binemic structure (loss of TADs halves) and 15% are explained by disorders connected with proofreading enzymology violations (clonal mutations). These disorders may be connected with clonal mutations in one of the multiple cohesin subunits (SMC1/3 complex). These may be some other proteins interacting with Topo 2B. These may be protein subunits of SMC5/6 complex interacting with Topo 2A (Deiss et al., 2019; Diaz & Pecinka, 2018; Verver et al., 2016). These may be clonal mutations in CTCF protein itself, not necessarily affecting its target sites. At last these could be some disorders with CTCF-like protein (BORIS) which often occupies one of the two neighboring unidirectional tandem CTCF-G sites embracing Topo 2A/B site. BORIS has a great number of functional splice variants as well as CTCF itself has great number of functional splice variants (Voutsadakis, 2018). Shift in balance between variants of BORIS may also lead to formation of cancer stem cells (Loukinov, 2018). In addition, BORIS determines stemness of cancer stem cells (CSC) (reviewed in Soltanian and Dehghani (2018)).
Whatever could be particular reason for TAD anchor plaque splitting it always should lead to cancer through formation of genetically unstable SAT areas (Fig. 5) generating RNA and DNA (by TERT) and spoiling other SATs in the genome by erroneous insertions leading to chromosome aberrations.
Thus the best way for diagnosis of cancer at the early stage seems to be determining of the level of SAT transcription (including centromeres and telomeres). The level of SAT transcription in non-dividing cells is not changed in spite of some primary events that may lead to TAD anchor plate splitting in dormant cells. In cycling cells only TAD anchor splitting, whatsoever its reason, conceivably leads to elevated level of SAT transcription in cancer cells (Bersani et al., 2015; Ting et al., 2011; Zhu et al., 2018). This way of diagnostics may prove to be inapplicable for the ALT cancers (15%).
The ways and approaches for to cure cancer in the context of the above said in this article may be only two in my opinion. Both of them concern necrotic cell death releasing chemokine CXCL12 from dying cancer cells and stimulating growth of both tumor and stroma cells, as a main driver of expansion.
First, not to allow cancer cells to enter mitosis for to prevent inevitable necrotic death of a portion of them (no more than 50%). Second, enhance necrotic death of cancer cells in mitosis above 50%; then tumor diminishes during subsequent cell divisions more and more if medical mutagenic treatment is repeated several times.
The second approach is currently applied through chemotherapy and radiotherapy. These mutagens induce DNA double-strand breaks (DSB) that are repaired in normal cells and not repaired in “lonely TADs” in cancer cells devoid of second copy of DNA in some chromosome sites. DNA DSB are also induced by some topoisomerase 2 inhibitors inducing DSB at TAD anchors. “Lonely TAD’s” anchors are more susceptible to Topo2 inhibitors inducing DNA DSB than normal ones. However, these inhibitors themselves may induce novel “lonely TADs” and thus may induce novel tumor clones. In addition, chemotherapy in general may prove dangerous—it may cause some new secondary cancer clones even from normal cells.
Seeking for ‘golden bullets’ to hit cancer at mutated TAD anchor sites is a task in vain. It is very difficult to imagine such bullets that reverse mutations at central adenine or flanking nucleotides around – 5 to + 5 positions from adenine in some of the mutated CTCF target sites frequently found in all cancer genomes (Guo et al., 2018; Hnisz et al., 2016; Kaiser et al., 2016; Katainen et al., 2015; Pinoli et al., 2020). Deletions at these sites found in cancers (Hnisz et al., 2016) are also impossible to revert.
The first approach I mentioned is not to allow cancer cells to enter mitosis. Some non-mutagenic inhibitors of DNA replication are used in cancer therapy in combination with other treatments. Apoptosis would be better.
So, the best way to prevent mitosis in cancer cells is to induce their apoptosis in interphase. In fact, tumors arise in every one of us every 3–5–10 years (depending on person’s genetic background and spontaneous mutagenic exposure). However, when these tumors are small they are recognized by neuro-humoral system and signals for targeted apoptosis are subsequently sent from hypothalamus–pituitary system. Only a small percent of humans suffers from indistinguishing of tumors by their own body surveillance. Why tumors are self-destroyed through apoptosis in the majority of people and not destroyed in some rare persons? Answering this question gives the best way to cure cancer, in my opinion.
References
Aksenova, A. Y., & Mirkin, S. M. (2019). At the beginning of the end and in the middle of the beginning: Structure and maintenance of telomeric DNA repeats and interstitial telomeric sequences. Genes, 10, 118. https://doi.org/10.3390/genes10020118
Altemose, N., Miga, K. H., Miggioni, M., & Willard, H. F. (2014). Genomic characterization of Large heterochromatic gaps in the human genome assembly. PLoS Computational Biology, 10(5), e1003628. https://doi.org/10.1371/journal.pcbi.1003628
Amaral, N., Ryu, T., & Chiolo, I. (2017). Nuclear dynamics of heterochromatin repair. Trends in Genetics, 33, 86–100. https://doi.org/10.1016/j.tig.2016.12.004
Barnard, S., Bouffler, S., & Rothkamm, K. (2013). The shape of the radiation dose response for DNA double-strand break induction and repair. Genome Integrity, 4, 1. https://doi.org/10.1186/2041-9414-4-1
Bersani, F., Lee, E., Kharchenko, P. V., Xu, A. W., Liu, M., Xega, K., et al. (2015). Pericentromeric satellite repeat expansions through RNA-derived DNA intermediates in cancer. Proceedings of the National Academy of Sciences of the United States of America, 112, 15148–15153. https://doi.org/10.1073/pnas.1518008112
Bianchi, M. E., & Mezzapelle, R. (2020). The chemokine receptor CXCR4 in cell proliferation and tissue regeneration. Frontiers in Immunology, 11, 2109. https://doi.org/10.3389/fimmu.2020.02109
Biehs, R., Steinlage, M., Barton, O., Juhasz, S., Kunzel, J., Spies, J., et al. (2017). DNA double-strand break resection occurs during non-homologous end joining in G1 but is distinct from resection during homologous recombination. Molecular Cell, 65, 671–684. https://doi.org/10.1016/j.molcel.2016.12.016
Billen, D. (1990). Spontaneous DNA damage and its significance for the “negligible dose” controvercy in radiation protection. Radiation Research, 124, 242–245. https://doi.org/10.2307/3577872
Brandt, S., Samartzis, E. P., Zimmermann, A.-K., Fink, D., Moch, H., Noske, A., & Dedes, K. (2017). Lack of MRE11-RAD50-NBS1 (MRN) complex detection occurs frequently in low-grade epithelial ovarian cancer. BMC Cancer, 17, 44. https://doi.org/10.1186/s12885-016-3026-2
Brouwer, I., Sitters, G., Candelli, A., Heerema, S. J., Heller, I., de Mello, A. J., et al. (2016). Sliding sleeves of XRCC4-XLF bridge DNA and connect fragments of broken DNA. Nature, 235, 566–569. https://doi.org/10.1038/nature18643
Canela, A., Maman, Y., Jung, S., Wong, N., Callen, E., Day, A., et al. (2017). Genome organization drives chromosome fragility. Cell, 170, 507–521. https://doi.org/10.1016/j.cell.2017.06.034
Caridi, P. C., Delabaere, L., Zapotoczny, G., & Chiolo, I. (2017). And yet, it moves: Nuclear and chromatin dynamics of a heterochromatic double-strand break. Philosophical Transactions of the Royal Society b: Biological Sciences, 372, 20160291. https://doi.org/10.1098/rstb.2016.0291
Chen, L., Trujillo, K., Ramos, W., Sang, P., & Tomkinson, A. E. (2001). Promotion of Dnl4-catalyzed DNA end-joining by the Rad50/Mre11/Xrs2 and Hdf1/Hdf2 complexes. Molecular Cell, 8, 1105–1115. https://doi.org/10.1016/s1097-2765(01)00388-4
Countryman, P., Fan, Y., Gorthi, A., Pan, H., Strickland, J., Kaur, P., et al. (2018). Cohesin SA2 is a sequence-independent DNA-binding protein that recognizes DNA replication and repair intermediates. Journal of Biological Chemistry, 293, 1054–1069. https://doi.org/10.1074/jbc.M117.806406
Danilowicz, C., Lee, C. E., Kim, K., Hatch, K., Coljee, V. W., Kleckner, N., & Prentiss, M. (2009). Single molecule detection of direct, homologous, DNA/DNA pairing. Proceedings of the National Academy of Sciences of the United States of America, 106, 19824–19829. https://doi.org/10.1073/pnas.0911214106
Deiss, K., Lockwood, N., Howell, M., Segeren, H. A., Saunders, R. E., Chakravarty, P., et al. (2019). A genome-wide RNAi screen identifies the SMC5/6 complex as a non-redundant regulator of a Topo2a-dependent G2 arrest. Nucleic Acids Research, 47, 2906–2921. https://doi.org/10.1093/narlgky1295
Diaz, M., & Pecinka, A. (2018). Scaffolding for repair: Understanding molecular functions of the SMC5/6 complex. Genes, 9, 36. https://doi.org/10.3390/genes9010036
Dixon, J. R., Selvaraj, S., Yue, F., Kim, A., Li, Y., Shen, Y., et al. (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature, 485, 376–380. https://doi.org/10.1038/nature11082
Guo, F., Wang, Y., Liu, J., Mok, S. C., Xue, F., & Zhang, W. (2016). CXCL12/CXCR4: A symbiotic bridge linking cancer cells and their stromal neighbors in oncogenic communication networks. Oncogene, 35, 816–826. https://doi.org/10.1038/onc.2015.139
Guo, Y. A., Chang, M. M., Huang, W., Ooi, W. F., Xing, M., Tan, P., et al. (2018). Mutation hotspots at CTCF binding sites coupled to chromosomal instability in gastrointestinal cancers. Nature Com, 9, 1520. https://doi.org/10.1038/s41467-018-03828-2
Hill, V. K., Kim, J.-S., & Waldman, T. (2016). Cohesin mutations in human cancer. Biochimica Et Biophysica Acta, 1866, 1–11. https://doi.org/10.1016/j.bbcan.2016.05.002
Hnisz, D., Weintraub, A. S., Day, D. S., Valton, A.-L., Bak, R. O., Li, C. H., et al. (2016). Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science, 351(6280), 1454–1458. https://doi.org/10.1126/science.aad9024
Huang, J., & Dynan, W. S. (2002). Reconstitution of the mammalian DNA double-strand break end-joining reaction reveals a requirement for an Mre11/Rad50/NBS1-containing fraction. Nucleic Acids Research, 30, 667–674. https://doi.org/10.1093/nar/30.3.667
Kaiser, V. B., Taylor, M. S., & Semple, C. A. (2016). Mutational biases drive elevated rates of substitution at regulatory sites across cancer types. PLoS Genetics, 12(8), e1006207. https://doi.org/10.1371/journal.pgen.1006207
Kanke, M., Tahara, E., Huis in’t Veld, P. J., & Nishiyama, T. (2016). (2016) Cohesin acetylation and Wapl-Pds5 oppositely regulate translocation of cohesin along DNA. The EMBO Journal, 35, 2686–2698. https://doi.org/10.15252/embj.201695756
Kanno, T., Berta, D. G., & Sjogren, C. (2015). The SMC5/6 complex is an ATP-dependent intermolecular DNA linker. Cell Reports, 12, 1–12. https://doi.org/10.1016/j.celrep.2015.07.048
Katainen, R., Dave, K., Pitkanen, E., Palin, K., Kivioja, T., Valimaki, N., et al. (2015). CTCF/cohesin-binding sites are frequently mutated in cancer. Nature Genetics, 47, 818–821. https://doi.org/10.1038/ng.3335
Krepel, D., Davtyan, A., Schafer, N. P., Wolynes, P. G., & Onuchic, J. N. (2020). Braiding topology and the energy landscape of chromosome organization proteins. Proceedings of the National Academy of Sciences of the United States of America, 117, 1468–1477. https://doi.org/10.1073/pnas.1917750117
Lee, D. J., Danilowicz, C., Rochester, C., Kornyshev, A. A., & Prentiss, M. (2016). Evidence for protein-free homology recognition in magnetic dead force-extension experiments. Proceedings of the Royal Society a: Mathematical, Physical and Engineering Sciences, 472, 20160186. https://doi.org/10.1098/rspa.2016.0186
Lengauer, C., Kinzler, K. W., & Vogelstein, B. (1998). Genetic instabilities in human cancers. Nature, 396, 643–649. https://doi.org/10.1038/25292
Leylek, T. R., Jeusset, L. M., Lichtensztein, Z., & McManus, K. J. (2020). Reduced expression of genes regulating cohesion induces chromosome instability that may promote cancer and impact patient outcomes. Science and Reports, 10, 592. https://doi.org/10.1038/s41598-020-57530-9
Li, H., Vogel, H., Holcomb, V. B., Gu, Y., & Hasty, P. (2007). Deletion of Ku70, Ku80, or both causes early aging without substantially increased cancer. Molecular and Cellular Biology, 27, 8205–8214. https://doi.org/10.1128/MCB.00785-07
Li, Y., Muir, K. W., Bowler, M. W., Metz, J., Haering, C. H., & Panne, D. (2018). Structural basis for Scc3-dependent cohesin recruitment to chromatin. eLife, 7, e38356. https://doi.org/10.7554/eLife.38356
Liang, S., Chaplin, A. K., Stavridi, A. K., Appleby, R., & Hnizda, A. (2020). Stages, scaffolds and strings in the spatial organization of non-homologous end joining: Insights from X-ray diffraction and Cryo-EM. Progress in Biophysics and Molecular Biology. https://doi.org/10.1016/j.pbiomolbio.11.008
Lieberman-Aiden, E., van Berkum, N. L., Williams, L., Imakaev, M., Ragoczy, T., Telling, A., et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science, 326, 289–293. https://doi.org/10.1126/science.1181369
Lindahl, T., & Barnes, D. E. (2000). Repair of endogenous DNA damage. Cold Spring Harbor Symposia on Quantitative Biology, 65, 127–133. https://doi.org/10.1101/sqb.2000.65.127
Lobanenkov, V., & Zentner, G. (2018). Discovering a binary CTFC code with a little help from BORIS. Nucleus, 9, 33–41. https://doi.org/10.1080/19491034.2017.1394536
Lopez-Gil, J. C., Martin-Hijano, L., Hermann, P. C., & Sainz, B., Jr. (2021). The CXCL12 crossroads in cancer stem cells and their niche. Cancers, 13, 469. https://doi.org/10.3390/cancers13030469
Loukinov, D. (2018). Targeting CTCFL/BORIS for the immunotherapy of cancer. Cancer Immunology, Immunotherapy, 67(12), 1955–1965. https://doi.org/10.1007/s00262-018-2251-8
Luchnik, A. N. (1982). Eukaryote chromosome structure: Foundation of the bineme model. Journal of Theoretical Biology, 99, 135–157. https://doi.org/10.1016/0022-5193(82)90395-2
Luchnik, A. N. (2003). A model of self-maintenance of in vivo malignant growth not dependent on tumor type or origin. Syndrome of everlasting wound healing. Cancer Biology & Therapy, 2, 343–346. https://doi.org/10.4161/cbt2.4.476
Luchnik, A. N., Glaser, V. M., & Shestakov, S. V. (1977). Repair of DNA double-strand breaks requires two homologous DNA duplexes. Molecular Biology Reports, 3, 437–442. https://doi.org/10.1007/BF00808385
Luchnik, A. N., Slesinger, M., & Vetrova, L. N. (1995). Experimental evidence for the binemic structure of human chromosomes. Biochemistry and Molecular Biology International, 36, 723–732. PMID: 8528135.
Luchnik, N. V. (1971). The chromosome cycle of DNA. Studia Biophys, 27, 157–164.
Menolfi, D., & Zha, S. (2020). ATM, DNA-PKcs and ATR: Shaping development through the regulation of the DNA damage responses. Genome Instability Disease, 1, 47–68. https://doi.org/10.1007/s42764-019-00003-9
Miyaji, M., Furuta, R., Hosoya, O., Sano, K., Hara, N., Kuwano, R., Kang, J., Teteno, M., et al. (2020). Topoisomerase IIb targets DNA crossovers formed between distant homologous sites to induce chromatin opening. Science and Reports, 10, 18550. https://doi.org/10.1038/s41598-020-75004-w
Mondal, G., Stevers, M., Goode, B., Ashworth, A., & Solomon, D. A. (2019). A requirement for STAG2 in replication fork progression creates a targetable synthetic lethality in cohesin-mutant cancers. Nature Communications, 10, 1686. https://doi.org/10.1038/s41467-019-09659-z
Moore, J. K., & Haber, J. E. (1996). Cell cycle and genetic requirements of two pathways of nonhomologous end joining repair of double-strand breaks in Saccharomices cerevisiae. Molecular and Cellular Biology, 16, 2164–2173. https://doi.org/10.1128/MCB.16.5.2164
Mortezaee, K. (2020). CXCL12/CXCR4 axis in microenvironment of solid tumors: A critical mediator of metastases. Life Sciences, 249, 117534. https://doi.org/10.1016/j.lfs.2020.117534
Nakahashi, H., Kiefer Kwon, K.-R., Resch, W., Vian, L., Dose, M., Stavreva, D., et al. (2013). A genome-wide map of CTCF multivalency redefines the CTCF code. Cell Reports, 3, 1–12. https://doi.org/10.1016/j.celrep.2013.04024
Naughton, C., Avlonitis, N., Corless, S., Prendergast, J., Mati, I. K., Eijk, P. P., et al. (2013). Transcription forms and remodels supercoiling domains unfolding large-scale chromatin structures. Nature Structural & Molecular Biology, 20, 387–397. https://doi.org/10.1038/nsmb.25609
Neumaier, T., Swenson, J., Pham, C., Polyzos, A., Lo, A. T., Yang, P. A., et al. (2012). Evidence for formation of DNA repair centers and dose-response nonlinearity in human cells. Proceedings of the National Academy of Sciences of the United States of America, 109, 443–448. https://doi.org/10.1073/pnas.1117849108
Nirmala, J. G., & Lopus, M. (2020). Cell death mechanisms in eukaryotes. Cell Biology and Toxicology, 36, 145–164. https://doi.org/10.1007/s10565-019-09496-2
Nora, E. P., Lajoie, B. R., Schulz, E. G., Giorgetti, L., Okamoto, I., Servant, N., et al. (2012). Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature, 485, 381–385. https://doi.org/10.1038/nature11049
Oliveri, M., Cho, T., Alvarez-Quilon, A., Li, K., Schellenberg, M., Zimmermann, M., et al. (2020). A genetic map of the response to DNA damage in human cells. Cell, 182, 1–16. https://doi.org/10.1016/j.cell.2020.05.040
Pinoli, P., Stamoulakatou, E., Nguyen, A.-P., Martinez, M. R., & Ceri, S. (2020). Pan-cancer analysis of somatic mutations and epigenetic alterations in insulated neighbourhood boundaries. PLoS ONE, 15(1), e0227180. https://doi.org/10.1371/journal.pone.0227180
Pugacheva, E. M., Rivero-Hinojsa, S., Espinosa, C. A., Mendes-Catala, C. F., Kang, S., Susuki, T., et al. (2015). Comparative analyses of CTCF and BORIS occupancies uncover two distinct classes of CTCF binding genomic regions. Genome Biology, 16, 1–24. https://doi.org/10.1186/s13059-015-0736-8
Pustovalova, M., Grekhova, A., Astrelina, T., Nikitina, V., Dobrovolskaya, E., Suchkova, Y., et al. (2016). Accumulation of spontaneous gH2AX foci in long-term cultured mesenchymal stromal cells. Aging, 8, 3498–3506. https://doi.org/10.18632/aging.101142
Ramsden, D. A., & Gellert, M. (1998). Ku protein stimulates DNA end joining by mammalian DNA ligases: A direct role for Ku in repair of DNA double-strand breaks. The EMBO Journal, 17, 609–614. https://doi.org/10.1093/emboj/17.2.609
Rass, E., Grabarz, A., Plo, I., Gautier, J., Bertrand, P., & Lopez, B. S. (2009). Role of MRE11 in chromosomal nonhomologous end joining in mammalian cells. Nature Structural & Molecular Biology, 16, 819–824. https://doi.org/10.1038/nsmb.1641
Reginato, G., & Cejka, P. (2020). The MRE11 complex: A versatile toolkit for the repair of broken DNA. DNA Repair, 91–92, 102869. https://doi.org/10.1016/j.dnarep.2020.102869
Ryu, T., Bonner, M. R., & Chiolo, I. (2016). Cervantes and Quijote protect heterochromatin from aberrant recombination and lead the way to the nuclear periphery. Nucleus, 7, 485–497. https://doi.org/10.1080/19491034.2016.1239683
Sasanuma, H., Yamada, S., Tsuda, M., & Takeda, S. (2020). Restoration of ligatable “clean” double-strand break ends is the rate-limiting step in the rejoining of ionizing-radiation-induced DNA breakage. DNA Repair, 93, 102913. https://doi.org/10.1016/j.dnarep.2020.102913
Scild, D., Ananthaswamy, H. N., & Mortimer, R. K. (1981). An endomitotic effect of a cell cycle mutation of Saccharomyces cerevisiae. Genetics, 97, 551–562. PMID7028565.
Shi, D., Zhao, S., Zuo, M.-Q., Zhang, J., Hou, W., Dong, M.-Q., & Cao, Q. (2020). The acetyltransferase Eco1 elicits cohesin dimerization during S phase. Journal of Biological Chemistry, 295, 7554–7565. https://doi.org/10.1074/jbcRA120013102
Solomon, D. A., Kim, J. S., & Waldman, T. (2014). Cohesin gene mutations in tumorigenesis: from discovery to clinical significance. BMB Reports, 47, 299–310. https://doi.org/10.5483/BMBRep.2014.47.6.092
Soltanian, S., & Dehghani, H. (2018). BORIS: A key regulator of cancer stemness. Cancer Cell International, 18, 154. https://doi.org/10.1186/s12935-018-0650-8
Stigler, J., Camdere, G. O., Koshland, D. E., & Greene, E. C. (2016). Single-molecule imaging reveals a collapsed conformational state for DNA-bound cohesin. Cell Reports, 15, 988–998. https://doi.org/10.1016/j.celrep.2016.04.003
Sun, M., Nishino, T., & Marko, J. F. (2013). The SMC1-SMC3 cohesin heterodimer structures DNA through supercoiling-dependent loop formation. Nucleic Acids Research, 41, 6149–6160. https://doi.org/10.1093/nar/gkt303
Tatebe, H., Lim, C. T., Konno, H., Shiozaki, K., Shinohara, A., Uchihashi, T., & Furukohri, A. (2020). Rad50 zinc hook functions as a constitutive dimerization module interchangeable with SMC hinge. Nature Communications, 11, 370. https://doi.org/10.1038/s41467-019-14025-0
Ting, D. T., Lipson, D., Paul, S., Brannigan, B. W., Akhavanfard, S., Coffman, E. J., et al. (2011). Aberrant overexpression of satellite repeats in pancreatic and other epithelial cancers. Science, 331, 593–596. https://doi.org/10.1126/science.1200801
Uuskula-Reimand, L., Hou, H., Smavarchi-Tehrani, P., Rudan, M. V., Liang, M., Medina-Rivera, A., et al. (2016). Topoisomerase II beta interacts with cohesin and CTCF at topological domain borders. Genome Biology, 17, 182. https://doi.org/10.1186/s13059-016-1043-8
Verver, D. E., Zheng, Y., Speijer, D., Hoebe, R., Dekker, H. L., Repping, S., et al. (2016). Non-SMC element 2 (NSMCE2) of the SMC5/6 complex helps to resolve topological stress. International Journal of Molecular Sciences, 17, 1782. https://doi.org/10.3390/ijms17111782
Vian, L., Pekowska, A., Rao, S. S. P., Kieffer-Kwon, K. R., Jung, S., Baranello, L., et al. (2018). The energetics and physiological impact of cohesin extrusion. Cell, 173, 1–14. https://doi.org/10.1016/j.cell.2018.03.072
Vickridge, E., Planchenaut, C., Cockram, C., Juceda, I. G., & Espeli, O. (2017). Management of E. coli sister chromatid cohesion in response to genotoxic stress. Nature Communications, 8, 14618. https://doi.org/10.1038/ncomms14618
Vilenchik, M. M., & Knudson, A. G. (2003). Endogenous DNA double-strand breaks: Production, fidelity of repair, and induction of cancer. Proceedings of the National Academy of Sciences, 100, 12871–12876. https://doi.org/10.1073/pnas.2135498100
Voutsadakis, I. A. (2018). Molecular lesions of insulator CTCF and its paralogue CTCFL (BORIS) in cancer: An analysis from published genomic studies. High-Throughput, 7, 30. https://doi.org/10.3390/ht7040030
Wu, Q. (2019). Structural mechanism of DNA-end synapsis in the non-homologous end joining pathway for repairing double-strand breaks: Bridge over troubled ends. Biochemical Society Transactions, 47, 1609–1619. https://doi.org/10.1042/bst20180518
Xie, A., Kwok, A., & Scully, R. (2009). Role of mammalian MRE11 in classical and alternative non-homologous end joining. Nature Structural & Molecular Biology, 16, 814–818. https://doi.org/10.1038/nsmb.1640
Xu, X., Kanai, R., Nakazawa, N., Wang, L., Toyoshima, C., & Yanagida, M. (2018). Suppressor mutation analysis combined with 3D modeling explains cohesin’s capacity to hold and release DNA. Proceedings of the National Academy of Sciences of the United States of America, 115, E4833–E4842. https://doi.org/10.1073/pnas.1803564115
Xu, X., Zhu, F., Zhang, M., Zeng, D., Luo, D., Liu, G., et al. (2013). Stromal cell-derived factor-1 enhances wound healing through recruiting bone marrow-derived mesenchymal stem cells to the wound area and promoting neovascularization. Cells, Tissues, Organs, 197, 103–113. https://doi.org/10.1159/000342921
Zabolotnaya, E., Mela, I., Henderson, R. M., & Robinson, M. (2020). Turning the Mre11/Rad50 DNA repair complex on its head: Lessons from SMC protein hinges, dynamic coiled-coil movements and DNA loop-extrusion? Biochemical Society Transactions, 48, 2359–2376. https://doi.org/10.1042/BST20170168
Zabrady, K., Adamus, M., Vondrova, L., Liao, C., Skoupilova, H., Novakova, M., Jarcisinova, L., Alt, A., et al. (2016). Chromatin association of the SMC5/6 complex is dependent on binding of its NSE3 subunit to DNA. Nucleic Acids Research, 44, 1064–1079. https://doi.org/10.1093/nar/gkv1021
Zhao, B., Watanabe, G., Morten, M. J., Reid, D. A., Rothenberg, E., & Lieber, M. R. (2019). The essential elements for the noncovalent association of two DNA ends during NHEJ synapsis. Nature Communications, 10, 3588. https://doi.org/10.1038/s41467-019-11507-z
Zhu, Q., Hoong, N., Aslanian, A., Hara, T., Benner, C., Heinz, S., et al. (2018). Heterochromatin-encoded satellite RNAs induce breast cancer. Molecular Cell, 70, 842–853. https://doi.org/10.1016/j.molcel.2018.04.023
Acknowledgements
(1) This paper is dedicated to the memory of my late father, Prof. N.V. Luchnik. (2) I am thankful to Yuri Safonov for help in preparation of figures.
Author information
Affiliations
Koltzov Institute of Developmental Biology, Russian Academy of Sciences, Moscow, Russia
Andrey N. Luchnik
Corresponding author
Correspondence to Andrey N. Luchnik.
Ethics declarations
Conflict of interest
The conflict of interests is absent in present paper.
Rights and permissions
About this article
Cite this article
Luchnik, A.N. Chromosome instability induced by mutations in TAD anchors leads to tumors. GENOME INSTAB. DIS. (2021). https://doi.org/10.1007/s42764-021-00050-1
Received15 July 2021
Revised08 September 2021
Accepted17 September 2021
Published29 September 2021
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
Necrotic cell death
Topologically associated domains
Topoisomerase 2A/B
CTCF target sites
SMC complex
用户登录
还没有账号?
立即注册