






Genome instability in pathogenesis of tuberculosis
Review Article
Kehong Zhang, Yuping Ning, Fanhui Kong, Xinchun Chen & Yi Cai
Genome Instability & Disease 2, 331–338 (2021)
Abstract
Tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb), remains a serious global health problem that kills over 1 million people annually. Understanding the pathogenesis of TB and the emergence of drug-resistant strains of Mtb is a priority for the development of strategies against TB. Its DNA damage and repair systems are essential for maintaining genome stability in Mtb. Their aberrant work leads to hypermutability and is often associated with the emergence of resistant bacteria. On the other hand, Mtb infection also induces genome instability of host cells, which are involved in the pathogenesis of TB, including the formation of giant cells. Collectively, this review is an attempt to summarize our current understanding of the role of genome instability in the pathogenesis of TB and to shed light on the development of new strategies for TB treatment.
Introduction
Tuberculosis (TB) is a highly infectious disease caused by Mycobacterium tuberculosis (Mtb) and has led to great human suffering worldwide for centuries. According to the WHO report in 2019, approximately 10 million people (range: 8.9–11.0 million) were infected with TB, and 1 million deaths had occurred (Chakaya et al., 2021). While most drug-sensitive TB is curable in about 6 months of treatment, only 50% of multidrug-resistant TB (MDR-TB) cases can be cured using second-line drugs that are generally toxic over a longer usage period. To overcome these challenges, new drugs, including conventional antibiotics and host-directed therapy (HDT) drugs, are needed. One major challenge is the high frequency of spontaneous chromosomal mutations caused by the selective application of antibiotics, which has led to multiple drug resistance and drug efficacy reduction during TB treatment (Balganesh et al., 2012). However, HDT works via a different mechanism to conventional drugs, therefore, has been recognized as a promising strategy to overcome Mtb drug resistance.
Pathogenic bacteria have to cope with a variety of challenges, from host defense systems to antibiotics; they must develop the ability to survive and grow in various hostile environments. The inhalation of aerosol droplets containing intracellular pathogen Mtb into the alveoli of the lungs initiates the TB infection cycle (Smith, 2003). The biological niche of Mtb is the host macrophages. When Mtb enters the host cell, nucleotide damage occurs under various stress conditions, resulting in mutations in the genome and impairment of genomic integrity in the bacteria (Cadet & Wagner, 2014; Cadet et al., 2010). Genomic integrity is a prerequisite for the survival, development, and propagation of all organisms. The inability to rectify such damage is detrimental to the pathogen’s survival in the host. Therefore, Mtb’s ability to withstand various environments and cause disease suggests a strong DNA repair system is required to ensure efficient error-free transmission of genetic material. Years of studies have revealed that Mtb has a remarkably evolved and highly redundant DNA repair system, providing it with robust survivability in harsh environments, such as reactive oxygen species (ROS), reactive nitrogen species (Cole et al., 1998), and low pH generated by the host immune system (Ehrt & Schnappinger, 2009).
Genome instability is a major driver leading to the acquisition of drug resistance in bacteria (Booth et al., 2020). It describes a state of increased tendency to acquire distinct phenotypes based on genetic alterations and displays a process, such as DNA repair, as a consequence of deficient genome maintenance (Ben-David, 2015). Mtb has multiple DNA repair mechanisms, including base excision repair, nucleotide excision repair (NER), and other pathways to restore the damaged genome (Singh, 2017). Understanding these DNA systems is important to aid the development of therapeutic agents that can counteract Mtb evolutionary changes. Antibiotic tolerance is also involved in genome instability, including DNA damage repair and genetic mutations. However, at present, knowledge of genome instability in Mtb remains limited.
Here, we review the current knowledge of genomic instability, mainly targeting DNA damage and repair in mycobacteria in infection and pathogenicity, aiming to highlight their potential impact on the development of novel drugs against TB.
Mtb genomic instability and DNA repair systems
The DNA repair systems in Mtb play essential roles in the pathogenesis and maintenance of genome stability (Gorna et al., 2010). Recent studies have demonstrated that the DNA repair-related genes are expressed differentially at each stage of Mtb infection (Gorna et al., 2010). In the following sections, we briefly introduce some DNA repair systems in Mtb and aim to improve our understanding of how these systems impact the infection processes and pathogen evolution.
Base excision repair (BER)
BER is of interest due to its ability to repair uracil and 7, 8-dihydro-8-oxoguanine (8-oxoG) since mycobacteria possess a GC-rich genome (Mizrahi & Andersen, 1998; Naz et al., 2021). Uracil DNA glycosylase (MacMicking et al.) catalyzes the removal of uracil from DNA, which promotes the uracil excision repair pathway (Krokan et al., 2002; Kurthkoti & Varshney, 2011). Ung and UdgB, which belong to the UDG family and are involved in uracil excision repair of Mtb, were found not relevant to growth in vitro (Kurthkoti & Varshney, 2010). The simultaneous deletion of Ung and UdgB in Mycobacterium smegmatis (Msm) showed a synergistic effect on the accumulation of mutations (Malshetty et al., 2010). It would be of interest to generate similarly mutated strains in Mtb and look at the growth of pathogens in all conditions. In addition, two DNA glycosylases (Fpg/MutM and MutY) and a nucleotide hydrolase (MutT) involved in the GO repair pathway, minimized the chances of misincorporation into DNA (Jain et al., 2007; Kurthkoti et al., 2010). The GO system forms part of the BER pathway and specifically recognizes and repairs the oxidized form of 8-oxoG and associated mismatches (van der Veen & Tang, 2015). Mycobacterial DNA polymerases have different properties regarding the incorporation of 8-oxoG in DNA. Multiple homologs in Mtb mutated strains involved in the GO repair pathway are critical for the survival of Mtb in the host cell and require further exploration. Moreover, Khanam et al. found that Mtb class-II AP-endonuclease (XthA) engaged with NAD + -dependent DNA ligase (LigA) to counter futile cleavage and ligation cycles that might derail bacterial BER (Khanam et al., 2020).
Nucleotide excision repair (NER)
Nucleotide excision repair (NER) is an important DNA repair pathway that is used to remove many bulky DNA adducts, such as cisplatin interstrand cross-links, cyclobutene pyrimidine dimers, and alkylation adducts in bacteria (Grossman & Thiagalingam, 1993; Kisker et al., 2013). Similar to many other bacteria, NER plays a key role in the growth or virulence in Mtb, including Mtb uvrA, uvrB, or uvrC mutant strains induced by UV irradiation and mitomycin C (Darwin & Nathan, 2005). Mtb uvrA is implicated to be important for optimal growth in the presence of DNA damaging agents (Rossi et al., 2011; Springall et al., 2018). In uvrB-deficient Mtb infected mice, the virulence of Mtb is markedly reduced (Darwin & Nathan, 2005). Mtb UvrB possesses ATP-dependent DNA helicase activity and its formed dimers bound to UvrA in the absence of any ligands (Lahiri et al., 2018; Thakur et al., 2016). Mtb uvrC is essential for Mtb DNA repair system, particularly in response to DNA damage caused by UV irradiation (Prammananan et al., 2012). The pairwise relationships of the UvrABC incision complex has been reported to promote the development of new therapeutics (Mazloum et al., 2011; Thakur et al., 2020). Overall, the divergence of the bacterial NER machinery remains challenging.
Mismatch repair (MMR)
MMR recognizes mismatched bases and is widely conserved across prokaryotes, maintaining genetic stability and integrity (Li et al., 2016). The complete genome sequence of Mtb revealed that Mtb lacks the classical form of the MMR pathway, suggesting a high level of genetic variation occurred, and its loss has a pivotal role in the biological consequences (Cole et al., 1998; Mizrahi & Andersen, 1998). Recent reports discovered a non-canonical form of MMR in Msm and Mtb, NucS/EndoMS. This endonuclease reduces the hypermutator phenotype, avoids a transition-biased mutational spectrum, and decreases homologous recombination (Castaneda-Garcia et al., 2017; Cebrian-Sastreet al., 2021). MMR defects are often associated with the emergence of resistant bacteria; thus the NucS pathway seems to be a promising target on which to focus to prevent TB drug-resistant mutations in the future (McGrath et al., 2014).
Double-strand breaks (DSBs) repair
Cell death is inevitable when DSBs remain unrepaired. Common homologous recombination (HR), mycobacterial-specific non-homologous end-joining (NHEJ), and single-strand annealing (Gutierrez et al.) are considered to be the three main pathways for processing DSBs in Mtb (Dos Vultos et al., 2009). HR is an error-free mechanism of the DSB damage repair system when the chromosome is damaged, but the second copy is still available to use as a DNA template (Dos Vultos et al., 2009). RecA, a RecFOR pathway product, initiates the exchange between the damaged and homologous DNA strands with further help from the RuvABC (a helicase) and RecG (an endonuclease) complexes (Singh et al., 2016). A recent study established a link between DNA repair, drug efflux, and biofilm formation and validated RecA as an effective drug target in Mtb and Msm (Hans et al., 2020). Further studies are required to research the endonuclease involved in HR pathways. Repetitive DNA induces deletions and translocations and promotes speculation that NHEJ is the major pathway when there is no homologous DNA template (Kurahashi et al., 2006). The NHEJ apparatus is encoded by the evolutionarily conserved ku and ligD genes (Della et al., 2004; Kha et al., 2010; Pitcher et al., 2007). Ku is a DNA-end binding protein and stimulates LigD to seal the broken ends in NHEJ, but also activates the helicase activity of UvrD1 and UvrD2 (Brissett et al., 2007; Sinha et al., 2007; Williams et al., 2011). LigD acts as a multifunctional enzyme (Della et al., 2004). RecBCD, a helicase-nuclease complex, is associated with a SSA mechanism (Amundsen et al., 2012) and mediates non-recombination-based SSA in mycobacteria, and the recombination repair of DSBs in E. coli (Singh et al., 2016). Gupta et al. found that mycobacterial RecBCD is required for the RecA-independent SSA pathway. They further demonstrated that mycobacteria RecBCD and AdnAB helicase-nuclease machines dedicate to distinct repair pathway (Gupta et al., 2011). The RecA-dependent HR pathway is considered to be the major mechanism for repairing DSBs to date. Meanwhile, identification of orthologs of NHEJ and SSA in mycobacteria provide new insights (Heaton et al., 2014).
Antibiotic tolerance: Mtb genome instability in response to antibiotic stress
Tolerance is influenced by both environmental and genetic factors, and also refers to the bacterial host resisting deadly doses of antibiotics after a transient exposure (Balaban et al., 2019; Brauner et al., 2016). How does antibiotic tolerance occur in Mtb referring to the genetic aspects related to DNA repair?
The genetic basis of the balance of resistance development, including DNA damage, recombination, replication fidelity, mutation, and genome maintenance, help improve or extend the efficacy of anti-TB drugs. A novel notion of DNA damage repair interference might be an effective strategy to get rid of the persister cell population, as supported by the changes in post-antibiotic treatment (Mittal et al., 2020). For instance, inhibition of DNA gyrase by fluoroquinolone modulates Mtb growth and also contributes to the drug tolerance via RecA/LexA-mediated SOS response (Choudhary et al., 2019). Meanwhile, DNA gyrase knockdown Mtb decreased drug susceptibility to rifampin (RIF), isoniazid (INH), and ethambutol (EMB) post-treatment (Choudhary et al., 2019).
Many studies observed that bactericidal antibiotic-induced ROS could lead to cell death due to oxidative damages to DNA (Belenky et al., 2015; Kohanski et al., 2007; Nandakumar et al., 2014). In particular, lethal doses of bactericidal antibiotics inducing DSBs in a ROS-dependent manner were confirmed in Mtb and Msm (Belenky et al., 2015; Foti et al., 2012; Singh, 2017). Recent findings revealed the oxidized dGTP (8-oxo-dGTP) not only contributed to the generation of ROS but also incorporated DNA polymerase and generated DSBs through incomplete BER (Foti et al., 2012; Gutierrez et al., 2013). In an Mtb infected mice model, the deletion of mycobacterial mazG, which encodes 5-OH-dCTP (an oxidized form of dCTP), resulted in a high frequency of genomic mutation and caused attenuation of virulence (Lyu et al., 2013). A new model for 5-OH-dCTP was further provided to incorporate genomic DNA via DnaE2, and incomplete repair of 5-OH-dC lesions via Nth, resulting in the generation of lethal DSBs and provides a broad view of ROS-mediated antibiotic lethality in the stationary phase (Fan et al., 2018) (Fig. 1).
Fig. 1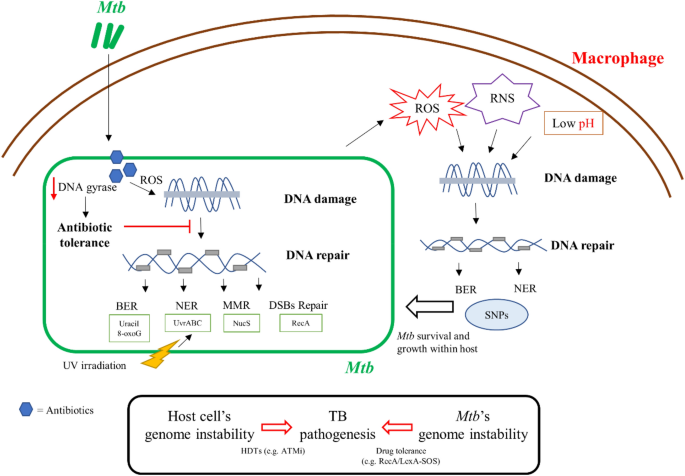
Schematics of genome instability in TB pathogenesis. Both of Mtb’s genome instability including DNA damage caused by antibiotics and host stress, and host cell’s genome instability induced by Mtb infection are involved in the pathogenesis of TB
Full size imagePhenotypic diversity usually arises from transient changes in gene expression during clinical infections and is associated with drug tolerance and genetic resistance (Veening et al., 2008). Mtb is a very smart and successful pathogen. In terms of drug resistance, Mtb shows considerable heterogeneity and diversification (Dhar et al., 2016). The heterogeneity in these processes is always of importance to the clinical implication, and DNA repair systems in Mtb contribute to its long-term survival under mutagenic stress (Ackermann, 2015; Dhar et al., 2016; Flentie et al., 2016). RecA, a known cornerstone of the DNA damage-related SOS response, helps resolve DNA lesions when DNA repair has failed (Baharoglu & Mazel, 2014; Singh, 2017). RecA serine 207, a multifunctional signaling hub involved in DNA damage, controls mutagenesis and antibiotic resistance in mycobacteria through phosphorylation and cardiolipin-mediated inhibition of RecA coprotease function (Wipperman et al., 2018). Interestingly, Manina et al. reported that the preexisting phenotypic variation in the DNA damage response is associated with differential susceptibility to fluoroquinolones (Manina et al., 2019). Furthermore, a recent whole-genome sequencing-based study showed the detected drug-resistant mutations and their frequencies in TB patients were pncA G132S (0.5%), embB M306L (0.53%), and katG A65T (0.51%), suggesting that heteroresistance less than 1% is not associated with poor treatment outcomes (Chen et al., 2021). Therefore, further exploration of the interplay between genome instability and anti-TB drug tolerance will facilitate the development of new drugs with improved outcomes in TB patients.
DNA repair in bacterial survival and within host adaption
The interaction between the host and the pathogen is extremely complex and is affected by anatomical, physiological, and immunological diversity in the microenvironment. As one of the most successful human pathogens, Mtb can reside within host macrophages and evade macrophage defenses. Macrophages produce ROS and RNS, which are essential to control mycobacterial infection (Adams et al., 1997; Darwin et al., 2003; MacMicking et al., 1997) (Fig. 1). Progress in mycobacterial DNA repair has identified several processes that are important for survival within the host. The NER system is reported to counteract the oxidative and nitrosative stress within macrophages (Darwin & Nathan, 2005; Darwin et al., 2003). Transposon sequencing and genetic deletion experiments suggest that the Uvr system is required for optimal growth of Mtb in macrophages and the mouse model (Darwin & Nathan, 2005; Rengarajan et al., 2005). Other studies have also confirmed the role of the BER system in Mtb survival within the host. Nfo, a BER enzyme, is required 1 week after infection, while ung and xthA, members of the BER system, are required for in vivo growth at the 2-week time point (Sassetti & Rubin, 2003). nei2 (Rv3297), a BER glycosylase, is required for successful infection and growth of Mtb in primate hosts (Dutta et al., 2010). Targeting the BER and NER pathways would be a strategy to develop new anti-TB drugs.
Continuous exposure to a multitude of DNA-damaging stresses within the host also compromises bacterial fitness by increasing genomic instability. A recent study revealed that an enhanced mutation rate is associated with ROS and related to the host environment (Poetsch, 2020). High rates of mutations were observed from HIV-negative but not HIV-positive individuals, suggesting that the mutations could be driven by immune pressure (Liu et al., 2020). Single nucleotide polymorphism (SNP) analysis of DNA repair, recombination, and replication genes of Mtb strains revealed a high level of SNPs compared to house-keeping genes (Dos Vultos et al., 2008) (Fig. 1). A recent study provided evidence that DNA repair systems are involved in Mtb within host adaptation. Liu et al. (2021) showed specific positive selection for mutations in DNA repair-associated genes (dnaE2, recB, and mfd) from Tibetan strains, and those mutations in dnaE2 and recB tended to be loss-of-function mutations (Liu et al., 2021). Warner et al., (2010) found that the imuA’-imuB/dnaE2 might drive the evolution of Mtb within its host (Warner et al., 2010)This study provides evidence that local pressures (cold stress and hypoxia) on the Tibetan Plateau selected for Mtb strains that had adapted to this environment (Liu et al., 2021). Using the guinea pig model of infection, Naz et al. found that double deletion of ung and udgB resulted in significant accumulation of SNPs and higher survival levels, suggesting that absence of a BER pathway leads to higher mutations and provides a survival advantage under stress within the host (Naz et al., 2021). Developing “anti-evolution” drugs to prevent the DNA repair pathways in Mtb within host may also be an attractive strategy.
Induction of host cell genome instability contributes to the pathogenesis of TB
It has been recognized that Mtb infection also induces host cell DNA damage. The level of DNA damage and chromosome damage was significantly higher in TB patients compared with healthy controls, suggesting that Mtb induces genome instability and leads to host DNA damage (Rao et al., 1990; Selek et al., 2012). da Silva et al. reported that TB patients presented with higher frequency of DNA damage and DNA repair dysfunction, but with a low frequency of permanent DNA damage (da Silva et al., 2015). Rv2346c, a member of ESAT-6 like family protein, and SecA2 secretome induce host DNA strand breaks (Lochab et al., 2020; Mohanty et al., 2016). DNA damage and genome instability induced by Mtb infection results in dysregulation of macrophage mitosis and consequently formation of polyploid cells, a hallmark of tuberculous granuloma. Mtb infection leads to activated TLR2-Myc signaling which resulted in inducing DNA damage response to promote the differentiation of polyploid macrophages in human granulomas (Herrtwich et al., 2016). The primary cellular component of granuloma is the macrophage, which can determine disease outcome for Mtb infections. Therefore targeting to host DNA damage-induced polyploid macrophage by Mtb could offer new opportunities for therapeutic strategy of TB. (Herrtwich et al., 2016). A recent study revealed that Mtb activates the host ATM-Chk2 pathway of DNA damage response (DDR) signaling instead of the classical ATM-Chk2 DDR to gain a survival advantage through the ATM-Akt signaling cascade. Combining ATM inhibitor, KU55933 with isoniazid promotes the clearance of Mtb in vivo and in vitro, suggesting the potential for targeting host ATM-Akt pathway for host-directed adjunct therapy (Lochab et al., 2020).
Conclusions
DNA repair pathways enable Mtb to survive DNA damage induced by antibiotic and host stress; therefore, targeting DNA repair pathways might prove efficacious when performed in combination with first and second-line TB drugs. As the DNA repair systems in Mtb are distinct from humans at the biochemical and structural level, drugs that inhibit Mtb DNA repair pathways have the potential advantage to be selective for Mtb with fewer side effects. For now, there is no approved TB drugs targeting DNA repair systems. Recently, several natural products were found to be effective in killing Mtb via targeting DNA repair-associated genes (Jadaun et al., 2015; Kling et al., 2015), which support the potential for new drug discovery in this area. However, it should be noted that antibiotic or host stress-induced mutations in DNA repair systems may also provide a mechanism for Mtb to develop resistance and evolve within the host. A detailed understanding of the different players, which are involved in mutagenesis can provide insights into the mechanism of drug resistance and allow better management of available therapeutics. Antibiotic tolerance in Mtb at the genetic level, such as DNA damage repair interference, has always been a research area worthy of further exploration and may shorten the duration of curative treatment. Besides, Mtb modulates the host genomic integrity for its survival and granuloma formation, targeting host cell DNA repair pathways might be a potential HDT strategy for TB treatment.
References
Ackermann, M. (2015). A functional perspective on phenotypic heterogeneity in microorganisms. Nature Reviews Microbiology, 13(8), 497–508. https://doi.org/10.1038/nrmicro3491
Adams, L. B., Dinauer, M. C., Morgenstern, D. E., & Krahenbuhl, J. L. (1997). Comparison of the roles of reactive oxygen and nitrogen intermediates in the host response to Mycobacterium tuberculosis using transgenic mice. Tubercle and Lung Disease, 78(5–6), 237–246. https://doi.org/10.1016/s0962-8479(97)90004-6
Amundsen, S. K., Spicer, T., Karabulut, A. C., Londono, L. M., Eberhart, C., Fernandez Vega, V., & Smith, G. R. (2012). Small-molecule inhibitors of bacterial AddAB and RecBCD helicase-nuclease DNA repair enzymes. ACS Chemical Biology, 7(5), 879–891. https://doi.org/10.1021/cb300018x
Baharoglu, Z., & Mazel, D. (2014). SOS, the formidable strategy of bacteria against aggressions. FEMS Microbiology Reviews, 38(6), 1126–1145. https://doi.org/10.1111/1574-6976.12077
Balaban, N. Q., Helaine, S., Lewis, K., Ackermann, M., Aldridge, B., Andersson, D. I., & Zinkernagel, A. (2019). Publisher correction: Definitions and guidelines for research on antibiotic persistence. Nature Reviews Microbiology, 17(7), 460. https://doi.org/10.1038/s41579-019-0207-4
Balganesh, M., Dinesh, N., Sharma, S., Kuruppath, S., Nair, A. V., & Sharma, U. (2012). Efflux pumps of Mycobacterium tuberculosis play a significant role in antituberculosis activity of potential drug candidates. Antimicrobial Agents and Chemotherapy, 56(5), 2643–2651. https://doi.org/10.1128/AAC.06003-11
Belenky, P., Ye, J. D., Porter, C. B., Cohen, N. R., Lobritz, M. A., Ferrante, T., & Collins, J. J. (2015). Bactericidal antibiotics induce toxic metabolic perturbations that lead to cellular damage. Cell Reports, 13(5), 968–980. https://doi.org/10.1016/j.celrep.2015.09.059
Ben-David, U. (2015). Genomic instability, driver genes and cell selection: Projections from cancer to stem cells. Biochimica Et Biophysica Acta, 1849(4), 427–435. https://doi.org/10.1016/j.bbagrm.2014.08.005
Booth, J. A., Spirek, M., Lobie, T. A., Skarstad, K., Krejci, L., & Bjoras, M. (2020). Antibiotic-induced DNA damage results in a controlled loss of pH homeostasis and genome instability. Science and Reports, 10(1), 19422. https://doi.org/10.1038/s41598-020-76426-2
Brauner, A., Fridman, O., Gefen, O., & Balaban, N. Q. (2016). Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nature Reviews Microbiology, 14(5), 320–330. https://doi.org/10.1038/nrmicro.2016.34
Brissett, N. C., Pitcher, R. S., Juarez, R., Picher, A. J., Green, A. J., Dafforn, T. R., & Doherty, A. J. (2007). Structure of a NHEJ polymerase-mediated DNA synaptic complex. Science, 318(5849), 456–459. https://doi.org/10.1126/science.1145112
Cadet, J., Douki, T., & Ravanat, J. L. (2010). Oxidatively generated base damage to cellular DNA. Free Radical Biology and Medicine, 49(1), 9–21. https://doi.org/10.1016/j.freeradbiomed.2010.03.025
Cadet, J., & Wagner, J. R. (2014). Oxidatively generated base damage to cellular DNA by hydroxyl radical and one-electron oxidants: Similarities and differences. Archives of Biochemistry and Biophysics, 557, 47–54. https://doi.org/10.1016/j.abb.2014.05.001
Castaneda-Garcia, A., Prieto, A. I., Rodriguez-Beltran, J., Alonso, N., Cantillon, D., Costas, C., & Blazquez, J. (2017). A non-canonical mismatch repair pathway in prokaryotes. Nature Communications, 8, 14246. https://doi.org/10.1038/ncomms14246
Cebrian-Sastre, E., Martin-Blecua, I., Gullon, S., Blazquez, J., & Castaneda-Garcia, A. (2021). Control of genome stability by EndoMS/NucS-mediated non-canonical mismatch repair. Cells. https://doi.org/10.3390/cells10061314
Chakaya, J., Khan, M., Ntoumi, F., Aklillu, E., Fatima, R., Mwaba, P., & Zumla, A. (2021). Global tuberculosis report 2020—reflections on the global TB burden, treatment and prevention efforts. International Journal of Infectious Diseases. https://doi.org/10.1016/j.ijid.2021.02.107
Chen, Y., Jiang, Q., Zou, J., Yang, T., Liu, Q., Luo, G., & Gao, Q. (2021). Deep whole-genome sequencing reveals no evidence for heteroresistance influencing treatment outcomes among drug-susceptible tuberculosis patients. Tuberculosis (edinburgh, Scotland), 130, 102120. https://doi.org/10.1016/j.tube.2021.102120
Choudhary, E., Sharma, R., Kumar, Y., & Agarwal, N. (2019). Conditional Silencing by CRISPRi reveals the role of DNA gyrase in formation of drug-tolerant persister population in Mycobacterium tuberculosis. Frontiers in Cellular and Infection Microbiology, 9, 70. https://doi.org/10.3389/fcimb.2019.00070
Cole, S. T., Brosch, R., Parkhill, J., Garnier, T., Churcher, C., Harris, D., & Barrell, B. G. (1998). Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature, 393(6685), 537–544. https://doi.org/10.1038/31159
da Silva, A. L., Bresciani, M. J., Karnopp, T. E., Weber, A. F., Ellwanger, J. H., Henriques, J. A., & Possuelo, L. G. (2015). DNA damage and cellular abnormalities in tuberculosis, lung cancer and chronic obstructive pulmonary disease. Multidisciplinary Respiratory Medicine, 10, 38. https://doi.org/10.1186/s40248-015-0034-z
Darwin, K. H., Ehrt, S., Gutierrez-Ramos, J. C., Weich, N., & Nathan, C. F. (2003). The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science, 302(5652), 1963–1966. https://doi.org/10.1126/science.1091176
Darwin, K. H., & Nathan, C. F. (2005). Role for nucleotide excision repair in virulence of Mycobacterium tuberculosis. Infection and Immunity, 73(8), 4581–4587. https://doi.org/10.1128/IAI.73.8.4581-4587.2005
Della, M., Palmbos, P. L., Tseng, H. M., Tonkin, L. M., Daley, J. M., Topper, L. M., & Doherty, A. J. (2004). Mycobacterial Ku and ligase proteins constitute a two-component NHEJ repair machine. Science, 306(5696), 683–685. https://doi.org/10.1126/science.1099824
Dhar, N., McKinney, J., & Manina, G. (2016). Phenotypic heterogeneity in Mycobacterium tuberculosis. Microbiology Spectrum. https://doi.org/10.1128/microbiolspec.TBTB2-0021-2016
Dos Vultos, T., Mestre, O., Rauzier, J., Golec, M., Rastogi, N., Rasolofo, V., & Gicquel, B. (2008). Evolution and diversity of clonal bacteria: The paradigm of Mycobacterium tuberculosis. PLoS ONE, 3(2), e1538. https://doi.org/10.1371/journal.pone.0001538
Dos Vultos, T., Mestre, O., Tonjum, T., & Gicquel, B. (2009). DNA repair in Mycobacterium tuberculosis revisited. FEMS Microbiology Reviews, 33(3), 471–487. https://doi.org/10.1111/j.1574-6976.2009.00170.x
Dutta, N. K., Mehra, S., Didier, P. J., Roy, C. J., Doyle, L. A., Alvarez, X., & Kaushal, D. (2010). Genetic requirements for the survival of tubercle bacilli in primates. Journal of Infectious Diseases, 201(11), 1743–1752. https://doi.org/10.1086/652497
Ehrt, S., & Schnappinger, D. (2009). Mycobacterial survival strategies in the phagosome: Defence against host stresses. Cellular Microbiology, 11(8), 1170–1178. https://doi.org/10.1111/j.1462-5822.2009.01335.x
Fan, X. Y., Tang, B. K., Xu, Y. Y., Han, A. X., Shi, K. X., Wu, Y. K., & Lyu, L. D. (2018). Oxidation of dCTP contributes to antibiotic lethality in stationary-phase mycobacteria. Proceedings of the National Academy of Sciences of the United States of America, 115(9), 2210–2215. https://doi.org/10.1073/pnas.1719627115
Flentie, K., Garner, A. L., & Stallings, C. L. (2016). Mycobacterium tuberculosis transcription machinery: Ready to respond to host attacks. Journal of Bacteriology, 198(9), 1360–1373. https://doi.org/10.1128/JB.00935-15
Foti, J. J., Devadoss, B., Winkler, J. A., Collins, J. J., & Walker, G. C. (2012). Oxidation of the guanine nucleotide pool underlies cell death by bactericidal antibiotics. Science, 336(6079), 315–319. https://doi.org/10.1126/science.1219192
Gorna, A. E., Bowater, R. P., & Dziadek, J. (2010). DNA repair systems and the pathogenesis of Mycobacterium tuberculosis: Varying activities at different stages of infection. Clinical Science (london, England), 119(5), 187–202. https://doi.org/10.1042/CS20100041
Grossman, L., & Thiagalingam, S. (1993). Nucleotide excision repair, a tracking mechanism in search of damage. Journal of Biological Chemistry, 268(23), 16871–16874. Accessed from https://www.ncbi.nlm.nih.gov/pubmed/8349576
Gupta, R., Barkan, D., Redelman-Sidi, G., Shuman, S., & Glickman, M. S. (2011). Mycobacteria exploit three genetically distinct DNA double-strand break repair pathways. Molecular Microbiology, 79(2), 316–330. https://doi.org/10.1111/j.1365-2958.2010.07463.x.
Gutierrez, A., Laureti, L., Crussard, S., Abida, H., Rodriguez-Rojas, A., Blazquez, J., & Matic, I. (2013). Beta-Lactam antibiotics promote bacterial mutagenesis via an RpoS-mediated reduction in replication fidelity. Nature Communications, 4, 1610. https://doi.org/10.1038/ncomms2607
Hans, S., Purkait, D., Nandan, S., Bansal, M., Hameed, S., & Fatima, Z. (2020). Rec A disruption unveils cross talk between DNA repair and membrane damage, efflux pump activity, biofilm formation in Mycobacterium smegmatis. Microbial Pathogenesis, 149, 104262. https://doi.org/10.1016/j.micpath.2020.104262
Heaton, B. E., Barkan, D., Bongiorno, P., Karakousis, P. C., & Glickman, M. S. (2014). Deficiency of double-strand DNA break repair does not impair Mycobacterium tuberculosis virulence in multiple animal models of infection. Infection and Immunity, 82(8), 3177–3185. https://doi.org/10.1128/IAI.01540-14
Herrtwich, L., Nanda, I., Evangelou, K., Nikolova, T., Horn, V., Sagar, A., & Triantafyllopoulou, A. (2016). DNA damage signaling instructs polyploid macrophage fate in granulomas. Cell, 167(5), 1264–1280. https://doi.org/10.1016/j.cell.2016.09.054
Jadaun, A., Sudhakar, D. R., Subbarao, N., & Dixit, A. (2015). In silico screening for novel inhibitors of DNA polymerase III alpha subunit of Mycobacterium tuberculosis (MtbDnaE2, H37Rv). PLoS ONE, 10(3), e0119760. https://doi.org/10.1371/journal.pone.0119760
Jain, R., Kumar, P., & Varshney, U. (2007). A distinct role of formamidopyrimidine DNA glycosylase (MutM) in down-regulation of accumulation of G, C mutations and protection against oxidative stress in mycobacteria. DNA Repair (amst), 6(12), 1774–1785. https://doi.org/10.1016/j.dnarep.2007.06.009
Kha, D. T., Wang, G., Natrajan, N., Harrison, L., & Vasquez, K. M. (2010). Pathways for double-strand break repair in genetically unstable Z-DNA-forming sequences. Journal of Molecular Biology, 398(4), 471–480. https://doi.org/10.1016/j.jmb.2010.03.035
Khanam, T., Afsar, M., Shukla, A., Alam, F., Kumar, S., Soyar, H., & Ramachandran, R. (2020). M. tuberculosis class II apurinic/apyrimidinic-endonuclease/3’-5’ exonuclease (XthA) engages with NAD+-dependent DNA ligase A (LigA) to counter futile cleavage and ligation cycles in base excision repair. Nucleic Acids Research, 48(8), 4325–4343. https://doi.org/10.1093/nar/gkaa188
Kisker, C., Kuper, J., & Van Houten, B. (2013). Prokaryotic nucleotide excision repair. Cold Spring Harbor Perspectives in Biology, 5(3), a012591. https://doi.org/10.1101/cshperspect.a012591
Kling, A., Lukat, P., Almeida, D. V., Bauer, A., Fontaine, E., Sordello, S., & Muller, R. (2015). Antibiotics. Targeting DnaN for tuberculosis therapy using novel griselimycins. Science, 348(6239), 1106–1112. https://doi.org/10.1126/science.aaa4690
Kohanski, M. A., Dwyer, D. J., Hayete, B., Lawrence, C. A., & Collins, J. J. (2007). A common mechanism of cellular death induced by bactericidal antibiotics. Cell, 130(5), 797–810. https://doi.org/10.1016/j.cell.2007.06.049
Krokan, H. E., Drablos, F., & Slupphaug, G. (2002). Uracil in DNA–occurrence, consequences and repair. Oncogene, 21(58), 8935–8948. https://doi.org/10.1038/sj.onc.1205996
Kurahashi, H., Inagaki, H., Ohye, T., Kogo, H., Kato, T., & Emanuel, B. S. (2006). Palindrome-mediated chromosomal translocations in humans. DNA Repair (amst), 5(9–10), 1136–1145. https://doi.org/10.1016/j.dnarep.2006.05.035
Kurthkoti, K., Srinath, T., Kumar, P., Malshetty, V. S., Sang, P. B., Jain, R., & Varshney, U. (2010). A distinct physiological role of MutY in mutation prevention in mycobacteria. Microbiology (reading), 156(Pt 1), 88–93. https://doi.org/10.1099/mic.0.033621-0
Kurthkoti, K., & Varshney, U. (2010). Detrimental effects of hypoxia-specific expression of uracil DNA glycosylase (Ung) in Mycobacterium smegmatis. Journal of Bacteriology, 192(24), 6439–6446. https://doi.org/10.1128/JB.00679-10
Kurthkoti, K., & Varshney, U. (2011). Base excision and nucleotide excision repair pathways in mycobacteria. Tuberculosis (edinburgh, Scotland), 91(6), 533–543. https://doi.org/10.1016/j.tube.2011.06.005
Lahiri, S., Rizzi, M., Rossi, F., & Miggiano, R. (2018). Mycobacterium tuberculosis UvrB forms dimers in solution and interacts with UvrA in the absence of ligands. Proteins, 86(1), 98–109. https://doi.org/10.1002/prot.25412
Li, Z., Pearlman, A. H., & Hsieh, P. (2016). DNA mismatch repair and the DNA damage response. DNA Repair (amst), 38, 94–101. https://doi.org/10.1016/j.dnarep.2015.11.019
Liu, Q., Liu, H., Shi, L., Gan, M., Zhao, X., Lyu, L. D., & Gao, Q. (2021). Local adaptation of Mycobacterium tuberculosis on the Tibetan Plateau. Proceedings of the National Academy of Sciences of the United States of America. https://doi.org/10.1073/pnas.2017831118
Liu, Q., Wei, J., Li, Y., Wang, M., Su, J., Lu, Y., & Gao, Q. (2020). Mycobacterium tuberculosis clinical isolates carry mutational signatures of host immune environments. Science Advances, 6(22), eaba4901. https://doi.org/10.1126/sciadv.aba4901
Lochab, S., Singh, Y., Sengupta, S., & Nandicoori, V. K. (2020). Mycobacterium tuberculosis exploits host ATM kinase for survival advantage through SecA2 secretome. eLife. https://doi.org/10.7554/eLife.51466
Lyu, L. D., Tang, B. K., Fan, X. Y., Ma, H., & Zhao, G. P. (2013). Mycobacterial MazG safeguards genetic stability via housecleaning of 5-OH-dCTP. PLoS Pathogens, 9(12), e1003814. https://doi.org/10.1371/journal.ppat.1003814
MacMicking, J. D., North, R. J., LaCourse, R., Mudgett, J. S., Shah, S. K., & Nathan, C. F. (1997). Identification of nitric oxide synthase as a protective locus against tuberculosis. Proceedings of the National Academy of Sciences of the United States of America, 94(10), 5243–5248. https://doi.org/10.1073/pnas.94.10.5243
Malshetty, V. S., Jain, R., Srinath, T., Kurthkoti, K., & Varshney, U. (2010). Synergistic effects of UdgB and Ung in mutation prevention and protection against commonly encountered DNA damaging agents in Mycobacterium smegmatis. Microbiology (reading), 156(Pt 3), 940–949. https://doi.org/10.1099/mic.0.034363-0
Manina, G., Griego, A., Singh, L. K., McKinney, J. D., & Dhar, N. (2019). Preexisting variation in DNA damage response predicts the fate of single mycobacteria under stress. EMBO Journal, 38(22), e101876. https://doi.org/10.15252/embj.2019101876
Mazloum, N., Stegman, M. A., Croteau, D. L., Van Houten, B., Kwon, N. S., Ling, Y., & Nathan, C. (2011). Identification of a chemical that inhibits the mycobacterial UvrABC complex in nucleotide excision repair. Biochemistry, 50(8), 1329–1335. https://doi.org/10.1021/bi101674c
McGrath, M., Gey van Pittius, N. C., van Helden, P. D., Warren, R. M., & Warner, D. F. (2014). Mutation rate and the emergence of drug resistance in Mycobacterium tuberculosis. Journal of Antimicrobial Chemotherapy, 69(2), 292–302. https://doi.org/10.1093/jac/dkt364
Mittal, P., Sinha, R., Kumar, A., Singh, P., Ngasainao, M. R., Singh, A., & Singh, I. K. (2020). Focusing on DNA repair and damage tolerance mechanisms in Mycobacterium tuberculosis: an emerging therapeutic theme. Current Topics in Medicinal Chemistry, 20(5), 390–408. https://doi.org/10.2174/1568026620666200110114322
Mizrahi, V., & Andersen, S. J. (1998). DNA repair in Mycobacterium tuberculosis. What have we learnt from the genome sequence? Molecular Microbiology, 29(6), 1331–1339. https://doi.org/10.1046/j.1365-2958.1998.01038.x
Mohanty, S., Dal Molin, M., Ganguli, G., Padhi, A., Jena, P., Selchow, P., & Sonawane, A. (2016). Mycobacterium tuberculosis EsxO (Rv2346c) promotes bacillary survival by inducing oxidative stress mediated genomic instability in macrophages. Tuberculosis (edinburgh, Scotland), 96, 44–57. https://doi.org/10.1016/j.tube.2015.11.006
Nandakumar, M., Nathan, C., & Rhee, K. Y. (2014). Isocitrate lyase mediates broad antibiotic tolerance in Mycobacterium tuberculosis. Nature Communications, 5, 4306. https://doi.org/10.1038/ncomms5306
Naz, S., Dabral, S., Nagarajan, S. N., Arora, D., Singh, L. V., Kumar, P., & Nandicoori, V. K. (2021). Compromised base excision repair pathway in Mycobacterium tuberculosis imparts superior adaptability in the host. PLoS Pathogens, 17(3), e1009452. https://doi.org/10.1371/journal.ppat.1009452
Pitcher, R. S., Green, A. J., Brzostek, A., Korycka-Machala, M., Dziadek, J., & Doherty, A. J. (2007). NHEJ protects mycobacteria in stationary phase against the harmful effects of desiccation. DNA Repair (amst), 6(9), 1271–1276. https://doi.org/10.1016/j.dnarep.2007.02.009
Poetsch, A. R. (2020). The genomics of oxidative DNA damage, repair, and resulting mutagenesis. Computational and Structural Biotechnology Journal, 18, 207–219. https://doi.org/10.1016/j.csbj.2019.12.013
Prammananan, T., Phunpruch, S., Jaitrong, S., & Palittapongarnpim, P. (2012). Mycobacterium tuberculosis uvrC essentiality in response to UV-induced cell damage. The Southeast Asian Journal of Tropical Medicine and Public Health, 43(2), 370–375. Accessed on https://www.ncbi.nlm.nih.gov/pubmed/23082589
Rao, V. V., Gupta, E. V., & Thomas, I. M. (1990). Chromosome damage in untreated tuberculosis patients. Tubercle, 71(3), 169–172. https://doi.org/10.1016/0041-3879(90)90070-o
Rengarajan, J., Bloom, B. R., & Rubin, E. J. (2005). Genome-wide requirements for Mycobacterium tuberculosis adaptation and survival in macrophages. Proceedings of the National Academy of Sciences of the United States of America, 102(23), 8327–8332. https://doi.org/10.1073/pnas.0503272102
Rossi, F., Khanduja, J. S., Bortoluzzi, A., Houghton, J., Sander, P., Guthlein, C., & Rizzi, M. (2011). The biological and structural characterization of Mycobacterium tuberculosis UvrA provides novel insights into its mechanism of action. Nucleic Acids Research, 39(16), 7316–7328. https://doi.org/10.1093/nar/gkr271
Sassetti, C. M., & Rubin, E. J. (2003). Genetic requirements for mycobacterial survival during infection. Proceedings of the National Academy of Sciences of the United States of America, 100(22), 12989–12994. https://doi.org/10.1073/pnas.2134250100
Selek, S., Aslan, M., Horoz, M., Celik, H., Cosar, N., Gunak, F., & Kocyigit, A. (2012). Peripheral DNA damage in active pulmonary tuberculosis. Environmental Toxicology, 27(6), 380–384. https://doi.org/10.1002/tox.20674
Singh, A. (2017). Guardians of the mycobacterial genome: A review on DNA repair systems in Mycobacterium tuberculosis. Microbiology (reading), 163(12), 1740–1758. https://doi.org/10.1099/mic.0.000578
Singh, A., Bhagavat, R., Vijayan, M., & Chandra, N. (2016). A comparative analysis of the DNA recombination repair pathway in mycobacterial genomes. Tuberculosis (edinburgh, Scotland), 99, 109–119. https://doi.org/10.1016/j.tube.2016.04.011
Sinha, K. M., Stephanou, N. C., Gao, F., Glickman, M. S., & Shuman, S. (2007). Mycobacterial UvrD1 is a Ku-dependent DNA helicase that plays a role in multiple DNA repair events, including double-strand break repair. Journal of Biological Chemistry, 282(20), 15114–15125. https://doi.org/10.1074/jbc.M701167200
Smith, I. (2003). Mycobacterium tuberculosis pathogenesis and molecular determinants of virulence. Clinical Microbiology Reviews, 16(3), 463–496. https://doi.org/10.1128/CMR.16.3.463-496.2003
Springall, L., Hughes, C. D., Simons, M., Azinas, S., Van Houten, B., & Kad, N. M. (2018). Recruitment of UvrBC complexes to UV-induced damage in the absence of UvrA increases cell survival. Nucleic Acids Research, 46(3), 1256–1265. https://doi.org/10.1093/nar/gkx1244
Thakur, M., Badugu, S., & Muniyappa, K. (2020). UvrA and UvrC subunits of the Mycobacterium tuberculosis UvrABC excinuclease interact independently of UvrB and DNA. FEBS Letters, 594(5), 851–863. https://doi.org/10.1002/1873-3468.13671
Thakur, M., Kumar, M. B., & Muniyappa, K. (2016). Mycobacterium tuberculosis UvrB Is a robust DNA-stimulated ATPase that also possesses structure-specific ATP-dependent DNA helicase activity. Biochemistry, 55(41), 5865–5883. https://doi.org/10.1021/acs.biochem.6b00558
van der Veen, S., & Tang, C. M. (2015). The BER necessities: The repair of DNA damage in human-adapted bacterial pathogens. Nature Reviews Microbiology, 13(2), 83–94. https://doi.org/10.1038/nrmicro3391
Veening, J. W., Smits, W. K., & Kuipers, O. P. (2008). Bistability, epigenetics, and bet-hedging in bacteria. Annual Review of Microbiology, 62, 193–210. https://doi.org/10.1146/annurev.micro.62.081307.163002
Warner, D. F., Ndwandwe, D. E., Abrahams, G. L., Kana, B. D., Machowski, E. E., Venclovas, C., & Mizrahi, V. (2010). Essential roles for imuA’- and imuB-encoded accessory factors in DnaE2-dependent mutagenesis in Mycobacterium tuberculosis. Proceedings of the National Academy of Sciences of the United States of America, 107(29), 13093–13098. https://doi.org/10.1073/pnas.1002614107
Williams, A., Guthlein, C., Beresford, N., Bottger, E. C., Springer, B., & Davis, E. O. (2011). UvrD2 is essential in Mycobacterium tuberculosis, but its helicase activity is not required. Journal of Bacteriology, 193(17), 4487–4494. https://doi.org/10.1128/JB.00302-11
Wipperman, M. F., Heaton, B. E., Nautiyal, A., Adefisayo, O., Evans, H., Gupta, R., & Glickman, M. S. (2018). Mycobacterial mutagenesis and drug resistance are controlled by phosphorylation- and cardiolipin-mediated inhibition of the RecA coprotease. Molecular Cell, 72(1), 152–161. https://doi.org/10.1016/j.molcel.2018.07.037
Acknowledgements
This work was supported by the Natural Science Foundation of China (grant nos. 82072252 and 91942315) and Guangdong Provincial Key Laboratory of Regional Immunity and Diseases (grant no. 2019B030301009). We thank Dr. Jessica Tamanini at Shenzhen University Health Science Center for editing the manuscript prior to submission.
Author information
Affiliations
Guangdong Provincial Key Laboratory of Regional Immunity and Diseases, Department of Pathogen Biology, Shenzhen University School of Medicine, Shenzhen, China
Kehong Zhang, Yuping Ning, Xinchun Chen & Yi Cai
Department of Pharmaceutical/Medicinal Chemistry, Institute of Pharmacy, Friedrich-Schiller-University, Philosophenweg 14, Jena, Germany
Kehong Zhang & Yuping Ning
Harbin Thoracic Hospital, Harbin, China
Fanhui Kong
Corresponding author
Correspondence to Yi Cai.
Rights and permissions
About this article
Cite this article
Zhang, K., Ning, Y., Kong, F. et al. Genome instability in pathogenesis of tuberculosis. GENOME INSTAB. DIS. 2, 331–338 (2021). https://doi.org/10.1007/s42764-021-00057-8
Received08 October 2021
Revised18 November 2021
Accepted18 November 2021
Published23 November 2021
Issue DateDecember 2021
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
Genome instability
Mycobacterium tuberculosis
DNA damage and repair
Drug-resistance
Pathogenesis
用户登录
还没有账号?
立即注册