






Nature of spontaneously arising single base substitutions in normal cells
Review Article
Genome Instability & Disease 2, 339–357 (2021)
Abstract
Next-generation sequencing (NGS) of single cells and micro-dissected tissues has demonstrated that the number of somatic mutations in normal cells increases linearly with age. The majority of somatic mutations are single base substitutions (SBSs). NGS has revealed the mutagenic effect of various metabolic and environmental genotoxic agents on different cells, such as normal colonic epithelial cells, hematopoietic cells, hepatocytes, and neurons. NGS has also uncovered the clonal expansion of cells in normal tissues of the elderly driven by oncogenic mutations. There are two main mechanisms for the generation of mutations, namely, replication errors and DNA damage, with both being followed by inaccurate repair. NGS studies of normal tissues highlight the substantial contribution of the latter mechanism by revealing the significant accumulation of somatic mutations in mitotically inactive hepatocytes and neurons. This review describes the nature of spontaneously arising SBSs in normal tissues, the causes of mutagenesis, cellular pathways of the DNA damage response that suppress mutagenesis, as well as unsolved questions on the matter.
Preamble
DNA damage arises from both endogenous (spontaneous) and exogenous (environmental) sources to then drive mutagenesis. Previous studies have identified a wide array of exogenous chemicals that cause genetic alterations as well as their precise genotoxic mechanisms, owing to the development of sensitive genotoxicity testing. However, the characterization of spontaneous mutations has been technically challenging since normal cell populations harbor various clones with distinct mutational profiles. NGS technology has been employed for the identification of small numbers of unique SBSs in single cells, their expanded populations, as well as micro-dissected tissues. The more recently developed error-reduced NGS strategies have sequencing error rates much lower than 5 × 10–8 base, which is comparable to the spontaneous mutation frequencies of mammalian genomes (10–9–10–7 base per cell division). Through these advances, NGS has been employed to reveal the mutagenic effects of environmental genotoxic agents in a variety of cells, such as normal colonic epithelial cells, hematopoietic cells, hepatocytes, and neurons. Sequencing has also revealed the clonal expansion of cells in normal tissues that occurs with aging. Subclonal multi-tissue analyses of somatic mutations have offered major insights into cell lineage commitment during embryonic development as well as into tissue-specific aspects of mutagenesis. This review summarizes basic concepts, introduces new insights, and discusses unresolved questions on the nature of mutations in normal tissues, the causes of mutagenesis, and cellular pathways that suppress mutagenesis.
The nature of mutations
Classification of mutations
Mutations are permanently transmissible changes in the amount and structure of genomic DNA in cells. These changes may involve a single base, gene, a block of genes, and whole chromosomes. In general, mutations include single base substitutions (SBSs), base deletions and insertions, gene amplification, and chromosomal rearrangements. Large-scale mutations also include changes in the number of chromosomes, resulting in aneuploidy (OECD, 2010).
SBS signatures were previously defined based on 96 mutation types. There are only six different possible substitutions that can occur between DNA bases: C>A, C>G, C>T, T>A, T>C, and T>G. These six nucleotide substitution types are further stratified based on the nucleotides immediately adjacent to the affected base pair, leading to the spectra of 96 mutation types (Fig. 1A) (Alexandrov et al., 2013, 2020; Nik-Zainal et al., 2012). Mutational signatures are defined based on the 96 mutation types and may arise as a result of multiple mutagenic processes, including the infidelity of DNA replication, exogenous or endogenous mutagen exposure, enzymatic modification of DNA, and defective DNA repair. Mutagenic signatures are presented in Fig. 1B. Mutational signature reference data are publicly available in the Catalogue of Somatic Mutation In Cancer (COSMIC) database (Alexandrov & Stratton, 2014; Alexandrov et al., 2013; Forbes et al., 2017) (see https://cancer.sanger.ac.uk/signatures/). Specific mutational signatures link endogenous and exogenous DNA damage and repair mechanisms (Table 1).
Fig. 1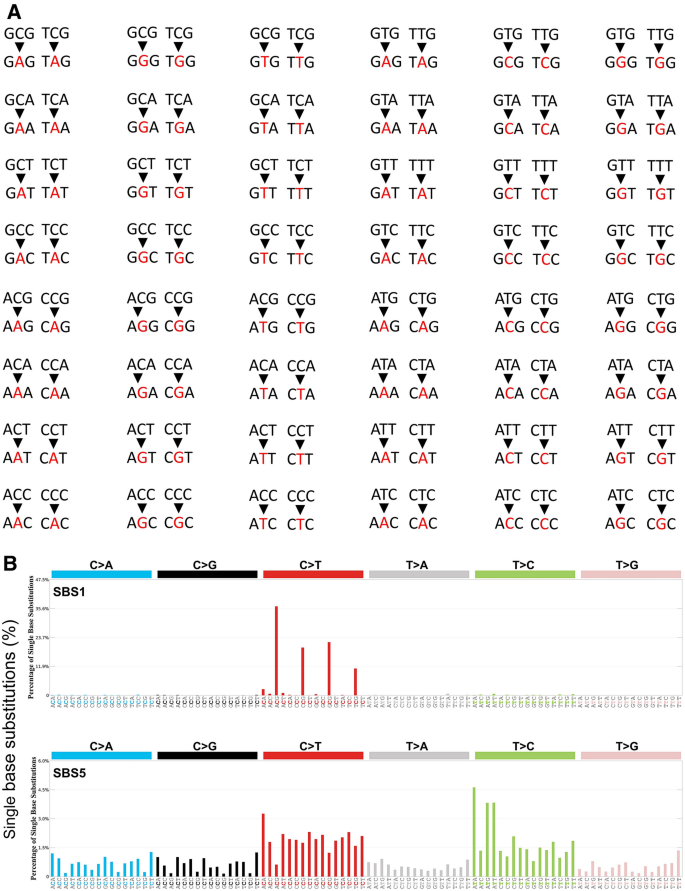
Mutational signatures. A 96 single base substitution (SBS) types. Red letters indicate the mutated nucleotides of the six substitution subtypes: C to A (the leftmost two columns), C to G (the third and fourth columns from the left), C to T (the fifth and sixth columns), T to A (the seventh and eighth columns), T to C (the ninth and tenth columns), and T to G (the rightmost two columns). These six SBSs are further subdivided, depending on the nucleotides immediately 5′ and 3′ to each mutated nucleotide. This subdivision generates all together 96 possible mutation types (six types of substitution × four types of 5′ base × four types of 3′ base). B Examples of mutation signatures, SBS1 and SBS5, which are known to be ‘clock’ mutations
Full size image
Mutations occurring in the coding region of genes are classified depending on their effect on the functionality of protein products. This division includes silent mutations, missense mutations, nonsense mutations, and frameshift mutations, which result from nucleotide deletions and insertions. Mutations occurring in splicing signals often completely disrupt gene function. There is a 50% probability that loss of heterozygosity (LOH) will inactivate the function of affected genes with heterozygous mutations. LOH results from mutations in the intact allele, heteroallelic recombination between homologous chromosomes, and abnormal chromosome segregation in the M phase (Fig. 2). LOH in tumor suppressor genes with heterozygous mutations leads to clonal expansion and increases the risk of cancer. Clonal expansion of blood cells is commonly seen with age, increases the risk of leukemia, and is caused by copy-neutral loss of heterozygosity (CN-LOH) (Fig. 2, right bottom) (Loh et al., 2020; Terao et al., 2020). A typical example of LOH-dependent carcinogenesis is the presence of mutations in one of the two RB allelic genes. Carriers suffer from retinoblastoma with an extremely high penetrance due to the frequent loss of the intact RB gene (Knudson, 1971). LOH is also responsible for Hereditary Breast and Ovarian Cancer (HBOC) syndrome in women having mutations in the breast cancer gene 1 (BRCA1) and BRCA2 genes (see https://www.cancer.gov/about-cancer/causes-prevention/genetics/brca-fact-sheet). LOH also accounts for enhanced carcinogenesis in carriers of ATM and TP53 mutations (see “DNA damage checkpoints”) (Huang et al., 2018).
Fig. 2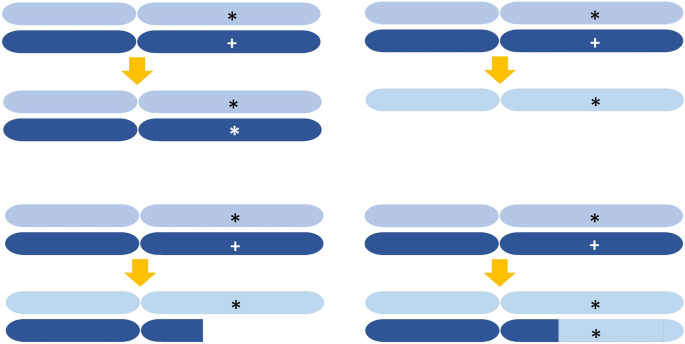
Schematic views showing four types of LOH. Assume a locus heterozygous for a wild-type allelic gene (marked with +) and a mutant allelic gene (marked with *). LOH can result from the loss-of-function mutation of the wild-type gene (left top), a long deletion (left bottom), and whole chromosome loss (right top). Recent NGS studies revealed copy-neutral loss of heterozygosity (CN-LOH) (right bottom). The molecular mechanism of generating CN-LOH is unknown
Full size imageOrigin of SBSs
SBSs are generated de novo during germ cell development as well as in somatic cells. NGS technology allows for the unbiased systematic investigation of mutations throughout the whole genome in large populations of people. Such investigations have revealed mutations present at the population level (see “Germline mutations”), mutations newly generated during germ cell development (see “De novo mutations (DNMs)”) (Goldmann et al., 2019), somatic cell mutations (see “Somatic mutations”), as well as the multi-step process of oncogenesis from normal tissues to malignant cancers (see “Multi-step oncogenesis”).
Germline mutations
Germline mutations are distinct from somatic mutations and are inherited by every somatic and germline cell of the offspring. An open database, gnomAD (Collins et al., 2020; Karczewski et al., 2020) (https://gnomad.broadinstitute.org/), stores germline mutations detected in a cohort of healthy individuals. Healthy individuals have an average of 140 germline truncating mutations in the coding region that significantly impair the function of encoded proteins (Lek et al., 2016). Surprisingly, healthy individuals harbor an average of 35 homozygous mutations that disrupt protein function, indicating that these mutated proteins are dispensable for embryogenesis and human health. Healthy people also have approximately 20 mutations in the germline that disrupt the function of encoded protein on one of the alleles, but these mutations are not homozygous, in which case they could cause prenatal lethality, developmental anomalies, and autosomal recessive diseases. In other words, healthy individuals harbor an average of 20 autosomal recessive mutations that would cause embryonic lethality and severe diseases if homozygous. Typical examples include mutations in the RB, BRCA1, and BRCA2 genes.
De novo mutations (DNMs)
DNMs are mutations that arise during germ cell development within a single generation and thus are not observed in the parents. DNMs act as drivers of evolution and are a major cause of genetic disease. A typical example is Down syndrome, caused by trisomy of chromosome 21, which develops as a result of missegregation during oocyte meiosis. NGS of parent–offspring trios allows for accurately measuring the frequency of DNMs in humans (Koboldt et al., 2013). The number of DNMs in offspring is positively correlated with the age of parents at conception, and thus the children of elderly parents more frequently suffer from conditions, such as intellectual disability, autism, and epilepsy (Krupp et al., 2017; McRae et al., 2017; Taylor et al., 2019; Veltman & Brunner, 2012; Wiener-Megnazi et al., 2012). Maternal and paternal nucleotide substitutions per year of aging have been estimated at 0.4 and 1.7 × 10–8, respectively (Goldmann et al., 2016, 2019; Kong et al., 2012; Rahbari et al., 2016). The de novo mutation rate of C57BL/6 laboratory mice is 0.54 × 10–8 per nucleotide per generation (Uchimura et al., 2015).
The paternal nucleotide substitution rate is several times higher than the maternal one, probably due to differences between sperm and oocyte development. During oogenesis, primordial germ cells (pGCs) are generated and enter meiosis I prior to the birth of female babies, subsequently completing it during ovulation to differentiate into mature oocytes. Thus, meiosis I of individual oocytes takes 15–50 years, with oocyte proliferation halted for extended periods. In contrast to the prenatal termination of oocyte generation, sperm precursors undergo active genome replication from puberty until approximately 50 years of age. Spermatogonia start cell division after birth and maintain it to supply mature sperm over an extended period. Accordingly, the number of DNA replication cycles necessary for preparing fertilization-ready gametes is greater in spermatogenesis than in oogenesis. Thus, more paternal SBSs are generated during aging relative to maternal ones. The precise mechanisms underlying maternal DNMs remain to be elucidated (Goldmann et al., 2019).
In a recently reported cohort study (Kaplanis et al., 2021), 12 hypermutated individuals were identified from over 20,000 parent offspring sequenced trios, with a two to sevenfold increase in the number of de novo single nucleotide variants (SNVs) in the genome. Both genetic and environmental exposures (e.g. chemotherapeutics) contributed to germline mutation rate variation. They found germline mutation rates were increased by the defects in DNA repair genes, XPC and MPG were identified from paternal mutator variants in the study. However, defects in DNA repair pathways do not always behave similarly in the soma and the germline, they found somatic mutator gene MBD4 did not have a detectable effect in the germline ((Kaplanis et al., 2021).
Somatic mutations
Somatic mutations occur in small groups of cells and even in single cells, depending on the timing and source of mutagenesis. NGS only recently elucidated the rate of somatic mutagenesis in various tissues. Methods for identifying mutations in normal tissues include sequencing DNA derived from (a) clonally expanded cells derived from single stem cells, (b) micro-dissected tissues derived from a small number of cells, and (c) single cells (Table 2). NGS revealed that somatic mutations increase linearly with age, similar to DNMs in germ cells (Goldmann et al., 2019). The mutation rate of actively cycling sperm precursors is approximately 27-fold lower than that of the colorectal epithelium (Moore et al., 2021). The comparison of various tissue samples across organs from the same individual indicated 2.4 mutations per cell division during the first few embryonic divisions (Table 2) (Coorens et al., 2021; Park et al., 2021). The rate of mutagenesis is subsequently reduced, presumably due to enhanced DNA repair activity.

NGS studies have demonstrated that cells in mitotically inactive tissues, such as hepatocytes and neurons, accumulate somatic mutations at rates similar to those in cells from mitotically active tissues, such as colon epithelial and hematopoietic cells (Table 2). Thus, somatic mutation rates vary only moderately between tissues, indicating that the mutagenesis of somatic cells is largely independent of division frequency (Abascal et al., 2021; Blokzijl et al., 2016). This finding was remarkable as DNA replication has long been considered the primary cause of somatic mutations due to DNA polymerase errors (Kunkel & Erie, 2015; Tomasetti & Vogelstein, 2015) and error-prone translesion DNA synthesis (TLS) (see the 5th paragraph of “Spontaneously occurring base damage”) (Quinet et al., 2021; Vaisman & Woodgate, 2020). The similar somatic mutation rates between mitotically inactive and active tissues suggest that endogenously arising DNA lesions followed by faulty repair are the significant source of somatic mutations. Further, the inaccurate repair of DNA damage accounts for the time-dependent, rather than cell cycle-dependent accumulation of somatic mutations (Abascal et al., 2021). The molecular mechanisms protecting cycling sperm precursors from the accumulation of somatic mutations and underlying time-dependent mutagenesis remain unresolved.
Multi-step oncogenesis
The constant accumulation of oncogenic mutations drives multi-step oncogenesis, which transforms normal tissues into malignant cancers (Ciriello et al., 2013; Futreal et al., 2004; Hanahan & Weinberg, 2011). Epidemiological studies indicate that the risk of developing cancer increases exponentially with age (Doll, 1971). That is, a twofold increase in age leads to an increase from 32 (25) to 256 (28) in the risk of developing cancer. This dramatic increase may be attributed to the rapid increases in mutation rates with age. However, the linear increase in somatic mutations with age (Table 2) suggests that the accumulation of five to eight oncogenic driver mutations in cells is more likely to account for the above-presented epidemiological data (Campbell et al., 2020). This assumption is also supported by a recent meta-analysis of 6904 tumor samples (Anandakrishnani et al., 2019).
The identification of genes whose mutant variants drive oncogenesis in individual malignant tumors is a major goal. Whole-genome NGS in normal and precancerous regions of the esophageal epithelia (Yokoyama et al., 2019), skin (Martincorena et al., 2015), colon (Lee-Six et al., 2019), ulcerative colitis tissue (Kakiuchi & Ogawa, 2021; Kakiuchi et al., 2020), hematopoietic cells (Collins et al., 2020; Jacobs et al., 2012; Jaiswal & Ebert, 2019; Karczewski et al., 2020; Saiki et al., 2021), bronchi (Yoshida et al., 2020), and urinary bladder (Lawson et al., 2020; Li et al., 2020) have identified driver mutations that promote multistep carcinogenesis. The number of mutations in the dividing cells of normal tissues is 1.14–1.37 mutations per division and is 4–100 times lower than that in adult malignant cells (Werner et al., 2020). The number of mutations is 14 times lower in childhood cancers than in adult cancers (Bandopadhayay & Meyerson, 2018; Gröbner et al., 2018; Kandoth et al., 2013; Ma et al., 2018). A high mutation frequency is associated with loss-of-function mutations in DNA damage response genes encoding p53, DNA-dependent protein kinase, mismatch repair factors (MSH2, MSH6, and PMS2), DNA polymerase ε (Polε), and DNA polymerase Q (PolQ) in adult and childhood cancers (Bandopadhayay & Meyerson, 2018; Gröbner et al., 2018; Kandoth et al., 2013).
The cause of spontaneous mutagenesis
Non-dividing cells in mitotically inactive tissues accumulate considerable somatic mutations, as discussed in “Somatic mutations”, indicating the presence of DNA replication-independent mutagenesis in addition to mutagenic mechanisms dependent on replication (Fig. 3). Replication-dependent mutagenesis is induced by the activity of replicative DNA polymerases (Fig. 3A) and translesion DNA synthesis (TLS) polymerases (Fig. 3B, C). Replication-independent mutagenesis is presumably initiated by DNA damage and subsequent inaccurate DNA repair. This chapter summarizes replication errors and DNA damage, while the next chapter describes DNA repair pathways.
Fig. 3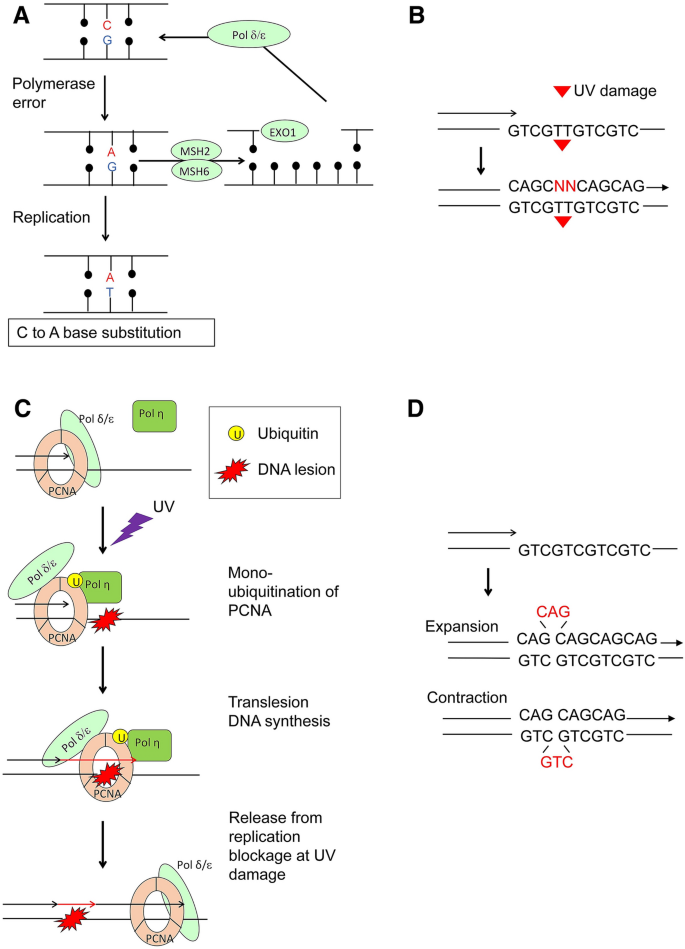
Mechanism for the mutagenesis during DNA replication. A Replicative DNA polymerases occasionally misincorporate nucleotides; for example, G opposite to A. MMR repairs the resulting G: A mispair and inserts T opposite to A. If MMR fails to repair the G: A mispair, the second DNA replication causes C to A substitution. B Translesion DNA synthesis (TLS) past a thymidine dimer, UV damage. The vast majority of the DNA lesions block the DNA synthesis by replicative DNA polymerases d and e (Pol d and Pol e). TLS polymerases insert a few nucleotides, shown as ‘NN’ in red, opposite DNA damage in an error-prone manner. Replicative DNA polymerases then extend DNA synthesis. C Regulation of TLS by mono-ubiquitination of the clamp protein, PCNA. UV damage stops Pol d and Pol e, leading to the mono-ubiquitination of PCNA. Ubiquitinated PCNA provides the docking site for TLS polymerase, Pol h, which inserts a few nucleotides opposite DNA lesions. D The slippage of replicative DNA polymerases changes the copy number of short tandem repeats (STRs) such as CAG repeats. The slippage towards the opposite direction of DNA synthesis increases the copy number, resulting in the expansion of repeats. The slippage can decrease the copy number, leading to the contraction of repeats
Full size imageReplication errors
The fidelity of DNA replication is primarily determined by the nucleotide selectivity and proofreading activity of replicative DNA polymerases as well as mismatch repair (MMR) machinery (Fig. 3A) (Ganai & Johansson, 2016). The replication error rate per division is once per 107 bases in yeast, Saccharomyces cerevisiae (St Charles et al., 2015), versus 1.14–1.37 mutations per 6 × 109 bases in normal human cells (Werner et al., 2020). Proofreading mechanisms and MMR remove misincorporated dNTPs, increasing the fidelity of DNA replication 250-fold in yeast (Lujan et al., 2012; Morrison & Sugino, 1994). MMR removes different types of mismatches with variable efficiency (St Charles et al., 2015). For example, C–T mismatches are corrected with 80 times greater efficiency than T–T mismatches. Thus, the variable efficiency of MMR is likely to determine SBS signatures in replicating cells. In addition to MMR, RecQ DNA helicase replication errors also arise when replicative DNA polymerases incorporate ribonucleotide triphosphates (rNTPs) into genomic DNA. The concentration of rNTPs is much higher than that of dNTPs in vivo (Nick McElhinny et al., 2010), and replicative DNA polymerases incorporate one rNTP for every 103–104 dNTPs (Ashour & Mosammaparast, 2021; McElhinny et al., 2010; Nick McElhinny et al., 2010). While most misincorporated rNMPs are removed by ribonuclease H2, rNTPs incorporated in the DNA can trap DNA topoisomerase I (TOP1), leading to the deletion of 2–5 nucleotides (Clark et al., 2011; Kim et al., 2011; Zimmermann et al., 2018).
Roughly 3% of the human genome is comprised of simple sequence repeats (Lander et al., 2001). Short tandem repeats (STRs) are defined by motifs with a length of six or fewer bases repeated many times (5–50), including repeats composed of (A)n, (CA)n, (TA)n, (CAG)n, and (CTG)n (Ellegren, 2004). STRs are unstable and prone to changes in copy numbers with a frequency as high as 10−4–10−3 per generation (Ellegren, 2004). The copy number change results from the slippage of replicative DNA polymerases during replication (Fig. 3D) (Viguera et al., 2001). WRN stabilizes TA-dinucleotide repeats in replicating cells (van Wietmarschen et al., 2020). Copy number changes can also occur independently of DNA replication, as the expansion of trinucleotide repeats is observed in non-dividing neurons and oocytes (Gonitel et al., 2008; Kovtun & McMurray, 2001; Kovtun et al., 2000). However, the molecular mechanism underlying the expansion of quiescent cells remains unclear (Polyzos & McMurray, 2017).
Spontaneously occurring base damage
Approximately 70,000 DNA lesions per cell are generated daily via hydrolysis, alkylation, and oxidation of nucleotides as well as the subsequent generation of single-strand breaks (SSBs) (Lindahl, 1993; Lindahl & Barnes, 2000; Tubbs & Nussenzweig, 2017). The base excision repair (BER) pathway repairs base damage, with SSBs being formed as intermediate repair substrates (Fig. 4A, B). DNA replication past unrepaired damaged nucleotides on template strands causes mutations (Fig. 4A–C).
Fig. 4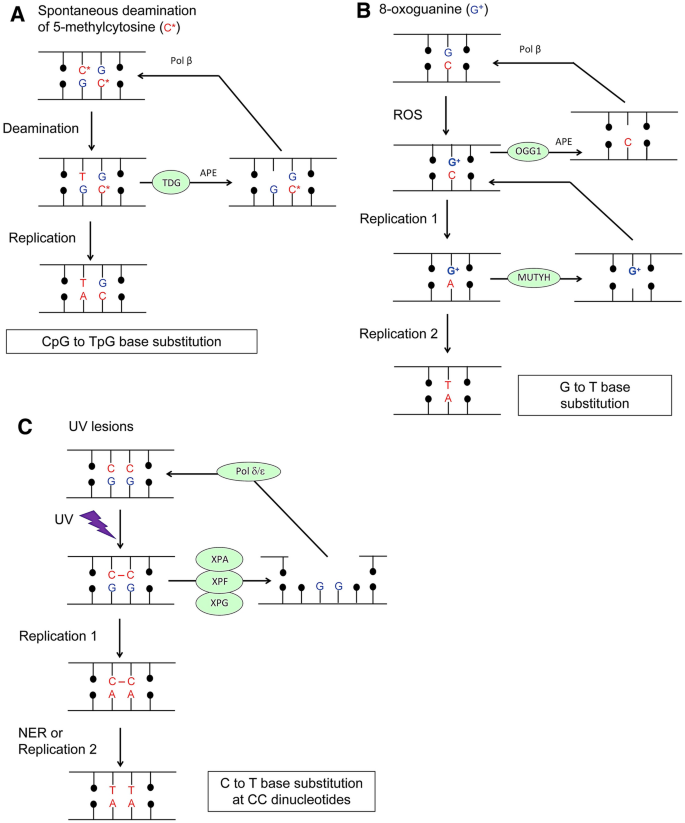
Molecular mechanisms for spontaneous and UV-induced mutagenesis. A Spontaneous deamination of 5-methylcytosine. Its deamination generates T resulting in a T: G mispair. BER involving TDG, APE, and Pol b changes T to C. B Oxidation of G by ROS generates 8-oxoguanine, and BER involving OGG1, APE, and Pol b repairs the damage. If replication occurs before BER, replicative DNA polymerases often incorporate A opposite to 8-oxoguanine. An MMR factor, MUTYH, removes A and restores an 8-oxoguanine: C pair. If the second round of replication occurs before MMR, it leads to G to T base substitution. C UV irradiation generates crosslinks between two neighboring pyrimidines. NER involving XPA, XPF, and XPG removes the resulting cytosine-cytosine photodimers. Replicative DNA polymerases are unable to undergo DNA synthesis past UV lesions. TLS polymerases then erroneously perform DNA synthesis and insert, for example, two adenines opposite to cytosine-cytosine photodimers. NER and the second round of replication using the AA-containing template strands cause C to T base substitution at UV-damaged CC dinucleotides
Full size imageIn neurons, mutation hotspots are localized at DNase-I-hypersensitive sites (Dileep & Tsai, 2021; Lodato et al., 2015; Wu et al., 2021), presumably because genomic DNA in euchromatin is more susceptible to base damage than that in heterochromatin. Similarly to ROS, formaldehyde is generated from various biochemical reactions and environmental factors (Burgos-Barragan et al., 2017). Formaldehyde considerably damages genomic DNA, particularly in people lacking the appropriate formaldehyde-detoxifying enzymes (Dingler et al., 2020; Garaycoechea et al., 2018). Acetaldehyde also damages DNA, with alcohol intake increasing esophageal cancer risk (Matejcic et al., 2017). APOBEC family enzymes damage both viral genomes and human genomic DNA through cytidine deaminase activity, causing C>T and C>G mutations at TCN sequences of SBS2 and SBS13 in normal tissues (Table 1).The activation-induced deaminase (AID) catalyzes cytidine deamination at immunoglobulin V (Ig V) segments and is responsible for their hypermutation (Di Noia & Neuberger, 2007). Unlike APOBEC enzymes, AID causes hypermutation at A:T pairs and C:G pairs within Ig V segments, yet the molecular mechanisms underlying mutagenesis at A:T pairs remain unknown.
The CpG>TpG transition occurs during DNA replication as well as independently of the replication process. 75% of CpG cytosines are methylated in human cells, and deamination of 5-methylcytosine generates thymine (Fig. 4A). If the resulting T:G mismatch is left unrepaired, DNA replication over the thymine leads to CpG>TpG transition mutations (Table 1, SBS1) (Abascal et al., 2021; Fryxell & Zuckerkandl, 2000; Lindahl, 1993). CpG>TpG transition mutations accumulate 2.5 substitutions per year in human post-mitotic neurons, indicating that this transition can occur independently of DNA replication (Abascal et al., 2021). The mechanism underlying mutagenesis outside the S phase remains an open question, and the very frequent CpG>TpG transition likely stems from inefficient recognition of the T:G mismatch (Tubbs & Nussenzweig, 2017). SBS1 is the second most common base substitution signature after SBS5 (Abascal et al., 2021; Moore et al., 2021). CpG>TpG transitions account for the majority of C>T mutations, reflecting the inherent mutability when cytosine is methylated at CpG sequences (Lindahl, 1993). In addition to 5-methylcytosine, hydroxymethylated cytosines are associated with increased C to G mutation rates (Supek et al., 2014).
NGS studies suggest that ROS only modestly contribute to time-dependent mutagenesis. ROS generated via cellular respiration have been proposed as drivers of aging and oncogenesis. Daily, ROS induce approximately 2800 8-oxoguanine (8-oxoG) alterations per cell (Tubbs & Nussenzweig, 2017). 8-oxoG can then mispair with adenine, and replicative DNA polymerases often misincorporate adenine opposite to 8-oxoG, resulting in G>T substitutions (Fig. 4B). 8-oxoG is widely believed to be a primary driver of mutagenesis. However, NGS has revealed that mutations resulting from oxidative DNA lesions, including 8-oxoG-dependent SBS18 (Table 1), do not significantly increase with age neither in genomic nor in mitochondrial DNA (Helleday et al., 2014; Kennedy et al., 2013). These data highlight the efficient repair of oxidative damage.
Base damage is converted to SBSs during DNA replication through the TLS mechanism. DNA lesions on template strands often block DNA synthesis by replicative DNA polymerases (Figs. 3B, C, 4C). The DNA damage tolerance (DDT) mechanism restores the progression of DNA replication forks without repairing these lesions. Instead, DDT employs TLS (Fig. 3B, C), template switching, and priming DNA synthesis via PrimPol (Ashour & Mosammaparast, 2021; Quinet et al., 2021; Wong et al., 2021) to proceed over lesions. TLS is carried out by DNA polymerases Pol η, Pol ι, Pol κ, Rev1, and Pol ζ. Pol β, Pol λ, Pol θ, Pol ν, and PrimPol are also involved in TLS (Bainbridge et al., 2021; Goodman & Woodgate, 2013). These polymerases can insert a few nucleotides opposite to various lesions, followed by a restart of DNA synthesis by replicative DNA polymerases (Fig. 3B). TLS polymerases have a fidelity that is several orders of magnitude lower than that of replicative DNA polymerases during the copying of undamaged DNA substrates (McCulloch & Kunkel, 2008). Yeast Pol ζ is responsible for over 95% of UV-induced mutagenesis and 50–75% of spontaneously arising mutations (Lawrence & Maher, 2001). The contribution of TLS to spontaneous somatic mutations in cycling mammalian cells is unclear because of the complex functional interactions between TLS polymerases. TLS DNA polymerases also play a role in excision DNA repair (Kawamoto et al., 2005; Ogi et al., 2010; Yoshimura et al., 2006). Of note, their involvement in somatic mutations in non-dividing cells remains unresolved.
Spontaneously occurring SSBs
An estimated 55,000 SSBs arise per day in each cell (Tubbs & Nussenzweig, 2017). The common triggers of SSBs are endogenous ROS. Unlike alkylating agents, such as methyl methanesulfonate (MMS), the exposure of cultured cells to H2O2 efficiently induces SSBs even on ice (Demin et al., 2021), indicative of direct cleavage of DNA strands by H2O2. SSBs occur during BER and as a consequence of erroneous enzyme catalysis. The most relevant enzyme in this regard is TOP1, which is essential for every transcription and replication event. TOP1 constantly repeats the cycle of SSB formation immediately followed by its religation. When TOP1 cleaves DNA to relieve torsional stress, broken ends rotate along the other intact strand, and TOP1 religates the SSBs only after stress relief. TOP1 cannot efficiently religate if it cleaves DNA sites near DNA damage, including SSBs, UV damage, ribonucleotides, abasic sites, DNA-RNA hybrids (R-loops), and G-quadruplex (G4 DNA) because these lesions dislocate the two ends of the breakage (Jakobsen et al., 2019; Kim et al., 2011; Pommier et al., 2016; Saha et al., 2020). The TOP1 trap at rNTPs embedded in DNA can lead to 2–5-nucleotide deletions (Clark et al., 2011; Kim et al., 2011; Zimmermann et al., 2018). Nonetheless, it is generally believed that SSBs are accurately repaired due to the presence of the other intact strand.
Spontaneously occurring DSBs in non-dividing cells and during DNA replication
DNA double-strand breaks (DSBs) cause indels (short deletions and insertions), copy number variation (long deletion and gene amplification), inversion, and chromosome translocation. Lethal DSBs are generated by environmental factors, such as X-irradiation and endogenous cellular processes. Recent studies have uncovered the role of DNA topoisomerase II (TOP2) in spontaneously occurring DSBs in non-dividing cells (Fig. 5A) (Morimoto et al., 2019; Tubbs & Nussenzweig, 2017). There are two TOP2 isoforms, TOP2A and TOP2B, which are essential for transcriptional elongation, particularly in long genes (Austin et al., 2018; Pommier et al., 2016). TOP2 enzymes resolve DNA catenanes by generating transient ‘gated’ DSBs, where TOP2 homodimers are covalently bound to the two ends of individual DSBs (Fig. 5A). ‘Abortive’ catalysis seems to occur frequently, leaving gated DSBs unsealed (Bunch et al., 2015; Gómez-Herreros et al., 2014; Haffner et al., 2010; Hoa et al., 2016; Ju et al., 2006; Madabhushi et al., 2015; Sasanuma et al., 2018; Williamson & Lees-Miller, 2011). TOP2-dependent DSBs may represent the recurrent DSBs seen in long transcribed gene bodies in neural progenitor cells (Wei et al., 2016) and cause over one megabase-scale de novo copy number variation in human neurons (McConnell et al., 2013). TOP2-dependent DSBs may be responsible for the unusual enrichment of indels in highly expressed genes (Abascal et al., 2021).
Fig. 5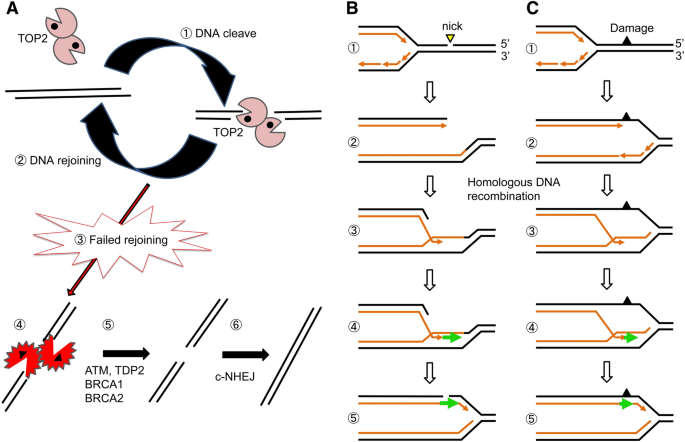
Two significant sources of double-strand DNA breakage, the abortive catalysis of TOP2 and replication blockage at damaged template strands. A TOP2 continuously generates a considerable number of double-strand breaks (DSBs) called TOP2 cleavage complexes (TOP2ccs), where TOP2 covalently binds to 5′ end of DSBs (Step 1). Intrinsic ligation activity of TOP2 seals DSBs (Step 2). TOP2 often fails to religate TOP2ccs, called abortive catalysis of TOP2 (Step 3), generating stalled TOP2ccs (Step 4). Their religation is carried out by the removal of 5′ TOP2 adducts (Step 5) followed by canonical nonhomologous end-joining (c-NHEJ) (Step 6). B When replication forks encounter an SSB (Step 1), it is converted to a breakage called one-end breakage (Step 2). Homologous DNA recombination (HR) repairs breaks using the other sister chromatids as a template (Steps 3–5). C The vast majority of base lesions stop DNA replication (Step 1), and the replication blockage is released by HR (Steps 2–5) as well as TLS. Note that neither HR nor TLS repairs the DNA lesions that block the progression of replication forks. HR releases replication blockage in an error-free manner while TLS is error-prone
Full size imageDNA replication forks stall at SSBs on template strands, generating one-end DSBs in one of the two sister chromatids (Fig. 5B). Homologous DNA recombination (HR) accurately repairs DSBs using the other sister chromatid as a template (Fig. 5B). Similarly to TLS (Fig. 3B, C), HR releases replication blockage at damaged nucleotides without repairing the damage (Fig. 5C). The complete loss of HR activity generates multiple DSBs and immediately kills dividing cells (Sonoda et al., 1998). The number of spontaneously occurring DSBs is much lower than that of spontaneously arising SSBs, with individual cells generating approximately 25 DSBs per day, yet DSBs are considerably more dangerous than SSBs (Tubbs & Nussenzweig, 2017). Active transcription often generates R-loops, and their collision with replication forks induces DSBs (García-Muse & Aguilera, 2019). Prolonged replication blockage at damaged nucleotides can also cause DSBs due to endonucleolytic cleavage by Mus81 (Ashour & Mosammaparast, 2021).
Inaccurate repair of DNA damage in spontaneous mutagenesis
Excision repair
Cells have three excision repair pathways, BER (Fig. 4A, B), nucleotide excision repair (NER) (Fig. 4C), and MMR (Figs. 3A, 4B) (Lindahl et al., 1997). BER and MMR are mainly responsible for correcting errors generated via spontaneous mutagenesis, while NER is involved in the repair of damage induced by exogenous chemicals and UV irradiation. BER is essential for genomic stability in mammalian cells (see “Spontaneously occurring base damage”). Mice deficient in key BER enzymes, such as apurinic/apyrimidinic endonuclease 1 (APE1) and DNA polymerase β (Pol β) (Fig. 4A, B), are embryonic lethal and die immediately after birth, respectively (Gu et al., 1994; Ludwig et al., 1998).The severe phenotype of BER-deficient animals contrasts with humans deficient in NER and MMR, who develop normally. MMR defects cause carcinogenesis in the colon epithelium in humans due to the accumulation of point mutations and changes in the copy number of STRs during DNA replication (see “Replication errors”) (Peltomäki et al., 2020). If excision repair fails to remove base damage before DNA replication, error-prone TLS past damaged template strands converts DNA damage to mutations (see “Spontaneously occurring DSBs in non-dividing cells and during DNA replication” and Fig. 3B, C).
BER constantly repairs large numbers of spontaneously arising damaged bases in both mitotically active and inactive tissues. Thus, inaccurate BER events are most likely to cause somatic mutations that accumulate linearly with age in mitotically inactive tissues. BER is initiated via the excision of damaged DNA bases by DNA glycosylases generating abasic sites, and human cells possess 11 glycosylases (Svilar et al., 2011). Increased expression of DNA glycosylases can enhance spontaneous mutagenesis in bacteria, yeast, and mammalian cells (Fu et al., 2012). Repair synthesis in BER is performed by replicative DNA polymerases δ and ε (Pol δ and Pol ε), Pol β, Pol λ, as well as Pol Θ (Braithwaite et al., 2010; Yoshimura et al., 2006) (Fig. 4A, B). However, the relative contribution of these polymerases in normal tissues is unclear. The latter three enzymes lack proofreading activity, and their fidelity is a few orders of magnitude lower than that of Pol δ and Pol ε. The actual mutation frequency of BER in vivo remains poorly understood.
The loss of MMR promotes tolerance, rather than sensitization, to alkylating agents. The molecular mechanisms underlying this tolerance are not yet fully understood. One identified mechanism is that alkylating agents form O6-methylguanine (O6meG), and DNA replication past the damaged nucleotide generates O6meG:T mispairs. MMR recognizes these, initiating futile cycles of DNA resection and the restoration of O6meG:T mispairs, eventually leading to mutagenic repair, DNA breakage, and apoptosis (Cejka & Jiricny, 2008; Fu et al., 2012; Mojas et al., 2007). It is not yet clear why BER cannot efficiently remove O6meG from the O6meG:T mispair. The functional interaction between the BER and MMR is discussed in the 4th section. BER can repair UV-induced lesions in NER-deficient cells when UV lesions irreversibly trap stable TOP1-SSB complexes near UV damage, with the resulting SSBs activating BER (Saha et al., 2020).
The contribution of BER, NER, and MMR to mutagenesis in mitotically inactive tissues has not yet been clarified. The neurodegeneration observed in mice deficient for XRCC1, a key BER factor, is primarily attributable to the depletion of NAD+, a substrate for poly (ADP ribose) polymerase, rather than the accumulation of mutations (Demin et al., 2021; Hoch et al., 2017). In NER-deficient patients, who have a mild neurodegenerative phenotype from puberty, neurons exhibit a several times greater accumulation of somatic mutations (Lodato et al., 2018). In summary, future studies are required to determine whether BER generates mutations in post-mitotic cells. Another unsolved question is whether the loss of MMR increases de novo mutations in non-dividing cells.
SSB and DSB repair
BER repairs smaller SSBs with extremely high efficiency within 60 min after pulse exposure to H2O2 (Hoch et al., 2017). Ligase-dependent rejoining of SSBs requires the preceding restoration of a phosphate group at their 5′ end and a hydroxy group at their 3′ end. Various enzymes are involved in the restoration, including polynucleotide kinase-phosphatase (PNKP) (Kalasova et al., 2020; Plo et al., 2003), the lyase function of Pol β removing a 5′-deoxyribose phosphate (5′-dRP) moiety of SSBs (Beard & Wilson, 2019; Sobol et al., 1996), DNA-apurinic–apyrimidinic (AP) endonuclease I (APE1, APEX1) (Caldecott, 2008), AP endonuclease 2 (APE2, APEX2) (Álvarez-Quilón et al., 2020; Li et al., 2019), aprataxin (APTX, AOA1) (Ahel et al., 2006; El-Khamisy et al., 2009; Grundy et al., 2013), and tyrosyl-DNA phosphodiesterase 1 (TDP1, SCAN1) (El-Khamisy et al., 2005; Sun et al., 2020a; Zeng et al., 2012). Homozygous mutations in the APTX, TDP1, and PNKP genes cause neurodegenerative diseases (Date et al., 2001; Moreira et al., 2001; Shen et al., 2010; Takashima et al., 2002). Further, the repair of TOP1-SSB complexes requires the coordinated action of TDP1 and PNKP (Sun et al., 2020b). No report has provided compelling evidence for the accumulation of mutations or defective SSB repair as the cause of the diseases.
Mammalian cells employ three DSB repair pathways, namely, HR, canonical non-homologous end-joining (c-NHEJ), and alternative end-joining (alt-EJ) (Ceccaldi et al., 2016). HR repairs DSBs when intact sister chromatids are available as template DNA (Fig. 5B, C) in the S to G2 phases, while NHEJ and alt-EJ operate throughout the cell cycle as well as in post-mitotic cells (Ceccaldi et al., 2016). alt-EJ has not yet been thoroughly characterized, operating as a backup to abrogated HR and c-NHEJ, with major contribution to the formation of chromosomal translocations (Bennardo et al., 2008; Ranjha et al., 2018). Pol Θ seems to play a key role in alt-EJ independently of canonical c-NHEJ (Ceccaldi et al., 2015; Saito et al., 2017).
DSBs generated during DNA replication are accurately repaired exclusively via HR between broken sister chromatids and intact chromatids (Dias et al., 2021; Thomas & Pommier, 2019) (Fig. 5B). In contrast, c-NHEJ plays a dominant role in the repair of TOP2-dependent DSBs (Maede et al., 2013) (Fig. 5A). c-NHEJ requires the removal of blocking adducts (Akagawa et al., 2020; Chappell et al., 2002). TOP2 adducts and 5′-blocking adducts are removed via TDP2 and MRE11 endonucleases (Deshpande et al., 2016; Hoa et al., 2016; Ledesma et al., 2009; Paull, 2018). It remains unclear whether MRE11-dependent repair can accurately repair DSBs covalently associated with TOP2 (Zagnoli-Vieira & Caldecott, 2020). DSBs generated via endogenous sources and ionizing-radiation (IR) generally carry blocking adducts at their ends and are termed ‘dirty’ DSBs (Woodbine et al., 2011). A recent study reported that ARTEMIS, EXD2, EXO1, and MRE11 nucleases contribute to ‘cleaning up’ dirty ends in preparation for subsequent c-NHEJ (Akagawa et al., 2020). It is unclear whether the cleaning up followed by c-NHEJ results in the deletion of nucleotides.
DNA damage checkpoints
DNA damage checkpoint (DDCs) sense DNA damage and initiate signal transduction through a kinase cascade, leading to transient cell cycle arrests at the G1/S transition, S phase, and G2/M transition (Paulovich & Hartwell, 1995; Weinert & Hartwell, 1988). The DDC is fully activated by a small number of unrepaired DSBs, HR intermediates, and stalled replication forks, providing cells with time to repair DNA (Lecona & Fernandez-Capetillo, 2018; Paulovich & Hartwell, 1995; Waterman et al., 2020; Weinert & Hartwell, 1988). During transient cell cycle arrest at the G1/S checkpoint, cells repair DNA damage to prevent the collapse of replication forks at broken DNA. DSB repair at the G2/M checkpoint prevents long deletions distal to DSBs and whole-chromosome loss during mitosis. In addition to cell cycle arrest, DDC promotes the HR-dependent repair of DSBs, thereby suppressing error-prone c-NHEJ when DSBs are generated due to DNA replication at broken template strands (Balmus et al., 2019). In cycling cells, the above mechanisms suppress DSB-induced mutagenesis, including deletion, chromosome translocation, and chromosome loss. If a single DSB is left unrepaired within a time window, activated DDCs eventually trigger apoptosis and eliminate cells carrying unrepaired DSBs from the body (Trenner & Sartori, 2019). The key players in DDC signaling are the ATM kinase and its substrate, the TP53 transcription factor (Banin et al., 1998; Lee & Paull, 2021). ATM and TP53 prevent oncogenesis by suppressing DSB-induced mutagenesis (Bailey et al., 2018; Honma & Hayashi, 2011), and carriers of ATM and TP53 mutations are predisposed to cancer (Huang et al., 2018). Although TP53 suppresses DSB-induced mutagenesis, TP53 does not seem to suppress SBSs, as indicated by TP53-deficient mice not showing a significant increase in SBSs neither in somatic tissues nor in the germline (Burr et al., 2005; Sands et al., 1995).
Unsolved questions and future prospect
Previous studies have examined the molecular mechanisms of mutagenesis by exposing cycling cells to exogenous DNA damage. Only recently did NGS studies reveal the accumulation of somatic mutations in normal tissues. Nevertheless, the mechanisms underlying mutagenesis in normal tissues are poorly understood, particularly in post-mitotic cells. Mutagenesis in G0/G1 phases is considered to result from erroneous BER of endogenous DNA damage, considering that BER repairs approximately 70,000 DNA lesions per cell daily (Lindahl, 1993; Lindahl & Barnes, 2000; Tubbs & Nussenzweig, 2017). Mutations occur at a rate of less than one per week in humans (Table 2), and it is a formidable challenge to understand the mechanism underlying such extremely rare events of mutagenesis. The number of SBSs in maternal and paternal germ cells is only 0.4 and 1.7, respectively, per year during aging (Goldmann et al., 2016, 2019; Kong et al., 2012; Rahbari et al., 2016) and over ten times lower than that in somatic cells (Table 2). The mechanism underlying such a low frequency in germ cells represents an essential topic for future studies.
B lymphocytes undergo programmed SBSs specifically at the variable region of the Ig V gene, increasing the affinity of the immunoglobulin to antigens. This Ig V hypermutation is initiated by the deamination of dC residues via AID, generating dU:dG mispairs (Di Noia & Neuberger, 2007; Muramatsu et al., 2000). BER and MMR are complementary in processing dU:dG mispairs and are required for Ig V hypermutation (Rada et al., 2004). The removal of dU by uracil-DNA glycosylase and TLS over the resulting abasic site causes hypermutation at dC:dG pairs (Nakahara et al., 2009; Sale, 2013). Overall, the molecular mechanism of MMR-dependent mutagenesis at Ig V is poorly understood. Another unsolved question is the molecular mechanism underlying hypermutation at dA:dT pairs. Addressing these questions in the future may further our understanding of the molecular mechanisms underlying mutagenesis, which involve BER and MMR in normal tissues.
It is well established that the accumulation of SBSs cause carcinogenesis. However, it is unclear whether accumulated SBSs promote aging and progressive neurodegenerative diseases as evidenced by the phenotype of MMR-deficient people. MMR-deficiency neither acerates aging nor cause neurodegenerative diseases although the number of newly generated SBSs per division increases nearly 100 times (Peltomäki et al., 2020). Thus, it is an issue of future study to clarify whether a few times increase in the number of SBSs (Table 2) causes neurodegenerative symptom in NER-deficient CS an d XP patients (Lodato et al., 2018). Likewise, patients deficient in BER factors, APTX, TDP1, and PNKP, suffer neurodegenerative diseases (Date et al., 2001; Moreira et al., 2001; Shen et al., 2010; Takashima et al., 2002), the contribution of increased SBSs to the disease is an important question for future research.
References
Abascal, F., Harvey, L. M. R., Mitchell, E., Lawson, A. R. J., Lensing, S. V., Ellis, P., et al. (2021). Somatic mutation landscapes at single-molecule resolution. Nature, 593, 405–410. https://doi.org/10.1038/s41586-021-03477-4
Ahel, I., Rass, U., El-Khamisy, S. F., Katyal, S., Clements, P. M., McKinnon, P. J., Caldecott, K. W., & West, S. C. (2006). The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature, 443, 713–716. https://doi.org/10.1038/nature05164
Akagawa, R., Trinh, H. T., Saha, L. K., Tsuda, M., Hirota, K., Yamada, S., et al. (2020). UBC13-mediated ubiquitin signaling promotes removal of blocking adducts from DNA double-strand breaks. Iscience. https://doi.org/10.1016/j.isci.2020.101027
Alexandrov, L. B., Jones, P. H., Wedge, D. C., Sale, J. E., Campbell, P. J., Nik-Zainal, S., & Stratton, M. R. (2015). Clock-like mutational processes in human somatic cells. Nature Genetics, 47, 1402–1407. https://doi.org/10.1038/ng.3441
Alexandrov, L. B., Ju, Y. S., Haase, K., Van Loo, P., Martincorena, I., Nik-Zainal, S., et al. (2016). Mutational signatures associated with tobacco smoking in human cancer. Science, 354, 618–622. https://doi.org/10.1126/science.aag0299
Alexandrov, L. B., Kim, J., Haradhvala, N. J., Huang, M. N., Tian Ng, A. W., Wu, Y., et al. (2020). The repertoire of mutational signatures in human cancer. Nature, 578, 94–101. https://doi.org/10.1038/s41586-020-1943-3
Alexandrov, L. B., Nik-Zainal, S., Wedge, D. C., Aparicio, S. A. J. R., Behjati, S., Biankin, A. V., et al. (2013). Signatures of mutational processes in human cancer. Nature, 500, 415–421. https://doi.org/10.1038/nature12477
Alexandrov, L. B., & Stratton, M. R. (2014). Mutational signatures: the patterns of somatic mutations hidden in cancer genomes. Current Opinion in Genetics & Development, 24, 52–60. https://doi.org/10.1016/j.gde.2013.11.014
Álvarez-Quilón, A., Wojtaszek, J. L., Mathieu, M. C., Patel, T., Appel, C. D., Hustedt, N., et al. (2020). Endogenous DNA 3′ blocks are vulnerabilities for BRCA1 and BRCA2 deficiency and are reversed by the APE2 nuclease. Molecular Cell, 78, 1152-1165.e8. https://doi.org/10.1016/j.molcel.2020.05.021
Anandakrishnani, R., Varghese, R. T., Kinneyi, N. A., & Garner, H. R. (2019). Estimating the number of genetic mutations (HITS) required for carcinogenesis based on the distribution of somatic mutations. PLoS Computational Biology. https://doi.org/10.1371/journal.pcbi.1006881
Ashour, M. E., & Mosammaparast, N. (2021). Mechanisms of damage tolerance and repair during DNA replication. Nucleic Acids Research, 49, 3033–3047. https://doi.org/10.1093/nar/gkab101
Austin, C. A., Lee, K. C., Swan, R. L., Khazeem, M. M., Manville, C. M., Cridland, P., Treumann, A., Porter, A., Morris, N. J., & Cowell, I. G. (2018). TOP2B: the first thirty years. International Journal of Molecular Sciences. https://doi.org/10.3390/ijms19092765
Bailey, M. H., Tokheim, C., Porta-Pardo, E., Sengupta, S., Bertrand, D., Weerasinghe, A., et al. (2018). Comprehensive characterization of cancer driver genes and mutations. Cell, 173, 371-385.e18. https://doi.org/10.1016/j.cell.2018.02.060
Bainbridge, L. J., Teague, R., & Doherty, A. J. (2021). Repriming DNA synthesis: an intrinsic restart pathway that maintains efficient genome replication. Nucleic Acids Research, 49, 4831–4847. https://doi.org/10.1093/nar/gkab176
Balmus, G., Pilger, D., Coates, J., Demir, M., Sczaniecka-Clift, M., Barros, A. C., et al. (2019). ATM orchestrates the DNA-damage response to counter toxic non-homologous end-joining at broken replication forks. Nature Communications. https://doi.org/10.1038/s41467-018-07729-2
Bandopadhayay, P., & Meyerson, M. (2018). Landscapes of childhood tumours. Nature, 555, 316–317. https://doi.org/10.1038/d41586-018-01648-4
Banin, S., Moyal, L., Shieh, S. Y., Taya, Y., Anderson, C. W., Chessa, L., et al. (1998). Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science, 281, 1674–1677. https://doi.org/10.1126/science.281.5383.1674
Beard, W. A., & Wilson, S. H. (2019). DNA polymerase beta and other gap-filling enzymes in mammalian base excision repair. The Enzymes. https://doi.org/10.1016/bs.enz.2019.08.002
Bennardo, N., Cheng, A., Huang, N., & Stark, J. M. (2008). Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genetics. https://doi.org/10.1371/journal.pgen.1000110
Blokzijl, F., De Ligt, J., Jager, M., Sasselli, V., Roerink, S., Sasaki, N., et al. (2016). Tissue-specific mutation accumulation in human adult stem cells during life. Nature, 538, 260–264. https://doi.org/10.1038/nature19768
Boot, A., Huang, M. N., Ng, A. W. T., Ho, S. C., Lim, J. Q., Kawakami, Y., Chayama, K., Teh, B. T., Nakagawa, H., & Rozen, S. G. (2018). In-depth characterization of the cisplatin mutational signature in human cell lines and in esophageal and liver tumors. Genome Research, 28, 654–665. https://doi.org/10.1101/gr.230219.117
Braithwaite, E. K., Kedar, P. S., Stumpo, D. J., Bertocci, B., Freedman, J. H., Samson, L. D., & Wilson, S. H. (2010). DNA polymerases β and λ mediate overlapping and independent roles in base excision repair in mouse embryonic fibroblasts. PLoS One. https://doi.org/10.1371/journal.pone.0012229
Brazhnik, K., Sun, S., Alani, O., Kinkhabwala, M., Wolkoff, A. W., Maslov, A. Y., Dong, X., & Vijg, J. (2020). Single-cell analysis reveals different age-related somatic mutation profiles between stem and differentiated cells in human liver. Science Advances. https://doi.org/10.1126/sciadv.aax2659
Bunch, H., Lawney, B. P., Lin, Y. F., Asaithamby, A., Murshid, A., Wang, Y. E., Chen, B. P. C., & Calderwood, S. K. (2015). Transcriptional elongation requires DNA break-induced signalling. Nature Communications. https://doi.org/10.1038/ncomms10191
Burgos-Barragan, G., Wit, N., Meiser, J., Dingler, F. A., Pietzke, M., Mulderrig, L., et al. (2017). Mammals divert endogenous genotoxic formaldehyde into one-carbon metabolism. Nature, 548, 549–554. https://doi.org/10.1038/nature23481
Burr, K. L. A., Smith, A. G., & Dubrova, Y. E. (2005). p53 deficiency does not affect mutation rate in the mouse germline. Oncogene, 24, 4315–4318. https://doi.org/10.1038/sj.onc.1208604
Caldecott, K. W. (2008). Single-strand break repair and genetic disease. Nature Reviews Genetics, 9, 619–631. https://doi.org/10.1038/nrg2380
Campbell, P. J., Getz, G., Korbel, J. O., Stuart, J. M., Jennings, J. L., Stein, L. D., et al. (2020). Pan-cancer analysis of whole genomes. Nature, 578, 82–93. https://doi.org/10.1038/s41586-020-1969-6
Ceccaldi, R., Liu, J. C., Amunugama, R., Hajdu, I., Primack, B., Petalcorin, M. I. R., et al. (2015). Homologous-recombination-deficient tumours are dependent on Polθ -mediated repair. Nature, 518, 258–262. https://doi.org/10.1038/nature14184
Ceccaldi, R., Rondinelli, B., & D’Andrea, A. D. (2016). Repair pathway choices and consequences at the double-strand break. Trends in Cell Biology, 26, 52–64. https://doi.org/10.1016/j.tcb.2015.07.009
Cejka, P., & Jiricny, J. (2008). Interplay of DNA repair pathways controls methylation damage toxicity in Saccharomyces cerevisiae. Genetics, 179, 1835–1844. https://doi.org/10.1534/genetics.108.089979
Chappell, C., Hanakahi, L. A., Karimi-Busheri, F., Weinfeld, M., & West, S. C. (2002). Involvement of human polynucleotide kinase in double-strand break repair by non-homologous end joining. EMBO Journal, 21, 2827–2832. https://doi.org/10.1093/emboj/21.11.2827
Ciriello, G., Miller, M. L., Aksoy, B. A., Senbabaoglu, Y., Schultz, N., & Sander, C. (2013). Emerging landscape of oncogenic signatures across human cancers. Nature Genetics, 45, 1127–1133. https://doi.org/10.1038/ng.2762
Clark, A. B., Lujan, S. A., Kissling, G. E., & Kunkel, T. A. (2011). Mismatch repair-independent tandem repeat sequence instability resulting from ribonucleotide incorporation by DNA polymerase e{open}. DNA Repair, 10, 476–482. https://doi.org/10.1016/j.dnarep.2011.02.001
Collins, R. L., Brand, H., Karczewski, K. J., Zhao, X., Alföldi, J., Francioli, L. C., et al. (2020). A structural variation reference for medical and population genetics. Nature, 581, 444–451. https://doi.org/10.1038/s41586-020-2287-8
Coorens, T. H. H., Moore, L., Robinson, P. S., Sanghvi, R., Christopher, J., Hewinson, J., et al. (2021). Extensive phylogenies of human development inferred from somatic mutations. Nature, 597, 387–392. https://doi.org/10.1038/s41586-021-03790-y
Date, H., Onodera, O., Tanaka, H., Iwabuchi, K., Uekawa, K., Igarashi, S., et al. (2001). Early-onset ataxia with ocular motor apraxia and hypoalbuminemia is caused by mutations in a new HIT superfamily gene. Nature Genetics, 29, 184–188. https://doi.org/10.1038/ng1001-184
Demin, A. A., Hirota, K., Tsuda, M., Adamowicz, M., Hailstone, R., Brazina, J., et al. (2021). XRCC1 prevents toxic PARP1 trapping during DNA base excision repair. Molecular Cell, 81, 3018-3030.e5. https://doi.org/10.1016/j.molcel.2021.05.009
Deshpande, R. A., Lee, J. H., Arora, S., & Paull, T. T. (2016). Nbs1 converts the human Mre11/Rad50 nuclease complex into an endo/exonuclease machine specific for protein-DNA adducts. Molecular Cell, 64, 593–606. https://doi.org/10.1016/j.molcel.2016.10.010
Di Noia, J. M., & Neuberger, M. S. (2007). Molecular mechanisms of antibody somatic hypermutation. Annual Review of Biochemistry, 76, 1–22. https://doi.org/10.1146/annurev.biochem.76.061705.090740
Dias, M. P., Moser, S. C., Ganesan, S., & Jonkers, J. (2021). Understanding and overcoming resistance to PARP inhibitors in cancer therapy. Nature Reviews Clinical Oncology. https://doi.org/10.1038/s41571-021-00532-x
Dileep, V., & Tsai, L. H. (2021). Neuronal enhancers get a break. Neuron, 109, 1766–1768. https://doi.org/10.1016/j.neuron.2021.05.008
Dingler, F. A., Wang, M., Mu, A., Millington, C. L., Oberbeck, N., Watcham, S., et al. (2020). Two aldehyde clearance systems are essential to prevent lethal formaldehyde accumulation in mice and humans. Molecular Cell, 80, 996-1012.e9. https://doi.org/10.1016/j.molcel.2020.10.012
Doll, R. (1971). The age distribution of cancer: implications for models of carcinogenesis. Journal of the Royal Statistical Society: Series A (statistics in Society), 134, 133–166. https://doi.org/10.2307/2343871
El-Khamisy, S. F., Katyal, S., Patel, P., Ju, L., McKinnon, P. J., & Caldecott, K. W. (2009). Synergistic decrease of DNA single-strand break repair rates in mouse neural cells lacking both Tdp1 and aprataxin. DNA Repair, 8, 760–766. https://doi.org/10.1016/j.dnarep.2009.02.002
El-Khamisy, S. F., Saifi, G. M., Weinfeld, M., Johansson, F., Helleday, T., Lupski, J. R., & Caldecott, K. W. (2005). Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1. Nature, 434, 108–113. https://doi.org/10.1038/nature03314
Ellegren, H. (2004). Microsatellites: simple sequences with complex evolution. Nature Reviews Genetics, 5, 435–445. https://doi.org/10.1038/nrg1348
Forbes, S. A., Beare, D., Boutselakis, H., Bamford, S., Bindal, N., Tate, J., et al. (2017). COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Research, 45, D777–D783. https://doi.org/10.1093/nar/gkw1121
Franco, I., Johansson, A., Olsson, K., Vrtačnik, P., Lundin, P., Helgadottir, H. T., et al. (2018). Somatic mutagenesis in satellite cells associates with human skeletal muscle aging. Nature Communications. https://doi.org/10.1038/s41467-018-03244-6
Fryxell, K. J., & Zuckerkandl, E. (2000). Cytosine deamination plays a primary role in the evolution of mammalian isochores. Molecular Biology and Evolution, 17, 1371–1383. https://doi.org/10.1093/oxfordjournals.molbev.a026420
Fu, D., Calvo, J. A., & Samson, L. D. (2012). Balancing repair and tolerance of DNA damage caused by alkylating agents. Nature Reviews Cancer, 12, 104–120. https://doi.org/10.1038/nrc3185
Futreal, P. A., Coin, L., Marshall, M., Down, T., Hubbard, T., Wooster, R., Rahman, N., & Stratton, M. R. (2004). A census of human cancer genes. Nature Reviews Cancer, 4, 177–183. https://doi.org/10.1038/nrc1299
Ganai, R. A., & Johansson, E. (2016). DNA replication-a matter of fidelity. Molecular Cell, 62, 745–755. https://doi.org/10.1016/j.molcel.2016.05.003
Garaycoechea, J. I., Crossan, G. P., Langevin, F., Mulderrig, L., Louzada, S., Yang, F., et al. (2018). Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature, 553, 171–177. https://doi.org/10.1038/nature25154
García-Muse, T., & Aguilera, A. (2019). R loops: from physiological to pathological roles. Cell, 179, 604–618. https://doi.org/10.1016/j.cell.2019.08.055
Goldmann, J. M., Veltman, J. A., & Gilissen, C. (2019). De novo mutations reflect development and aging of the human germline. Trends in Genetics, 35, 828–839. https://doi.org/10.1016/j.tig.2019.08.005
Goldmann, J. M., Wong, W. S. W., Pinelli, M., Farrah, T., Bodian, D., Stittrich, A. B., et al. (2016). Parent-of-origin-specific signatures of de novo mutations. Nature Genetics, 48, 935–939. https://doi.org/10.1038/ng.3597
Gómez-Herreros, F., Schuurs-Hoeijmakers, J. H. M., McCormack, M., Greally, M. T., Rulten, S., Romero-Granados, R., et al. (2014). TDP2 protects transcription from abortive topoisomerase activity and is required for normal neural function. Nature Genetics, 46, 516–521. https://doi.org/10.1038/ng.2929
Gonitel, R., Moffitt, H., Sathasivam, K., Woodman, B., Detloff, P. J., Faull, R. L. M., & Bates, G. P. (2008). DNA instability in postmitotic neurons. Proceedings of the National Academy of Sciences of the United States of America, 105, 3467–3472. https://doi.org/10.1073/pnas.0800048105
Goodman, M. F., & Woodgate, R. (2013). Translesion DNA polymerases. Cold Spring Harbor Perspectives in Biology. https://doi.org/10.1101/cshperspect.a010363
Gröbner, S. N., Worst, B. C., Weischenfeldt, J., Buchhalter, I., Kleinheinz, K., Rudneva, V. A., et al. (2018). The landscape of genomic alterations across childhood cancers. Nature, 555, 321–327. https://doi.org/10.1038/nature25480
Grundy, G. J., Rulten, S. L., Zeng, Z., Arribas-Bosacoma, R., Iles, N., Manley, K., Oliver, A., & Caldecott, K. W. (2013). APLF promotes the assembly and activity of non-homologous end joining protein complexes. EMBO Journal, 32, 112–125. https://doi.org/10.1038/emboj.2012.304
Gu, H., Marth, J. D., Orban, P. C., Mossmann, H., & Rajewsky, K. (1994). Deletion of a DNA polymerase β gene segment in T cells using cell type-specific gene targeting. Science, 265, 103–106. https://doi.org/10.1126/science.8016642
Haffner, M. C., Aryee, M. J., Toubaji, A., Esopi, D. M., Albadine, R., Gurel, B., et al. (2010). Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nature Genetics, 42, 668–675. https://doi.org/10.1038/ng.613
Hanahan, D., & Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell, 144, 646–674. https://doi.org/10.1016/j.cell.2011.02.013
Helleday, T., Eshtad, S., & Nik-Zainal, S. (2014). Mechanisms underlying mutational signatures in human cancers. Nature Reviews Genetics, 15, 585–598. https://doi.org/10.1038/nrg3729
Hoa, N. N., Shimizu, T., Zhou, Z. W., Wang, Z. Q., Deshpande, R. A., Paull, T. T., et al. (2016). Mre11 is essential for the removal of lethal topoisomerase 2 covalent cleavage complexes. Molecular Cell, 64, 580–592. https://doi.org/10.1016/j.molcel.2016.10.011
Hoch, N. C., Hanzlikova, H., Rulten, S. L., Tétreault, M., Komulainen, E., Ju, L., et al. (2017). XRCC1 mutation is associated with PARP1 hyperactivation and cerebellar ataxia. Nature, 541, 87–91. https://doi.org/10.1038/nature20790
Honma, M., & Hayashi, M. (2011). Comparison of in vitro micronucleus and gene mutation assay results for p53-competent versus p53-deficient human lymphoblastoid cells. Environmental and Molecular Mutagenesis, 52, 373–384. https://doi.org/10.1002/em.20634
Huang, K. L., Mashl, R. J., Wu, Y., Ritter, D. I., Wang, J., Oh, C., et al. (2018). Pathogenic germline variants in 10,389 adult cancers. Cell, 173, 355-370.e14. https://doi.org/10.1016/j.cell.2018.03.039
Jacobs, K. B., Yeager, M., Zhou, W., Wacholder, S., Wang, Z., Rodriguez-Santiago, B., et al. (2012). Detectable clonal mosaicism and its relationship to aging and cancer. Nature Genetics, 44, 651–658. https://doi.org/10.1038/ng.2270
Jaiswal, S., & Ebert, B. L. (2019). Clonal hematopoiesis in human aging and disease. Science. https://doi.org/10.1126/science.aan4673
Jakobsen, K. P., Andersen, A. H., & Bjergbæk, L. (2019). Abortive activity of topoisomerase I: a challenge for genome integrity? Current Genetics, 65, 1141–1144. https://doi.org/10.1007/s00294-019-00984-w
Ju, B. G., Lunyak, V. V., Perissi, V., Garcia-Bassets, I., Rose, D. W., Glass, C. K., & Rosenfeld, M. G. (2006). A topoisomerase IIβ-mediated dsDNA break required for regulated transcription. Science, 312, 1798–1802. https://doi.org/10.1126/science.1127196
Kakiuchi, N., & Ogawa, S. (2021). Clonal expansion in non-cancer tissues. Nature Reviews Cancer, 21, 239–256. https://doi.org/10.1038/s41568-021-00335-3
Kakiuchi, N., Yoshida, K., Uchino, M., Kihara, T., Akaki, K., Inoue, Y., et al. (2020). Frequent mutations that converge on the NFKBIZ pathway in ulcerative colitis. Nature, 577, 260–265. https://doi.org/10.1038/s41586-019-1856-1
Kalasova, I., Hailstone, R., Bublitz, J., Bogantes, J., Hofmann, W., Leal, A., Hanzlikova, H., & Caldecott, K. W. (2020). Pathological mutations in PNKP trigger defects in DNA single-strand break repair but not DNA double-strand break repair. Nucleic Acids Research, 48, 6672–6684. https://doi.org/10.1093/nar/gkaa489
Kandoth, C., McLellan, M. D., Vandin, F., Ye, K., Niu, B., Lu, C., et al. (2013). Mutational landscape and significance across 12 major cancer types. Nature, 502, 333–339. https://doi.org/10.1038/nature12634
Kaplanis, J., Ide, B., Sanghvi, R., Neville, M., Danecek, P., Coorens, T., et al. (2021). Genetic and chemotherapeutic causes of germline hypermutation. bioRxiv. https://doi.org/10.1101/2021.06.01.446180
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581, 434–443. https://doi.org/10.1038/s41586-020-2308-7
Kawamoto, T., Araki, K., Sonoda, E., Yamashita, Y. M., Harada, K., Kikuchi, K., et al. (2005). Dual roles for DNA polymerase η in homologous DNA recombination and translesion DNA synthesis. Molecular Cell, 20, 793–799. https://doi.org/10.1016/j.molcel.2005.10.016
Kennedy, S. R., Salk, J. J., Schmitt, M. W., & Loeb, L. A. (2013). Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genetics, 9, e1003794. https://doi.org/10.1371/journal.pgen.1003794
Kim, N., Huang, S. Y. N., Williams, J. S., Li, Y. C., Clark, A. B., Cho, J. E., Kunkel, T. A., Pommier, Y., & Jinks-Robertson, S. (2011). Mutagenic processing of ribonucleotides in DNA by yeast topoisomerase I. Science, 332, 1561–1564. https://doi.org/10.1126/science.1205016
Knudson, A. G. (1971). Mutation and cancer: statistical study of retinoblastoma. Proceedings of the National Academy of Sciences of the United States of America, 68, 820–823. https://doi.org/10.1073/pnas.68.4.820
Koboldt, D. C., Steinberg, K. M., Larson, D. E., Wilson, R. K., & Mardis, E. R. (2013). The next-generation sequencing revolution and its impact on genomics. Cell, 155, 27. https://doi.org/10.1016/j.cell.2013.09.006
Kong, A., Frigge, M. L., Masson, G., Besenbacher, S., Sulem, P., Magnusson, G., et al. (2012). Rate of de novo mutations and the importance of father-s age to disease risk. Nature, 488, 471–475. https://doi.org/10.1038/nature11396
Kovtun, I. V., & McMurray, C. T. (2001). Trinucleotide expansion in haploid germ cells by gap repair. Nature Genetics, 27, 407–411. https://doi.org/10.1038/86906
Kovtun, I. V., Therneau, T. M., & McMurray, C. T. (2000). Gender of the embryo contributes to CAG instability in transgenic mice containing a Huntington’s disease gene. Human Molecular Genetics, 9, 2767–2775. https://doi.org/10.1093/hmg/9.18.2767
Krupp, D. R., Barnard, R. A., Duffourd, Y., Evans, S. A., Mulqueen, R. M., Bernier, R., Rivière, J. B., Fombonne, E., & O’Roak, B. J. (2017). Exonic mosaic mutations contribute risk for autism spectrum disorder. American Journal of Human Genetics, 101, 369–390. https://doi.org/10.1016/j.ajhg.2017.07.016
Kuijk, E., Jager, M., van der Roest, B., Locati, M. D., Van Hoeck, A., Korzelius, J., et al. (2020). The mutational impact of culturing human pluripotent and adult stem cells. Nature Communications. https://doi.org/10.1038/s41467-020-16323-4
Kunkel, T. A., & Erie, D. A. (2015). Eukaryotic mismatch repair in relation to DNA replication. Annual Review of Genetics, 49, 291–313. https://doi.org/10.1146/annurev-genet-112414-054722
Lander, E. S., Linton, L. M., Birren, B., Nusbaum, C., Zody, M. C., Baldwin, J., et al. (2001). Initial sequencing and analysis of the human genome. Nature, 409, 860–921. https://doi.org/10.1038/35057062
Lawrence, C. W., & Maher, V. M. (2001). Mutagenesis in eukaryotes dependent on DNA polymerase zeta and Rev1p. Philosophical Transactions of the Royal Society b: Biological Sciences. https://doi.org/10.1098/rstb.2000.0001
Lawson, A. R. J., Abascal, F., Coorens, T. H. H., Hooks, Y., O’Neill, L., Latimer, C., et al. (2020). Extensive heterogeneity in somatic mutation and selection in the human bladder. Science, 370, 75–82. https://doi.org/10.1126/science.aba8347
Lecona, E., & Fernandez-Capetillo, O. (2018). Targeting ATR in cancer. Nature Reviews Cancer, 18, 586–595. https://doi.org/10.1038/s41568-018-0034-3
Ledesma, F. C., El Khamisy, S. F., Zuma, M. C., Osborn, K., & Caldecott, K. W. (2009). A human 5′-tyrosyl DNA phosphodiesterase that repairs topoisomerase-mediated DNA damage. Nature, 461, 674–678. https://doi.org/10.1038/nature08444
Lee, J.-H., & Paull, T. T. (2021). Cellular functions of the protein kinase ATM and their relevance to human disease. Nature Reviews Molecular Cell Biology. https://doi.org/10.1038/s41580-021-00394-2
Lee-Six, H., Olafsson, S., Ellis, P., Osborne, R. J., Sanders, M. A., Moore, L., et al. (2019). The landscape of somatic mutation in normal colorectal epithelial cells. Nature, 574, 532–537. https://doi.org/10.1038/s41586-019-1672-7
Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536, 285–291. https://doi.org/10.1038/nature19057
Li, F., Wang, Q., Seol, J. H., Che, J., Lu, X., Shim, E. Y., Lee, S. E., & Niu, H. (2019). Apn2 resolves blocked 3′ ends and suppresses Top1-induced mutagenesis at genomic rNMP sites. Nature Structural & Molecular Biology, 26, 155–163. https://doi.org/10.1038/s41594-019-0186-1
Li, R., Di, L., Li, J., Fan, W., Liu, Y., Guo, W., et al. (2021). A body map of somatic mutagenesis in morphologically normal human tissues. Nature, 597, 398–403. https://doi.org/10.1038/s41586-021-03836-1
Li, R., Du, Y., Chen, Z., Xu, D., Lin, T., Jin, S., et al. (2020). Macroscopic somatic clonal expansion in morphologically normal human urothelium. Science, 370, 82–89. https://doi.org/10.1126/science.aba7300
Li, X. C., Wang, M. Y., Yang, M., Dai, H. J., Zhang, B. F., Wang, W., et al. (2018). A mutational signature associated with alcohol consumption and prognostically significantly mutated driver genes in esophageal squamous cell carcinoma. Annals of Oncology, 29, 938–944. https://doi.org/10.1093/annonc/mdy011
Lindahl, T. (1993). Instability and decay of the primary structure of DNA. Nature, 362, 709–715. https://doi.org/10.1038/362709a0
Lindahl, T., & Barnes, D. E. (2000). Repair of endogenous DNA damage. Cold Spring Harbor Symposia on Quantitative Biology. https://doi.org/10.1101/sqb.2000.65.127
Lindahl, T., Karran, P., & Wood, R. D. (1997). DNA excision repair pathways. Current Opinion in Genetics & Development, 7, 158–169. https://doi.org/10.1016/S0959-437X(97)80124-4
Lodato, M. A., Rodin, R. E., Bohrson, C. L., Coulter, M. E., Barton, A. R., Kwon, M., et al. (2018). Aging and neurodegeneration are associated with increased mutations in single human neurons. Science, 359, 555–559. https://doi.org/10.1126/science.aao4426
Lodato, M. A., Woodworth, M. B., Lee, S., Evrony, G. D., Mehta, B. K., Karger, A., et al. (2015). Somatic mutation in single human neurons tracks developmental and transcriptional history. Science, 350, 94–98. https://doi.org/10.1126/science.aab1785
Loh, P. R., Genovese, G., & McCarroll, S. A. (2020). Monogenic and polygenic inheritance become instruments for clonal selection. Nature, 584, 136–141. https://doi.org/10.1038/s41586-020-2430-6
Ludwig, D. L., MacInnes, M. A., Takiguchi, Y., Purtymun, P. E., Henrie, M., Flannery, M., Meneses, J., Pedersen, R. A., & Chen, D. J. (1998). A murine AP-endonuclease gene-targeted deficiency with post-implantation embryonic progression and ionizing radiation sensitivity. Mutation Research/DNA Repair, 409, 17–29. https://doi.org/10.1016/S0921-8777(98)00039-1
Lujan, S. A., Williams, J. S., Pursell, Z. F., Abdulovic-Cui, A. A., Clark, A. B., Nick McElhinny, S. A., & Kunkel, T. A. (2012). Mismatch repair balances leading and lagging strand DNA replication fidelity. PLoS Genetics. https://doi.org/10.1371/journal.pgen.1003016
Ma, X., Liu, Y., Liu, Y., Alexandrov, L. B., Edmonson, M. N., Gawad, C., et al. (2018). Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature, 555, 371–376. https://doi.org/10.1038/nature25795
Madabhushi, R., Gao, F., Pfenning, A. R., Pan, L., Yamakawa, S., Seo, J., et al. (2015). Activity-induced DNA breaks govern the expression of neuronal early-response genes. Cell, 161, 1592–1605. https://doi.org/10.1016/j.cell.2015.05.032
Maede, Y., Shimizu, H., Fukushima, T., Kogame, T., Nakamura, T., Miki, T., Takeda, S., Pommier, Y., & Murai, J. (2013). Differential and common DNA repair pathways for topoisomerase I- and II-targeted drugs in a genetic DT40 repair cell screen panel. Molecular Cancer Therapeutics, 13, 214–220. https://doi.org/10.1158/1535-7163.MCT-13-0551
Martincorena, I., Roshan, A., Gerstung, M., Ellis, P., Van Loo, P., McLaren, S., et al. (2015). High burden and pervasive positive selection of somatic mutations in normal human skin. Science, 348, 880–886. https://doi.org/10.1126/science.aaa6806
Matejcic, M., Gunter, M. J., & Ferrari, P. (2017). Alcohol metabolism and oesophageal cancer: a systematic review of the evidence. Carcinogenesis, 38, 859–872. https://doi.org/10.1093/carcin/bgx067
McConnell, M. J., Lindberg, M. R., Brennand, K. J., Piper, J. C., Voet, T., Cowing-Zitron, C., et al. (2013). Mosaic copy number variation in human neurons. Science, 342, 632–637. https://doi.org/10.1126/science.1243472
McCulloch, S. D., & Kunkel, T. A. (2008). The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Research, 18, 148–161. https://doi.org/10.1038/cr.2008.4
McElhinny, S. A. N., Kumar, D., Clark, A. B., Watt, D. L., Watts, B. E., Lundström, E. B., Johansson, E., Chabes, A., & Kunkel, T. A. (2010). Genome instability due to ribonucleotide incorporation into DNA. Nature Chemical Biology, 6, 774–781. https://doi.org/10.1038/nchembio.424
McRae, J. F., Clayton, S., Fitzgerald, T. W., Kaplanis, J., Prigmore, E., Rajan, D., et al. (2017). Prevalence and architecture of de novo mutations in developmental disorders. Nature, 542, 433–438. https://doi.org/10.1038/nature21062
Mojas, N., Lopes, M., & Jiricny, J. (2007). Mismatch repair-dependent processing of methylation damage gives rise to persistent single-stranded gaps in newly replicated DNA. Genes & Development, 21, 3342–3355. https://doi.org/10.1101/gad.455407
Moore, L., Cagan, A., Coorens, T. H. H., Neville, M. D. C., Sanghvi, R., Sanders, M. A., et al. (2021). The mutational landscape of human somatic and germline cells. Nature, 597, 381–386. https://doi.org/10.1038/s41586-021-03822-7
Moreira, M. C., Barbot, C., Tachi, N., Kozuka, N., Uchida, E., Gibson, T., et al. (2001). The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nature Genetics, 29, 189–193. https://doi.org/10.1038/ng1001-189
Morimoto, S., Tsuda, M., Bunch, H., Sasanuma, H., Austin, C., & Takeda, S. (2019). Type II DNA topoisomerases cause spontaneous double-strand breaks in genomic DNA. Genes. https://doi.org/10.3390/genes10110868
Morrison, A., & Sugino, A. (1994). The 3′ → 5′ exonucleases of both DNA polymerases δ and ε participate in correcting errors of DNA replication in Saccharomyces cerevisiae. MGG Molecular and General Genetics, 242, 289–296. https://doi.org/10.1007/BF00280418
Muramatsu, M., Kinoshita, K., Fagarasan, S., Yamada, S., Shinkai, Y., & Honjo, T. (2000). Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell, 102, 553–563. https://doi.org/10.1016/S0092-8674(00)00078-7
Nakahara, M., Sonoda, E., Nojima, K., Sale, J. E., Takenaka, K., Kikuchi, K., et al. (2009). Genetic evidence for single-strand lesions initiating Nbs1-dependent homologous recombination in diversification of Ig V in chicken B lymphocytes. PLoS Genetics. https://doi.org/10.1371/journal.pgen.1000356
Nick McElhinny, S. A., Watts, B. E., Kumar, D., Watt, D. L., Lundström, E. B., Burgers, P. M. J., Johansson, E., Chabes, A., & Kunkel, T. A. (2010). Abundant ribonucleotide incorporation into DNA by yeast replicative polymerases. Proceedings of the National Academy of Sciences of the United States of America, 107, 4949–4954. https://doi.org/10.1073/pnas.0914857107
Nik-Zainal, S., Alexandrov, L. B., Wedge, D. C., Van Loo, P., Greenman, C. D., Raine, K., et al. (2012). Mutational processes molding the genomes of 21 breast cancers. Cell, 149, 979–993. https://doi.org/10.1016/j.cell.2012.04.024
OECD. (2010). Overview on genetic toxicology TGs, OECD Series on Testing and Assessment, No. 238, OECDPublishing, Paris. https://doi.org/10.1787/9789264274761-en.
Ogi, T., Limsirichaikul, S., Overmeer, R. M., Volker, M., Takenaka, K., Cloney, R., et al. (2010). Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Molecular Cell, 37, 714–727. https://doi.org/10.1016/j.molcel.2010.02.009
Park, S., Mali, N. M., Kim, R., Choi, J. W., Lee, J., Lim, J., et al. (2021). Clonal dynamics in early human embryogenesis inferred from somatic mutation. Nature, 597, 393–397. https://doi.org/10.1038/s41586-021-03786-8
Paull, T. T. (2018). 20 Years of Mre11 biology: no end in sight. Molecular Cell, 71, 419–427. https://doi.org/10.1016/j.molcel.2018.06.033
Paulovich, A. G., & Hartwell, L. H. (1995). A checkpoint regulates the rate of progression through S phase in S. cerevisiae in response to DNA damage. Cell, 82, 841–847. https://doi.org/10.1016/0092-8674(95)90481-6
Peltomäki, P., Olkinuora, A., & Nieminen, T. T. (2020). Updates in the field of hereditary nonpolyposis colorectal cancer. Expert Review of Gastroenterology & Hepatology, 14, 707–720. https://doi.org/10.1080/17474124.2020.1782187
Pleguezuelos-Manzano, C., Puschhof, J., Rosendahl Huber, A., van Hoeck, A., Wood, H. M., Nomburg, J., et al. (2020). Mutational signature in colorectal cancer caused by genotoxic pks + E. coli. Nature, 580, 269–273. https://doi.org/10.1038/s41586-020-2080-8
Plo, I., Liao, Z. Y., Barceló, J. M., Kohlhagen, G., Caldecott, K. W., Weinfeld, M., & Pommier, Y. (2003). Association of XRCC1 and tyrosyl DNA phosphodiesterase (Tdp1) for the repair of topoisomerase I-mediated DNA lesions. DNA Repair, 2, 1087–1100. https://doi.org/10.1016/S1568-7864(03)00116-2
Polyzos, A. A., & McMurray, C. T. (2017). Close encounters: moving along bumps, breaks, and bubbles on expanded trinucleotide tracts. DNA Repair, 56, 144–155. https://doi.org/10.1016/j.dnarep.2017.06.017
Pommier, Y., Sun, Y., Huang, S. Y. N., & Nitiss, J. L. (2016). Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nature Reviews Molecular Cell Biology, 17, 703–721. https://doi.org/10.1038/nrm.2016.111
Quinet, A., Tirman, S., Cybulla, E., Meroni, A., & Vindigni, A. (2021). To skip or not to skip: choosing repriming to tolerate DNA damage. Molecular Cell, 81, 649–658. https://doi.org/10.1016/j.molcel.2021.01.012
Rada, C., Di Noia, J. M., & Neuberger, M. S. (2004). Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T-focused phase of somatic mutation. Molecular Cell, 16, 163–171. https://doi.org/10.1016/j.molcel.2004.10.011
Rahbari, R., Wuster, A., Lindsay, S. J., Hardwick, R. J., Alexandrov, L. B., Al Turki, S., et al. (2016). Timing, rates and spectra of human germline mutation. Nature Genetics, 48, 126–133. https://doi.org/10.1038/ng.3469
Ranjha, L., Howard, S. M., & Cejka, P. (2018). Main steps in DNA double-strand break repair: an introduction to homologous recombination and related processes. Chromosoma, 127, 187–214. https://doi.org/10.1007/s00412-017-0658-1
Rouhani, F. J., Nik-Zainal, S., Wuster, A., Li, Y., Conte, N., Koike-Yusa, H., Kumasaka, N., Vallier, L., Yusa, K., & Bradley, A. (2016). Mutational history of a human cell lineage from somatic to induced pluripotent stem cells. PLoS Genetics. https://doi.org/10.1371/journal.pgen.1005932
Saha, L. K., Wakasugi, M., Akter, S., Prasad, R., Wilson, S. H., Shimizu, N., et al. (2020). Topoisomerase I-driven repair of UV-induced damage in NER-deficient cells. Proceedings of the National Academy of Sciences of the United States of America, 117, 14412–14420. https://doi.org/10.1073/pnas.1920165117
Saiki, R., Momozawa, Y., Nannya, Y., Nakagawa, M. M., Ochi, Y., Yoshizato, T., et al. (2021). Combined landscape of single-nucleotide variants and copy number alterations in clonal hematopoiesis. Nature Medicine, 27, 1239–1249. https://doi.org/10.1038/s41591-021-01411-9
Saito, S., Maeda, R., & Adachi, N. (2017). Dual loss of human POLQ and LIG4 abolishes random integration. Nature Communications. https://doi.org/10.1038/ncomms16112
Sale, J. E. (2013). Translesion DNA synthesis and mutagenesis in eukaryotes. Cold Spring Harbor Perspectives in Biology. https://doi.org/10.1101/cshperspect.a012708
Sands, A. T., Suraokar, M. B., Sanchez, A., Marth, J. E., Donehower, L. A., & Bradley, A. (1995). p53 Deficiency does not affect the accumulation of point mutations in a transgene target. Proceedings of the National Academy of Sciences of the United States of America, 92, 8517–8521. https://doi.org/10.1073/pnas.92.18.8517
Sasanuma, H., Tsuda, M., Morimoto, S., Saha, L. K., Rahman, M. M., Kiyooka, Y., et al. (2018). BRCA1 ensures genome integrity by eliminating estrogen-induced pathological topoisomerase II-DNA complexes. Proceedings of the National Academy of Sciences of the United States of America, 115, E10642–E10651. https://doi.org/10.1073/pnas.1803177115
Shen, J., Gilmore, E. C., Marshall, C. A., Haddadin, M., Reynolds, J. J., Eyaid, W., et al. (2010). Mutations in PNKP cause microcephaly, seizures and defects in DNA repair. Nature Genetics, 42, 245–249. https://doi.org/10.1038/ng.526
Sobol, R. W., Horton, J. K., Kühn, R., Gu, H., Singhal, R. K., Prasad, R., Rajewsky, K., & Wilson, S. H. (1996). Requirement of mammalian DNA polymerase-β in base-excision repair. Nature, 379, 183–186. https://doi.org/10.1038/379183a0
Sonoda, E., Sasaki, M. S., Buerstedde, J. M., Bezzubova, O., Shinohara, A., Ogawa, H., Takata, M., Yamaguchi-Iwai, Y., & Takeda, S. (1998). Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO Journal, 17, 598–608. https://doi.org/10.1093/emboj/17.2.598
St Charles, J. A., Liberti, S. E., Williams, J. S., Lujan, S. A., & Kunkel, T. A. (2015). Quantifying the contributions of base selectivity, proofreading and mismatch repair to nuclear DNA replication in Saccharomyces cerevisiae. DNA Repair, 31, 41–51. https://doi.org/10.1016/j.dnarep.2015.04.006
Sun, Y., Saha, L. K., Saha, S., Jo, U., & Pommier, Y. (2020a). Debulking of topoisomerase DNA-protein crosslinks (TOP-DPC) by the proteasome, non-proteasomal and non-proteolytic pathways. DNA Repair. https://doi.org/10.1016/j.dnarep.2020.102926
Sun, Y., Saha, S., Wang, W., Saha, L. K., Huang, S. Y. N., & Pommier, Y. (2020b). Excision repair of topoisomerase DNA-protein crosslinks (TOP-DPC). DNA Repair. https://doi.org/10.1016/j.dnarep.2020.102837
Supek, F., Lehner, B., Hajkova, P., & Warnecke, T. (2014). Hydroxymethylated cytosines are associated with elevated C to G transversion rates. PLoS Genetics. https://doi.org/10.1371/journal.pgen.1004585
Svilar, D., Goellner, E. M., Almeida, K. H., & Sobol, R. W. (2011). Base excision repair and lesion-dependent subpathways for repair of oxidative DNA damage. Antioxidants Redox Signaling, 14, 2491–2507. https://doi.org/10.1089/ars.2010.3466
Takashima, H., Boerkoel, C. F., John, J., Saifi, G. M., Salih, M. A. M., Armstrong, D., et al. (2002). Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nature Genetics, 32, 267–272. https://doi.org/10.1038/ng987
Taylor, J. L., Debost, J. C. P. G., Morton, S. U., Wigdor, E. M., Heyne, H. O., Lal, D., et al. (2019). Paternal-age-related de novo mutations and risk for five disorders. Nature Communications. https://doi.org/10.1038/s41467-019-11039-6
Terao, C., Suzuki, A., Momozawa, Y., Akiyama, M., Ishigaki, K., Yamamoto, K., et al. (2020). Chromosomal alterations among age-related haematopoietic clones in Japan. Nature, 584, 130–135. https://doi.org/10.1038/s41586-020-2426-2
Thomas, A., & Pommier, Y. (2019). Targeting topoisomerase I in the era of precision medicine. Clinical Cancer Research, 25, 6581–6589. https://doi.org/10.1158/1078-0432.CCR-19-1089
Tomasetti, C., & Vogelstein, B. (2015). Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science, 347, 78–81. https://doi.org/10.1126/science.1260825
Trenner, A., & Sartori, A. A. (2019). Harnessing DNA double-strand break repair for cancer treatment. Frontiers in Oncology. https://doi.org/10.3389/fonc.2019.01388
Tubbs, A., & Nussenzweig, A. (2017). Endogenous DNA damage as a source of genomic instability in cancer. Cell, 168, 644–656. https://doi.org/10.1016/j.cell.2017.01.002
Uchimura, A., Higuchi, M., Minakuchi, Y., Ohno, M., Toyoda, A., Fujiyama, A., & Yagi, T. (2015). Germline mutation rates and the long-term phenotypic effects of mutation accumulation in wild-type laboratory mice and mutator mice. Genome Research, 25(8), 1125–1134. https://doi.org/10.1101/gr.186148.114
Vaisman, A., & Woodgate, R. (2020). Mysterious and fascinating: DNA polymerase ɩ remains enigmatic 20 years after its discovery. DNA Repair. https://doi.org/10.1016/j.dnarep.2020.102914
van Wietmarschen, N., Sridharan, S., Nathan, W. J., Tubbs, A., Chan, E. M., Callen, E., et al. (2020). Repeat expansions confer WRN dependence in microsatellite-unstable cancers. Nature, 586, 292–298. https://doi.org/10.1038/s41586-020-2769-8
Veltman, J. A., & Brunner, H. G. (2012). De novo mutations in human genetic disease. Nature Reviews Genetics, 13, 565–575. https://doi.org/10.1038/nrg3241
Viguera, E., Canceill, D., & Ehrlich, S. D. (2001). Replication slippage involves DNA polymerase pausing and dissociation. EMBO Journal, 20, 2587–2595. https://doi.org/10.1093/emboj/20.10.2587
Waterman, D. P., Haber, J. E., & Smolka, M. B. (2020). Checkpoint responses to DNA double-strand breaks. Annual Review of Biochemistry, 89, 103–133. https://doi.org/10.1146/annurev-biochem-011520-104722
Wei, P. C., Chang, A. N., Kao, J., Du, Z., Meyers, R. M., Alt, F. W., & Schwer, B. (2016). Long neural genes harbor recurrent DNA break clusters in neural stem/progenitor cells. Cell, 164, 644–655. https://doi.org/10.1016/j.cell.2015.12.039
Weinert, T. A., & Hartwell, L. H. (1988). The RAD9 gene controls the cell cycle response to DNA damage in saccharomyces cerevisiae. Science, 241, 317–322. https://doi.org/10.1126/science.3291120
Werner, B., Case, J., Williams, M. J., Chkhaidze, K., Temko, D., Fernández-Mateos, J., et al. (2020). Measuring single cell divisions in human tissues from multi-region sequencing data. Nature Communications. https://doi.org/10.1038/s41467-020-14844-6
Wiener-Megnazi, Z., Auslender, R., & Dirnfeld, M. (2012). Advanced paternal age and reproductive outcome. Asian Journal of Andrology, 14, 69–76. https://doi.org/10.1038/aja.2011.69
Williamson, L. M., & Lees-Miller, S. P. (2011). Estrogen receptor α-mediated transcription induces cell cycle-dependent DNA double-strand breaks. Carcinogenesis, 32, 279–285. https://doi.org/10.1093/carcin/bgq255
Wong, R. P., Petriukov, K., & Ulrich, H. D. (2021). Daughter-strand gaps in DNA replication – substrates of lesion processing and initiators of distress signalling. DNA Repair. https://doi.org/10.1016/j.dnarep.2021.103163
Woodbine, L., Brunton, H., Goodarzi, A. A., Shibata, A., & Jeggo, P. A. (2011). Endogenously induced DNA double strand breaks arise in heterochromatic DNA regions and require ataxia telangiectasia mutated and Artemis for their repair. Nucleic Acids Research, 39, 6986–6997. https://doi.org/10.1093/nar/gkr331
Wu, W., Hill, S. E., Nathan, W. J., Paiano, J., Callen, E., Wang, D., et al. (2021). Neuronal enhancers are hotspots for DNA single-strand break repair. Nature, 593, 440–444. https://doi.org/10.1038/s41586-021-03468-5
Xing, D., Tan, L., Chang, C. H., Li, H., & Xie, X. S. (2021). Accurate SNV detection in single cells by transposon-based whole-genome amplification of complementary strands. Proceedings of the National Academy of Sciences of the United States of America, 118, e2013106118. https://doi.org/10.1073/pnas.2013106118
Yokoyama, A., Kakiuchi, N., Yoshizato, T., Nannya, Y., Suzuki, H., Takeuchi, Y., et al. (2019). Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature, 565, 312–317. https://doi.org/10.1038/s41586-018-0811-x
Yoshida, K., Gowers, K. H. C., Lee-Six, H., Chandrasekharan, D. P., Coorens, T., Maughan, E. F., et al. (2020). Tobacco smoking and somatic mutations in human bronchial epithelium. Nature, 578, 266–272. https://doi.org/10.1038/s41586-020-1961-1
Yoshimura, M., Kohzaki, M., Nakamura, J., Asagoshi, K., Sonoda, E., Hou, E., et al. (2006). Vertebrate POLQ and POLβ cooperate in base excision repair of oxidative DNA damage. Molecular Cell, 24, 115–125. https://doi.org/10.1016/j.molcel.2006.07.032
Zagnoli-Vieira, G., & Caldecott, K. W. (2020). Untangling trapped topoisomerases with tyrosyl-DNA phosphodiesterases. DNA Repair. https://doi.org/10.1016/j.dnarep.2020.102900
Zeng, Z., Sharma, A., Ju, L., Murai, J., Umans, L., Vermeire, L., et al. (2012). TDP2 promotes repair of topoisomerase I-mediated DNA damage in the absence of TDP1. Nucleic Acids Research, 40, 8371–8380. https://doi.org/10.1093/nar/gks622
Zhang, L., Dong, X., Lee, M., Maslov, A. Y., Wang, T., & Vijg, J. (2019). Single-cell whole-genome sequencing reveals the functional landscape of somatic mutations in B lymphocytes across the human lifespan. Proceedings of the National Academy of Sciences of the United States of America, 116, 9014–9019. https://doi.org/10.1073/pnas.1902510116
Zimmermann, M., Murina, O., Reijns, M. A. M., Agathanggelou, A., Challis, R., Tarnauskaite, Ž, et al. (2018). CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature, 559, 285–289. https://doi.org/10.1038/s41586-018-0291-z
Acknowledgements
This work is supported by JSPS KAKENHI (16H12595 and 16H06306) (to ST), and by the JSPS Core-to-Core Program, A. Advanced Research Networks (to ST), and by the Major Program of the National Natural Science Foundation of China, grant number 41991314.
Author information
Affiliations
Shenzhen University School of Medicine, Guangdong, Shenzhen, 518071, China
Shunichi Takeda
School of Medicine, Kyoto University, Yoshida Konoe, Kyoto, 606-8501, Japan
Shunichi Takeda
Shanghai Jiao Tong University School of Medicine, Shanghai, 200025, China
Yang Luan
Corresponding authors
Correspondence to Shunichi Takeda or Yang Luan.
Rights and permissions
About this article
Cite this article
Takeda, S., Luan, Y. Nature of spontaneously arising single base substitutions in normal cells. GENOME INSTAB. DIS. 2, 339–357 (2021). https://doi.org/10.1007/s42764-021-00056-9
Received19 October 2021
Revised09 November 2021
Accepted17 November 2021
Published26 November 2021
Issue DateDecember 2021
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
Spontaneous mutagenesis
Error-reduced NGS technology
Replication errors
Inaccurate repair of DNA damage
de novo mutation
用户登录
还没有账号?
立即注册