






MRE11-RAD50-NBS1-CtIP: one key nuclease ensemble functions in the maintenance of genome stability
Review Article
Tao Zhang, Zenan Zhou, Han Yang & Weibin Wang Genome Instability & Disease (2022)
Abstract
To safeguard genome integrity, cells channel DNA double-strand breaks (DSBs) to repair pathway of either DNA end-joining or homology-directed repair. The MRE11-RAD50-NBS1 (MRN) complex functions in both pathways, teaming up with the right co-factors to ensure the correct repair of DSBs. The MRN complex is well known as a nuclease capable of resecting DNA in both endonucleolytic and exonucleolytic manners. At occupied DSB ends, DNA end resection is initiated by the “endonucleolytic cleavage followed by exonucleolytic digestion” action of MRN, with endonucleolytic cleavage specifically requiring phosphorylated CtIP being present. Previous studies by us and the other groups in the field help to uncover the mechanistic details of the MRN-CtIP nuclease ensemble functioning in DNA end resection and homologous recombination (HR), although many unclear parts still exist. Besides DSB repair, replication fork processing, R-loop, and transcription–replication conflict (TRC) resolution also have been suggested by accumulating studies to be relevant to MRN and CtIP. In this review, we will summarize current knowledge about the MRN-CtIP nuclease ensemble, its functions in DNA processing in various contexts that could generate genome instability, how this complex is regulated and its relevance to diseases like cancers.
Introduction
Genome integrity is constantly challenged by various types of insults, including those from the cellular environment such as chemicals and ionizing radiation, and endogenous metabolites such as reactive oxygen species (ROS) and toxic aldehydes (Cannan & Pederson, 2016). These genotoxins can result in different DNA damage outcomes, and cells have evolved a specific repair pathway for each type of DNA damage. Among these DNA damage types, DNA double-strand breaks (DSBs) are the most deleterious DNA lesions. If left unrepaired or repaired inappropriately, DSBs could lead to the loss of DNA fragments or chromosome translocation, which may result in cell death or transformation toward tumor. Therefore, understanding the mechanistic details of DSB repair is highly relevant for genome stability maintenance and cancer prevention.
When DSBs occur, cells rely on two main pathways to deal with this type of DNA lesion: non-homologous end-joining (NHEJ) and homologous recombination (HR) (Fig. 1) (Ranjha et al., 2018; Symington & Gautier, 2011). The NHEJ repair pathway can be further divided into two pathways including canonical non-homologous end-joining (c-NHEJ) and alternative non-homologous end-joining (alt-NHEJ; also termed as microhomology-mediated end-joining, MMEJ). Unlike HR, NHEJ does not rely on genetically identical sister chromatids to template DNA break repair. During NHEJ, two broken DSB ends are directly ligated/annealed together, with no or limited DNA end processing. Thus, the NHEJ repair pathway is error-prone and usually results in genetic information missing at the DSB site. In comparison, HR repairs DSBs with high fidelity as the missing DNA sequence is copied from its homology.
Fig. 1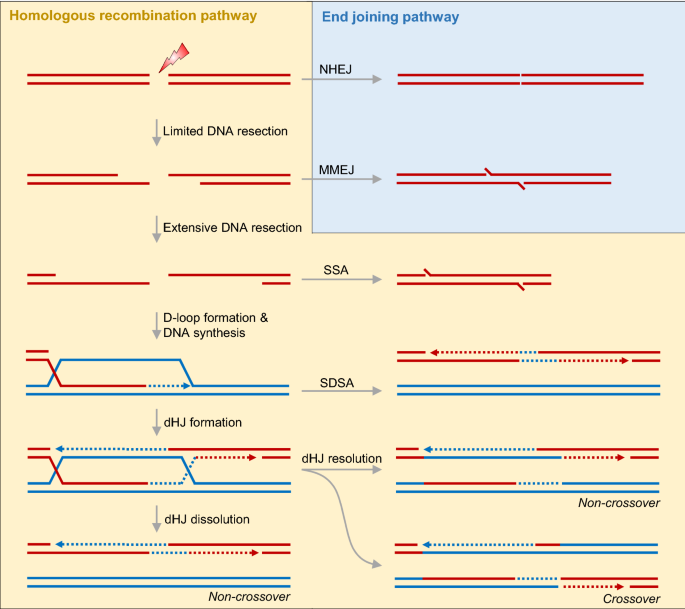
The pathways to repair DNA double-strand breaks (DSBs). There are two main pathways to deal with DSBs: one is non-homologous end-joining (NHEJ) and the other one is homologous recombination (HR). The NHEJ repair pathway can be further divided into two pathways including canonical non-homologous end-joining (c-NHEJ) and alternative non-homologous end-joining (alt-NHEJ; also termed as microhomology-mediated end-joining, MMEJ). Single-strand annealing (SSA) involves DNA end resection to reveal DNA strands which are subsequently annealed. At the later stage of HR, extended D-loop can be channeled to synthesis-dependent strand annealing (SDSA) pathway, in which D-loop can be unwound and annealed to the second DSB end, followed by further DNA synthesis. Alternatively, D-loop captures the second DSB end to form dHJ, which is either dissolved into non-crossover product, or resolved into non-crossover or crossover product
Full size imageThe HR pathway is initiated by a process termed DNA end resection, in which the 5’ DNA strand at DSB end is specifically digested, leading to the formation of 3’ single-strand DNA. Previous studies by us and the other groups in the field propose a “three-step” DNA end resection model (Fig. 2) (Cannavo & Cejka, 2014; Garcia et al., 2011; Wang et al., 2017). Following the Ku70-Ku80 complex recruitment to “clean” DSB end or at protein-conjugated “dirty” DSB end, DNA end resection is initiated with the endonucleolytic cleavage carried out by the MRE11-RAD50-NBS1 (MRN) complex and its co-factor CtIP. Subsequently, the short-range DNA end resection is performed by MRN 3’-5’ exonuclease activity. Then, at the entry site produced by MRN-CtIP, the long-range DNA end resection is carried out in the 5’-3’ direction by either EXO1 or DNA2 (Wang et al., 2018). Besides these findings, accumulating studies also expand the roles of MRN-CtIP in other more contexts such as replication fork processing, R-loop and transcription–replication conflict (TRC) resolution. The MRN-CtIP nuclease ensemble integrating multifaceted functions into itself contributes to the maintenance of genome stability. In this following review, we mainly focus on MRN-CtIP, summarize current knowledge about the molecular details of this complex, its involvement in different DNA damage contexts, the regulatory mechanisms of this complex, and the relevance to genome instability and disease like tumors.
Fig. 2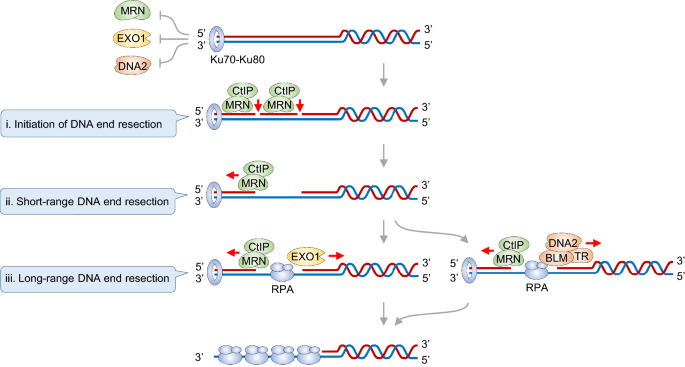
The three-step bidirectional DNA end resection model. Following Ku70-Ku80 recruitment to the DSB end, DNA end resection is initiated with the endonucleolytic cleavage carried out by the MRN complex and its co-factor CtIP. Subsequently, the short-range DNA end resection is performed by MRN 3’-5’ exonuclease activity. Then, at the entry site produced by MRN-CtIP, the long-range DNA end resection is carried out in the 5’-3’ direction by either EXO1 or DNA2
Full size imageBiochemical properties of MRN-CtIP
In mammalian cells, the MRN complex exists as a hetero-hexameric assembly, with the MRE11 subunit forming a homodimer and interacting with two molecules of RAD50 and NBS1 via different domains (Fig. 3) (Casari et al., 2019; Syed & Tainer, 2018). The counterpart of MRN in budding yeast (Saccharomyces cerevisiae) is the Mre11-Rad50-Xrs2 (MRX) complex. In 1998, the Sung group first established the biochemistry methods to purify MRN to near homogeneity and identify its nuclease activity (Trujillo et al., 1998). In this study, when MRE11-RAD50 was purified from human Burkitt’s lymphoma cells, a 95 kDa protein species (P95, equal to NBS1) was found coeluting precisely with MRE11-RAD50 during the gel filtration step. This MRE11-RAD50-P95 (aka MRN) complex was proved to have divalent manganese ion (Mn2+) dependent 3’-5’ exonuclease activity on double-strand DNA (dsDNA) and endonuclease activity on circular single-strand DNA (ssDNA). In the same year, the Gellert group also successfully purified the MRE11-RAD50 subcomplex using an insect cell-baculovirus expression system, and proved this subcomplex capable of exonucleolytically digesting dsDNA and endonucleolytically cleaving the hairpin DNA loop (Paull & Gellert, 1998). In the following years, the MRX complex was also successfully purified and proved to have similar nuclease activities to human MRN (Chen et al., 2001; Trujillo & Sung, 2001; Trujillo et al., 2003). The endonuclease activity of MRN/MRX toward ssDNA with secondary structures has the properties that (1) the NBS1/Xrs2 subunit is dispensable, (2) ATP hydrolysis by RAD50/Rad50 is not absolutely required, and (3) the co-factors such as CtIP (Sae2 in Saccharomyces cerevisiae) are unnecessary. However, these endonuclease properties are different from those observed with dsDNA substrates, which we will discuss in the following.
Fig. 3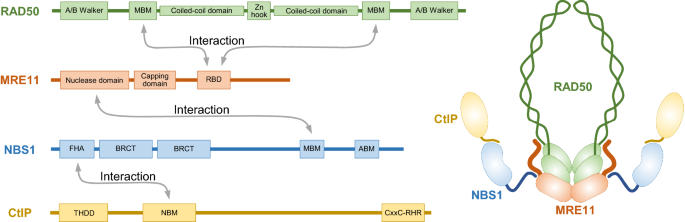
The assembly of the MRN-CtIP complex. For each subunit in the MRN-CtIP complex, protein–protein binding domain was found to mediate its interaction with another subunit. The MRN complex exists as a hetero-hexameric assembly, with the MRE11 subunit forming a homodimer and interacting with two molecules of RAD50 and NBS1 via different domains. CtIP binds to the MRN complex through NBS1. MBM, MRE11-binding motif; RBD, RAD50-binding domain; ABM, ATM-binding motif; NBM, NBS1-binding motif
Full size imageIn DSB repair by HR, the 5’-terminated DNA strand must first be resected to generate the 3’-ssDNA tail, and this process is initiated by the MRN/MRX complex. However, the MRN/MRX complex exhibits 3’-5’ exonuclease activity to dsDNA, which is opposite from that required for DNA end resection. Subsequently, to explain this polarity paradox, several genetic studies proposed a bidirectional model (Garcia et al., 2011; Mimitou & Symington, 2008), whereby MRN/MRX first cleaves DNA strand endonucleolytically and then carries out resection in the 3’-5’ direction, the resulting DNA nick or gap serves as an entry site for EXO1 or DNA2 to perform long-range DNA end resection in the 5’-3’ direction. In 2014, the Cejka group first provided the biochemical evidence to support the above model (Cannavo & Cejka, 2014). Using purified budding yeast proteins, the authors show that when DSB ends are blocked by streptavidin, MRX is able to specifically cleave the 5’-terminated strand of dsDNA at the internal site, and this endonuclease activity is triggered by the highly phosphorylated Sae2. Subsequently, the mammalian counterpart MRN and its co-factor CtIP also have been proved capable of cleaving streptavidin-blocked dsDNA endonucleolytically (Anand et al., 2016; Deshpande et al., 2016). In 2017, the study by our laboratory expanded this observation to contexts with more physiological relevance (Wang et al., 2017). We were able to reconstitute the endo- and exonuclease activities of MRX-Sae2 on dsDNA engaged by physiological protein obstacles, such as the Ku70-Ku80 complex, ssDNA binding protein RPA, or the nucleosome. Based on these findings, we proposed a “nicking and chew-back” model for the initiation and short-range DNA end resection (Fig. 2). Meanwhile, another work by us further provided evidence that the DNA nick and gap (generated by MRX-Sae2 endo- and exonuclease activities, respectively) can serve as entry sites for Exo1 or Dna2 (complexed with Sgs1 and RPA) to carry out long-range DNA end resection (Wang et al., 2018). These studies provide more mechanistic details to support the bidirectional model mentioned above.
The nuclease catalytic center of the MRN complex lies in MRE11. In addition to functioning as a nuclease, MRN also possesses ATPase activity. Purified RAD50 protein is capable of binding and hydrolyzing ATP, which fuels the endonucleolytic resection of DSB end by MRE11. Recent studies suggest that the MRN complex may also has ATP-dependent translocase and DNA unwinding activities (Ghodke & Muniyappa, 2013; Myler et al., 2017), but with much lower efficiency. Thus, a safe description could be MRN capable of “diffusing” along dsDNA in the 3’-5’ direction, and “melting” dsDNA at an internal site or short dsDNA fragment. These concepts may help to explain and understand how DNA is processed by MRN. Unlike MRE11 and RAD50, the NBS1 subunit is more likely to play a structural role in the MRN complex. For example, budding yeast Xrs2 has DNA-binding activity. Using the biochemical reconstitution system with purified proteins, the Sung group showed that Xrs2 helps target MRE11 and RAD50 to DNA ends (Trujillo et al., 2003). In addition, the ATM-binding domain in NBS1 functions to mediate the interaction between MRN and ATM. The Paull group provided the biochemical evidence that NBS1 is critical for MRN-mediated stimulation of ATM kinase activity (Lee & Paull, 2004). During endonucleolytic resection by MRN, NBS1 is indispensable and it promotes this endonuclease activity by sensing CtIP phosphorylation (Anand et al., 2019). But for MRX endonuclease activity, Xrs2 is not absolutely required although its presence can dramatically stimulate the endonucleolytic cleavage (Wang et al., 2017). CtIP/Sae2 is the key partner for MRN/MRX during the DNA end resection step in HR. Biochemical experiments by us and the other groups proved that highly phosphorylated form of CtIP/Sae2 stimulates the endonuclease activity of MRN/MRX on dsDNA substrates, which is in accord with the genetic observations that CtIP/Sae2 is phosphorylated by cyclin-dependent kinase (CDK) in S/G2 phase of the cell cycle when DSB is prone to be repaired by HR.
Structural features of MRN-CtIP
Both MRN/MRX and CtIP/Sae2 can be purified to near homogeneous form, which favors the studies to investigate the biochemical and structural properties of these proteins. In the past 20 years, many key findings were published in decent journals to decipher the architecture of the MRN/MRX-CtIP/Sae2 nuclease ensemble using techniques such as atomic force microscopy (AFM), electron microscopy (EM), X-ray scattering (SAXS) and crystallography. In 2001 and 2005, three articles were published to reveal multiple conformations of MRN/MRX by AFM (Chen et al., 2001; de Jager et al., 2001; Moreno-Herrero et al., 2005). Budding yeast MRX complex is prone to bind DNA end and mediates the alignment between two DSB ends. For human MRN, similar images were captured by AFM and more conformation details were provided. MRN architecture consists of a globular head for DNA binding and two 50-nm-long coiled coil antennae. Upon DNA binding, the coiled coils adopt a parallel orientation which is favorable for the interaction between two molecules. Almost at the same time, EM graphs of the MR subcomplex from multiple species including Homo sapiens, Saccharomyces cerevisiae and Pyrococcus furiosus were revealed by two articles (Anderson et al., 2001; Hopfner et al., 2002). The conformational properties revealed by electron micrographs are similar to those by AFM images.
Due to the resolution limits of AFM and EM, more structural details of the MRN complex need techniques such as SAXS and crystallography to be revealed. From 2000 to recent years, several papers were published mainly by the Tainer and Cho groups to resolve the structure of the MRN complex (Fig. 3) (Hopfner et al., 2000, 2002; Lim et al., 2011; Park et al., 2011; Williams et al., 2008, 2009). RAD50 is a member of the ABC ATPase superfamily. Its N- and C-termini gather to form the ABC ATPase domain (the head) harboring the Walker A/B motifs. Between the two terminal ends, there are anti-parallel coiled coil domains with the length of ~ 500 Å. At the tip of the coiled coil part, there is a CxxC motif creating a zinc-hook. MRE11 is the catalytic center of MRN nuclease. One MRE11 molecule dimerizes with another one through their N-terminal core domains. Both the endo- and exonuclease activities of MRE11 lie in the core phosphodiesterase domain. Biochemical data show that Mn2+ supports the nuclease activity of MRE11. Consistently, five histidines, two aspartic acids and one asparagine in nuclease active site were revealed to be capable of coordinating Mn2+. Besides the core domain in MRE11, there are capping and C-terminal domains which are thought to mediate protein–DNA and protein–protein interactions. Different from archaea and bacteria, eukaryotes have NBS1 (Xrs2 in Saccharomyces cerevisiae). At the N-terminal of NBS1, there are one forkhead-associated (FHA) domain and two breast cancer C-terminal (BRCT) domains. The FHA and BRCT domains are responsible for NBS1 interaction with phosphorylated proteins like CtIP. At the C-terminus of NBS1, there are motifs mediating the interactions with MRE11 and ATM.
CtIP (Ctp1 in Schizosaccharomyces pombe, Sae2 in Saccharomyces cerevisiae) collaborates with MRN to initiate the endonucleolytic resection in HR. Its architecture also has been deciphered using techniques such as AFM, EM, SAXS and crystallography. In 2015, the Williams group revealed the X-ray crystal structure of Ctp1 (Andres et al., 2015). At the N-terminus, the interlocking tetrameric helical dimer-of-dimers (THDD) domain mediates the assembly of Ctp1 dimer/tetramer. On the other hand, the C-terminus of Ctp1 harbors RHR motif which is responsible for protein–DNA interaction. Between the N- and C-termini, the central region is intrinsically disordered (termed as IDR). These structural details help to understand how Ctp1 performs DNA-binding and DNA-bridging activities. In 2018, paper published by the Cejka group showed EM and AFM images of phosphorylated Sae2 (Cannavo et al., 2018). In phosphorylated form, Sae2 is prone to assemble into oligomer (equal to tetramer) instead of multimer. Sae2 oligomerization is important for its stimulatory effect on MRX endonuclease activity. In the following year, another study using AFM revealed the architectural details of Ctp1-mediated DNA binding and bridging (Andres et al., 2019). For human CtIP, similar observations were made when carrying out EM and AFM analyses (Wilkinson et al., 2019). CtIP also exists as a tetrameric protein and adopts a dumbbell architecture with DNA-binding domains connected by long coiled coils, thus providing the structural basis for the DNA-bridging function of CtIP at DSBs.
HR, DNA end resection and the MRN-CtIP nuclease ensemble
DSB repair by HR is initiated by a nucleolytic process termed DNA end resection. The MRN/MRX-CtIP/Sae2 complex mainly functions as a nuclease ensemble in this process. Based on the extensive genetic and biochemical studies regarding DNA end resection, a three-step bidirectional model has been proposed: the initiation of DNA end resection, short-range DNA end resection, and the long-range DNA end resection (Cannavo & Cejka, 2014; Garcia et al., 2011; Wang et al., 2017). At clean DSB ends bound by the Ku70-Ku80 heterodimer or dirty DSB ends covalently conjugated with proteins, the initiation of DNA end resection is carried out by MRN/MRX and phosphorylated CtIP/Sae2. Instead of digesting DNA strand from DSB end, MRN/MRX-CtIP/Sae2 makes at least two endonucleolytic nicks following the rules that: (1) the nicks are created on 5’-terminated DNA strand specifically; (2) the first nick is ~ 35–45 nt away from DSB end with the following nicks appearing in increments of ~ 20 nt. After the endonucleolytic cleavage, MRN/MRX uses its 3’-5’ exonuclease activity to carry out short-range (< ~ 100–200 nt) DNA end resection. In this step, CtIP/Sae2 may not be required, but MRN/MRX exonuclease is more active with CtIP/Sae2 being present. Subsequently, the resulting DNA nick or gap generated by MRN/MRX-CtIP/Sae2 can serve as an entry site for EXO1 or DNA2 nuclease to carry out long-range DNA end resection in the 5’-3’ direction. EXO1 alone can digest DNA strand efficiently, whereas DNA2 needs to be complexed with BLM (Sgs1 in Saccharomyces cerevisiae) and RPA.
During DNA end resection, the 5’-terminated DNA strand is specifically digested by MRE11/EXO1/DNA2 nuclease, leaving the 3’ ssDNA tail coated by RPA. With the help of the mediator BRCA2 and its co-factor DSS1, RPA is replaced by the recombinase RAD51 to form the RAD51-ssDNA nucleoprotein filament (Zhao et al., 2015). Subsequently, RAD51 filament catalyzes the key step in HR pathway, which includes homology search, DNA pairing and strand invasion. This ultimately leads to the formation of a displacement loop (termed as D-loop). Multiple factors were reported to regulate RAD51-mediated D-loop formation, such as RAD51 paralogs or the BRCA1-BARD1 complex (Taylor et al., 2015; Zhao et al., 2017). Following D-loop formation, DNA synthesis is catalyzed by DNA polymerase δ (Pol δ) using the homology as a template. After the missing DNA sequence at DSB site is restored, the extended D-loop structure is channeled to one of three mechanistically distinct pathways: synthesis-dependent strand annealing (SDSA), double Holliday junction (dHJ) dissolution and dHJ resolution (Ranjha et al., 2018). In SDSA, the extended invading DNA strand is dissociated from the template DNA by helicases (such as RTEL1, RECQ1 and BLM in humans; Mph1 in budding yeast) and annealed to the second end of the broken DNA. Then, the repair is completed by further DNA synthesis and ligation. In canonical double-strand break repair (DSBR), the second DNA end is captured and annealed to the displaced ssDNA strand of the D-loop, thus forming a dHJ structure. Subsequently, dHJ is either dissolved by the BTR (BLM-Topo IIIα-RMI1/2) complex to generate the non-crossover product, or resolved by multiple nucleases (such as MUS81-EME1, SLX4-SLX1 and GEN1) to result in the crossover/non-crossover product.
Stressed replication fork and MRN-CtIP
Accurate DNA synthesis is the key event in whole-genome doubling in eukaryotic cells and largely depends on replication fork stability. Exogenous and intrinsic obstacles to DNA replication lead to fork slowdown, stalling, collapse, or remodeling into the structure of four-way junctions, also known as fork reversal, which anneals the parental strands to suppress replication progression and prevent ssDNA accumulation under replication stress, but the reversed arms are easily attacked by nucleolytic cleavage (Atkinson & McGlynn, 2009). Therefore, stalled or reversed forks need to be protected appropriately and restart promptly. Otherwise, they can cause poisonous lesions to result in gene mutations or cell death. The MRN-CtIP complex, as the genomic guardian, plays multifaceted roles in dealing with replication fork stress.
The arrested and collapsed replication fork restart needs appropriate MRN-mediated resection (Bryant et al., 2009). The MRX complex (MRN in humans) with the chromatin modifiers Gcn5 and Set1 enhance chromatin accessibility to facilitate DNA2- and EXO1-mediated cleavage of the stalled fork and the subsequent fork restart (Delamarre et al., 2020). MRE11 cooperates with SAMHD1 to promote resection-mediated fork restart, which also prevents interferon induction by limiting the release of ssDNA from stalled replication forks (Coquel et al., 2018). If the replication fork cannot restart under long-term stress, it collapses to form a one-ended DSB, which is commonly fixed by the break-induced replication pathway, and here, the function of the MRN-CtIP complex is similar to that in HR.
On the other hand, MRE11, as the catalytic core subunit of the MRN complex, must be tightly regulated to prevent excessive resection of the replication fork. MRE11 is rapidly recruited to the stalled replication fork under stresses such as hydroxyurea (HU, leading to inhibition of DNA replication via depletion of the deoxyribonucleotide triphosphate (dNTP) pool level) or low-dose CPT treatment. The protection mechanisms of the replication fork are mainly involved in safeguard of ssDNA and fork reversal and inhibition of MRE11 nucleolytic attack (by the CST complex, MRNIP, etc.) (Bennett et al., 2020; Lyu et al., 2021). Under replication stress, RPA rapidly binds to naked ssDNA and is phosphorylated to protect the replication fork. NBS1 is crucial for the ATR-mediated phosphorylation of RPA and fork restart (Manthey et al., 2007).
In BRCA1/2-deficient cells, the replication fork is more vulnerable, because it can still form fork reversal but fails to effectively maintain RAD51 on the naked ssDNA and reversal strands. Thus, reversed fork in BRCA1/2-deficient cells turns out to be an unstable structure, and the cytotoxicity caused by MRE11 is more lethal than that in wild-type cells. Here, there are multiple mechanisms to curtail MRE11-mediated reversed fork digestion to sustain replication fork stability, and it could be used in synthetic lethal strategies in BRCA1/2-deficient cancer therapy. For example, PTIP absence inhibits the recruitment of MRE11 to stalled replication forks and enhances its stability in BRCA1/2-deficient cells (Ray Chaudhuri et al., 2016). PCAF can acetylate histones at H4K8 sites that can be recognized by MRE11 at the stalled fork (Kim et al., 2020). PCAF knockout inhibits MRE11 recruitment and reverses fork degradation phenotype in BRCA1/2-deficient cells.
Taken together, the MRN complex and its co-factor CtIP play dual roles in the maintenance of replication fork stability. The nuclease activity of MRN-CtIP is regulated tightly to ensure appropriate DNA resection required for arrested replication fork restart, and on the other hand, to avoid replication fork from excessive DNA resection.
R-loop, TRC and MRN-CtIP
R-loop is a nucleic acid structure composed of an RNA–DNA hybrid and a displaced ssDNA (Thomas et al., 1976). This structure has been reported to be detectable and have positive physiological functions in multiple specific genomic regions (Costantino & Koshland, 2015; Santos-Pereira & Aguilera, 2015). For example, at active transcriptional sites, R-loop regulates transcription proceeding and termination; at telomeres, R-loop is required for the elongation of impaired telomeres; and at DSB sites, R-loop can facilitate the recruitment of DNA repair proteins and homologous DNA capture in concert with D-loop. However, R-loops also represent the fragile sites in the genome. Persistent R-loops can either develop into DNA breaks directly, or present an obstacle to DNA replication machinery, which may cause DSBs ultimately. Among the R-loop induced DSBs, the majority are generated via TRC.
MRN/MRX-CtIP/Sae2 is tightly involved in DSB repair by HR and replication fork processing, thus this nuclease ensemble is most likely to function in R-loop/TRC resolution as well. In 2018, the Paull group published a paper revealing that CtIP/Sae2 depletion leads to RNA polymerase stalling and the accumulation of RNA–DNA hybrids at highly transcribing sites (Makharashvili et al., 2018). However, the underlying mechanistic details of this phenomenon still need further investigation. In 2019, another study from the Stirling group showed that the MRN complex suppresses R-loops and associated DNA damages at TRC sites (Chang et al., 2019). MRN-mediated R-loop suppression at TRC is independent of its nuclease activity, but relies on Fanconi anemia (FA) factors such as FANCM. This work uncovers the connections between MRN and DNA damages at R-loop/TRC sites, which to some extent is in line with the following study that MRN is recruited to highly transcribing genes by RNA polymerase II (RNAPII) and protects these sites from genome instability (Salifou et al., 2021). However, whether these two studies can use the same mechanism to interpret their findings still needs further investigation.
In addition to highly transcribing sites, RNA–DNA hybrid/R-loop is also detected at DSB sites. In 2016, an article in Cell revealed that following MRN-initiated DNA end resection in Schizosaccharomyces pombe, transient RNA–DNA hybrids were generated by RNAPII and ensured DSB repair by HR (Ohle et al., 2016). In the following year, another article showed a similar result in mammalian cells (Michelini et al., 2017). RNAPII binds to MRN, is recruited to DSBs and synthesizes damage-induced long non-coding RNAs (dilncRNAs), which promote DNA damage response and repair. In 2021, the Kong group found that RNAPIII also binds to MRN (Liu et al., 2021). During DNA end resection, RNAPIII is recruited by MRN to synthesize RNA. The resulting RNA–DNA hybrids protect the 3’ overhangs in DSB repair. Recently, another article in Nature revealed that during homologous DNA pairing of HR, R-loop facilitates D-loop formation via a specific DNA structure termed DR-loops (Ouyang et al., 2021). Thus, in addition to DNA end resection, the later stage of HR also involves RNA–DNA hybrid/R-loop.
MRN-CtIP’s regulation
MRN and CtIP-related co-factors in DNA damage response
So far, a series of co-factors have been found to regulate the MRN complex assembly, DNA-binding ability, nuclease activity, etc., which maintain DNA damage response (DDR) progression in order and promote resistance to DNA damage stress. The MRN and CtIP-related co-factors and their functions are summarized in Table 1.

Post-translational modifications of MRN and CtIP
Post-translational modification (PTM) of proteins is a reversible biological process for rapidly regulating protein functions, which allows proteins to exhibit different activities to deal with diverse cellular stresses. In DDR, the functions of MRN and CtIP are broadly regulated by various types of PTMs as reviewed in the following.
Phosphorylation
Phosphorylation plays an important role in signal transduction during DDR. MRN and CtIP are both substrates of ATM kinase (Lavin et al., 2015). When DSBs occur, MRN is recruited to damaged chromatin and transforms ATM dimers into activated monomers. Conversely, ATM can phosphorylate both MRN and CtIP, which facilitates DNA repair. Specifically, MRE11 is phosphorylated at S676/S678 by ATM and this helps MRN turnover from damaged chromatin. Phosphorylation of MRE11 is also regulated by other kinases, such as PLK1 and CK2, and has effects on MRE11 DNA binding and MRN disassembly (Studach et al., 2010; von Morgen et al., 2017). ATM phosphorylates RAD50 at S635, and the S635G mutant causes IR sensitivity, HR impairment and S-phase checkpoint inactivation but without affecting MRN assembly. NBS1 can be phosphorylated by ATM and CDK (Falck et al., 2012). Phosphorylation mutant of NBS1 sensitizes cells to DNA damaging drugs and inhibits stalled replication fork restart. For CtIP (Huertas & Jackson, 2009), its phosphorylation enhances MRE11 endonuclease activity, facilitates DNA end resection, and significantly boosts HR efficiency.
Methylation
Methylation is a process of transferring methyl from active donors to substrates and is catalyzed by methyltransferases. PRMT1 was reported to catalyze asymmetric dimethylation of MRE11 at its conserved GAR motif (Boisvert et al., 2005). The MRE11 methylation mutant has IR hypersensitivity in both cell and mouse models, and exhibits a robust decrease in RPA and RAD51 foci formation at DSB sites. Meanwhile, some upstream proteins, such as TIS21, GFI1 and USP11 (Choi et al., 2012; Sanchez-Bailon et al., 2021; Vadnais et al., 2018), can affect MRE11 methylation via regulating PRMT1, which ultimately regulates DNA end resection. These results suggest that methylation of the GAR motif could alter MRE11 nuclease activity, but the mechanism is still unclear.
Ubiquitylation
During DNA damage repair, ubiquitylation of MRE11 is stringently mediated by the proteasome-degradation mechanism. p97/VCP is a key component of the proteasome system, which interacts with MRN and maintains MRE11 physiological protein levels (Kilgas et al., 2021). The inhibition of p97 results in excessive MRE11 accumulation on damaged chromatin and DNA repair defects. Ubiquitylated MRE11 is regulated by the proteasomal shuttle factor UBQLN4, which removes ubiquitylated MRE11 from damaged chromatin to curtail DSB resection (Jachimowicz et al., 2019). However, neither UBQLN4 knockout nor overexpression affected the initiation of DNA end resection, indicating that MRE11 is ubiquitylated at the later stage. For NBS1, its ubiquitylation can be triggered by Skp2 E3 ligase with the K63 chain, which enhances DNA repair by facilitating the NBS1–ATM interaction (Wu et al., 2012). NBS1 is also ubiquitylated by Pellino1 in ATM- and γH2AX-dependent manners at DSB sites and further promotes feedback activation of ATM and HR repair (Ha et al., 2019). CtIP can be ubiquitylated by BRCA1 following DNA damage, which plays an essential role in the regulation of the G2/M checkpoint but not for its degradation (Yu et al., 2006). Besides, USP52 removes the ubiquitylation of CtIP after IR to promote phosphorylation of CtIP and ensure appropriate DNA end resection (Gao et al., 2020).
SUMOylation
Small ubiquitin-related modifiers (SUMOs) are ubiquitin-like peptides that are covalently modified to substrates with extensive functions in the cellular stress response. For SUMOylation of MRN, the Hearing group first revealed that human MRE11 and NBS1 are modified by SUMO1/2 by viral E4-ORF3 during adenovirus infections and then degraded by E1B-55K and E4-ORF6 via the ubiquitin–proteasome system (Sohn & Hearing, 2012). Interestingly, SUMOylation of MRE11 was still increased under infection by E4-ORF3-deficient adenoviruses, indicating that MRE11 SUMOylation has multiple functions in antiviral activities (Castillo-Villanueva et al., 2014).
In contrast to MRN, the function of CtIP SUMOylation is clearly shown in HR (Han et al., 2021). After DSB induction, CtIP is phosphorylated by ATM and promotes DNA end resection. Then, phosphorylated CtIP is SUMOylated at K578 by PIAS4, subsequently ubiquitylated by SUMO-targeted ubiquitin ligase (STUbL) RNF4, and finally degraded by the proteasome system. This mechanism controls the extent of DNA end resection through CtIP turnover from damaged chromatin to prevent excessive resection-mediated HR deficiency.
Acetylation
Acetylation is significant in DDR as well. NBS1 is a hyperacetylated protein, and its acetylation is strictly regulated by the deacetylase SIRT1 (Yuan et al., 2007). SIRT1 can directly interact with the MRN complex and maintain NBS1 in a low-acetylation state, which is necessary for the phosphorylation of NBS1 at S343 induced by IR, indicating that phosphorylation-acetylation/deacetylation crosstalk of NBS1 controls dynamic regulation of the intra-S-phase checkpoint and DDR. In addition, MRE11, RAD50 and CtIP also have been found to possess many acetylation sites, but their functions remain unclear.
UFMylation
UFMylation, a ubiquitin-like protein modification, also participates in DNA repair. MRE11 UFMylation promoted the recruitment of MRE11 at DSBs and MRN complex assembly, accelerated the activation of ATM, and further enhanced HR repair and genomic stability (Wang et al., 2019). Another study showed that UFMylation regulates the phosphorylation of NBS1 via recruitment of the phosphatase PP1-α, and then affects MRN complex recruitment to telomeres (Lee et al., 2021). MRE11 UFMylation is responsible for the MRN–TRF2 interaction, which contributes to the maintenance of telomere length. However, in the second study, MRN complex formation was not found affected in UFL1-deficient cells. This discrepancy may be due to that different experimental conditions like cell lines and gene silencing techniques were used.
In conclusion, the different modifications of the MRN complex and CtIP play key roles in multiple processes including signal cascade transmission, DNA end resection, protein turnover from chromatin, checkpoint regulation, and so forth. Although many PTMs have been identified in the MRN complex and CtIP, most of their specific functions are still unknown.
MRN-CtIP-related genome instability phenotypes and diseases
As MRN-CtIP has critical functions in HR, replication fork processing, R-loop/TRC resolution as reviewed above, impairments of this nuclease ensemble may lead to severe genome instability phenotypes, which could result in multiple diseases including cancers (Syed & Tainer, 2018). For example, in cells deficient for MRE11, RAD50, NBS1 or CtIP, chromosomes exhibit various types of abnormalities such as chromosomal breaks and translocations. Studies using mouse models also revealed that homozygous mutation in any one subunit of the MRN-CtIP complex can result in early embryonic lethality likely due to severe chromosome abnormalities.
In humans, the functional impairments of MRN-CtIP have also been suggested to be connected to genome instability-associated syndromes (Syed & Tainer, 2018). Responding to DSBs, the kinase ATM (ataxia-telangiectasia mutated) is activated by MRN. Thus, the hereditary MRE11 mutations cause a syndrome similar to ataxia-telangiectasia (termed ataxia-telangiectasia-like disorder, ATLD). The ATLD patients show clinical features of progressive cerebellar ataxia, abnormal eye movement, etc. Nijmegen breakage syndrome (NBS) is caused by mutations in the gene encoding the NBS1 protein. This syndrome is characterized by microcephaly, growth retardation and immunodeficiency. For RAD50, the mutations at its gene locus lead to a disorder similar to NBS, termed as NBS-like disorder (NBSLD). Furthermore, both Seckel and Jawad syndromes are characterized by microcephaly and mental retardation. It has been reported that CtIP mutations are one of the molecular causes for Seckel and Jawad syndromes. At the cellular level, cells derived from patients with the above disorders show increased levels of chromosomal abnormalities and hypersensitivity to DNA damaging agents like ionizing radiation (IR). MRN-CtIP mutation carriers are more likely to exhibit neurological disorders, which highlights the specific neuroimpairments caused by unrepaired DNA damage, but the molecular details connecting MRN-CtIP and neurological disorders deserve further investigation.
Genome instability is thought to tightly bond to cell death or transformation, which could result in tumorigenesis. Therefore, the correlations between MRN-CtIP and multiple types of cancers also have been extensively studied (Paudyal & You, 2016). For example, MRE11 expression levels were analyzed in gastric and breast cancers, and found positively correlated with the overall survival of these patients. In samples from aneuploid acute myeloid leukemia patients (A-ALL), both RAD50 protein levels and DDR were down-regulated. Besides, leukemia is also common in NBS1 germline-mutated carriers, such as the NBS1 E185Q polymorphism was found to be associated with ALL, suggesting this polymorphism may serve as a genetic marker for the potential to develop ALL. For CtIP, its status was also assessed in early-onset breast cancers without BRCA1/2 mutations. A number of CtIP variants were revealed to potentiate tumorigenesis via destabilizing RAD51 filament at the replication fork, a manner similar to that seen with BRCA1/2. Most of the above studies and those not reviewed here highlight tight connections between MRN-CtIP and cancers, but whether all these can be explained from the DNA damage repair layer and the exact molecular basis remain unclear.
Conclusions and perspectives
Investigating how DNA end resection is initiated is important, as it is the first step of the HR pathway. Multiple nucleases including MRE11, EXO1 and DNA2 team up with the right partners, and work successively to complete DNA end resection. Based on previous genetic and biochemical studies (Cannavo & Cejka, 2014; Garcia et al., 2011; Wang et al., 2017), a three-step bidirectional model is proposed: (1) the endonucleolytic resection by MRN/MRX-CtIP/Sae2 initiates DNA end resection; (2) the short-range DNA end resection is carried out by MRN/MRX in the 3’-5’ direction; (3) the long-range DNA end resection is performed by EXO1 or DNA2 in the 5’-3’ direction. The resulting 3’ ssDNA tail coated by RPA is handed over to RAD51 to form a nucleoprotein filament. Then, the following steps like D-loop formation and dHJ dissolution/resolution are successively performed to complete DSB repair by HR. Besides the well-studied roles of MRN/MRX-CtIP/Sae2 in DNA end resection of HR, their functions in replication fork stability have also being intensively investigated, and the emerging involvement in R-loop/TRC resolution was also reported in recent years. These above functions of MRN/MRX-CtIP/Sae2 and those not reviewed here collectively contribute to the maintenance of genome stability. Their functional impairments or misregulation can lead to severe genome instability phenotypes such as chromosome abnormalities, and ultimately result in the occurrence of diseases like AT-like syndrome and Nijmegen syndrome, most of which predispose their carriers to various types of cancers.
Although MRN/MRX-CtIP/Sae2 has been extensively studied, unsolved questions regarding this nuclease ensemble remain existing. For example, at the later stage of the dirty DSB end resection, how the protein–DNA tip is dissociated is puzzling. To this end, one study proposed a model wherein MRN-CtIP endonucleolytically chops off the 3’-terminated single-strand tip (Deshpande et al., 2016). However, for budding yeast MRX-Sae2, no similar observation was obtained. Besides, other unknown endonucleases/stimulators, or helicases may collaborate with MRN/MRX-CtIP/Sae2 to get rid of the protein–DNA tip. Regarding the crystal structure studies of the MRN complex, previous work used either bacterial or truncated proteins, thus solving the structure of human full-length MRN is still a big challenge. In the context of replication fork stress, nearly all the studies involving MRN are linked to MRE11-mediated DNA degradation, and further work could help to uncover more non-catalytic functions of MRN at the replication fork. Furthermore, although several studies link MRN/MRX-CtIP/Sae2 to R-loop/TRC resolution, the underlying molecular details deserve further investigation. Overall, studying the biochemical and structural features of MRN/MRX-CtIP/Sae2, and defining the biological functions of this nuclease ensemble will help to understand the connections between genome instability and diseases such as AT-like syndrome, Nijmegen syndrome and cancers.
References
Anand, R., Jasrotia, A., Bundschuh, D., Howard, S. M., Ranjha, L., Stucki, M., & Cejka, P. (2019). NBS1 promotes the endonuclease activity of the MRE11-RAD50 complex by sensing CtIP phosphorylation. EMBO Journal. https://doi.org/10.15252/embj.2018101005
Anand, R., Ranjha, L., Cannavo, E., & Cejka, P. (2016). Phosphorylated CtIP functions as a Co-factor of the MRE11-RAD50-NBS1 endonuclease in DNA end resection. Molecular Cell, 64(5), 940–950. https://doi.org/10.1016/j.molcel.2016.10.017
Anderson, D. E., Trujillo, K. M., Sung, P., & Erickson, H. P. (2001). Structure of the Rad50 x Mre11 DNA repair complex from Saccharomyces cerevisiae by electron microscopy. Journal of Biological Chemistry, 276(40), 37027–37033. https://doi.org/10.1074/jbc.M106179200
Andres, S. N., Appel, C. D., Westmoreland, J. W., Williams, J. S., Nguyen, Y., Robertson, P. D., & Williams, R. S. (2015). Tetrameric Ctp1 coordinates DNA binding and DNA bridging in DNA double-strand-break repair. Nature Structural & Molecular Biology, 22(2), 158–166. https://doi.org/10.1038/nsmb.2945
Andres, S. N., Li, Z. M., Erie, D. A., & Williams, R. S. (2019). Ctp1 protein–DNA filaments promote DNA Bridging and DNA double-strand break repair. Journal of Biological Chemistry. https://doi.org/10.1074/jbc.RA118.006759
Atkinson, J., & McGlynn, P. (2009). Replication fork reversal and the maintenance of genome stability. Nucleic Acids Research, 37(11), 3475–3492. https://doi.org/10.1093/nar/gkp244
Bai, Y., Wang, W., Li, S., Zhan, J., Li, H., Zhao, M., & Wang, J. (2019). C1QBP promotes homologous recombination by stabilizing MRE11 and controlling the assembly and activation of MRE11/RAD50/NBS1 complex. Molecular Cell, 75(6), 1299–1314. https://doi.org/10.1016/j.molcel.2019.06.023
Bennett, L. G., Wilkie, A. M., Antonopoulou, E., Ceppi, I., Sanchez, A., Vernon, E. G., & Staples, C. J. (2020). MRNIP is a replication fork protection factor. Science Advances, 6(28), eaba5974. https://doi.org/10.1126/sciadv.aba5974
Boisvert, F. M., Dery, U., Masson, J. Y., & Richard, S. (2005). Arginine methylation of MRE11 by PRMT1 is required for DNA damage checkpoint control. Genes and Development, 19(6), 671–676. https://doi.org/10.1101/gad.1279805
Broderick, R., Nieminuszczy, J., Baddock, H. T., Deshpande, R., Gileadi, O., Paull, T. T., & Niedzwiedz, W. (2016). EXD2 promotes homologous recombination by facilitating DNA end resection. Nature Cell Biology, 18(3), 271–280. https://doi.org/10.1038/ncb3303
Bryant, H. E., Petermann, E., Schultz, N., Jemth, A. S., Loseva, O., Issaeva, N., & Helleday, T. (2009). PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO Journal, 28(17), 2601–2615. https://doi.org/10.1038/emboj.2009.206
Cannan, W. J., & Pederson, D. S. (2016). Mechanisms and consequences of double-strand DNA break formation in chromatin. Journal of Cellular Physiology, 231(1), 3–14. https://doi.org/10.1002/jcp.25048
Cannavo, E., & Cejka, P. (2014). Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature, 514(7520), 122–125. https://doi.org/10.1038/nature13771
Cannavo, E., Johnson, D., Andres, S. N., Kissling, V. M., Reinert, J. K., Garcia, V., & Cejka, P. (2018). Regulatory control of DNA end resection by Sae2 phosphorylation. Nature Communications, 9(1), 4016. https://doi.org/10.1038/s41467-018-06417-5
Casari, E., Rinaldi, C., Marsella, A., Gnugnoli, M., Colombo, C. V., Bonetti, D., & Longhese, M. P. (2019). Processing of DNA double-strand breaks by the MRX complex in a chromatin context. Frontiers in Molecular Biosciences, 6, 43. https://doi.org/10.3389/fmolb.2019.00043
Castillo-Villanueva, E., Ballesteros, G., Schmid, M., Hidalgo, P., Schreiner, S., Dobner, T., & Gonzalez, R. A. (2014). The Mre11 cellular protein is modified by conjugation of both SUMO-1 and SUMO-2/3 during adenovirus infection. ISRN Virology, 2014, 1–14. https://doi.org/10.1155/2014/989160
Chang, E. Y., Tsai, S., Aristizabal, M. J., Wells, J. P., Coulombe, Y., Busatto, F. F., & Stirling, P. C. (2019). MRE11-RAD50-NBS1 promotes Fanconi anemia R-loop suppression at transcription-replication conflicts. Nature Communications, 10(1), 4265. https://doi.org/10.1038/s41467-019-12271-w
Chen, L., Trujillo, K., Ramos, W., Sung, P., & Tomkinson, A. E. (2001). Promotion of Dnl4-catalyzed DNA end-joining by the Rad50/Mre11/Xrs2 and Hdf1/Hdf2 complexes. Molecular Cell, 8(5), 1105–1115. http://www.ncbi.nlm.nih.gov/pubmed/11741545
Choi, K. S., Kim, J. Y., Lim, S. K., Choi, Y. W., Kim, Y. H., Kang, S. Y., & Lim, I. K. (2012). TIS21(/BTG2/PC3) accelerates the repair of DNA double strand breaks by enhancing Mre11 methylation and blocking damage signal transfer to the Chk 2(T68)-p53(S20) pathway. DNA Repair (amst), 11(12), 965–975. https://doi.org/10.1016/j.dnarep.2012.09.009
Coquel, F., Silva, M. J., Techer, H., Zadorozhny, K., Sharma, S., Nieminuszczy, J., & Pasero, P. (2018). SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature, 557(7703), 57–61. https://doi.org/10.1038/s41586-018-0050-1
Costantino, L., & Koshland, D. (2015). The Yin and Yang of R-loop biology. Current Opinion in Cell Biology, 34, 39–45. https://doi.org/10.1016/j.ceb.2015.04.008
de Jager, M., van Noort, J., van Gent, D. C., Dekker, C., Kanaar, R., & Wyman, C. (2001). Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Molecular Cell, 8(5), 1129–1135. https://doi.org/10.1016/s1097-2765(01)00381-1
Delamarre, A., Barthe, A., de la Roche Saint-Andre, C., Luciano, P., Forey, R., Padioleau, I., & Lengronne, A. (2020). MRX increases chromatin accessibility at stalled replication forks to promote nascent DNA resection and cohesin loading. Molecular Cell, 77(2), 395–410. https://doi.org/10.1016/j.molcel.2019.10.029
Deshpande, R. A., Lee, J. H., Arora, S., & Paull, T. T. (2016). Nbs1 converts the human Mre11/Rad50 nuclease complex into an endo/exonuclease machine specific for protein-DNA adducts. Molecular Cell, 64(3), 593–606. https://doi.org/10.1016/j.molcel.2016.10.010
Falck, J., Forment, J. V., Coates, J., Mistrik, M., Lukas, J., Bartek, J., & Jackson, S. P. (2012). CDK targeting of NBS1 promotes DNA-end resection, replication restart and homologous recombination. EMBO Reports, 13(6), 561–568. https://doi.org/10.1038/embor.2012.58
Gao, M., Guo, G., Huang, J., Kloeber, J. A., Zhao, F., Deng, M., & Lou, Z. (2020). USP52 regulates DNA end resection and chemosensitivity through removing inhibitory ubiquitination from CtIP. Nature Communications, 11(1), 5362. https://doi.org/10.1038/s41467-020-19202-0
Garcia, V., Phelps, S. E., Gray, S., & Neale, M. J. (2011). Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature, 479(7372), 241–244. https://doi.org/10.1038/nature10515
Ghodke, I., & Muniyappa, K. (2013). Processing of DNA double-stranded breaks and intermediates of recombination and repair by Saccharomyces cerevisiae Mre11 and its stimulation by Rad50, Xrs2, and Sae2 proteins. Journal of Biological Chemistry, 288(16), 11273–11286. https://doi.org/10.1074/jbc.M112.439315
Ha, G. H., Ji, J. H., Chae, S., Park, J., Kim, S., Lee, J. K., & Lee, C. W. (2019). Pellino1 regulates reversible ATM activation via NBS1 ubiquitination at DNA double-strand breaks. Nature Communications, 10(1), 1577. https://doi.org/10.1038/s41467-019-09641-9
Han, J., Wan, L., Jiang, G., Cao, L., Xia, F., Tian, T., & Huang, J. (2021). ATM controls the extent of DNA end resection by eliciting sequential posttranslational modifications of CtIP. Proceedings of the National Academy of Sciences, 118, 12. https://doi.org/10.1073/pnas.2022600118
He, Y. J., Meghani, K., Caron, M. C., Yang, C., Ronato, D. A., Bian, J., & Chowdhury, D. (2018). DYNLL1 binds to MRE11 to limit DNA end resection in BRCA1-deficient cells. Nature, 563(7732), 522–526. https://doi.org/10.1038/s41586-018-0670-5
Hopfner, K. P., Craig, L., Moncalian, G., Zinkel, R. A., Usui, T., Owen, B. A., & Tainer, J. A. (2002). The Rad50 zinc-hook is a structure joining Mre11 complexes in DNA recombination and repair. Nature, 418(6897), 562–566. https://doi.org/10.1038/nature00922
Hopfner, K. P., Karcher, A., Shin, D. S., Craig, L., Arthur, L. M., Carney, J. P., & Tainer, J. A. (2000). Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily. Cell, 101(7), 789–800. https://doi.org/10.1016/s0092-8674(00)80890-9
Huertas, P., & Jackson, S. P. (2009). Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. Journal of Biological Chemistry, 284(14), 9558–9565. https://doi.org/10.1074/jbc.M808906200
Jachimowicz, R. D., Beleggia, F., Isensee, J., Velpula, B. B., Goergens, J., Bustos, M. A., & Shiloh, Y. (2019). UBQLN4 represses homologous recombination and is overexpressed in aggressive tumors. Cell, 176(3), 505–519. https://doi.org/10.1016/j.cell.2018.11.024
Kilgas, S., Singh, A. N., Paillas, S., Then, C. K., Torrecilla, I., Nicholson, J., & Ramadan, K. (2021). p97/VCP inhibition causes excessive MRE11-dependent DNA end resection promoting cell killing after ionizing radiation. Cell Reports, 35(8), 109153. https://doi.org/10.1016/j.celrep.2021.109153
Kim, J. J., Lee, S. Y., Choi, J. H., Woo, H. G., Xhemalce, B., & Miller, K. M. (2020). PCAF-mediated histone acetylation promotes replication fork degradation by MRE11 and EXO1 in BRCA-deficient cells. Molecular Cell, 80(2), 327–344. https://doi.org/10.1016/j.molcel.2020.08.018
Lavin, M. F., Kozlov, S., Gatei, M., & Kijas, A. W. (2015). ATM-dependent phosphorylation of all three members of the MRN complex: from sensor to adaptor. Biomolecules, 5(4), 2877–2902. https://doi.org/10.3390/biom5042877
Lee, J. H., & Paull, T. T. (2004). Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science, 304(5667), 93–96. https://doi.org/10.1126/science.1091496
Lee, L., Perez Oliva, A. B., Martinez-Balsalobre, E., Churikov, D., Peter, J., Rahmouni, D., & Lachaud, C. (2021). UFMylation of MRE11 is essential for telomere length maintenance and hematopoietic stem cell survival. Science Advance, 7(39), 7371. https://doi.org/10.1126/sciadv.abc7371
Lim, H. S., Kim, J. S., Park, Y. B., Gwon, G. H., & Cho, Y. (2011). Crystal structure of the Mre11-Rad50-ATPgammaS complex: Understanding the interplay between Mre11 and Rad50. Genes and Development, 25(10), 1091–1104. https://doi.org/10.1101/gad.2037811
Liu, H., Zhang, H., Wang, X., Tian, Q., Hu, Z., Peng, C., & Pei, H. (2015). The deubiquitylating enzyme USP4 cooperates with CtIP in DNA double-strand break end resection. Cell Reports, 13(1), 93–107. https://doi.org/10.1016/j.celrep.2015.08.056
Liu, S., Hua, Y., Wang, J., Li, L., Yuan, J., Zhang, B., & Kong, D. (2021). RNA polymerase III is required for the repair of DNA double-strand breaks by homologous recombination. Cell, 184(5), 1314–1329. https://doi.org/10.1016/j.cell.2021.01.048
Lou, J., Chen, H., Han, J., He, H., Huen, M. S. Y., Feng, X. H., & Huang, J. (2017). AUNIP/C1orf135 directs DNA double-strand breaks towards the homologous recombination repair pathway. Nature Communications, 8(1), 985. https://doi.org/10.1038/s41467-017-01151-w
Lyu, X., Lei, K. H., Biak Sang, P., Shiva, O., Chastain, M., Chi, P., & Chai, W. (2021). Human CST complex protects stalled replication forks by directly blocking MRE11 degradation of nascent-strand DNA. EMBO Journal, 40(2), e103654. https://doi.org/10.15252/embj.2019103654
Makharashvili, N., Arora, S., Yin, Y., Fu, Q., Wen, X., Lee, J. H., & Paull, T. T. (2018). Sae2/CtIP prevents R-loop accumulation in eukaryotic cells. eLife. https://doi.org/10.7554/eLife.42733
Manthey, K. C., Opiyo, S., Glanzer, J. G., Dimitrova, D., Elliott, J., & Oakley, G. G. (2007). NBS1 mediates ATR-dependent RPA hyperphosphorylation following replication-fork stall and collapse. Journal of Cell Science, 120(Pt 23), 4221–4229. https://doi.org/10.1242/jcs.004580
Michelini, F., Pitchiaya, S., Vitelli, V., Sharma, S., Gioia, U., Pessina, F., & d’Adda di Fagagna, F. (2017). Damage-induced lncRNAs control the DNA damage response through interaction with DDRNAs at individual double-strand breaks. Nature Cell Biology, 19(12), 1400–1411. https://doi.org/10.1038/ncb3643
Mimitou, E. P., & Symington, L. S. (2008). Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature, 455(7214), 770–774. https://doi.org/10.1038/nature07312
Moreno-Herrero, F., de Jager, M., Dekker, N. H., Kanaar, R., Wyman, C., & Dekker, C. (2005). Mesoscale conformational changes in the DNA-repair complex Rad50/Mre11/Nbs1 upon binding DNA. Nature, 437(7057), 440–443. https://doi.org/10.1038/nature03927
Myler, L. R., Gallardo, I. F., Soniat, M. M., Deshpande, R. A., Gonzalez, X. B., Kim, Y., & Finkelstein, I. J. (2017). Single-molecule imaging reveals how Mre11-Rad50-Nbs1 initiates DNA break repair. Molecular Cell. https://doi.org/10.1016/j.molcel.2017.08.002
Nath, S., & Nagaraju, G. (2020). FANCJ helicase promotes DNA end resection by facilitating CtIP recruitment to DNA double-strand breaks. PLoS Genetics, 16(4), e1008701. https://doi.org/10.1371/journal.pgen.1008701
Ohle, C., Tesorero, R., Schermann, G., Dobrev, N., Sinning, I., & Fischer, T. (2016). Transient RNA-DNA hybrids are required for efficient double-strand break repair. Cell, 167(4), 1001–1013. https://doi.org/10.1016/j.cell.2016.10.001
Ouyang, J., Yadav, T., Zhang, J. M., Yang, H., Rheinbay, E., Guo, H., & Zou, L. (2021). RNA transcripts stimulate homologous recombination by forming DR-loops. Nature, 594(7862), 283–288. https://doi.org/10.1038/s41586-021-03538-8
Park, Y. B., Chae, J., Kim, Y. C., & Cho, Y. (2011). Crystal structure of human Mre11: Understanding tumorigenic mutations. Structure, 19(11), 1591–1602. https://doi.org/10.1016/j.str.2011.09.010
Paudyal, S. C., & You, Z. (2016). Sharpening the ends for repair: Mechanisms and regulation of DNA resection. Acta Biochimica Et Biophysica Sinica (shanghai), 48(7), 647–657. https://doi.org/10.1093/abbs/gmw043
Paull, T. T., & Gellert, M. (1998). The 3' to 5' exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Molecular Cell, 1(7), 969–979. http://www.ncbi.nlm.nih.gov/pubmed/9651580
Ranjha, L., Howard, S. M., & Cejka, P. (2018). Main steps in DNA double-strand break repair: An introduction to homologous recombination and related processes. Chromosoma, 127(2), 187–214. https://doi.org/10.1007/s00412-017-0658-1
Ray Chaudhuri, A., Callen, E., Ding, X., Gogola, E., Duarte, A. A., Lee, J. E., & Nussenzweig, A. (2016). Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature, 535(7612), 382–387. https://doi.org/10.1038/nature18325
Salifou, K., Burnard, C., Basavarajaiah, P., Grasso, G., Helsmoortel, M., Mac, V., & Kiernan, R. (2021). Chromatin-associated MRN complex protects highly transcribing genes from genomic instability. Science Advance, 7, 21. https://doi.org/10.1126/sciadv.abb2947
Sanchez-Bailon, M. P., Choi, S. Y., Dufficy, E. R., Sharma, K., McNee, G. S., Gunnell, E., & Davies, C. C. (2021). Arginine methylation and ubiquitylation crosstalk controls DNA end-resection and homologous recombination repair. Nature Communications, 12(1), 6313. https://doi.org/10.1038/s41467-021-26413-6
Santos-Pereira, J. M., & Aguilera, A. (2015). R loops: New modulators of genome dynamics and function. Nature Reviews Genetics, 16(10), 583–597. https://doi.org/10.1038/nrg3961
Sohn, S. Y., & Hearing, P. (2012). Adenovirus regulates sumoylation of Mre11-Rad50-Nbs1 components through a paralog-specific mechanism. Journal of Virology, 86(18), 9656–9665. https://doi.org/10.1128/JVI.01273-12
Staples, C. J., Barone, G., Myers, K. N., Ganesh, A., Gibbs-Seymour, I., Patil, A. A., & Collis, S. J. (2016). MRNIP/C5orf45 interacts with the MRN complex and contributes to the DNA damage response. Cell Reports, 16(10), 2565–2575. https://doi.org/10.1016/j.celrep.2016.07.087
Studach, L., Wang, W. H., Weber, G., Tang, J., Hullinger, R. L., Malbrue, R., & Andrisani, O. (2010). Polo-like kinase 1 activated by the hepatitis B virus X protein attenuates both the DNA damage checkpoint and DNA repair resulting in partial polyploidy. Journal of Biological Chemistry, 285(39), 30282–30293. https://doi.org/10.1074/jbc.M109.093963
Syed, A., & Tainer, J. A. (2018). The MRE11-RAD50-NBS1 complex conducts the orchestration of damage signaling and outcomes to stress in DNA replication and repair. Annual Review of Biochemistry, 87, 263–294. https://doi.org/10.1146/annurev-biochem-062917-012415
Symington, L. S., & Gautier, J. (2011). Double-strand break end resection and repair pathway choice. Annual Review of Genetics, 45, 247–271. https://doi.org/10.1146/annurev-genet-110410-132435
Taylor, M. R., Spirek, M., Chaurasiya, K. R., Ward, J. D., Carzaniga, R., Yu, X., & Boulton, S. J. (2015). Rad51 paralogs remodel pre-synaptic Rad51 filaments to stimulate homologous recombination. Cell, 162(2), 271–286. https://doi.org/10.1016/j.cell.2015.06.015
Thomas, M., White, R. L., & Davis, R. W. (1976). Hybridization of RNA to double-stranded DNA: Formation of R-loops. Proceedings of the National Academy of Sciences of the United States of America, 73(7), 2294–2298. https://doi.org/10.1073/pnas.73.7.2294
Trujillo, K. M., Yuan, S. S., Lee, E. Y., & Sung, P. (1998). Nuclease activities in a complex of human recombination and DNA repair factors Rad50, Mre11, and p95. Journal of Biological Chemistry, 273(34), 21447–21450. http://www.ncbi.nlm.nih.gov/pubmed/9705271
Trujillo, K. M., Roh, D. H., Chen, L., Van Komen, S., Tomkinson, A., & Sung, P. (2003). Yeast Xrs2 binds DNA and helps target Rad50 and Mre11 to DNA ends. Journal of Biological Chemistry, 278(49), 48957–48964. https://doi.org/10.1074/jbc.M309877200
Trujillo, K. M., & Sung, P. (2001). DNA structure-specific nuclease activities in the Saccharomyces cerevisiae Rad50-Mre11 complex. Journal of Biological Chemistry, 276(38), 35458–35464. https://doi.org/10.1074/jbc.M105482200
Vadnais, C., Chen, R., Fraszczak, J., Yu, Z., Boulais, J., Pinder, J., & Moroy, T. (2018). GFI1 facilitates efficient DNA repair by regulating PRMT1 dependent methylation of MRE11 and 53BP1. Nature Communications, 9(1), 1418. https://doi.org/10.1038/s41467-018-03817-5
von Morgen, P., Burdova, K., Flower, T. G., O’Reilly, N. J., Boulton, S. J., Smerdon, S. J., & Horejsi, Z. (2017). MRE11 stability is regulated by CK2-dependent interaction with R2TP complex. Oncogene, 36(34), 4943–4950. https://doi.org/10.1038/onc.2017.99
Wang, W., Daley, J. M., Kwon, Y., Krasner, D. S., & Sung, P. (2017). Plasticity of the Mre11-Rad50-Xrs2-Sae2 nuclease ensemble in the processing of DNA-bound obstacles. Genes and Development, 31(23–24), 2331–2336. https://doi.org/10.1101/gad.307900.117
Wang, W., Daley, J. M., Kwon, Y., Xue, X., Krasner, D. S., Miller, A. S., & Sung, P. (2018). A DNA nick at Ku-blocked double-strand break ends serves as an entry site for exonuclease 1 (Exo1) or Sgs1-Dna2 in long-range DNA end resection. Journal of Biological Chemistry, 293(44), 17061–17069. https://doi.org/10.1074/jbc.RA118.004769
Wang, Z., Gong, Y., Peng, B., Shi, R., Fan, D., Zhao, H., & Xu, X. (2019). MRE11 UFMylation promotes ATM activation. Nucleic Acids Research, 47(8), 4124–4135. https://doi.org/10.1093/nar/gkz110
Wijnhoven, P., Konietzny, R., Blackford, A. N., Travers, J., Kessler, B. M., Nishi, R., & Jackson, S. P. (2015). USP4 auto-deubiquitylation promotes homologous recombination. Molecular Cell, 60(3), 362–373. https://doi.org/10.1016/j.molcel.2015.09.019
Wilkinson, O. J., Martin-Gonzalez, A., Kang, H., Northall, S. J., Wigley, D. B., Moreno-Herrero, F., & Dillingham, M. S. (2019). CtIP forms a tetrameric dumbbell-shaped particle which bridges complex DNA end structures for double-strand break repair. eLife. https://doi.org/10.7554/eLife.42129
Williams, R. S., Dodson, G. E., Limbo, O., Yamada, Y., Williams, J. S., Guenther, G., & Tainer, J. A. (2009). Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair. Cell, 139(1), 87–99. https://doi.org/10.1016/j.cell.2009.07.033
Williams, R. S., Moncalian, G., Williams, J. S., Yamada, Y., Limbo, O., Shin, D. S., & Tainer, J. A. (2008). Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell, 135(1), 97–109. https://doi.org/10.1016/j.cell.2008.08.017
Wu, J., Zhang, X., Zhang, L., Wu, C. Y., Rezaeian, A. H., Chan, C. H., & Lin, H. K. (2012). Skp2 E3 ligase integrates ATM activation and homologous recombination repair by ubiquitinating NBS1. Molecular Cell, 46(3), 351–361. https://doi.org/10.1016/j.molcel.2012.02.018
Yu, X., Fu, S., Lai, M., Baer, R., & Chen, J. (2006). BRCA1 ubiquitinates its phosphorylation-dependent binding partner CtIP. Genes and Development, 20(13), 1721–1726. https://doi.org/10.1101/gad.1431006
Yuan, Z., Zhang, X., Sengupta, N., Lane, W. S., & Seto, E. (2007). SIRT1 regulates the function of the Nijmegen breakage syndrome protein. Molecular Cell, 27(1), 149–162. https://doi.org/10.1016/j.molcel.2007.05.029
Zhao, W., Steinfeld, J. B., Liang, F., Chen, X., Maranon, D. G., Jian Ma, C., & Sung, P. (2017). BRCA1-BARD1 promotes RAD51-mediated homologous DNA pairing. Nature, 550(7676), 360–365. https://doi.org/10.1038/nature24060
Zhao, W., Vaithiyalingam, S., San Filippo, J., Maranon, D. G., Jimenez-Sainz, J., Fontenay, G. V., & Sung, P. (2015). Promotion of BRCA2-dependent homologous recombination by DSS1 via RPA targeting and DNA mimicry. Molecular Cell, 59(2), 176–187. https://doi.org/10.1016/j.molcel.2015.05.032
Zhao, W., Wiese, C., Kwon, Y., Hromas, R., & Sung, P. (2019). The BRCA Tumor Suppressor Network in Chromosome Damage Repair by Homologous Recombination. Annual Review of Biochemistry. https://doi.org/10.1146/annurev-biochem-013118-111058
Acknowledgements
This review was supported by grants from the National Natural Science Foundation of China (81972608 and 82172951).
Author information
Affiliations
Department of Radiation Medicine, School of Basic Medical Sciences, Peking University Health Science Center, Beijing, 100191, China
Tao Zhang, Zenan Zhou, Han Yang & Weibin Wang
Corresponding author
Correspondence to Weibin Wang.
Rights and permissions
About this article
Cite this article
Zhang, T., Zhou, Z., Yang, H. et al. MRE11-RAD50-NBS1-CtIP: one key nuclease ensemble functions in the maintenance of genome stability. GENOME INSTAB. DIS. (2022). https://doi.org/10.1007/s42764-022-00065-2
Received10 December 2021
Revised04 February 2022
Accepted08 February 2022
Published23 February 2022
DOIhttps://doi.org/10.1007/s42764-022-00065-2
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
DSB
HR
DNA end resection
MRN
CtIP
Replication fork
R-loop
TRC
Genome instability
Cancer
用户登录
还没有账号?
立即注册