






Centrosome, microtubule and DNA damage response
Review Article
Genome Instability & Disease (2022)
Abstract
Centrosomes are the major microtubule-organizing center (MTOC), which are important cytoplasmic organelles regulating cell cycle, cell morphology, genomic stability, etc. The role of centrosomes on genomic stability has been reported in different conditions. Centrosomal proteins such as centrin2, pericentrin (PCNT), CEP164 have been reported to facilitate DNA damage repair. Various DNA damage response (DDR) proteins locate on centrosomes or microtubules, such as ATM, ATR, DNA-PKcs, 53BP1, etc. Meanwhile, microtubules serve as the “highway” for the transportation of DNA damage proteins into the nucleus. Microtubules have also been discovered to regulate the DNA double strands breaks (DSBs) mobility in DSBs repair. In this review, we first summarize the association between centrosome, microtubules, and DDR. Further, we discuss the new progression on how cells coordinate DDR with microtubule dynamics to facilitate DSBs repair.
Introduction
Various physical or chemical sources could induce DNA damage. To survive from these genomic stresses, cells have evolved the DNA damage response (DDR) to counteract the deleterious effects of DNA damage. As DNA damage mainly occurs in the nucleus, numerous nuclear factors have been involved in DDR. Meanwhile, cytoplasmic organelles have also been found to be of significance for DDR, such as centrosomes (Mullee & Morrison, 2016), microtubules (Lottersberger et al., 2015), Golgi apparatus (Farber-Katz et al., 2014), endoplasmic reticulum (ER) (Zheng et al., 2018), mitochondria (Fang et al., 2016), etc. Centrosomes are small, membrane-free organelles, which serve as the principal microtubule-organizing center (MTOC) in animal somatic cells (Fu et al., 2015). Centrosomes also regulate cell adhesion, motility, polarity, intracellular trafficking through microtubules (Nigg & Holland, 2018). Microtubules are the major cytoskeletal components, which are highly dynamic tubular structures composed of α- and β-tubulin dimers. In recent years, the centrosome and microtubules have been implicated in the maintenance of genome stability in different aspects (Lottersberger et al., 2015; Mullee & Morrison, 2016; Shokrollahi & Mekhail, 2021). Given the pivotal roles of centrosome and microtubules dynamics in regulating DNA repair, plenty of efforts has been devoted to understanding the underlying mechanisms. Here, we focus on the associations between centrosomes, microtubules, and DDR and point out some new progression in this field.
Centrosome and DNA damage response
The centrosomes are micron-sized organelles and comprise two principal components: a pair of orthogonally arranged centrioles and the pericentriolar material (PCM) (Fu et al., 2015). The two centrioles are hollow structures with a ninefold symmetric arrangement of microtubule (MT) triplets, which are organized by an internal cartwheel-like scaffold (Tian et al., 2021). During each cell cycle, the centriole pair disengages at the mitotic exit, allowing the daughter centriole to assemble next to the mother centriole (Nigg, 2007). The mother centriole serves as a scaffold for the recruitment of PCM proteins, leading to centrosome maturation (Fu et al., 2015). PCM is a dynamic collection of protein complexes but not membrane-bound. PCM serves as a scaffold to recruit MT regulators and nucleators, such as gamma-tubulin ring complex (γ-TuRC) (Moritz et al., 2000; Zheng et al., 1995), NEDD1 and pericentrin (PCNT) (Woodruff et al., 2014; Zhang et al., 2009), which accounts for the primary organizing center for microtubule nucleation in somatic cells (Conduit et al., 2015). PCM also serves as a platform for protein complexes that regulate organelle trafficking, protein degradation, and spindle assembly. In interphase cells, PCM contains relatively small amounts of proteins, which is organized as a layered nanometer-sized toroid (Lawo et al., 2012; Mennella et al., 2012). When centrosome maturation starts in late G2 and mitosis, the PCM expands dramatically into a micron-sized ensemble, which accounts for the increased microtubule-organizing activity of centrosomes (Khodjakov & Rieder, 1999; Mennella et al., 2014; Palazzo et al., 2000).
Centrosomes are involved in several interdependent pathways, including DDR (Mullee & Morrison, 2016). The communication between centrosome and DNA damage could be classified into different categories. First, genetic mutation of centrosomal proteins such as centrin2, Cep164, and PCNT affect DDR. Second, DDR proteins, such as ATM, ATR, DNA-PKcs, BRCA1, BRCA2, CHK1, CHK2, have also been reported to localize on centrosome. Third, centrosome structure and numbers can be affected by DNA damage.
Some centrosomal proteins have been directly linked to DDR. Centrin2, a component of the proximal centriolar lumen, could stimulate in vitro DNA repair and is required for efficient nucleotide excision repair (NER) (Araki et al., 2001; Nishi et al., 2005). CEP164, a centriole distal appendage protein, has been described as a substrate and interactor of ATM/ATR. CEP164 depletion affects DDR activation and G2/M checkpoint (Sivasubramaniam et al., 2008). CEP164 could also be recruited to UV-induced DNA damage sites through interaction with a NER factor, Xeroderma pigmentosum group A (XPA), and coordinate checkpoint pathways with repair systems. (Pan & Lee, 2009). PCNT is the first identified PCM protein, and cell lines from PCNT-mutant patients exhibit impaired ATR-dependent phosphorylation and impaired G2/M checkpoint (Griffith et al., 2008). Proteomics studies reveal an increasing list of centrosomal proteins, and more centrosomal proteins are reported to link with DDR (Johnson & Collis, 2016; Morrison, 2021). However, given that only part of these proteins is associated with the centrosome and the bulk of them is widely distributed in the cytoplasm or even in nucleus, whether the functions of these proteins on DDR are directly centrosome related is still elusive.
Various DDR proteins locate on the centrosomes, such as ATM, ATR, DNA-PKcs, CHK1 and CHK2 kinases, the BRCA1 ubiquitin ligase complex (Mullee & Morrison, 2016). ATM and ATR kinase have been described at mitotic centrosomes in different human cell lines, which may be involved in regulating spindle assembly or centrosome amplification (Brown & Costanzo, 2009; Fan et al., 2021; Shen et al., 2006). CHK1, a kinase regulating DNA damage checkpoint, is reported to localize at the centrosome (Tibelius et al., 2009), although the underlying mechanism and the real function of centrosomal CHK1 are still controversial (Mullee & Morrison, 2016). BRCA1 and BRCA2, which are vital factors regulating DSBs repair choice and facilitating homologous recombination (HR), localize on centrosomes (Brodie & Henderson, 2012; Brodie et al., 2012; Zhang et al., 2016). BRCA1 interacts with OLA1 and BARD1, which ubiquitinates γ-tubulin and regulates centrosome number and centrosome-dependent microtubule nucleation (Matsuzawa et al., 2014; Yoshino et al., 2018). Defects of BRCA1 induce centrosome amplification, which may lead to tumorigenesis. However, whether the nuclear function of BRCA1 and BRCA2 communicates with the role of centrosomal BRCA1 and BRCA2 during DNA damage still needs further study.
DNA damage could also modulate the centrosome number and structure (Antonczak et al., 2016; Bourke et al., 2007; Dodson et al., 2004). DNA damage promotes centrosome duplication, resulting in multi-centrosomes in interphase cells (Bourke et al., 2007; Dodson et al., 2007; Loffler et al., 2013). S-phase delay could induce centrosome amplification, which depended on CHK1 and CDK kinase (Bourke et al., 2007). CHK1 associates with centrosome proteins PCNT and microcephalin (MCPH1) to regulate centrosome amplification. After DNA damage, CHK1 and the PCM form a positive feedback loop in which CHK1 regulates PCNT-dependent PCM expansion to control its own activation (Antonczak et al., 2016). CHK2 is also involved in HU-induced centrosome overduplication in U2OS cells (Wang et al., 2015). As to the centrosome structure, DNA damage could lead to the splitting of centrosomes into individual centrioles and disruption of PCM (Conroy et al., 2012; Saladino et al., 2009). These reports revealed the effect of DNA damage stress on centrosome function and duplication. However, these findings are mainly discovered from long-term DNA damage stress (> 24 h), and the short-term effect of DNA damage on centrosome structure and function is still not well studied. The recent progression on the short-term effect of DNA damage on centrosomes and microtubules will be discussed later.
Primary cilia and DNA damage response
In quiescence cells, the mother centriole provides a template for cilium or flagellum assembly, which is a crucial organelle for signal transduction, locomotion, etc. (Breslow & Holland, 2019; Chavali et al., 2014). Cilia are found on most eukaryotic cells and contain two categories: primary and motile. Motile cilia appear in larger numbers and move the surrounding fluid by beating asymmetrically (Dawe et al., 2007). For primary cilia, there is only one primary cilium in each cell, which have primarily sensory functions, such as receptors for external chemical, mechanical or other signals (Brown & Zhang, 2020). Primary cilia are anchored to the cell membrane via the distal appendages. The core of primary cilia (axoneme) is membrane-bounded, antenna-like structures, which is composed of microtubules connected to the centriole. The microtubules in primary cilia are arranged in a 9 + 0 fashion, i.e., nine outer microtubule doublets and no central singlets (Sun et al., 2019). Primary cilium biogenesis is a cell cycle-dependent event, which solely depends on the status of the centrosome (Breslow & Holland, 2019; Wang, Chen, et al., 2013). During the G1/G0 phase, primary cilium is assembled from the plus end of the mother centriole. In S/G2 cells, primary cilium disassembles, which is tightly controlled by various proteins, such as Aurora A, Plk1, HDAC6, etc. (Pugacheva et al., 2007; Wang, Chen, et al., 2013). In multiple contexts, primary cilia contain components of different signaling pathways and serve as signaling centers for the Hedgehog (Hh), Wnt, TGF-β, and calcium signaling pathways (Alvarez-Satta & Matheu, 2018; Clement et al., 2013; Goetz & Anderson, 2010; Monnich et al., 2018). Defects of primary cilia dynamics have been associated with many diseases known as ciliopathies (Reiter & Leroux, 2017).
Primary cilia have also been linked to DDR, which was extensively discussed in recent reviews (Johnson & Collis, 2016; Morrison, 2021). Various ciliopathy-related genes have known roles in DDR, such as centrosomal protein CEP63, CEP152, CEP164, CEP290, MCPH1, etc. Meanwhile, some established DDR proteins are linked to ciliogenesis or ciliopathy, including ATR, MRE11, ORC1, etc. Furthermore, primary cilia are linked to some specific kind of DNA damage. For instance, UV treatment but not IR reorganizes PCM proteins and leads to ciliogenesis through p38/MAP14 (Villumsen et al., 2013). A recent study describes that genotoxic drug treatment, such as etoposide, neocarzinostatin, and cisplatin, leads to ciliogenesis through DNA-PK-p53 cascade (Chen et al., 2021).
Microtubule-nucleus connection
In interphase cells, centrosomes and microtubules could modulate nuclear functions, which are mediated by the connection between microtubules and the nucleus. Microtubules could affect nuclear organization, chromatin dynamics, and gene transcription. These functions of microtubules on the nucleus are also involved in DDR and contribute to maintaining genomic stability.
Microtubules facilitate DDR by transporting DDR proteins into the nucleus. Microtubule serves as a “highway" for transporting specific molecular cargo to the nucleus by microtubule-associated minus-end motor proteins (Shokrollahi & Mekhail, 2021). This cargo includes transcription factors, DNA damage response proteins, etc. The active transportation of specific proteins facilitates the nuclear importation, promoting the communication between cytoplasm and nucleus under DNA damage. For instance, DDR proteins such as MRE11, NBS1, and 53BP1 localize on microtubules and are transported into the nucleus by microtubules in a dynein-dependent manner after DNA damage (Poruchynsky et al., 2015). The promoted nuclear translocation of these DDR proteins may contribute to the rapid nuclear response to DNA damage.
Besides contributing to DDR protein transportation, microtubules directly interact with nuclear envelope proteins, which transduces the force of microtubule dynamics to the nucleus. These interactions depend on specific molecular complexes and can regulate the chromatin dynamics and the global organization of the 3-dimensional genome (Dobrzynska et al., 2016). This direct interaction is mediated through the linker of the nucleoskeleton and cytoskeleton (LINC) complex. The LINC complex comprises Sad1 UNC-84 (SUN) and Klarsicht-ANC-1-Sync homology (KASH) domain proteins (Starr, 2011). SUN proteins are embedded in the inner nuclear membrane (INM) through transmembrane motif and interact with nuclear lamina-associated proteins. KASH proteins link SUN protein complex to microtubules, which are extended into the cytoplasm from SUN protein complex (Rothballer & Kutay, 2013). Thus, LINC complex formed by SUN and KASH proteins directly couples microtubules with the nuclear lamina. This interaction enables the transduction of forces generated by microtubule dynamics to the nucleus, which can be applied globally or locally to the nucleus. For instance, microtubules promote DSBs mobility through LINC complex (Lottersberger et al., 2015). DSBs mobility has been described to facilitate DSBs repair (Hauer & Gasser, 2017; Marnef & Legube, 2017). DSBs mobility is regulated by several factors, such as microtubules, LINC complex, nuclear actin, Lamin A/C, and IFFO1 (Caridi et al., 2018; Lawrimore et al., 2017; Li et al., 2019; Lottersberger et al., 2015; Schrank et al., 2018). Among these factors, only microtubules localize in the cytoplasm in vertebrate cells. Microtubules contribute to uncapped telomere or irradiation-induced DSBs mobility (Lawrimore et al., 2017; Lottersberger et al., 2015). Within this context, the LINC complex and the motor proteins kinesin-1 and 2 are required for microtubules to fulfill its role on DSBs mobility (Lottersberger et al., 2015). Thus, microtubules could facilitate mobility-dependent DNA repair through promoting DSBs mobility. Furthermore, SUN1 and SUN2 double-knockout embryonic fibroblasts exhibit excessive DNA damage, confirming that either SUN1 or SUN2 is required for the DDR (Lei et al., 2012). The kinesin motor protein Kif2C has also been reported to promote the mobility of damaged DNA and facilitate DNA repair (Zhu et al., 2020). All these reports reveal that the force of microtubule dynamics is linked to DSBs mobility and repair (Fig. 1A).
Fig. 1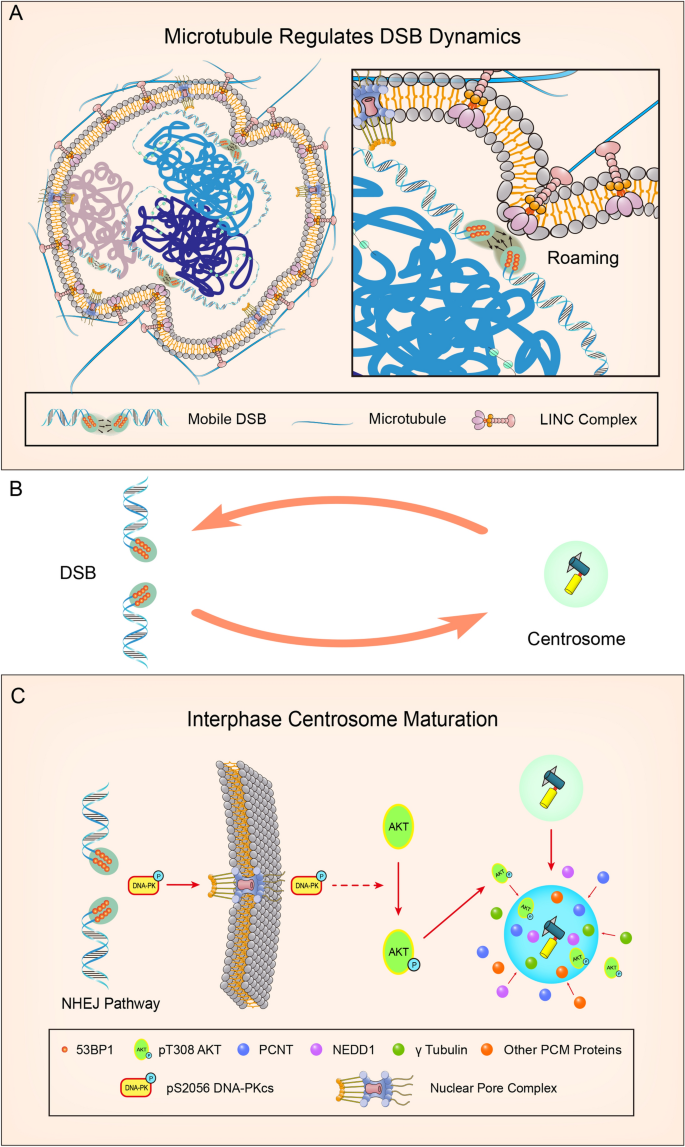
The interplay between DSBs and centrosomes during DSBs repair. A The forces of microtubule dynamics can exert on the nuclear envelope, which reshape the nucleus to facilitate DSBs mobility-dependent repair. The connection between microtubule and DSBs mobility is mediated by LINC complex. B A closed feedback loop between DSBs and microtubule dynamics after DNA damage. C DSBs promote centrosome-dependent microtubule nucleation through interphase centrosome maturation. In normal G1/G0 cells, fewer PCM proteins are recruited on the centrosome. After DNA damage, DSBs lead to DNA-PK autophosphorylation and activation, which may be exported from the nucleus to the cytoplasm. In cytoplasm, DNA-PK promotes phosphorylation of T308 AKT, which accumulates on the centrosome and induces recruitment of PCM protein. The accumulated PCM proteins on the centrosome leads to interphase centrosome maturation and enhanced microtubule nucleation
Full size imageDNA damage-induced short-term microtubule dynamics stress response
Microtubule dynamics regulate DSBs mobility and facilitate DSBs repair. However, whether DSBs affect microtubule organization is an interesting point. Although DNA damage may regulate centrosome duplication and maturation in the long-term (> 24 h), these effects might arise from DNA damage-induced cell cycle defects. Recently, the short-term (< 8 h) effect of DNA damage on centrosomes was revealed in interphase cells (Ma et al., 2021), which indicates that DSBs promoted centrosome-dependent microtubule nucleation and polymerization in G1 or G0 phase cells. This specific microtubule response was named DSBs-induced Microtubule-dynamics Stress Response (DMSR). DMSR defects induced by PCM protein depletion led to decreased DSBs mobility, indicating that DMSR contributes to the high mobility of DSBs sites. As microtubules dynamics promote DSBs mobility-dependent repair, this finding revealed a closed feedback loop between DSBs and microtubule dynamics during DSBs repair (Fig. 1B).
DMSR happens in both no-transformed cell line (RPE-1) and transformed cancer cell lines and only occurs in G0/G1 cells. In normal G0/G1 cells, fewer PCM proteins are recruited on the centrosome. However, DSBs lead to the accumulation of PCM proteins on centrosome within 6 h after DNA damage in G0/G1 cells, which induces interphase centrosome maturation. The interphase centrosome maturation happens 1 h after DSB, peaks at 4 h, and diminishes at around 6 h after DNA damage, which leads to the increase in centrosome dependent microtubule nucleation and microtubule polymerization (Ma et al., 2021). DSB-induced interphase centrosome maturation is distinct from physiological centrosome maturation that happened on G2 or mitosis since Plk1 is dispensable for DSB-related interphase maturation. For the underlying mechanism, the DNA-PK-AKT axis is responsible for DMSR. DNA-PK is activated in response to DSBs in G0/G1 cells, which initiates NHEJ process. DNA-PK inhibition leads to DMSR defects, revealing that DNA-PK is responsible for DMSR. For the downstream factors, AKT inhibition leads to DMSR defect, and the accumulation of pT308 AKT on interphase centrosomes is identified during DMSR. Although the localization of AKT and pT308 AKT on mitotic centrosomes has been reported (Buttrick & Wakefield, 2008; Buttrick et al., 2008; Wakefield et al., 2003), the interphase localization of pT308 AKT on centrosomes is a novel finding. Furthermore, DNA-PK but not ATM or PDK1 is dispensable for DSBs-induced interphase centrosomal accumulation of pThr308 AKT. Thus, DNA-PK and its downstream factor pThr308 AKT are responsible for the DMSR-related interphase centrosome maturation (Fig. 1C). AKT could regulate interphase or mitotic centrosomes functions through phosphorylating several substrates, including GSK-3 (Buttrick & Wakefield, 2008; Buttrick et al., 2008; Wakefield et al., 2003), TEIF (Zhao et al., 2014), Inversin (Suizu et al., 2016). But, whether these AKT substrates are involved in DMSR still needs further studies.
Collectively, there are four interesting characters in DMSR. (1) DMSR only happens in G1 or G0 cells. (2) DMSR is a short-term stress response, which starts at 1 h after DSBs, peaks at around 4 h, and gradually decreases from 6 h. (3) The extent of DMSR is positively correlated with the extent of DNA damage stress. (4) DMSR is accompanied with interphase centrosome maturation.
Effect of DMSR on DSBs repair
In contrast to HR, which happens in S and G2 phase cells, c-NHEJ is a relatively fast and efficient process and functions throughout the cell cycle (Jackson & Bartek, 2009; Lukas & Lukas, 2013). Key components in c-NHEJ are Ku70/Ku80 heterodimer which could form a complex at DNA breaks with the DNA-dependent protein kinase catalytic subunit (DNA-PKcs), generating the DNA-PK holoenzyme (Jette & Lees-Miller, 2015). The DNA ligase 4 complex, including XRCC4, XLF, and PAXX, carries out the direct ligation step of the two broken DNA ends in the later stages of c-NHEJ (Biehs et al., 2017; Ochi et al., 2015). Intriguingly, recent reports indicated that c-NHEJ in G1 shows biphasic kinetics involving a fast and a slow process in response to IR-induced DSBs (Biehs et al., 2017; Lobrich & Jeggo, 2017). Fast NHEJ is an end-resection independent process, and slow NHEJ process is end-resection dependent (Lobrich & Jeggo, 2017). Mre11 exonuclease, EXD2, and Exo1 are required for this end-resection dependent slow NHEJ in G1 (Riballo et al., 2004). Recent findings suggest that Plk3 mediated phosphorylation of CtIP in G1 could promote CtIP-BRCA1 interaction and regulate end resection-dependent slow c-NHEJ (Barton et al., 2014; Biehs et al., 2017; Lobrich & Jeggo, 2017).
DMSR appears at around 1 h after DNA damage and diminishes at around 6 h in G1, which overlaps with the fast NHEJ repair period (1–4 h after DNA damage) (Lobrich & Jeggo, 2017; Ma et al., 2021). DMSR requires activation of DNA-PK and exists only in G1. 53BP1 depletion leads to the disappearance of DMSR and depletion of Ligase 4, XLF or XRCC4, which are crucial for fast-NHEJ, leads to persistent DMSR in G1. These facts indicate that DMSR is closely related to fast NHEJ. Furthermore, G1 cells with PCM protein depletion show increased end-resection in G1 as shown by the increase of RPA2 foci. Thus, DMSR may inhibit end-resection process in slow-NHEJ through maintaining DSBs mobility.
Another interesting point of DMSR is that the extent of DMSR is positively correlated with the extent of DNA damage. A high dosage of IR leads to a significant change of centrosome-mediated microtubule organization. There are two possible explanations for this phenomenon. First, to repair a high level of DSBs, fast-NHEJ should be kept in high efficiency. DMSR is activated to elevate microtubule dynamics. Enhanced microtubule dynamics promote DSBs mobility, which facilitates the high efficiency of fast NHEJ. When only low level of DNA damage happens, the basal level of microtubule dynamics could maintain the DSBs mobility required for fast NHEJ. Thus, DMSR is not activated at a low level of DSBs. Second, fast-NHEJ could efficiently repair most DSBs when cells are treated with a low dosage of IR. In this case, DMSR is not activated due to the low activation of DNA-PK. On the contrary, when fast-NHEJ failed to repair a high level of DSBs properly in G1 cells, accumulated activation of DNA-PK leads to DMSR, which inhibits end-resection through increasing DSBs mobility and avoids the occurrence of slow-NHEJ.
Moreover, several reports discovered that the mobility of DSBs should be tightly regulated. For instance, high mobility of DSBs can lead to genomic translocation (Li et al., 2019; Roukos et al., 2013). The duration and extent of DMSR are tightly regulated, which may avoid the harmful effect of prolonged high mobility of DSBs. Intriguingly, excessive changes of microtubule dynamics are also detrimental to cells, which may rupture the nuclear envelope (Chow et al., 2012; Estrem & Moore, 2020; Penfield et al., 2018). The tightly regulated DMSR could also avoid the prolonged overactivation of microtubule dynamics, preventing the bad effect of microtubule dynamics on the nuclear envelope and nuclear organization.
Remaining questions about DMSR
One remaining question is how signals are transported from the nucleus to the cytoplasmic centrosome during DMSR. It is possible that after NHEJ is started, activated DNA-PKcs are exported from the nucleus to the cytoplasm, where DNA-PK promotes pT308 AKT. Subsequently, pT308 AKT accumulates at the centrosome, which leads to interphase centrosome maturation. In fact, DNA-PKcs could locate in both the nucleus and cytoplasm (Poruchynsky et al., 2015; Saji et al., 2005; Wang, Kao, et al., 2013) and microtubules localization of DNA-PKcs has also been described (Poruchynsky et al., 2015). Previous reports showed that both DNA-PK and ATM may contribute to Ser473 AKT phosphorylation, which mainly exists in the nucleus (Bozulic et al., 2008; Fraser et al., 2011). However, whether DNA-PK directly phosphorylates AKT on Thr308 is also unknown. More experiments are needed to elucidate the relationship between DNA-PK and pT308 AKT during DDR in the future.
Centrosome is responsible for the assembly of the primary cilium in G1/G0 cells, and primary cilia are associated with DDR. As DMSR happens only in G1/G0 cells, whether DMSR affects the assembly or disassembly of primary cilia is an interesting question. In quiescent cells, gene transcription or exposure to DNA damage agents could induce massive endogenous DSBs (Dellino et al., 2019), suggesting that DMSR may happen in physiological conditions. Thus, DMSR may affect the proper assembly or disassembly of primary cilia. As primary cilia serve as an antenna for signaling transduction, DMSR-related defective primary cilia formation may affect the normal communication of external signals. Furthermore, Ligase 4 or XRCC4 depletion leads to defective NHEJ and accumulated DSBs in G1/G0 cells, which induce the overactivation of DMSR (Ma et al., 2021). Interestingly, patients with Ligase 4 or XRCC4 mutant always exhibit microcephaly (Park et al., 2016; Rosin et al., 2015; Woodbine et al., 2014), which is related to defective primary cilia formation. Thus, whether DMSR contributes to the diseases related to Ligase 4 or XRCC4 mutant deserves further studies.
Conclusion
This review summarizes the interplay between centrosome, microtubule, and DNA damage repair. We highlight the short-term effects of DNA damage on centrosomes and microtubules (DMSR) and discuss the underlying mechanism regulating DMSR. DMSR and microtubule-dependent regulation of DSBs mobility form a closed feedback loop between DSBs and microtubule dynamics, which facilitates DSBs mobility-dependent repair (Fig. 1).
References
Alvarez-Satta, M., & Matheu, A. (2018). Primary cilium and glioblastoma. Ther Adv Med Oncol, 10, 1758835918801169.
Antonczak, A. K., Mullee, L. I., Wang, Y., Comartin, D., Inoue, T., Pelletier, L., & Morrison, C. G. (2016). Opposing effects of pericentrin and microcephalin on the pericentriolar material regulate CHK1 activation in the DNA damage response. Oncogene, 35, 2003–2010.
Araki, M., Masutani, C., Takemura, M., Uchida, A., Sugasawa, K., Kondoh, J., Ohkuma, Y., & Hanaoka, F. (2001). Centrosome protein centrin 2/caltractin 1 is part of the xeroderma pigmentosum group C complex that initiates global genome nucleotide excision repair. The Journal of Biological Chemistry, 276, 18665–18672.
Barton, O., Naumann, S. C., Diemer-Biehs, R., Kunzel, J., Steinlage, M., Conrad, S., Makharashvili, N., Wang, J., Feng, L., Lopez, B. S., et al. (2014). Polo-like kinase 3 regulates CtIP during DNA double-strand break repair in G1. The Journal of Cell Biology, 206, 877–894.
Biehs, R., Steinlage, M., Barton, O., Juhasz, S., Kunzel, J., Spies, J., Shibata, A., Jeggo, P. A., & Lobrich, M. (2017). DNA double-strand break resection occurs during non-homologous end joining in G1 but is distinct from resection during homologous recombination. Molecular Cell, 65(671–684), e675.
Bourke, E., Dodson, H., Merdes, A., Cuffe, L., Zachos, G., Walker, M., Gillespie, D., & Morrison, C. G. (2007). DNA damage induces Chk1-dependent centrosome amplification. EMBO Reports, 8, 603–609.
Bozulic, L., Surucu, B., Hynx, D., & Hemmings, B. A. (2008). PKBalpha/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Molecular Cell, 30, 203–213.
Breslow, D. K., & Holland, A. J. (2019). Mechanism and regulation of centriole and cilium biogenesis. Annual Review of Biochemistry, 88, 691–724.
Brodie, K. M., & Henderson, B. R. (2012). Characterization of BRCA1 protein targeting, dynamics, and function at the centrosome: A role for the nuclear export signal, CRM1, and Aurora A kinase. The Journal of Biological Chemistry, 287, 7701–7716.
Brodie, K. M., Mok, M. T., & Henderson, B. R. (2012). Characterization of BARD1 targeting and dynamics at the centrosome: The role of CRM1, BRCA1 and the Q564H mutation. Cellular Signalling, 24, 451–459.
Brown, A., & Zhang, R. (2020). Primary cilia: A closer look at the antenna of cells. Current Biology : CB, 30, R1494–R1496.
Brown, N., & Costanzo, V. (2009). An ATM and ATR dependent pathway targeting centrosome dependent spindle assembly. Cell Cycle, 8, 1997–2001.
Buttrick, G. J., Beaumont, L. M., Leitch, J., Yau, C., Hughes, J. R., & Wakefield, J. G. (2008). Akt regulates centrosome migration and spindle orientation in the early Drosophila melanogaster embryo. The Journal of Cell Biology, 180, 537–548.
Buttrick, G. J., & Wakefield, J. G. (2008). PI3-K and GSK-3: Akt-ing together with microtubules. Cell Cycle, 7, 2621–2625.
Caridi, C. P., D’Agostino, C., Ryu, T., Zapotoczny, G., Delabaere, L., Li, X., Khodaverdian, V. Y., Amaral, N., Lin, E., Rau, A. R., et al. (2018). Nuclear F-actin and myosins drive relocalization of heterochromatic breaks. Nature, 559, 54–60.
Chavali, P. L., Putz, M., & Gergely, F. (2014). Small organelle, big responsibility: The role of centrosomes in development and disease (p. 369). Philosophical transactions of the Royal Society of London.
Chen, T. Y., Huang, B. M., Tang, T. K., Chao, Y. Y., Xiao, X. Y., Lee, P. R., Yang, L. Y., & Wang, C. Y. (2021). Genotoxic stress-activated DNA-PK-p53 cascade and autophagy cooperatively induce ciliogenesis to maintain the DNA damage response. Cell Death and Differentiation, 28, 1865–1879.
Chow, K. H., Factor, R. E., & Ullman, K. S. (2012). The nuclear envelope environment and its cancer connections. Nature Reviews Cancer, 12, 196–209.
Clement, C. A., Ajbro, K. D., Koefoed, K., Vestergaard, M. L., Veland, I. R., Henriques de Jesus, M. P., Pedersen, L. B., Benmerah, A., Andersen, C. Y., Larsen, L. A., et al. (2013). TGF-beta signaling is associated with endocytosis at the pocket region of the primary cilium. Cell Reports, 3, 1806–1814.
Conduit, P. T., Wainman, A., & Raff, J. W. (2015). Centrosome function and assembly in animal cells. Nature Reviews. Molecular Cell Biology, 16, 611–624.
Conroy, P. C., Saladino, C., Dantas, T. J., Lalor, P., Dockery, P., & Morrison, C. G. (2012). C-NAP1 and rootletin restrain DNA damage-induced centriole splitting and facilitate ciliogenesis. Cell Cycle, 11, 3769–3778.
Dawe, H. R., Farr, H., & Gull, K. (2007). Centriole/basal body morphogenesis and migration during ciliogenesis in animal cells. Journal of Cell Science, 120, 7–15.
Dellino, G. I., Palluzzi, F., Chiariello, A. M., Piccioni, R., Bianco, S., Furia, L., De Conti, G., Bouwman, B. A. M., Melloni, G., Guido, D., et al. (2019). Release of paused RNA polymerase II at specific loci favors DNA double-strand-break formation and promotes cancer translocations. Nature Genetics, 51, 1011.
Dobrzynska, A., Gonzalo, S., Shanahan, C., & Askjaer, P. (2016). The nuclear lamina in health and disease. Nucleus, 7, 233–248.
Dodson, H., Bourke, E., Jeffers, L. J., Vagnarelli, P., Sonoda, E., Takeda, S., Earnshaw, W. C., Merdes, A., & Morrison, C. (2004). Centrosome amplification induced by DNA damage occurs during a prolonged G2 phase and involves ATM. EMBO Journal, 23, 3864–3873.
Dodson, H., Wheatley, S. P., & Morrison, C. G. (2007). Involvement of centrosome amplification in radiation-induced mitotic catastrophe. Cell Cycle, 6, 364–370.
Estrem, C., & Moore, J. K. (2020). Help or hindrance: How do microtubule-based forces contribute to genome damage and repair? Current Genetics, 66, 303–311.
Fan, G., Sun, L., Meng, L., Hu, C., Wang, X., Shi, Z., Hu, C., Han, Y., Yang, Q., Cao, L., et al. (2021). The ATM and ATR kinases regulate centrosome clustering and tumor recurrence by targeting KIFC1 phosphorylation. Nature Communications, 12, 20.
Fang, E. F., Scheibye-Knudsen, M., Chua, K. F., Mattson, M. P., Croteau, D. L., & Bohr, V. A. (2016). Nuclear DNA damage signalling to mitochondria in ageing. Nature Reviews. Molecular Cell Biology, 17, 308–321.
Farber-Katz, S. E., Dippold, H. C., Buschman, M. D., Peterman, M. C., Xing, M., Noakes, C. J., Tat, J., Ng, M. M., Rahajeng, J., Cowan, D. M., et al. (2014). DNA damage triggers Golgi dispersal via DNA-PK and GOLPH3. Cell, 156, 413–427.
Fraser, M., Harding, S. M., Zhao, H., Coackley, C., Durocher, D., & Bristow, R. G. (2011). MRE11 promotes AKT phosphorylation in direct response to DNA double-strand breaks. Cell Cycle, 10, 2218–2232.
Fu, J., Hagan, I. M., & Glover, D. M. (2015). The centrosome and its duplication cycle. Cold Spring Harbor Perspectives in Biology, 7, a015800.
Goetz, S. C., & Anderson, K. V. (2010). The primary cilium: A signalling centre during vertebrate development. Nature Reviews. Genetics, 11, 331–344.
Griffith, E., Walker, S., Martin, C. A., Vagnarelli, P., Stiff, T., Vernay, B., Al Sanna, N., Saggar, A., Hamel, B., Earnshaw, W. C., et al. (2008). Mutations in pericentrin cause Seckel syndrome with defective ATR-dependent DNA damage signaling. Nature Genetics, 40, 232–236.
Hauer, M. H., & Gasser, S. M. (2017). Chromatin and nucleosome dynamics in DNA damage and repair. Genes & Development, 31, 2204–2221.
Jackson, S. P., & Bartek, J. (2009). The DNA-damage response in human biology and disease. Nature, 461, 1071–1078.
Jette, N., & Lees-Miller, S. P. (2015). The DNA-dependent protein kinase: A multifunctional protein kinase with roles in DNA double strand break repair and mitosis. Progress in Biophysics and Molecular Biology, 117, 194–205.
Johnson, C. A., & Collis, S. J. (2016). Ciliogenesis and the DNA damage response: A stressful relationship. Cilia, 5, 19.
Khodjakov, A., & Rieder, C. L. (1999). The sudden recruitment of gamma-tubulin to the centrosome at the onset of mitosis and its dynamic exchange throughout the cell cycle, do not require microtubules. The Journal of Cell Biology, 146, 585–596.
Lawo, S., Hasegan, M., Gupta, G. D., & Pelletier, L. (2012). Subdiffraction imaging of centrosomes reveals higher-order organizational features of pericentriolar material. Nature Cell Biology, 14, 1148–1158.
Lawrimore, J., Barry, T. M., Barry, R. M., York, A. C., Friedman, B., Cook, D. M., Akialis, K., Tyler, J., Vasquez, P., Yeh, E., et al. (2017). Microtubule dynamics drive enhanced chromatin motion and mobilize telomeres in response to DNA damage. Molecular Biology of the Cell, 28, 1701–1711.
Lei, K., Zhu, X., Xu, R., Shao, C., Xu, T., Zhuang, Y., & Han, M. (2012). Inner nuclear envelope proteins SUN1 and SUN2 play a prominent role in the DNA damage response. Current Biology : CB, 22, 1609–1615.
Li, W., Bai, X., Li, J., Zhao, Y., Liu, J., Zhao, H., Liu, L., Ding, M., Wang, Q., Shi, F. Y., et al. (2019). The nucleoskeleton protein IFFO1 immobilizes broken DNA and suppresses chromosome translocation during tumorigenesis. Nature Cell Biology, 21, 1273–1285.
Lobrich, M., & Jeggo, P. (2017). A process of resection-dependent nonhomologous end joining involving the goddess artemis. Trends in Biochemical Sciences, 42, 690–701.
Loffler, H., Fechter, A., Liu, F. Y., Poppelreuther, S., & Kramer, A. (2013). DNA damage-induced centrosome amplification occurs via excessive formation of centriolar satellites. Oncogene, 32, 2963–2972.
Lottersberger, F., Karssemeijer, R. A., Dimitrova, N., & de Lange, T. (2015). 53BP1 and the LINC complex promote microtubule-dependent DSB mobility and DNA repair. Cell, 163, 880–893.
Lukas, J., & Lukas, C. (2013). Molecular biology. Shielding broken DNA for a quick fix. Science, 339, 652–653.
Ma, S., Rong, Z., Liu, C., Qin, X., Zhang, X., & Chen, Q. (2021). DNA damage promotes microtubule dynamics through a DNA-PK-AKT axis for enhanced repair. Journal of Cell Biology. https://doi.org/10.1083/jcb.201911025
Marnef, A., & Legube, G. (2017). Organizing DNA repair in the nucleus: DSBs hit the road. Current Opinion in Cell Biology, 46, 1–8.
Matsuzawa, A., Kanno, S., Nakayama, M., Mochiduki, H., Wei, L., Shimaoka, T., Furukawa, Y., Kato, K., Shibata, S., Yasui, A., et al. (2014). The BRCA1/BARD1-interacting protein OLA1 functions in centrosome regulation. Molecular Cell, 53, 101–114.
Mennella, V., Agard, D. A., Huang, B., & Pelletier, L. (2014). Amorphous no more: Subdiffraction view of the pericentriolar material architecture. Trends in Cell Biology, 24, 188–197.
Mennella, V., Keszthelyi, B., McDonald, K. L., Chhun, B., Kan, F., Rogers, G. C., Huang, B., & Agard, D. A. (2012). Subdiffraction-resolution fluorescence microscopy reveals a domain of the centrosome critical for pericentriolar material organization. Nature Cell Biology, 14, 1159–1168.
Monnich, M., Borgeskov, L., Breslin, L., Jakobsen, L., Rogowski, M., Doganli, C., Schroder, J. M., Mogensen, J. B., Blinkenkjaer, L., Harder, L. M., et al. (2018). CEP128 localizes to the subdistal appendages of the mother centriole and regulates TGF-beta/BMP signaling at the primary cilium. Cell Reports, 22, 2584–2592.
Morrison, C. G. (2021). Primary cilia and the DNA damage response: Linking a cellular antenna and nuclear signals. Biochemical Society Transactions, 49, 829–841.
Mullee, L. I., & Morrison, C. G. (2016). Centrosomes in the DNA damage response–the hub outside the centre. Chromosome Research : An International Journal on the Molecular, Supramolecular and Evolutionary Aspects of Chromosome Biology, 24, 35–51.
Nigg, E. A. (2007). Centrosome duplication: Of rules and licenses. Trends in Cell Biology, 17, 215–221.
Nigg, E. A., & Holland, A. J. (2018). Once and only once: Mechanisms of centriole duplication and their deregulation in disease. Nature Reviews. Molecular Cell Biology, 19, 297–312.
Nishi, R., Okuda, Y., Watanabe, E., Mori, T., Iwai, S., Masutani, C., Sugasawa, K., & Hanaoka, F. (2005). Centrin 2 stimulates nucleotide excision repair by interacting with xeroderma pigmentosum group C protein. Molecular and Cellular Biology, 25, 5664–5674.
Ochi, T., Blackford, A. N., Coates, J., Jhujh, S., Mehmood, S., Tamura, N., Travers, J., Wu, Q., Draviam, V. M., Robinson, C. V., et al. (2015). DNA repair. PAXX, a paralog of XRCC4 and XLF, interacts with Ku to promote DNA double-strand break repair. Science, 347, 185–188.
Palazzo, R. E., Vogel, J. M., Schnackenberg, B. J., Hull, D. R., & Wu, X. (2000). Centrosome maturation. Current Topics in Developmental Biology, 49, 449–470.
Pan, Y. R., & Lee, E. Y. (2009). UV-dependent interaction between Cep164 and XPA mediates localization of Cep164 at sites of DNA damage and UV sensitivity. Cell Cycle, 8, 655–664.
Park, J., Welner, R. S., Chan, M. Y., Troppito, L., Staber, P. B., Tenen, D. G., & Yan, C. T. (2016). The DNA ligase IV syndrome R278H mutation impairs B lymphopoiesis via error-prone nonhomologous end-joining. Journal of Immunology, 196, 244–255.
Penfield, L., Wysolmerski, B., Mauro, M., Farhadifar, R., Martinez, M. A., Biggs, R., Wu, H. Y., Broberg, C., Needleman, D., & Bahmanyar, S. (2018). Dynein pulling forces counteract lamin-mediated nuclear stability during nuclear envelope repair. Molecular Biology of the Cell, 29, 852–868.
Poruchynsky, M. S., Komlodi-Pasztor, E., Trostel, S., Wilkerson, J., Regairaz, M., Pommier, Y., Zhang, X., Kumar Maity, T., Robey, R., Burotto, M., et al. (2015). Microtubule-targeting agents augment the toxicity of DNA-damaging agents by disrupting intracellular trafficking of DNA repair proteins. Proceedings of the National Academy of Sciences of the United States of America, 112, 1571–1576.
Pugacheva, E. N., Jablonski, S. A., Hartman, T. R., Henske, E. P., & Golemis, E. A. (2007). HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell, 129, 1351–1363.
Reiter, J. F., & Leroux, M. R. (2017). Genes and molecular pathways underpinning ciliopathies. Nature Reviews. Molecular Cell Biology, 18, 533–547.
Riballo, E., Kuhne, M., Rief, N., Doherty, A., Smith, G. C., Recio, M. J., Reis, C., Dahm, K., Fricke, A., Krempler, A., et al. (2004). A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Molecular Cell, 16, 715–724.
Rosin, N., Elcioglu, N. H., Beleggia, F., Isguven, P., Altmuller, J., Thiele, H., Steindl, K., Joset, P., Rauch, A., Nurnberg, P., et al. (2015). Mutations in XRCC4 cause primary microcephaly, short stature and increased genomic instability. Human Molecular Genetics, 24, 3708–3717.
Rothballer, A., & Kutay, U. (2013). The diverse functional LINCs of the nuclear envelope to the cytoskeleton and chromatin. Chromosoma, 122, 415–429.
Roukos, V., Voss, T. C., Schmidt, C. K., Lee, S., Wangsa, D., & Misteli, T. (2013). Spatial dynamics of chromosome translocations in living cells. Science, 341, 660–664.
Saji, M., Vasko, V., Kada, F., Allbritton, E. H., Burman, K. D., & Ringel, M. D. (2005). Akt1 contains a functional leucine-rich nuclear export sequence. Biochemical and Biophysical Research Communications, 332, 167–173.
Saladino, C., Bourke, E., Conroy, P. C., & Morrison, C. G. (2009). Centriole separation in DNA damage-induced centrosome amplification. Environmental and Molecular Mutagenesis, 50, 725–732.
Schrank, B. R., Aparicio, T., Li, Y., Chang, W., Chait, B. T., Gundersen, G. G., Gottesman, M. E., & Gautier, J. (2018). Nuclear ARP2/3 drives DNA break clustering for homology-directed repair. Nature, 559, 61–66.
Shen, K., Wang, Y., Brooks, S. C., Raz, A., & Wang, Y. A. (2006). ATM is activated by mitotic stress and suppresses centrosome amplification in primary but not in tumor cells. Journal of Cellular Biochemistry, 99, 1267–1274.
Shokrollahi, M., & Mekhail, K. (2021). Interphase microtubules in nuclear organization and genome maintenance. Trends in Cell Biology, 31, 721–731.
Sivasubramaniam, S., Sun, X., Pan, Y. R., Wang, S., & Lee, E. Y. (2008). Cep164 is a mediator protein required for the maintenance of genomic stability through modulation of MDC1, RPA, and CHK1. Genes & Development, 22, 587–600.
Starr, D. A. (2011). KASH and SUN proteins. Current Biology : CB, 21, R414-415.
Suizu, F., Hirata, N., Kimura, K., Edamura, T., Tanaka, T., Ishigaki, S., Donia, T., Noguchi, H., Iwanaga, T., & Noguchi, M. (2016). Phosphorylation-dependent Akt-Inversin interaction at the basal body of primary cilia. The EMBO Journal, 35, 1346–1363.
Sun, S., Fisher, R. L., Bowser, S. S., Pentecost, B. T., & Sui, H. (2019). Three-dimensional architecture of epithelial primary cilia. Proceedings of the National Academy of Sciences of the United States of America, 116, 9370–9379.
Tian, Y., Wei, C., He, J., Yan, Y., Pang, N., Fang, X., Liang, X., & Fu, J. (2021). Superresolution characterization of core centriole architecture. The Journal of Cell Biology. https://doi.org/10.1083/jcb.202005103
Tibelius, A., Marhold, J., Zentgraf, H., Heilig, C. E., Neitzel, H., Ducommun, B., Rauch, A., Ho, A. D., Bartek, J., & Kramer, A. (2009). Microcephalin and pericentrin regulate mitotic entry via centrosome-associated Chk1. The Journal of Cell Biology, 185, 1149–1157.
Villumsen, B. H., Danielsen, J. R., Povlsen, L., Sylvestersen, K. B., Merdes, A., Beli, P., Yang, Y. G., Choudhary, C., Nielsen, M. L., Mailand, N., et al. (2013). A new cellular stress response that triggers centriolar satellite reorganization and ciliogenesis. The EMBO Journal, 32, 3029–3040.
Wakefield, J. G., Stephens, D. J., & Tavare, J. M. (2003). A role for glycogen synthase kinase-3 in mitotic spindle dynamics and chromosome alignment. Journal of Cell Science, 116, 637–646.
Wang, C. Y., Huang, E. Y., Huang, S. C., & Chung, B. C. (2015). DNA-PK/Chk2 induces centrosome amplification during prolonged replication stress. Oncogene, 34, 1263–1269.
Wang, C. Y., Kao, Y. H., Lai, P. Y., Chen, W. Y., & Chung, B. C. (2013). Steroidogenic factor 1 (NR5A1) maintains centrosome homeostasis in steroidogenic cells by restricting centrosomal DNA-dependent protein kinase activation. Molecular and Cellular Biology, 33, 476–484.
Wang, G., Chen, Q., Zhang, X., Zhang, B., Zhuo, X., Liu, J., Jiang, Q., & Zhang, C. (2013). PCM1 recruits Plk1 to the pericentriolar matrix to promote primary cilia disassembly before mitotic entry. Journal of Cell Science, 126, 1355–1365.
Woodbine, L., Gennery, A. R., & Jeggo, P. A. (2014). The clinical impact of deficiency in DNA non-homologous end-joining. DNA Repair, 16, 84–96.
Yoshino, Y., Qi, H., Fujita, H., Shirota, M., Abe, S., Komiyama, Y., Shindo, K., Nakayama, M., Matsuzawa, A., Kobayashi, A., et al. (2018). BRCA1-interacting protein OLA1 requires interaction with BARD1 to regulate centrosome number. Molecular Cancer Research, 16, 1499–1511.
Zhang, J., Xu, J., Wang, G., Sun, P., Yan, T., & Zhao, X. (2016). WTIP interacts with BRCA2 and is essential for BRCA2 centrosome localization in cervical cancer cell. Archives of Gynecology and Obstetrics, 294, 1311–1316.
Zhao, J., Zou, Y., Liu, H., Wang, H., Zhang, H., Hou, W., Li, X., Jia, X., Zhang, J., Hou, L., et al. (2014). TEIF associated centrosome activity is regulated by EGF/PI3K/Akt signaling. Biochimica Et Biophysica Acta, 1843, 1851–1864.
Zheng, P., Chen, Q., Tian, X., Qian, N., Chai, P., Liu, B., Hu, J., Blackstone, C., Zhu, D., Teng, J., et al. (2018). DNA damage triggers tubular endoplasmic reticulum extension to promote apoptosis by facilitating ER-mitochondria signaling. Cell Research, 28, 833–854.
Zhu, S., Paydar, M., Wang, F., Li, Y., Wang, L., Barrette, B., Bessho, T., Kwok, B. H., & Peng, A. (2020). Kinesin Kif2C in regulation of DNA double strand break dynamics and repair. eLife. https://doi.org/10.7554/eLife.53402
Funding
Q. Chen is supported by Grants from the National Key Research and Development Program of China (2018YFC1003400), the National Natural Science Foundation of China (32170698, 31770868).
Author information
Dingwei Li and Xiuwen Liu have contributed equally.
Affiliations
Department of Radiation and Medical Oncology, Medical Research Institute, Zhongnan Hospital of Wuhan University, Wuhan University, Wuhan, 430071, People’s Republic of China
Dingwei Li & Qiang Chen
Frontier Science Center for Immunology and Metabolism, Medical Research Institute, Wuhan University, Wuhan, People’s Republic of China
Dingwei Li & Qiang Chen
Department of Intensive Care Medicine, Dongping County People’s Hospital, Dongping County, Tai’an, 271500, Shandong Province, People’s Republic of China
Xiuwen Liu
Corresponding author
Correspondence to Qiang Chen.
Rights and permissions
About this article
Cite this article
Li, D., Liu, X. & Chen, Q. Centrosome, microtubule and DNA damage response. GENOME INSTAB. DIS. (2022). https://doi.org/10.1007/s42764-022-00068-z
Received24 January 2022
Revised17 February 2022
Accepted25 February 2022
Published22 March 2022
DOIhttps://doi.org/10.1007/s42764-022-00068-z
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable link
用户登录
还没有账号?
立即注册