






DNA damage accumulation in aging brain and its links to Alzheimer’s disease progression
Review Article
Genome Instability & Disease (2022)
Abstract
DNA, which carries information to build the entire human body, is constantly challenged by a variety of endogenous and environmental agents, ultimately leading to mutations and genomic instability, triggering a wide range of cellular responses and causing diverse human diseases. Cells have evolved a variety of DNA repair mechanisms to respond to DNA damage. Recent findings have linked DNA damage in the brain with many neurological diseases. In this review, we discuss how DNA damage in the brain correlates with Alzheimer’s disease and how the absence of repair mechanisms accelerates age-related Alzheimer’s disease progression. We also review the potential sources of DNA damage, which may be closely associated with cognitive decline in Alzheimer’s disease.
Introduction
The global population aging trend is increasingly obvious. Aging has caused many health and societal problems, which have in turn caused wide concern among all of society. Alzheimer’s disease (AD) is an aging-related neurodegenerative disease with insidious onset and progressive development. Clinically, AD is characterized by memory impairment, aphasia, apraxia, agnosia, visuospatial skill impairment, executive dysfunction, and personality and behavior changes (Masters et al., 2015). Approximately, 50 million people worldwide suffer from AD (Gauthier et al., 2021). As society advances and life expectancy increases, the prevalence of AD will continue to rise, and the number of people with AD is expected to reach 150 million by 2050, which will bring heavy financial and care burdens to families and society (Ren Rujing & Zhihui, 2021).
Aging is well known to be closely related to DNA damage. When DNA is replicated, fragment loss, fractures and other errors inevitably occur that lead to DNA sequence changes. Endogenous chemicals and exogenous environmental factors continue to threaten the stability of cellular genetic material, resulting in a variety of DNA damage events. These DNA damage events may be caused by the effects of ultraviolet radiation, ionizing radiation, plant toxins, toxic agents and reactive oxygen species (ROS) (Finkel & Holbrook, 2000). Up to 105 DNA lesions occur daily in active mammalian cells, and it is estimated that spontaneous hydrolysis alone produces approximately 104 abasic (mostly apurinic) sites (Lindahl, 1993). Researchers found that DNA damage caused by ionizing radiation can induce senescence in human fibroblasts in cell culture (Wahl & Carr, 2001), suggesting that DNA damage may be directly related with senescence and aging processes.
With increasing age, the DNA scaffolding proteins that help stabilize the genome become less effective, which leads to increased DNA damage (Chen et al., 2007). To combat DNA damage, cells have evolved complex and finely regulated DNA damage repair (DDR) mechanisms to repair the different types of DNA damage, such as base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), non-homologous end joining (NHEJ) and homologous recombination (HR). However, DDR also declines with aging. Impaired DDR makes small mistakes into a large mistake, and the temporary errors become permanent. These errors accumulate to a certain degree and can cause substantial damage, eventually leading to a decline in human body function and further inhibiting DNA repair ability, thus leading to a vicious cycle. This vicious cycle happens in many human systems. In this review, we discuss DNA damage in the aging brain and its relationship to AD as well as the intrinsic interaction between DDR malfunctions and AD.
DNA damage in the aging brain and its correlation with age-related neurodegenerative diseases
Aging affects all organs of the body as the basis for neurodegeneration and dementia (Wyss-Coray, 2016). AD and Parkinson’s disease (PD) are the most common neurodegenerative diseases among elderly individuals. At the same time, with increasing age, the risk of these diseases is greatly increased (Wyss-Coray, 2016). With the aging of the human body and the declines in DDR, DNA damage gradually accumulates in human cells. Recent findings have suggested that a significant portion of aging phenotypes are associated with DNA damage and are potentially the ultimate cause of aging (Schumacher et al., 2021). DNA damage and genomic instability activate DDR response pathways, triggering cell cycle arrest, apoptosis, and cell senescence, thus accelerating the aging process of individual organisms. In all types of mouse tissue, a marker for DNA double-strand breaks (DSBs), phosphorylation of the Ser-139 residue of the histone variant H2AX (γH2AX), has been observed to accumulate in tissue with age (Sedelnikova et al., 2004b).
For over 20 years, 8-hydroxyguanine (8-OHG), a marker of DNA oxidation levels, has been observed to be elevated, while DNA repair levels are decreased, in the brain and cerebrospinal fluid of older adults and AD patients (Lovell et al., 1999; Mecocci et al., 1994). The oxidative DNA damage level was increased in patients with mild cognitive impairment (MCI) as well as in patients with severe dementia. In these patients, oxidative DNA damage to mitochondrial DNA (mtDNA) and nuclear DNA (nDNA) was observed in various brain regions of the patients (Wang, 2006). Meanwhile, as AD progresses, the level of proteins involved in DNA repair decreases. For instance, BER is one of the major DNA repair pathways and plays an important role in the development and maintenance of the nervous system. Significant BER defects were observed in the brains of AD patients compared with the brains of healthy controls, and the activity of BER-related proteins such as uracil DNA glycosylase, β-ogG1 glycosylase and DNA polymerase β (Polβ) was significantly reduced and inversely proportional to disease severity (Weissman et al., 2007a, 2007b). The levels of γH2AX in astrocytes and neurons in the hippocampus and frontal cortex of AD brains were significantly increased compared to those in normal brains, suggesting that more DSBs had accumulated in AD brains (Shanbhag et al., 2019).
Telomeres are the protective ends of DNA that are crucial to the integrity and stability of the genome. Cells with incomplete chromosomes tend to activate bypasses, leading to cell cycle arrest or death. Shortened telomere length is an indicator of aging cells and predicts an increased risk of age-related diseases. Reductions in telomere length have also been observed in patients with AD. Patients with slow-progressing late-onset AD had shorter telomeres than patients with fast-progressing disease or healthy elderly controls (Gauthier et al., 2021). These results suggest that DNA damage plays an important role in the progression of AD.
The accumulation of H3K4me2 can reportedly restore the balance of gene transcription and prolong the life span of C. elegans after restoring the blocked transcription of C. elegans (Wang et al., 2020). The poly-ADP ribosylation of histones plays a key role in DNA repair. Poly-ADP ribosylation can reduce the concentration of NAD + , induce apoptosis or indirectly inhibit sirtuin protein, which can affect genome-wide chromatin acetylation, senescence and DNA repair (Fang et al., 2014) and cause changes in gene expression in the brains of elderly mice (Oberdoerffer et al., 2008).
DNA DSB repair in AD
The accumulation of DNA double-strand damage has been observed in aging mice and human cells (Sedelnikova et al., 2004a). In AD model mice, it has been observed that exploring new environments leads to increased DNA double-strand damage in several brain regions, especially those involved in spatial learning and memory pairs (Suberbielle et al., 2013), and elevated levels of γ-H2AX in hippocampal and cortical regions in the brains of AD patients (Myung et al., 2008; Shanbhag et al., 2019), while a trend toward elevation has been found in the brains of patients with MCI in AD (Kirova et al., 2015; Korolev et al., 2016), which suggests that DNA double-strand damage may play an important role in the AD disease process, be an early event and lead to the development of neuronal tangles later in life (Su et al., 1997). In addition to the damage caused by ROS, it has been reported in recent years that DNA double-strand damage may also be associated with neuronal transcriptional activity (Marnef et al., 2017) and that these transcriptional activities occur during learning and memory (Cholewa-Waclaw et al., 2016; West & Greenberg, 2011), suggesting that DNA DSB may be associated with the loss of learning and memory capacity that occurs during AD.
Two major mechanistically distinct pathways exist for DSB repair in mammalian cells: HR and NHEJ. After DNA double-strand damage occurs, the MRN complex (MER11A-NBS1-RAD50) recognizes and binds to the damaged site and recruits ATM to activate DNA double-strand damage repair. BRCA1-dependent excision of 5′ DNA flanking DNA double-stranded damage sites exposes two single-stranded DNA regions where BRCA2 recruits the DNA recombinase RAD51, which binds to DNA to form nucleoprotein filaments and uses homologous DNA as a template, synthesize new DNA with the help of DNA ligase and endonuclease. Eventually, DNA double-strand damage is repaired (Lord & Ashworth, 2016). The protein expression levels and mRNA levels of key HR factors were observed to decrease in the brains of AD patients and AD mouse models (Shen et al., 2016), indicating HR deficiency may play a role in AD initiation and/or progression. In hAPP-J20 mice, the level of BRCA1 was significantly decreased in the DG region of the hippocampus and Aβ oligomers also could reduce the level of BRCA1 in mouse primary neurons (Suberbielle et al., 2013). DNA double-strand damage also recruits Ku70/Ku80 complexes, which in turn activate and recruit DNA-PKcs to the damage sites, initiating DSB repair via the 53BP1-mediated NHEJ repair pathway. The NHEJ key factor Ku subunit and DNA-PKcs and poly(ADP-ribose) polymerase-1 (PARP1) were found to be absent in cortical regions in the brains of AD (postmortem) patients and may be associated with synaptic loss and memory impairment in AD (Davydov et al., 2003). In PC12 cells, the aggregated Aβ short peptide inhibits DNA-PKcs enzymatic activity while disrupting DSB repair (Culmsee et al., 2001). In addition, the reduction in RAD51 in the hippocampal region of APP/PSEN1 mice was more significant than that in 53BP1 in the AD transgenic mouse model (Yu et al., 2018), suggesting that the HR pathway may play a more important role than other double-strand damage repair pathways in AD.
Defects in BER in AD
The daily production of single-strand breaks (SSBs) is much higher in mammalian cells than that of DSBs (Lindahl, 1993; Vilenchik & Knudson, 2003). The brain is protected from external or environmental genotoxins by the blood–brain barrier, while neurons in the central nervous system have a high metabolic rate (Hegde et al., 2012), and the brain accounts for up to 50% of systemic oxygen consumption as well as 10% of glucose consumption (Magistretti & Pellerin, 1999; Shulman et al., 2004); therefore, endogenous ROS may be a major source of DNA damage in the brains of AD patients (Barja, 2004; Chandrika Canugovi et al., 2014). Oxidative DNA damage has also been observed in the brains of MCI (Bradley-Whitman et al., 2014; Lyras et al., 1997; Wang et al., 2006), preclinical AD (PCAD) (Bradley-Whitman et al., 2014; Lovell et al., 2011) and AD (Gabbita et al., 1998; Lyras et al., 1997) patients.
The repair of oxidative damage to base pairs occurs mainly through the BER pathway. One of the pathways is short-patch BER, where DNA glycosylases recognize and excise bases at the damage site to obtain an abasic site, which is then excised by apurinic/apyrimidinic endonuclease (APE), causing an SSB. This site is excised by Polβ, which then introduces a normal nucleotide, and finally, the normal duplex is obtained by ligating it through the Lig III-XRCC1 complex. Another BER pathway is the NEIL1-3 DNA glycosylase long-patch BER repair pathway. ROS can produce SSBs, which are recognized by poly(ADP) ribose polymerase 1 (PARP1); the ends are then treated with PNKP, actinic dysfunctional ataxia (APTX) or tyrosyl DNA phosphodiesterase 1 (TDP1). Repair is then performed with short-patch BER or long-patch BER (Coppedè et al., 2007; Hart et al., 2001; Weissman, et al., 2007a, 2007b).
Impaired BER pathway function in MCI brains suggests that BER may have an impact on the AD disease process, and reduced 8-oxoguanine (8-oxoG) DNA glycosylase activity (Lovell et al., 2000) and POLβ expression levels (Weissman, et al., 2007a, 2007b) were found in different brain regions of AD brains compared to normal aged brains. For AD brains, the patients were found to carry OCG1 mutations, with different mutations leading to impaired 8-oxoG DNA glycosylase activity (Mao et al., 2007), altered PRP1 and XRCC1 interactions, reduced ability to bind substrates, and reduced AP lyase activity (Jacob et al., 2013).
PARP1, a key protein for SSB recognition, has also been found to be functionally impaired in AD brains and mouse models. In the CA3 region of the adult rat brain, Aβ can activate PARP1 by increasing oxidative stress (Strosznajder et al., 2000), and the introduction of exogenous NAD+ can alleviate Aβ-induced DNA damage (Wu et al., 2014)]. PARP1 can also promote AD disease development by affecting metabolism, and PARP1 activity has been found to be associated with elevated PAR levels in AD brains (Love et al., 1999; Strosznajder et al., 2012).
In 3xTg-AD mice, knocking down POLβ leads to neuronal death, memory loss and impaired synaptic plasticity accompanied by increased DNA damage (Sykora et al., 2015). Recent reports have also focused on the role of mitochondrial BER in AD (Canugovi et al., 2014; Santos et al., 2013).
Tau and DNA damage
Tau is a microtubule-associated protein encoded by MAPT (Houda Benhelli-Mokrani et al., 2018a, 2018b) and is thought to be localized in the cytoplasm. Tau was shown to react with nucleic acids and localize in the nucleus of neuronal cells (Brady et al., 1995; Bukar Maina et al., 2016; Frost et al., 2014; Greenwood & Johnson, 1995; Lu et al., 2014). In mouse primary neurons, tau can bind to chromatin, and this binding is dynamically variable and regulated by cellular stress (H. Benhelli-Mokrani et al., 2018a, 2018b). In addition, in primary neurons, Tau can participate in the DDR process, and in the absence of Tau, the level of γ-H2AX is elevated (Violet et al., 2014). Although the mechanism is not clear, these results suggest that Tau may facilitate the process of cellular DDR. A similar trend was found in the mouse brain, where both DNA DSBs and γ-H2AX were at higher levels and DNA repair processes were impaired in Tau knocked out mice (Violet et al., 2014). A shift in the localization of Tau to the nucleus along with a decrease in its phosphorylation level was also observed upon the induction of DNA damage in cells (Ulrich et al., 2018).
Tau oligomers may also affect DDR (Violet et al., 2015). In mouse models, the presence of Tau oligomers is accompanied by an increase in Polβ expression (Zheng et al., 2020). Tau multimers have also been reported to alter the localization of key proteins of the DDR pathway, such as BRCA1 and 53BP1 (Kurihara et al., 2019; Nakamura et al., 2020), but the exact mechanism is still unclear.
Conclusion
Here, we reviewed recent studies on DNA damage in age-related AD, which focused on DNA damage in AD and the impact of DDR on disease progression. Maintaining the integrity of the genome is crucial for the physiology of any nucleated cell. Oxidized DNA is believed to be the most common DNA lesion in neurons, which is unavoidable due to their high metabolic activity. Aside from oxidized DNA, a second major threat to neuronal genomes comes from DNA DSBs arising from the development and plasticity of the brain.
It is now increasingly clear that the DNA damage response plays an important role in the AD process. Mutations or deletions in core DDR proteins may accelerate the development of AD disease. On the other hand, the link between DDR and brain aging and neurodegenerative diseases remains complicated. Several similar results showed that oxidative DNA damage may originate from environmental neurotoxin threats (Migliore & Coppedè, 2009), may be associated with aging-related functional deficits (Coppedè & Migliore, 2010), or may be the result of neurodegeneration (Coppedè & Migliore, 2010; Taupin, 2011). DNA damage undoubtedly exacerbates the process of aging and neurodegenerative diseases; therefore, studies are necessary to explore the specific underlying mechanisms of damage and the repair involved (Fig. 1).
Fig. 1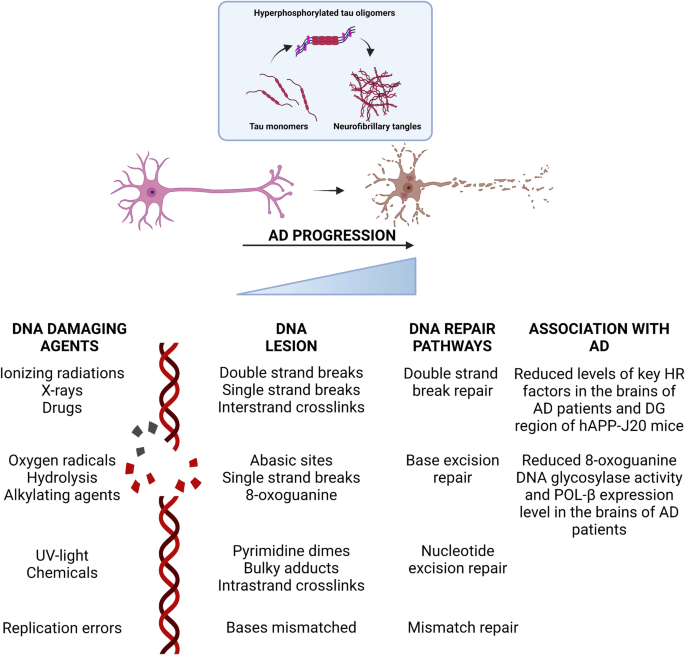
DNA damage repair pathways and their potential relationship with AD
Full size imageReferences
Barja, G. (2004). Free radicals and aging. Trends in Neurosciences, 27(10), 595–600. https://doi.org/10.1016/j.tins.2004.07.005
Benhelli-Mokrani, H., Mansuroglu, Z., Chauderlier, A., Albaud, B., Gentien, D., Sommer, S., Schirmer, C., Laqueuvre, L., Josse, T., Buée, L., Lefebvre, B., Galas, M.-C., Souès, S., & Bonnefoy, E. (2018a). Genome-wide identification of genic and intergenic neuronal DNA regions bound by Tau protein under physiological and stress conditions. Nucleic Acids Research. https://doi.org/10.1093/nar/gky929
Benhelli-Mokrani, H., Mansuroglu, Z., Chauderlier, A., Albaud, B., Gentien, D., Sommer, S., Schirmer, C., Laqueuvre, L., Josse, T., Buée, L., Lefebvre, B., Galas, M. C., Souès, S., & Bonnefoy, E. (2018b). Genome-wide identification of genic and intergenic neuronal DNA regions bound by Tau protein under physiological and stress conditions. Nucleic Acids Research, 46(21), 11405–11422. https://doi.org/10.1093/nar/gky929
Bradley-Whitman, M. A., Timmons, M. D., Beckett, T. L., Murphy, M. P., Lynn, B. C., & Lovell, M. A. (2014). Nucleic acid oxidation: An early feature of Alzheimer’s disease. Journal of Neurochemistry, 128(2), 294–304. https://doi.org/10.1111/jnc.12444
Brady, R. M., Zinkowski, R. P., & Binder, L. I. (1995). Presence of tau in isolated nuclei from human brain. Neurobiology of Aging, 16(3), 479–486. https://doi.org/10.1016/0197-4580(95)00023-8
BukarMaina, M., Al-Hilaly, Y. K., & Serpell, L. C. (2016). Nuclear Tau and its potential role in Alzheimer’s disease. Biomolecules, 6(1), 9. https://doi.org/10.3390/biom6010009
Canugovi, C., Shamanna, R. A., Croteau, D. L., & Bohr, V. A. (2014a). Base excision DNA repair levels in mitochondrial lysates of Alzheimer’s disease. Neurobiology of Aging, 35(6), 1293–1300. https://doi.org/10.1016/j.neurobiolaging.2014.01.004
Chen, J. H., Hales, C. N., & Ozanne, S. E. (2007). DNA damage, cellular senescence and organismal ageing: Causal or correlative? Nucleic Acids Research, 35(22), 7417–7428. https://doi.org/10.1093/nar/gkm681
Cholewa-Waclaw, J., Bird, A., von Schimmelmann, M., Schaefer, A., Yu, H. M., Song, H. J., Madabhushi, R., & Tsai, L. H. (2016). The role of epigenetic mechanisms in the regulation of gene expression in the nervous system. Journal of Neuroscience, 36(45), 11427–11434. https://doi.org/10.1523/JNEUROSCI.2492-16.2016
Coppedè, F., Mancuso, M., Lo Gerfo, A., Manca, M. L., Petrozzi, L., Migliore, L., Siciliano, G., & Murri, L. (2007). A Ser326Cys polymorphism in the DNA repair gene hOGG1 is not associated with sporadic Alzheimer’s disease. Neuroscience Letters, 414(3), 282–285. https://doi.org/10.1016/j.neulet.2006.12.035
Coppedè, F., & Migliore, L. (2010). DNA repair in premature aging disorders and neurodegeneration. Current Aging Science, 3(1), 3–19. https://doi.org/10.2174/1874609811003010003
Culmsee, C., Bondada, S., & Mattson, M. P. (2001). Hippocampal neurons of mice deficient in DNA-dependent protein kinase exhibit increased vulnerability to DNA damage, oxidative stress and excitotoxicity. Molecular Brain Research, 87(2), 257–262. https://doi.org/10.1016/S0169-328X(01)00008-0
Davydov, V., Hansen, L. A., & Shackelford, D. A. (2003). Is DNA repair compromised in Alzheimer’s disease? Neurobiology of Aging, 24(7), 953–968. https://doi.org/10.1016/S0197-4580(02)00229-4
Fang, E. F., Scheibye-Knudsen, M., Brace, L. E., Kassahun, H., SenGupta, T., Nilsen, H., Mitchell, J. R., Croteau, D. L., & Bohr, V. A. (2014). Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell, 157(4), 882–896. https://doi.org/10.1016/j.cell.2014.03.026
Finkel, T., & Holbrook, N. J. (2000). Oxidants, oxidative stress and the biology of ageing. Nature, 408(6809), 239–247. https://doi.org/10.1038/35041687
Frost, B., Hemberg, M., Lewis, J., & Feany, M. B. (2014). Tau promotes neurodegeneration through global chromatin relaxation. Nature Neuroscience, 17(3), 357–366. https://doi.org/10.1038/nn.3639
Gabbita, S. P., Lovell, M. A., & Markesbery, W. R. (1998). Increased nuclear DNA oxidation in the brain in Alzheimer’s disease. Journal of Neurochemistry, 71(5), 2034–2040. https://doi.org/10.1046/j.1471-4159.1998.71052034.x
Gauthier, S., Rosa-Neto, P., Morais, J. A., & Webster, C. (2021). World Alzheimer Report 2021: journey through the diagnosis of dementia. London, England: Alzheimer’s Disease International
Greenwood, J. A., & Johnson, G. V. (1995). Localization and in situ phosphorylation state of nuclear tau. Experimental Cell Research, 220(2), 332–337. https://doi.org/10.1006/excr.1995.1323
Hart, B. A., Potts, R. J., & Watkin, R. D. (2001). Cadmium adaptation in the lung—a double-edged sword? Toxicology, 160(1–3), 65–70. https://doi.org/10.1016/s0300-483x(00)00436-4
Hegde, M. L., Mantha, A. K., Hazra, T. K., Bhakat, K. K., Mitra, S., & Szczesny, B. (2012). Oxidative genome damage and its repair: Implications in aging and neurodegenerative diseases. Mechanisms of Ageing and Development, 133(4), 157–168. https://doi.org/10.1016/j.mad.2012.01.005
Jacob, K. D., Noren Hooten, N., Tadokoro, T., Lohani, A., Barnes, J., & Evans, M. K. (2013). Alzheimer’s disease-associated polymorphisms in human OGG1 alter catalytic activity and sensitize cells to DNA damage. Free Radical Biology and Medicine, 63, 115–125. https://doi.org/10.1016/j.freeradbiomed.2013.05.010
Kirova, A.-M., Bays, R. B., & Lagalwar, S. (2015). Working memory and executive function decline across normal aging, mild cognitive impairment, and Alzheimer’s disease. BioMed Research International, 2015, 1–9. https://doi.org/10.1155/2015/748212
Korolev, I. O., Symonds, L. L., & Bozoki, A. C. (2016). Predicting progression from mild cognitive impairment to alzheimer’s dementia using clinical, MRI, and plasma biomarkers via probabilistic pattern classification. PLoS ONE, 11(2), e0138866. https://doi.org/10.1371/journal.pone.0138866
Kurihara, M., Mano, T., Saito, Y., Murayama, S., Toda, T., & Iwata, A. (2019). Colocalization of BRCA1 with Tau aggregates in human tauopathies. Brain Science. https://doi.org/10.3390/brainsci10010007
Lindahl, T. (1993). Instability and decay of the primary structure of DNA. Nature, 362(6422), 709–715. https://doi.org/10.1038/362709a0
Lord, C. J., & Ashworth, A. (2016). BRCAness revisited. Nature Reviews Cancer, 16(2), 110–120. https://doi.org/10.1038/nrc.2015.21
Love, S., Barber, R., & Wilcock, G. K. (1999). Increased poly(ADP-ribosyl)ation of nuclear proteins in Alzheimer’s disease. Brain, 122, 247–253. https://doi.org/10.1093/brain/122.2.247
Lovell, M. A., Gabbita, S. P., & Markesbery, W. R. (1999). Increased DNA oxidation and decreased levels of repair products in Alzheimer’s disease ventricular CSF. Journal of Neurochemistry, 72(2), 771–776. https://doi.org/10.1046/j.1471-4159.1999.0720771.x
Lovell, M. A., Soman, S., & Bradley, M. A. (2011). Oxidatively modified nucleic acids in preclinical Alzheimer’s disease (PCAD) brain. Mechanisms of Ageing and Development, 132(8–9), 443–448. https://doi.org/10.1016/j.mad.2011.08.003
Lovell, M. A., Xie, C. S., & Markesbery, W. R. (2000). Decreased base excision repair and increased helicase activity in Alzheimer’s disease brain. Brain Research, 855(1), 116–123. https://doi.org/10.1016/S0006-8993(99)02335-5
Lu, J., Li, T., He, R., Bartlett, P. F., & Götz, J. (2014). Visualizing the microtubule-associated protein tau in the nucleus. Science China Life Science, 57(4), 422–431. https://doi.org/10.1007/s11427-014-4635-0
Lyras, L., Cairns, N. J., Jenner, A., Jenner, P., & Halliwell, B. (1997). An assessment of oxidative damage to proteins, lipids, and DNA in brain from patients with Alzheimer’s disease. Journal of Neurochemistry, 68(5), 2061–2069.
Magistretti, P. J., & Pellerin, L. (1999). Cellular mechanisms of brain energy metabolism and their relevance to functional brain imaging. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, 354(1387), 1155–1163. https://doi.org/10.1098/rstb.1999.0471
Mao, G. G., Pan, X. Y., Zhu, B. B., Zhang, Y. B., Yuan, F. H., Huang, J., Lovell, M. A., Lee, M. P., Markesbery, W. R., Li, G. M., & Gu, L. Y. (2007). Identification and characterization of OGG1 mutations in patients with Alzheimer’s disease. Nucleic Acids Research, 35(8), 2759–2766. https://doi.org/10.1093/nar/gkm189
Marnef, A., Cohen, S., & Legube, G. (2017). Transcription-coupled DNA double-strand break repair: active genes need special care. Journal of Molecular Biology, 429(9), 1277–1288. https://doi.org/10.1016/j.jmb.2017.03.024
Masters, C. L., Bateman, R., Blennow, K., Rowe, C. C., Sperling, R. A., & Cummings, J. L. (2015). Alzheimer’s disease. Nature Reviews Disease Primers, 1, 15056. https://doi.org/10.1038/nrdp.2015.56
Mecocci, P., MacGarvey, U., & Beal, M. F. (1994). Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Annals of Neurology, 36(5), 747–751. https://doi.org/10.1002/ana.410360510
Migliore, L., & Coppedè, F. (2009). Environmental-induced oxidative stress in neurodegenerative disorders and aging. Mutation Research, 674(1–2), 73–84. https://doi.org/10.1016/j.mrgentox.2008.09.013
Myung, N.-H., Zhu, X., Kruman, I. I., Castellani, R. J., Petersen, R. B., Siedlak, S. L., Perry, G., Smith, M. A., & Lee, H.-G. (2008). Evidence of DNA damage in Alzheimer disease: Phosphorylation of histone H2AX in astrocytes. Age, 30(4), 209–215. https://doi.org/10.1007/s11357-008-9050-7
Nakamura, M., Kaneko, S., Dickson, D. W., & Kusaka, H. (2020). Aberrant accumulation of BRCA1 in Alzheimer disease and other tauopathies. Journal of Neuropathology and Experimental Neurology, 79(1), 22–33. https://doi.org/10.1093/jnen/nlz107
Oberdoerffer, P., Michan, S., McVay, M., Mostoslavsky, R., Vann, J., Park, S. K., Hartlerode, A., Stegmuller, J., Hafner, A., Loerch, P., Wright, S. M., Mills, K. D., Bonni, A., Yankner, B. A., Scully, R., Prolla, T. A., Alt, F. W., & Sinclair, D. A. (2008). SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell, 135(5), 907–918. https://doi.org/10.1016/j.cell.2008.10.025
Ren Rujing, Y. P., Zhihui, Q., et al. (2021). China Alzheimer’s disease report 2021. China Alzheimer’s Disease Report 2021, 20(04), 317–337.
Santos, R. X., Correia, S. C., Zhu, X., Smith, M. A., Moreira, P. I., Castellani, R. J., Nunomura, A., & Perry, G. (2013). Mitochondrial DNA oxidative damage and repair in aging and Alzheimer’s disease. Antioxidants & Redox Signaling, 18(18), 2444–2457. https://doi.org/10.1089/ars.2012.5039
Schumacher, B., Pothof, J., Vijg, J., & Hoeijmakers, J. H. J. (2021). The central role of DNA damage in the ageing process. Nature, 592(7856), 695–703. https://doi.org/10.1038/s41586-021-03307-7
Sedelnikova, O. A., Horikawa, I., Zimonjic, D. B., Popescu, N. C., Bonner, W. M., & Barrett, J. C. (2004a). Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nature Cell Biology, 6(2), 168. https://doi.org/10.1038/ncb1095
Sedelnikova, O. A., Horikawa, I., Zimonjic, D. B., Popescu, N. C., Bonner, W. M., & Barrett, J. C. (2004b). Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nature Cell Biology, 6(2), 168–170. https://doi.org/10.1038/ncb1095
Shanbhag, N. M., Evans, M. D., Mao, W., Nana, A. L., Seeley, W. W., Adame, A., Rissman, R. A., Masliah, E., & Mucke, L. (2019). Early neuronal accumulation of DNA double strand breaks in Alzheimer’s disease. Acta Neuropathologica Communications. https://doi.org/10.1186/s40478-019-0723-5
Shen, X., Chen, J., Li, J., Kofler, J., & Herrup, K. (2016). Neurons in vulnerable regions of the Alzheimer’s disease brain display reduced ATM signaling. Eneuro. https://doi.org/10.1523/eneuro.0124-15.2016 ENEURO.0124-0115.
Shulman, R. G., Rothman, D. L., Behar, K. L., & Hyder, F. (2004). Energetic basis of brain activity: Implications for neuroimaging. Trends in Neurosciences, 27(8), 489–495. https://doi.org/10.1016/j.tins.2004.06.005
Strosznajder, J. B., Czapski, G. A., Adamczyk, A., & Strosznajder, R. P. (2012). Poly(ADP-ribose) polymerase-1 in amyloid beta toxicity and Alzheimer’s disease. Molecular Neurobiology, 46(1), 78–84. https://doi.org/10.1007/s12035-012-8258-9
Strosznajder, J. B., Jesko, H., & Strosznajder, R. P. (2000). Effect of amyloid beta peptide on poly(ADP-ribose) polymerase activity in adult and aged rat hippocampus. Acta Biochimica Polonica, 47(3), 847–854. https://doi.org/10.18388/abp.2000_4003
Su, J. H., Deng, G., & Cotman, C. W. (1997). Neuronal DNA damage precedes tangle formation and is associated with up-regulation of nitrotyrosine in Alzheimer’s disease brain. Brain Research, 774(1–2), 193–199. https://doi.org/10.1016/s0006-8993(97)81703-9
Suberbielle, E., Sanchez, P. E., Kravitz, A. V., Wang, X., Ho, K., Eilertson, K., Devidze, N., Kreitzer, A. C., & Mucke, L. (2013). Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-beta. Nature Neuroscience, 16(5), 613–621. https://doi.org/10.1038/nn.3356
Sykora, P., Misiak, M., Wang, Y., Ghosh, S., Leandro, G. S., Liu, D., Tian, J., Baptiste, B. A., Cong, W. N., Brenerman, B. M., Fang, E., Becker, K. G., Hamilton, R. J., Chigurupati, S., Zhang, Y. Q., Egan, J. M., Croteau, D. L., Wilson, D. M., Mattson, M. P., & Bohr, V. A. (2015). DNA polymerase beta deficiency leads to neurodegeneration and exacerbates Alzheimer disease phenotypes. Nucleic Acids Research, 43(2), 943–959. https://doi.org/10.1093/nar/gku1356
Taupin, P. (2011). Neurogenesis, NSCs, pathogenesis and therapies for Alzheimer’s disease. Frontiers in Bioscience (scholar Edition), 3, 178–190. https://doi.org/10.2741/s143
Ulrich, G., Salvadè, A., Boersema, P., Calì, T., Foglieni, C., Sola, M., Picotti, P., Papin, S., & Paganetti, P. (2018). Phosphorylation of nuclear Tau is modulated by distinct cellular pathways. Science and Reports, 8(1), 17702. https://doi.org/10.1038/s41598-018-36374-4
Vilenchik, M. M., & Knudson, A. G. (2003). Endogenous DNA double-strand breaks: Production, fidelity of repair, and induction of cancer. Proceedings of the National Academy of Sciences, 100(22), 12871–12876. https://doi.org/10.1073/pnas.2135498100
Violet, M., Chauderlier, A., Delattre, L., Tardivel, M., Chouala, M. S., Sultan, A., Marciniak, E., Humez, S., Binder, L., Kayed, R., Lefebvre, B., Bonnefoy, E., Buée, L., & Galas, M. C. (2015). Prefibrillar Tau oligomers alter the nucleic acid protective function of Tau in hippocampal neurons in vivo. Neurobiology of Diseases, 82, 540–551. https://doi.org/10.1016/j.nbd.2015.09.003
Violet, M., Delattre, L., Tardivel, M., Sultan, A., Chauderlier, A., Caillierez, R., Talahari, S., Nesslany, F., Lefebvre, B., Bonnefoy, E., Buée, L., & Galas, M. C. (2014). A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Frontiers in Cellular Neuroscience, 8, 84. https://doi.org/10.3389/fncel.2014.00084
Wahl, G. M., & Carr, A. M. (2001). The evolution of diverse biological responses to DNA damage: Insights from yeast and p53. Nature Cell Biology, 3(12), E277-286. https://doi.org/10.1038/ncb1201-e277
Wang, J., Markesbery, W. R., & Lovell, M. A. (2006). Increased oxidative damage in nuclear and mitochondrial DNA in mild cognitive impairment. Journal of Neurochemistry, 96(3), 825–832. https://doi.org/10.1111/j.1471-4159.2005.03615.x
Wang, S., Meyer, D. H., & Schumacher, B. (2020). H3K4me2 regulates the recovery of protein biosynthesis and homeostasis following DNA damage. Nature Structural & Molecular Biology, 27(12), 1165–1177. https://doi.org/10.1038/s41594-020-00513-1
Weissman, L., Jo, D. G., Sorensen, M. M., de Souza-Pinto, N. C., Markesbery, W. R., Mattson, M. P., & Bohr, V. A. (2007a). Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucleic Acids Research, 35(16), 5545–5555. https://doi.org/10.1093/nar/gkm605
Weissman, L., Jo, D. G., Sørensen, M. M., de Souza-Pinto, N. C., Markesbery, W. R., Mattson, M. P., & Bohr, V. A. (2007b). Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucleic Acids Research, 35(16), 5545–5555. https://doi.org/10.1093/nar/gkm605
West, A. E., & Greenberg, M. E. (2011). Neuronal activity-regulated gene transcription in synapse development and cognitive function. Cold Spring Harbor Perspectives in Biology, 3(6), a005744–a005744. https://doi.org/10.1101/cshperspect.a005744
Wu, M. F., Yin, J. H., Hwang, C. S., Tang, C. M., & Yang, D. I. (2014). NAD attenuates oxidative DNA damages induced by amyloid beta-peptide in primary rat cortical neurons. Free Radical Research, 48(7), 794–805. https://doi.org/10.3109/10715762.2014.907889
Wyss-Coray, T. (2016). Ageing, neurodegeneration and brain rejuvenation. Nature, 539(7628), 180–186. https://doi.org/10.1038/nature20411
Yu, H., Harrison, F. E., & Xia, F. (2018). Altered DNA repair; an early pathogenic pathway in Alzheimer’s disease and obesity. Scientific Reports. https://doi.org/10.1038/s41598-018-23644-4
Zheng, J., Akbari, M., Schirmer, C., Reynaert, M. L., Loyens, A., Lefebvre, B., Buée, L., Croteau, D. L., Galas, M. C., & Bohr, V. A. (2020). Hippocampal tau oligomerization early in tau pathology coincides with a transient alteration of mitochondrial homeostasis and DNA repair in a mouse model of tauopathy. Acta Neuropathologica Communications, 8(1), 25. https://doi.org/10.1186/s40478-020-00896-8
Author information
Affiliations
Key Laboratory of Structural Biology of Zhejiang Province, School of Life Sciences, Westlake University, Hangzhou, 310024, Zhejiang, China
Han Zhao, Shiyao Wang & Xu Li
Westlake Laboratory of Life Sciences and Biomedicine, Hangzhou, 310024, Zhejiang, China
Han Zhao, Shiyao Wang & Xu Li
Corresponding author
Correspondence to Xu Li.
Rights and permissions
About this article
Cite this article
Zhao, H., Wang, S. & Li, X. DNA damage accumulation in aging brain and its links to Alzheimer’s disease progression. GENOME INSTAB. DIS. (2022). https://doi.org/10.1007/s42764-022-00069-y
Received23 January 2022
Revised26 February 2022
Accepted27 February 2022
Published23 March 2022
DOIhttps://doi.org/10.1007/s42764-022-00069-y
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
DNA damage repair
Alzheimer’s disease
Tau
用户登录
还没有账号?
立即注册