






The impact of TOPBP1 mutations in human cancers on the DNA damage response
Original Research Paper
Zhenzhen Yan, Xiuling Ge, Jiaxu Wang, Feng Xu, Mengyao Li, Xiuhua Liu, Xiaochun Yu & Chen Wu
Genome Instability & Disease 3, 144–162 (2022)
Abstract
TOPBP1 (Topoisomerase IIβ-binding protein 1) mediates protein–protein interaction and DNA damage response (DDR) activation in DNA damage sensing and signaling to maintain genome integrity. However, the cancer-associated mutations of TOPBP1 have not been well studied. Here, 369 variants of TOPBP1 across 31 types of human cancer from three databases: The Cancer Genome Atlas (TCGA), International Cancer Genome Consortium (ICGC) and Catalogue of Somatic Mutations in Cancer (COSMIC), were analyzed. We found that six truncation mutations of TOPBP1 impaired its ability to repair DNA lesions and activate damage checkpoint, and ten missense mutations impaired the recruitment of TOPBP1 to DNA damage sites. Therefore, DNA damage repair capacity and cell cycle arrest in G2/M phase were disrupted. The structural modeling also confirms that missense mutations of TOPBP1 change the local spatial structure, which may further abolish the function of TOPBP1 in DDR. Taken together, our study reveals the functional defects of cancer-associated TOPBP1 mutations in DDR and may provide new therapy targets for cancer treatment.
Introduction
Human DNA is exposed daily to various exogenous and endogenous stimuli, which can lead to tens of thousands of DNA lesions in each human cell per day (Jackson & Bartek, 2009; Mooser et al., 2020). These DNA lesions will result in mutations or genome aberrations that are associated with a variety of human disorders (Lord & Ashworth, 2012). Fortunately, cells have evolved a precise network of signaling pathway that is generally referred as DNA damage response (DDR) to detect the presence of DNA lesions, transduce signals and promote the damage repair (Ciccia & Elledge, 2010; Jackson & Bartek, 2009). In response to DNA damage or replication stress, a series of events are activated to induce the DDR. The “sensors” (MRE11-RAD50-NBS1 complex and Ku70/Ku80) recognize damage signals and recruit other DDR proteins to DNA damage sites (Harper & Elledge, 2007). “Transducers” act as functional partners of DNA damage “sensors” (T. Li & Chen, 2018) and transduce the damage signals to downstream mediators and effectors to trigger biological processes, including DNA damage repair, cell cycle arrest or apoptosis (Huang & Zhou, 2020). Kinase activity is commonly found in DDR transducers, such as ataxia-telangiectasia mutated (ATM), ataxia-telangiectasia mutated- and Rad3-related (ATR) and DNA-PK. Several BRCT (BRCA1 carboxyl-terminal) domains containing factors are addressed as major “mediators” to mediate the protein–protein interaction in DNA damage sensing and DDR signaling (Huang & Zhou, 2020). Among these factors, TOPBP1 (Topoisomerase IIβ-binding protein 1) is famous for the role of its BRCT repeats in modulating phosphoprotein scaffold (Kumagai et al., 2006).
TOPBP1 contains eight BRCT (BRCT1-BRCT8) domains and an ATR activating domain (AAD) between the BRCT6 and BRCT7 (Blackford et al., 2015). BRCT domains of TOPBP1 bind with different phosphorylated proteins to modulate diverse cellular processes, including DNA replication, DNA damage repair and cell cycle checkpoint (Wardlaw et al., 2014) (Feng et al., 2020). BRCT1/2 domains of TOPBP1 interact with phosphorylated RAD9, Treslin and MDC1 to activate checkpoint signaling, initiate DNA replication and maintain genome integrity (Delacroix et al., 2007; Leimbacher et al., 2019). BRCT4/5 motifs bind to phosphorylated 53BP1 or BLM to regulate DDR or prevent chromosomal aberrations (Blackford et al., 2015; Cescutti et al., 2010). BRCT7/8 domains of TOPBP1 interact with PLK to phosphorylate RAD51, which promotes RAD51 IRIF formation at double strand break (DSB) sites (Moudry et al., 2016). TOPBP1 also binds to phosphorylated FANCJ to regulate replication checkpoint control in a BRCT7/8 domains dependent way (Gong et al., 2010). In addition, TOPBP1 regulates the function of p53, which is mediated by an interaction between BRCT7/8 domains of TOPBP1 and the DNA-binding domain of p53, leading to inhibition of p53 promoter binding activity and aberrant proliferation as well as increased chemotherapy resistance (Liu et al., 2009). TOPBP1 is also an activator of ATR (Kumagai et al., 2006). The interaction between TOPBP1 and ATRIP can facilitate TOPBP1 to associate with ATR and activate ATR-Chk1 axis, which contributes to DNA damage response and DNA replication (Kumagai et al., 2006). ATR activation by mTOPBP1 relies on a conserved tryptophan residue that lies within the AAD (W1147). A point mutation in mTOPBP1 AAD (W1147R) results in cellular senescence and early embryonic lethality in mice (Zhou et al., 2013).
Maintaining genomic stability contributes to reducing cancer susceptibility (Blackford et al., 2015; Mooser et al., 2020; Paulsen et al., 2009; Santos-Pereira & Aguilera, 2015). It has been proposed that TOPBP1 plays essential roles in DNA damage repair and checkpoint signaling that are important for preserving genome stability (Sokka et al., 2010). During cell division, TOPBP1 is recruited to DSBs to accelerate DSBs repair through its interaction with MDC1 and disrupting MDC1-TOPBP1 interaction leads to chromosome abnormalities (Leimbacher et al., 2019). Moreover, the interaction between phosphorylated BLM and TOPBP1 is also required for genome integrity, and the disruption of BLM-TOPBP1 interaction results in increased sister chromatid exchanges, replication origin firing and chromosomal aberrations (Blackford et al., 2015). In addition to genomic instability, increased mutagenesis has been addressed as the cause of cancer progression. Accumulating evidence has focused on the roles of TOPBP1 in DNA repair and DDR. A previous report showed that a missense variant (R309C) of TOPBP1 occurred at an elevated frequency in familial breast cancer cases compared to healthy controls from Finland (Karppinen et al., 2006). Thus, TOPBP1 has been suggested as a breast cancer susceptibility gene. However, another study showed that this R309C allele was a common polymorphic variant at least in Germany and that the R309C allele did not confer an increased risk for breast cancer (Magda et al., 2010). In view of these inconsistent conclusions, further study is needed to identify the function of cancer-associated mutants of TOPBP1. Here, by combining the data from three leading cancer mutation databases (The Cancer Genome Atlas-TCGA, International Cancer Genome Consortium-ICGC and Catalogue of Somatic Mutations in Cancer-COSMIC), we analyzed the types and proportions of the mutations on TOPBP1 and screened the mutations located in the important domains of TOPBP1, including both truncation mutation and missense mutation. We found several representative cancer-associated mutants of TOPBP1 that were deficient in DSB repair and checkpoint signaling, which might lead to genomic instability and further cause cancers. Taken together, our research may illustrate the mechanism by which TOPBP1 mutations cause cancers and provide potential targets for the treatment of related cancers in the future.
Materials and methods
Database of TOPBP1 mutations
TOPBP1 mutations in this study are collected from three cancer-associated databases including TCGA database (https://portal.gdc.cancer.gov/), ICGC database (https://icgc.org/) and COSMIC database (https://cancer.sanger.ac.uk/cosmic). The transcript annotation of TOPBP1 is ENSG00000163781. Human SNPs of TOPBP1 are searched from SNP database (dbSNP, https://www.ncbi.nlm.nih.gov/snp). Cancer-related TOPBP1 mutations are obtained by filtering out the benign polymorphisms.
Plasmids
TOPBP1 and its mutations were cloned into the SFB vector (S-FLAG-SBP-tagged at the N terminus). The correctly sequenced plasmids were used for subsequent experiments.
Cell lines
Human embryonic kidney 293T (HEK-293T) cell lines were obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). All cell lines were grown in a sterile cell culture environment and regularly tested for mycoplasma contamination. Cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, GIBCO, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS, GEMINI, 900–108), 100 μg/mL penicillin and 100 μg/mL streptomycin, at 37 ℃ with 5% CO2 in a humidified incubator.
Generation of stable cell lines
HEK-293T cells were transfected with lentiviral vector (PLKO.1) carrying an inducible short hairpin RNA (shRNA: 5′-gcacaaggtttaatgaggaag-3′) to target TOPBP1. Cells with stable integration of shRNA constructs were selected with puromycin (2 μg/mL) for approximately one week. Knockdown of TOPBP1 was confirmed by western blotting.
DNA damage-induced foci-forming assay
Cells were grown on coverslips in 6-well plates and transfected with indicated plasmids using Lipofectamine 2000 (Thermo, 11668019). After 24 h incubation, cells were treated with 1 mM MMS for 30 min. After recovery for 4 h, cells were fixed in 4% paraformaldehyde (Sigma, P6148), followed by being permeabilized with 1% Triton X-100 (Sigma, T8787) for 5 min. After blocking with 8% goat serum in PBS for an hour, cells were incubated with indicated primary antibodies for 1 h at room temperature. Coverslips were washed three times with PBS and secondary antibody incubation was performed for 1 h at room temperature in dark. After washing with PBS for three times, the nuclei were stained with DAPI (Sigma, D9542; 1:10000 in PBS). The coverslips were mounted onto glass slides and visualized with confocal laser scanning microscope (OLYMPUS, FV3000).
Colony formation assay
The plasmids of wild type TOPBP1 and its mutants were transfected into TOPBP1-knockdown cells. 24 h after transfection, 1 × 103 cells were spread in each well of the six-well plate. 24 h later, cells were treated with 1 mM MMS for 1 h, and then washed with PBS and replaced with DMEM containing 10% FBS and 1% penicillin–streptomycin. Colonies were grown for 14 days, the viable cells were then washed and fixed in methanol and stained with Giemsa. The number of colonies is divided by the number of seeded cells and normalized to the plating efficiency of the untreated controls to calculate the survival fraction.
Comet assay
TOPBP1 knockdown cells reconstituted with wild type or mutant TOPBP1 were treated with or without 1 mM MMS. After incubating in fresh medium for indicated time at 37 °C, cells were harvested and mixed with 1% LMP agarose at 42 °C and immediately pipetted onto slides. To perform neutral conditional assay, the slides were immersed in the neutral lysis buffer (2% sarkosyl, 0.5 M EDTA, 0.5 mg/mL proteinase K, pH 8.0) overnight at 4 °C. After lysis, the slides were washed with electrophoresis buffer (90 mM Tris–HCL at pH 8.5, 90 mM boric acid, 2 mM EDTA), and electrophored at 25 V for 30 min (0.6 V/cm) and stained with 10 μg/mL propidium iodide for 30 min in dark. All images were collected with a fluorescence microscope and analyzed by the Comet Assay IV software.
Cell lysis and western blotting
Cells were briefly rinsed with ice-cold PBS before lysed with NETN 420 lysis buffer (20 mM Tris–HCl (pH 7.4), 420 mM NaCl, 1 mM EDTA, 1% Triton X-100). The lysates were cleared by centrifugation (13,000 rpm for 10 min at 4 ℃). Samples were separated on 10% polyacrylamide Tris–Glycine gels and transferred to PVDF membranes (Merck Millipore). The membranes were blocked with 5% nonfat milk, then incubated with indicated antibodies and followed by incubation with horseradish peroxidase-labeled secondary antibody. The signals were tested using the ECL (Thermo, 34577). All western blotting experiments were performed according to standard procedures. The following antibodies were used at the indicated dilutions: TOPBP1 (rabbit, Cell Signaling Technology, D8G4L, 1:1000), Chk1 pS345 (rabbit, Cell Signaling Technology, 2348, 1:500), GAPDH (mouse, Cell Signaling Technology, 2118S, 1:1000).
Structural modeling
The mutated BRCT domains of TOPBP1 were built from the crystal structure of the BRCT domain of human TOPBP1 (PDB code: 2XNH, 3UEN, 3JVE, 3PD7 and 3AL3) using Open-Source PyMOL 2.6. version.
Prediction of damaging missense mutations of TOPBP1
We predict the impact of coding variants on the structure and function of TOPBP1 by the Sorting Intolerant from Tolerant (SIFT, https://sift.bii.a-star.edu.sg/) and Polymorphism Phenotyping 2 (PolyPhen2, http://genetics.bwh.harvard.edu/pph2/index.shtml). SIFT is a non-synonymous mutation prediction tool based on sequence homology. The normal score range is 0–1, and a smaller score is more harmful. The score at 0–0.05 is “damaging”, and the score at 0.05–1 is “tolerated”. A low PolyPhen2 score (0) indicates a benign mutation, and a high score (1) suggests a damaging mutation.
Cell cycle checkpoint assay
Asynchronous cells were exposed to 2 Gy of gamma irradiation and then incubated at 37 °C in 5% (vol/vol) CO2 for 1 h. Cells were fixed with 70% ethanol and stained with rabbit anti-phospho-histone H3 antibody (Phospho-Histone H3-Ser10, Cell Signaling Technology, 53348), followed by incubation with fluorescein isothiocyanate-conjugated goat anti-rabbit immuno-globulin G secondary antibody. The stained cells were treated with RNase, incubated with propidium iodide, and then analyzed by flow cytometry (Beckman, CytoFLEX).
Results
Multiple types of mutations on TOPBP1 exist in human cancers
To explore the mutations on TOPBP1 in human cancers, including frame-shift, missense mutation and nonsense mutation, data from 3 databases including TCGA, ICGC and COSMIC were analyzed. We found that missense mutation accounted for the largest proportion (79.62% in TCGA, 87.35% in ICGC and 79.66% in COSMIC) among these mutations (Fig. 1A–C). Frame-shift mutation caused by insertion or deletion made up to 11.46% in TCGA, 4.89% in ICGC and 11.03% in COSMIC (Fig. 1A–C). Nonsense mutation accounted for 8.92%, 7.76% and 9.31% in TCGA, ICGC and COSMIC, respectively (Fig. 1A–C). On the other hand, among 64,781 tumor samples from 31 different types of cancer tissues included in the 3 databases (Supplementary Table S1 and Fig. 1D), we found 369 variants of TOPBP1, which have not been identified in the SNP database from normal tissues, and 215 out of the 369 TOPBP1 variants were recurrent mutations (Supplementary Table S2 and S3). Moreover, by combining data from TCGA, ICGC and COSMIC databases, we conclude that TOPBP1 is more frequently mutated in liver cancer (LICA), skin cutaneous melanoma (SKCM) and uterine corpus endometrial carcinoma (UCEC) (Fig. 1D).
Fig. 1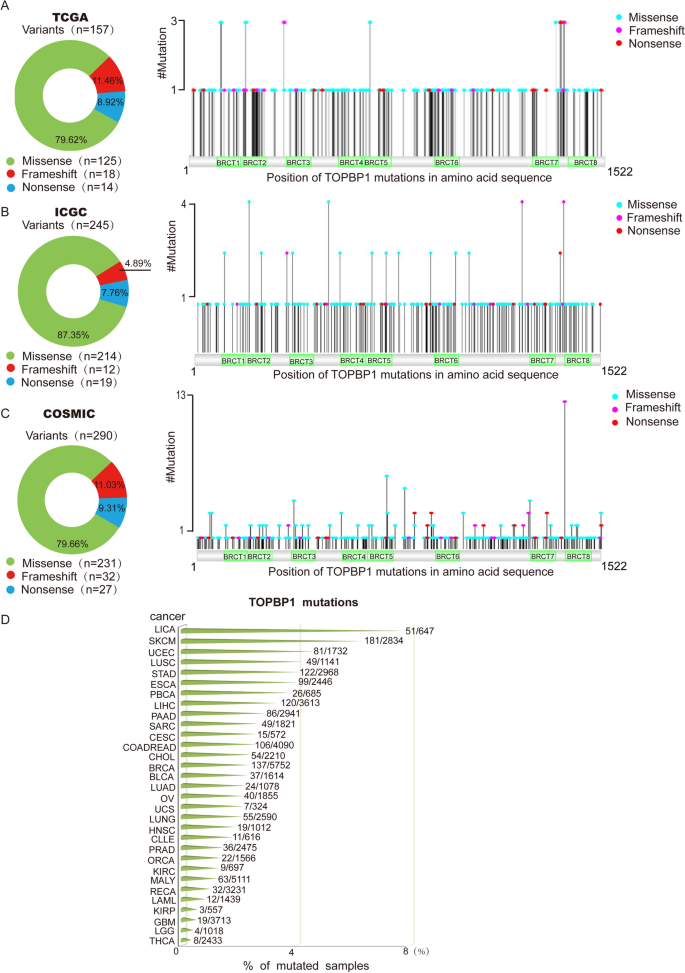
Landscape of TOPBP1 mutations in human cancers in TCGA, ICGC, and COSMIC databases. A–C An integrated analysis of types and proportions of TOPBP1 mutations in TCGA database (A), ICGC database (B), and COSMIC database (C). The pie charts indicating the proportion of different types of mutations of TOPBP1 were shown on the left. Positions of TOPBP1 mutations in three databases were shown on the right. D TOPBP1 mutations in different human cancer types. X axis represents the percentage of TOPBP1 mutations in each type of cancer. Y axis represents the cancer types. The cancer type abbreviation on Y axis is stated in Supplementary Table S1
Full size imageHomologous recombination (HR) repair and checkpoint control are defective in cells with TOPBP1 truncation mutations
Protein truncation mutation may partially or completely abolish the function of a protein due to the loss of the key motif. After screening the truncation mutations from the 3 databases by their recurrent frequency in different cancers and their location (whether the mutation located in key functional domains of TOPBP1), 6 truncation mutations, including N955Ifs*7, L1044Rfs*12, Q1271*, R1314*, I1381Nfs*11 and I1381Sfs*61 were selected for further study (Fig. 2A and Supplementary Table S4). N955Ifs*7 is a mutation located in BRCT6 domain of TOPBP1 that occurs in lung adenocarcinoma. L1044Rfs*12 is a recurrent mutation that has been found in breast cancer and bladder urothelial carcinoma. Q1271* and R1314* are mutations on BRCT7 motif of TOPBP1 that are recurrent mutations identified in colorectal adenocarcinoma and esophageal cancer. I1381Nfs*11 and I1381Sfs* frequently appear concurrently in colorectal adenocarcinoma and esophageal cancer.
Fig. 2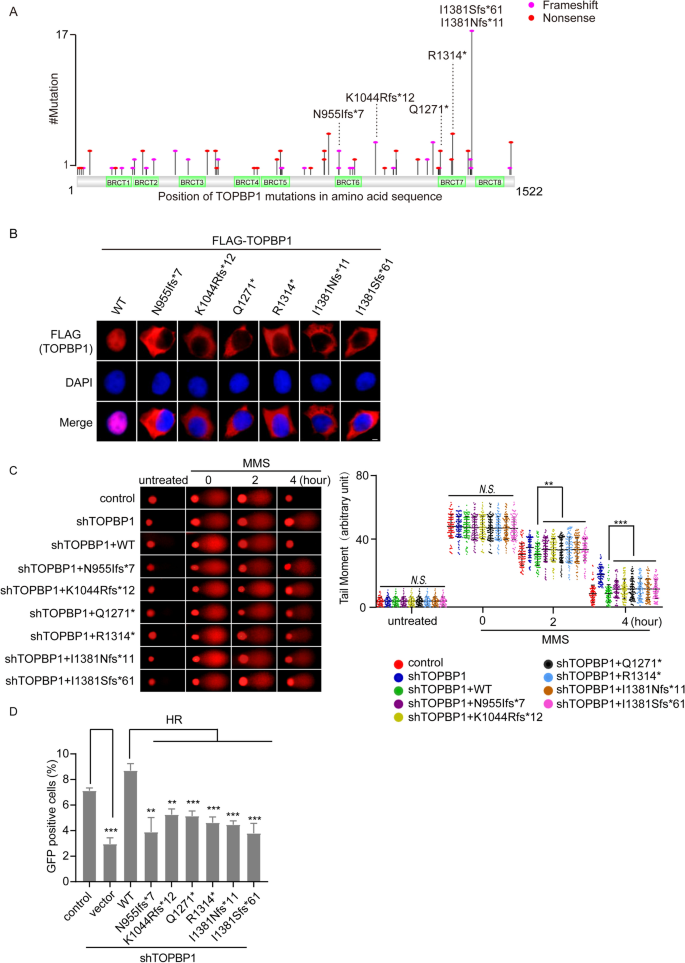
Truncation mutations of TOPBP1 affect localization of TOPBP1 and impair DNA damage repair. A Map of TOPBP1 truncation mutations in human cancers according to the three databases. B Localization of truncation mutations of TOPBP1 in cells. 24 h after transfection of FLAG-tagged TOPBP1 wild-type or truncation mutants, localization of TOPBP1 was examined by immunofluorescence staining with anti-FLAG antibody. Nucleus was stained with DAPI. Scale bar, 5 μm. C Truncation mutations of TOPBP1 result in DNA damage repair defects. TOPBP1-knockdown cells reconstituted with TOPBP1 wild type or TOPBP1 truncation mutants were treated with 1 mM MMS and recovered for the indicated time. Comet assays were performed under neutral conditions. Comet tail moments were examined to indicate the DNA damage repair kinetics. Data are shown as mean ± SEM from three independent experiments. *P < 0.05; ***P < 0.001, N.S. no significance. D The role of TOPBP1 mutants in HR repair. TOPBP1-knockdown cells were transfected with TOPBP1 wild-type or mutants, respectively, and cells expressing GFP, which represents for the HR repair efficacy, were assessed as indicated by flow cytometry. **P < 0.01; ***P < 0.001
Full size imageAs shown in Fig. 2A, the truncation mutations of TOPBP1 concentrated in BRCT6-8 domains. Given that these truncation mutations would abolish the nuclear localization signal of TOPBP1 (852–858 aa and 1517–1520 aa), we conjectured that all these truncation mutations might disrupt the correct localization of TOPBP1 in cells. As evidenced by immunofluorescence staining, all of these truncated mutants localized in cytoplasm instead of nucleus (Fig. 2B), which suggested that truncation mutations might abrogate the function of TOPBP1.
We then asked if these truncated mutants could affect DDR regulated by TOPBP1. To this end, we reconstituted TOPBP1 knockdown stable cell line with wild type or truncated mutants of TOPBP1 (Supplemental Fig. S1A, B) and evaluated DNA damage repair after methyl methanesulfonate (MMS) treatment by neutral comet assay. As is shown in Fig. 2C, the defect in DNA damage repair caused by TOPBP1 knockdown could only be rescued by wild type TOPBP1, while the truncated mutants of TOPBP1 could not (Fig. 2C). Previous studies have shown that TOPBP1 is involved in DNA DSB-induced HR repair (Moudry et al., 2016). We then examined HR repair by conventional GFP reporter assays (DR-GFP system) (Zhang et al., 2020) and found that truncation mutations of TOPBP1 significantly impaired HR pathway (Fig. 2D).
Chk1 is a crucial DNA damage checkpoint kinase and its activation, which requires ATR, inhibits DNA replication and cell cycle progression. Therefore, ATR-Chk1 axis modulates cell cycle arrest at certain points in response to DNA damage (Ashwell et al., 2008; Choi et al., 2009). TOPBP1 plays a critical role in Chk1 activation following DNA damage through its AAD domain and BRCT domains (Burrows & Elledge, 2008). AAD domain of TOPBP1 locates between BRCT6 and BRCT7 and is essential for ATR-Chk1 activation (Kumagai et al., 2006). The interaction between BRCT7/8 domains and FANCJ can also promote Chk1 activation (Gong et al., 2010). Since the truncated mutations of TOPBP1 might result in the loss of the normal C-terminal after BRCT6 domain or BRCT7/8 domains, we further explored the effects of these truncation mutations on Chk1 phosphorylation and cell cycle control. We found that phosphorylation of Chk1 at Ser345, which is a surrogate marker of Chk1 activation, remarkably decreased in TOPBP1 knockdown cells complemented with truncated mutants of TOPBP1 compared to the cells complemented with wild type TOPBP1, suggesting that these truncation mutations impaired the Chk1 activation (Fig. 3A). Consistently, as indicated by histone H3 pS10 staining, we found that the G2/M arrest defect in the TOPBP1-deficient cells could be rescued by wild-type TOPBP1, while all the truncated mutants of TOPBP1 failed to rescue it (Fig. 3B). Also, cells complemented with truncated TOPBP1 were hypersensitive to MMS treatment, which is revealed by colony formation assay (Fig. 3C). These discoveries support a conclusion that truncated mutants of TOPBP1 that can abolish the functional BRCT6-8 domains of TOPBP1 lead to compromised DNA repair and cell cycle checkpoint activation.
Fig. 3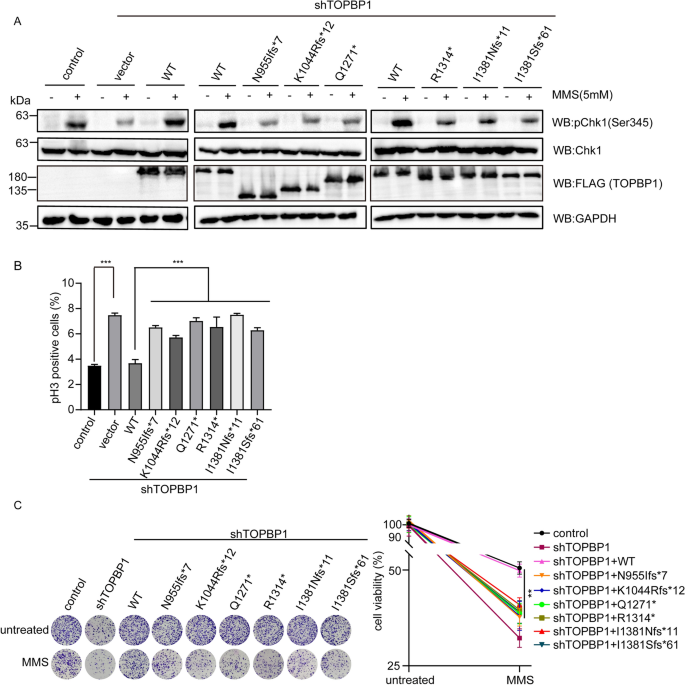
Truncation mutations of TOPBP1 impair G2/M checkpoint activation and reduce cell viability following DNA damage. A Truncation mutations of TOPBP1 impaired Chk1 activation in response to DNA damage. TOPBP1-depleted cells were rescued by TOPBP1 wild type or truncation mutants and then treated with 5 mM MMS. The cells were collected and the cell lysates were tested with the indicated antibodies by western blotting. 293T parental cells acted as the control in this experiment. The histogram (lower panel) shows the relative level of the pChk1 signal. B TOPBP1 truncation mutations impaired G2/M checkpoint activation. TOPBP1-knockdown cells reconstituted with TOPBP1 wild-type or TOPBP1 truncation mutants were treated with 2 Gy of IR, and cells were allowed to recover for 1 h before being fixed and stained with anti-phospho-H3 antibody, and analyzed by flow cytometry. 293T parental cells and TOPBP1-knockdown cells were included as positive or negative control. The results from triplicate experiments were represented as mean ± SEM. ***P < 0.001. C Truncation mutations of TOPBP1 reduced cell viability following DNA damage. Colony formation assay was performed with TOPBP1-depleted cells rescued with TOPBP1 wild-type or TOPBP1 truncation mutants following MMS treatment
Full size imageScreening missense mutations of TOPBP1 by bioinformatics analysis
Except for TOPBP1 truncation mutations, a series of missense mutations were also found across TOPBP1 gene sequence (Fig. 4A). Among these missense mutations, some may be benign mutations, whereas others may be highly associated with tumorigenesis. To predict the functional defect caused by missense mutations, we used PolyPhen-2 and SIFT, which are two online tools for predicting possible impact of an amino acid substitution on the structure and function of a human protein, to determine which missense mutations of TOPBP1 are likely to affect its function. As a result, we found that missense mutations were distributed in each of the 8 BRCT domains of TOPBP1 (Fig. 4A), which mediate protein–protein interaction to participate in DDR (Feng et al., 2020), and nearly 50% of the missense mutations on TOPBP1 are probably deleterious to cells (Fig. 4B and Supplementary Table S5). After screening by the high recurrence frequency, damaging prediction and location of these missense mutations, 10 potential mutations (C197Y, G202R, G248R, K250N, H694R, R730S, I968M, S1332F, G1418R and L1464F) are selected for further functional analysis (Fig. 4C).
Fig. 4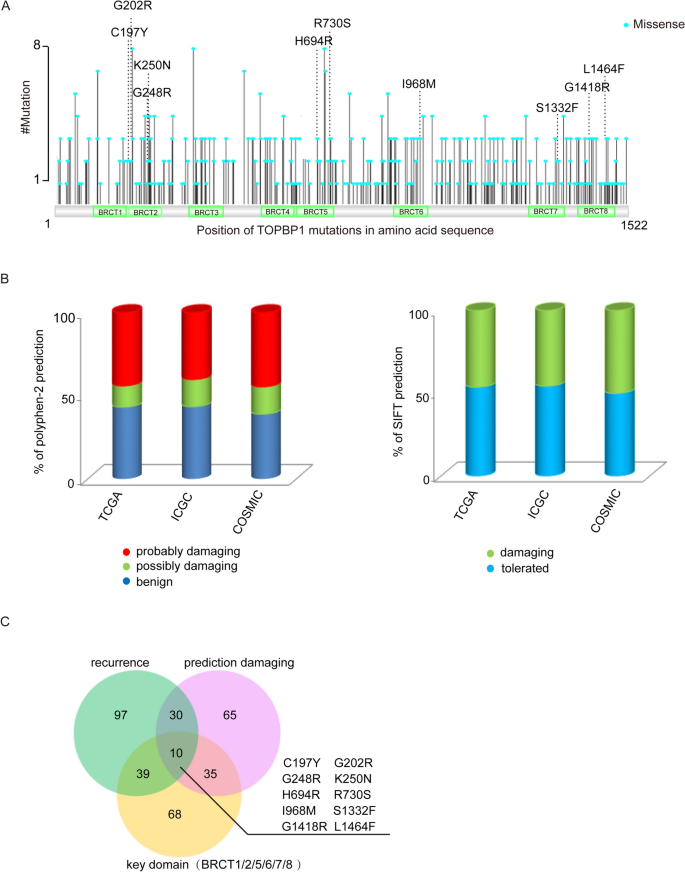
Screening TOPBP1 missense mutations by bioinformatic analysis. A Map of TOPBP1 missense mutations in human cancers. B The damaging prediction of missense mutations of TOPBP1 using PolyPhen-2 (left panel) and SIFT (right panel). The detailed results are also shown in Supplementary Table S5. C A Venn diagram of TOPBP1 missense mutations shared between different groups of mutations. Mutations that are recurrent in cancers are indicated in blue circles; mutations that are predicted to affect the structure of the proteins are indicated in pink circles; the mutations that locate in the key BRCT domains of TOPBP1 are indicated in orange circles
Full size imageMissense mutations of TOPBP1 cause conformational changes
To demonstrate the structural impacts of the 10 missense mutations on the biological function of TOPBP1, we first analyzed their structural changes caused by amino acid substitution. The 10 missense mutations of TOPBP1 are distributed in BRCT2, BRCT5/6 and BRCT7/8 domains, respectively (Fig. 4A). The structures of BRCT motifs of TOPBP1 have been determined by X-ray crystallography (Day et al., 2018; Leung et al., 2010, 2011; Sun et al., 2017). The individual BRCT domain includes a β1/α1/β2/β3/α2/β4/α3/α4 topology, which facilitates TOPBP1 phosphor-peptide binding with its partners (Rappas et al., 2011). Using computational modeling, the potential changes of structure caused by these missense mutations of TOPBP1 were analyzed.
TOPBP1 contains two consecutive BRCT domain at its N terminus, in which each of BRCT 1 and BRCT 2 adopts a compact β1/α1/β2/β3/α2/β4/α3 topology and is connected by a linker region (PDB code: 2XNH). TOPBP1 interacts with the phosphorylated tail of Rad9 through its BRCT1/2 domains, which can promote the activation of Chk1 (Delacroix et al., 2007; Rappas et al., 2011). C197 and G202 locate in the linker region between BRCT1 and BRCT2. C197 forms a hydrogen bond with the main chain of L168, and a hydrophobic core is generated between C197 and its surrounding residues A148, V151, Y156, I166, L168, P198 and L201, which stabilizes the two BRCT domains. C197Y mutation increases the length of side chain and leads to serious steric hindrance and abolishes its interaction with surrounding residues, which may disrupt the structure of BRCT domains (Fig. 5A). G202 is at turn between α4 of BRCT1 and β1 of BRCT2, and G202R mutation would generate electrostatic interaction with E150 located in BRCT1, and also forms hydrogen bonds with S115 and V151, both of which will destabilize the inter-domain linker and BRCT1, ultimately affecting the function of BRCT1 (Fig. 5B). G248 is at α2 of BRCT2, the G248R mutation may destabilize this secondary structure (Fig. 5C). K250 is the key amino acid for phosphor-peptide binding. The side chain of K250 forms a hydrogen bond with Q232, and hydrophobically interacts with L241 and L233, the K250N mutation abolishes these interactions, and may perturb the structure and function of BRCT2 domain (Fig. 5D).
Fig. 5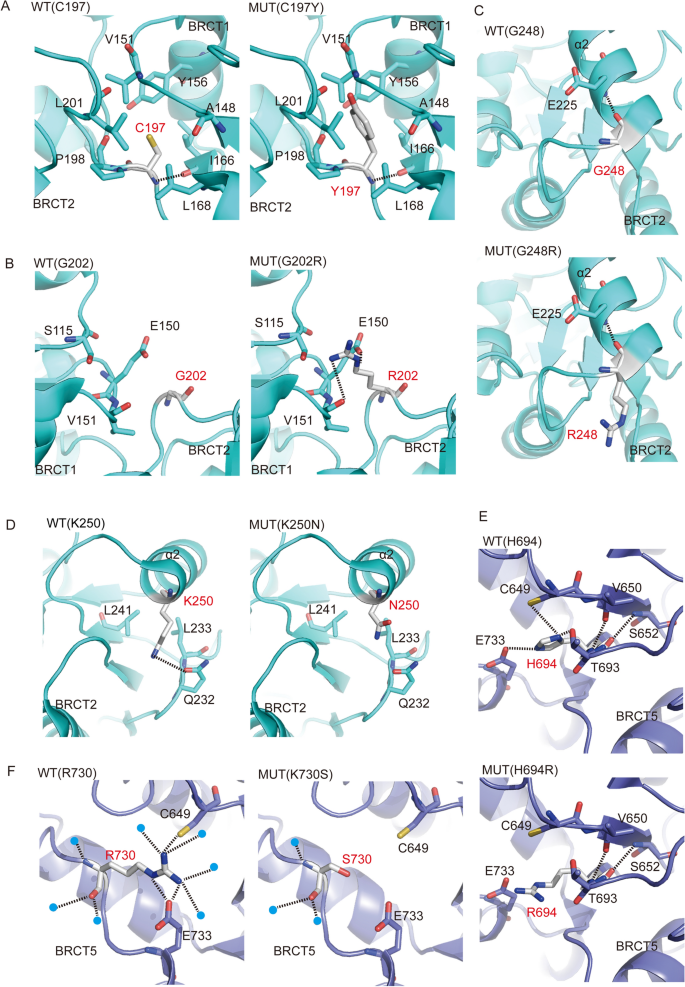
Missense mutations of TOPBP1 cause conformational changes. A–I Structural conformation changes caused by missense mutations of TOPBP1. The side-by-side alignments of BRCT domains in wild type (WT) and mutation (MUT) were shown, the amino-acid substitutions in BRCT1-2 domain (A–D), BRCT5 domain (E, F), BRCT6 domain (G) and BRCT7-8 domain (H, I). The amino acids are depicted as stick models with gray carbons. Selected amino acids that make side chain or main chain contacts to the mutant amino acids in BRCT domains are rendered with different color schemes, BRCT1-2 domain (PDB code: 2XNH, cyan), BRCT4-5 domain (PDB code: 3UEN, purple), BRCT6 domain (PDB code: 3JVE and 3PD7, magenta) and BRCT7-8 domain (PDB code: 3AL3, yellow). The tertiary structures of BRCT domains affected by missense mutations are labeled. Hydrogen bond contacts are denoted by black dashed lines. The waters are depicted as blue spheres
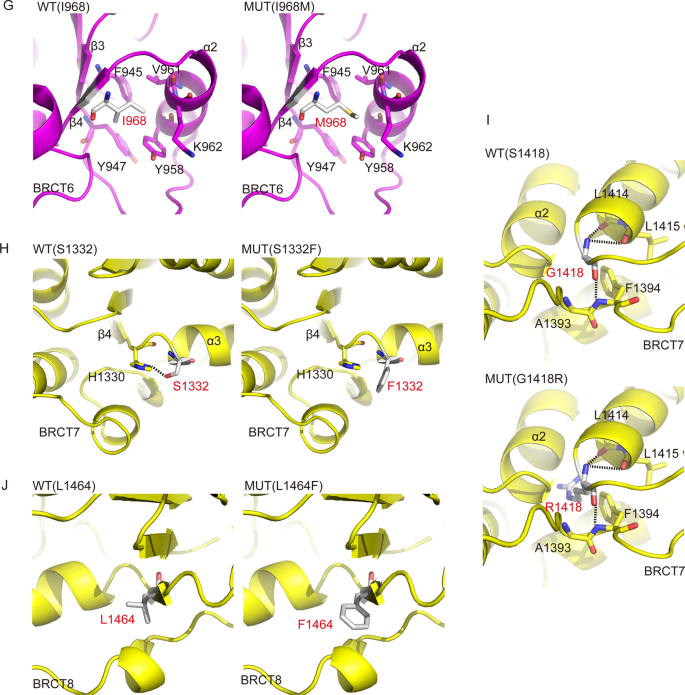
Full size imageThe phosphate binding pocket in BRCT5 domain of TOPBP1 is responsible for the association of TOPBP1 with the conserved Ser–Asp–Thr (SDT) repeats of MDC1. This interaction is required for TOPBP1 focus formation and ATR-Chk1 activation in response to replication stress (Wang et al., 2011). Moreover, BLM binding to TOPBP1 also requires BRCT5 domain. BLM-TOPBP1 binding is important for maintaining genome integrity (Blackford et al., 2015). Based on the crystal structure of BRCT4-BRCT5 domain of TOPBP1 (PDB code: 3UEN), as shown in Fig. 5E, the side chain of H694 forms hydrogen bonds with the side chains of E733, C649 and T693, meanwhile, the main chain of H694 forms hydrogen bonds with that of V650 and S652. These hydrogen bonds contribute to stabilization of BRCT5. The H694R mutation will abolish the hydrogen bonds with E733, C649 and T693, disrupting the conformation of BRCT5, and furthermore affecting its biological functions. The side chain of R730 forms hydrogen bonds network the side chains of E733 and C649 to stabilize the structure of BRCT5 (Leung et al., 2013). However, R730S mutation will disrupt theses hydrogen bonds and accordingly influence the function of BRCT5 domain (Fig. 5F). BRCT6 domain of TOPBP1 interacts with transcription factor E2F1 and represses E2F1 activity in induction of apoptosis in response to DNA damage (Leung et al., 2010). Crystal structure of TOPBP1 BRCT6 represents the overall fold consists of a central four-stranded parallel β-sheet flanked by a single helix (α2) on one side and by a pair of helices (α1and α3) on the other side (PDB code: 3PD7 and 3JVE). I968 forms the hydrophobic interactions with the aromatic rings of F945, Y947 and Y958, V961 and K962, forming a hydrophobic core to stabilize α2, β3 and β4 in BRCT6, the I968M mutation would destabilize the BTCR6 (Fig. 5G) and affect its biological function.
BRCT7/8 domains of TOPBP1 physically bind PLK1 and promote PLK1-mediated phosphorylation of RAD51, which is essential for RAD51 recruitment to chromatin (Moudry et al., 2016). BRCT7/8 domains also interact with BACH1 and accelerate Chk1 activation (Gong et al., 2010). Similar to other BRCT domain structures of TOPBP1 (19), BRCT7/8 packs in a head to tail manner via a hydrophobic interface between the two BRCT domains. Each BRCT domain consists of a central four-stranded parallel β-sheet packed on opposite sides by helical elements (PDB code: 3AL3). The side chain of S1332 forms a hydrogen bond with the side chain of H1330. And the main chain of S1332 also forms a hydrogen bond the main chain of H1330. The S1332F mutation has a longer side chain and abolishes the hydrogen bonds with H1330, which would destabilize β4 and α3 and affect the interaction with PLK1 or BACH1 (Fig. 5H). The G1418 carbonyl oxygen forms hydrogen bond with the main chain of F1394 and amino nitrogen with the main chain of L1414 and L1415 to stabilize the inter-domain linker. While the G1418R mutation generates a longer side chain, not only disrupting these hydrogen bonds, but also forming a steric hindrance with α2, and finally the G1418R mutation destabilizing inter-domain linker (Fig. 5I). L1464F mutation, with a large benzene ring, produces a steric hindrance and accordingly disrupts its interaction with surrounding residues, which yields subtle structural changes and might affect the interactions between TOPBP1 and its partners (Fig. 5J). Collectively, we conclude that the ten missense mutations of TOPBP1 change the original spatial structures of the residues and their surrounding environments, which may further affect the functions of TOPBP1 in DDR.
Missense mutations of TOPBP1 are defective in DNA damage repair
To explore whether these missense mutations of TOPBP1 will lead to its functional defect in DNA damage repair, TOPBP1 knockdown cells were reconstituted with wild type or 10 missense mutants of TOPBP1, respectively (Fig. 6A and Supplementary Fig. 2A). We found that, consistent with TOPBP1 wild type, all the missense mutants still localized in nucleus (Supplementary Fig. 2B). BRCT1/2, BRCT4/5 and BRCT7/8 domains of TOPBP1 are essential for the recruitment of TOPBP1 to sites of DNA damage (Cescutti et al., 2010; Montales et al., 2021; Yamane et al., 2002). Specifically, BRCT4/5 motifs mediate the interaction between 53BP1 and TOPBP1 and promote the relocation of TOPBP1 to the sites of DNA lesions (Cescutti et al., 2010). Thus, we examined the foci formation of missense mutants of TOPBP1 after MMS treatment. Intriguingly, the H694R mutation in BRCT5 domain completely abolished the foci formation of TOPBP1 and R730S partially disrupted TOPBP1 foci formation (Fig. 6B). The missense mutations located in BRCT2, BRCT6 and BRCT7/8 also impaired TOPBP1 foci formation to various extents (Fig. 6C, D). Abolished or impaired foci formation of these missense mutants of TOPBP1 were likely due to the weakened interaction between TOPBP1 and 53BP1 or other binding partners caused by the structural changes of TOPBP1, which needs to be further explored. In addition, the neutral comet assay indicated that all the missense mutations of TOPBP1 could not repair the DNA lesions caused by MMS (Fig. 6E, F). We further examined HR repair using conventional DR-GFP system assays. We found that missense mutations of TOPBP1 impaired HR repair (Fig. 6G). Collectively, these results indicate that missense mutations of TOPBP1 result in DNA damage repair defects.
Fig. 6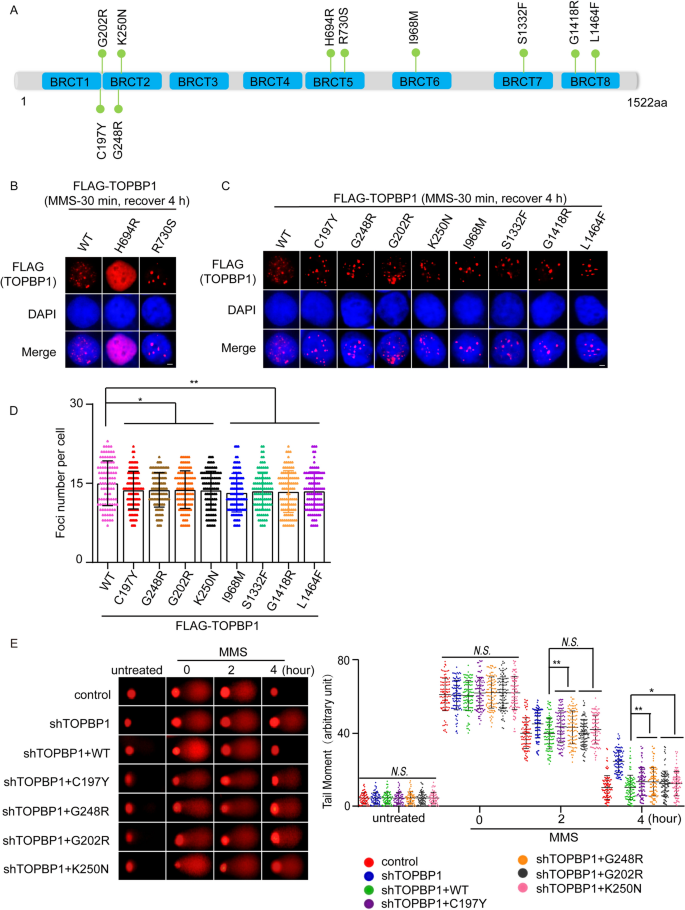
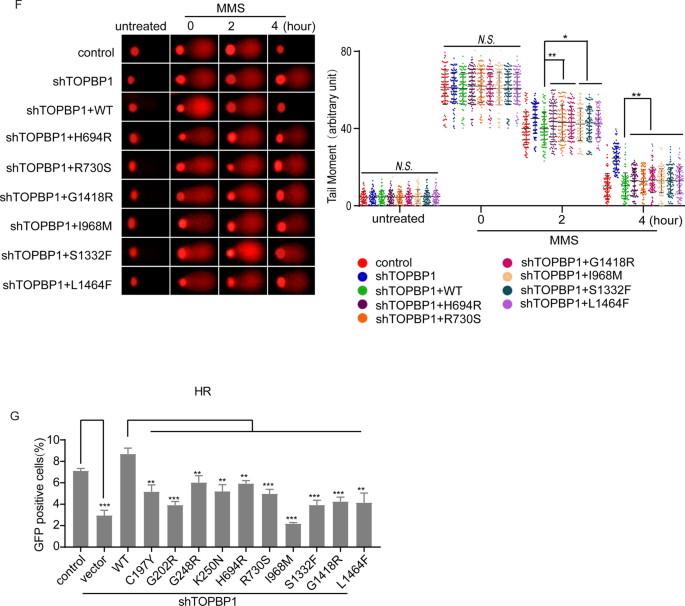
Missense mutations of TOPBP1 are defective in DNA damage repair. A The map that shows the positions of the 10 representative missense mutations of TOPBP1. B–D The DNA damage-induced foci formation of TOPBP1 wild type or missense mutants. 293T cells transfected with TOPBP1 wild-type or mutants were stimulated with 1 mM MMS for 30 min and recovered for 4 h to induce foci formation. Shown are the representative images and the statistical analysis. Scale bar, 5 μm. 100 cells with foci were randomly selected from three independent experiments. *P < 0.05; **P < 0.01. E, F All ten missense mutations of TOPBP1 lead to DNA damage repair defects. Comet assays under neutral condition were performed. Data are represented as mean ± SEM as indicated from three independent experiments. *P < 0.05; **P < 0.01; N.S. no significance. G The deficient effects of all ten missense mutants of TOPBP1 in HR repair pathway. TOPBP1-knockdown cells expressed TOPBP1 wild-type or missense mutants, respectively, were assessed for HR repair pathway as indicated by the percentage of cells that express GFP by flow cytometry. **P < 0.01; ***P < 0.001
Full size imageMissense mutations of TOPBP1 impair G2/M checkpoint activation
It has been reported that TOPBP1 acts as a bridge that links the independently recruited Rad9-Hus1-Rad1 (9-1-1) complex and ATRIP-ATR through its BRCT1/2 domains, resulting in ATR-Chk1 activation by TOPBP1 AAD domain (Delacroix et al., 2007). The C-terminal tandem BRCT domains of TOPBP1 also interact with phosphorylated BACH1, which is required for the efficient chromatin loading of RPA to further activate Chk1 (Ashwell et al., 2008; Gong et al., 2010). To further explore the functions of missense mutations of TOPBP1 in DNA damage checkpoints signaling, TOPBP1 knockdown cells reconstituted with wild type or missense mutants were treated with DNA damage and then Chk1 phosphorylation and cell cycle arrest were examined. We found that only wild type TOPBP1 could completely rescue the defective activation of Chk1 in TOPBP1-deficient cells, while missense mutants could not rescue activation of Chk1 (Fig. 7A). To examine whether missense mutations of TOPBP1 could affect the G2/M checkpoint activation, we measured the phosphorylated serine 10 of histone H3 by flow cytometry. The results indicated that phospho-histone H3 protein levels in TOPBP1-deficient cells were increased compared to those in control cells after IR exposure, suggesting inefficient G2/M checkpoint activation for blocking the M phase entry (Fig. 7B). Only the Wild type TOPBP1 could rescue the defects in G2/M checkpoint activation, whereas missense mutations of TOPBP1 were unable to rescue it (Fig. 7B). Moreover, colony formation assay showed that cells complemented with missense mutations of TOPBP1 were hypersensitive to MMS treatment (Fig. 7C, D). Taken together, all the 10 missense mutations of TOPBP1 that located in BRCT2, BRCT5/6 and BRCT7/8 domains impair the regulation of G2/M cell cycle checkpoint.
Fig. 7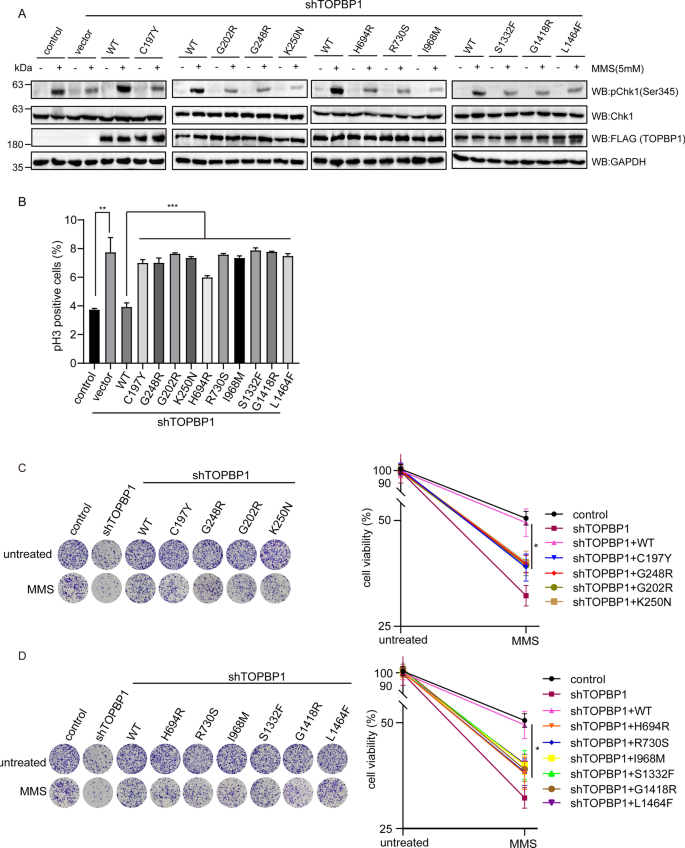
Missense mutations of TOPBP1 impair G2/M checkpoint activation. A The influence of missense mutations of TOPBP1 on Chk1 activation. TOPBP1-knockdown cells transfected with TOPBP1 wild-type or ten missense mutants were treated with 5 mM MMS for 1 h and harvested for examining phosphorylated Chk1 on Ser345. The other proteins were tested by indicated antibodies. GAPDH was the internal control. Relative pChk1 (Ser345) expression was evaluated by the histogram (lower panel). ***P < 0.001. B Impaired G2/M checkpoint control of missense mutations of TOPBP1. TOPBP1-knockdown cells transfected with TOPBP1 wild-type and ten missense mutations treated with 2 Gy of IR and then recovered for 1 h before being fixed and stained with anti-phospho-H3 antibody. GFP positive signals were examined by flow cytometry. **P < 0.01; ***P < 0.001. C, D Missense mutations of TOPBP1 reduced cell viability following DNA damage. Colony formation assay was performed with TOPBP1-depleted cells rescued with TOPBP1 wild-type or TOPBP1 missense mutants, which were treated with MMS. Colony formation assay was performed after proliferation of cells for 14 days. *P < 0.05
Full size imageDiscussion
TOPBP1 is a key player in DNA replication, DNA damage repair and cell cycle checkpoint control (Leimbacher et al., 2019). However, the cancer-associated mutations of TOPBP1 are not well explored in these cellular responses. Here, we summarized diversified mutations of TOPBP1 from three cancer-related databases (TCGA, ICGC and COSMIC) and characterized their functional defects of these mutants in DDR. We focused on 6 truncation mutations and 10 missense mutations in the key domains and confirmed that these mutations exhibit dysfunctions of TOPBP1. Among 8 BRCT domains of TOPBP1, BRCT 1/2/5/7 are essential for ATR-mediated phosphorylation of Chk1 and BRCT 6 and BRCT 8 facilitate the process to be more efficient (Montales et al., 2021).The recruitment of TOPBP1 to DSB sites dependents on BRCT domains 1/2, BRCT4/5 and BRCT7 (Cescutti et al., 2010; Montales et al., 2021; Yamane et al., 2002). 6 truncation mutations result in the loss of the C-terminal BRCT6 or BRCT7/8 domains. Abolishing these functional domains leads to compromised DDR. Missense mutations selected in this study are located in BRCT2/5/6/7/8, respectively. Intriguingly, H694R mutation in BRCT5 domain completely abolishes the recruitment of TOPBP1 to DSB sites, and other mutations also impaired its recruitment, which further disrupt the biological function of TOPBP1 in DNA damage repair. Moreover, we further explore the molecular basis which could help to explain how these mutations abolish the stable structure of each domain by structure modeling analyses. This is a comprehensive research for the dysfunctions of cancer-related mutations of TOPBP1 in DDR. Our results may provide an insight into understanding the mechanism of how TOPBP1 mutants are associated with tumor progression and potential clinical implications for cancer treatment.
TOPBP1 plays important roles in cell proliferation and maintenance of chromosomal integrity (Jeon et al., 2011). Cancer cells with highly expression of TOPBP1 could survive from radiation therapy. Disruption of TOPBP1 in radioresistant lung cancer cells (PC14PE6) exhibited increased sensitivity to radiation (Choi et al., 2014; Chowdhury et al., 2014; Forma et al., 2012). TOPBP1 is frequently overexpressed in primary breast cancer, which results in highly risk of relapse or death (Forma et al., 2012; Going et al., 2007; Liu et al., 2009). Moudry et al. also reported altered TOPBP1 protein expression in ovarian cancer, although at modest frequencies (Moudry et al., 2016). It is interesting to speculate on the implications of understanding the roles of TOPBP1 in HR-mediated repair for cancer research and treatment.
Previous study reported that TOPBP1 and BRCA1 have overlapping functions and both of them regulate the G2/M cell cycle checkpoint. BRCA1 play an important role in DDR and is a tumor suppressor gene for familial breast and ovarian cancers. Loss-of-function mutations in BRCA1 gene can increase the risk of breast, ovarian and pancreatic cancers. There are several common features shared by BRCA1 and TOPBP1 (Yamane et al., 2002). First, both are BRCT-containing proteins, and TOPBP1 also shares other sequence homology with BRCA1 at the C-terminal region (Yamane et al., 1997). Second, cancer-associated mutations of BRCA1 have been frequently identified in BRTC domain in ovarian and breast cancers (Li & Greenberg, 2012). TOPBP1 mutations have been found in ovarian and breast cancers (Forma et al., 2013; Rebbeck et al., 2009), although they are relatively infrequent. Third, both are phosphorylated by ATM in response to DNA damage and DNA replication stress and are involved in HR repair (Cortez et al., 1999). Fourth, PARP inhibitors selectively kill tumor cells bearing the BRCA1 mutations, and loss of TOPBP1 sensitized human cells to PARP inhibitors as well (Moudry et al., 2016). Based on these similarities between TOPBP1 and BRCA1, we hypothesized that tumor cells bearing TOPBP1 mutations might be also sensitive to DNA damage and the sensitivity might be likely caused by unrepaired DNA lesions. In our study, we discovered that TOPBP1 mutations frequently appeared in most cancers. The cancer-associated mutants of TOPBP1 showed the abolished or impaired TOPBP1 foci formation, increased sensitivity to DNA damage treatment, deficiency in HR repair, disrupted Chk1 activation and partial abrogation of G2/M checkpoint after IR. Overall, both of truncation and missense mutations examined in our study cause deficiency in DDR. Since synthetic lethality treatment has been applied to clinical therapy in ovarian or breast cancer patients with BRCA mutations (Golan et al., 2019), possibility for applying synthetic lethality treatment to treating cancers with TOPBP1 mutations warrants further investigation. Cancer cells with these TOPBP1mutations are likely to be the potential targets for PARP inhibitors, radiation therapy or other treatments to achieve synthetic lethality (Adam et al., 2021; Laine et al., 2021).
The mechanistic roles that cancer-associated mutants of TOPBP1 play in DDR are not yet completely clear. A previous study showed three nonsynonymous SNPs of TOPBP1, K370Q, S730L, or N955S, were not associated with ovarian cancer risk (Rebbeck et al., 2009). Thus, more cancer-driving mutations need to be further identified in TOPBP1. Here, we found the dysfunctions of cancer-associated mutants of TOPBP1. Based on our discovery, it is likely that, similar to BRCA1, these mutations are potential cancer-driving mutations through affecting the functions of TOPBP1 in DDR. We suspect that the functional deficiency may be due to the impaired binding ability of TOPBP1mutants to phosphorylated binding partners. Missense or truncation mutants destroy the “mediator” role of TOPBP1 in DDR through affecting its binding motif and influencing its ability to recruit other DDR factors (Cescutti et al., 2010; Moudry et al., 2016; Wardlaw et al., 2014). The mutants of TOPBP1 also probably impact phosphorylation-independent function of TOPBP1, which may destroy its recognition of other molecules such as poly(ADP)-ribose or DNA (Blackford et al., 2015; Leung & Glover, 2011). It is possible that a nonsense-mediated decay (NMD) may mediate partial mRNA decay of nonsense mutations and lead to the reduction of the expression of these mutants. However, to date, little evidence indicates that NMD is 100% penetrance for removal of all the nonsense mRNA. Moreover, NMD is even variable among individuals (Nguyen et al., 2014). Thus, it is likely that residual mutant mRNAs may still generate mutant TOPBP1 protein to deregulate DSB repair. The specific mechanism of TOPBP1 mutants in DDR needs further exploration.
In conclusion, we analyzed and characterized the mutations of TOPBP1 in various cancers and identified their functional defects in HR repair and cell cycle checkpoint control. Our data provide proof-of-concept evidence for the association between cancer progression and TOPBP1 mutations and may provide a promising therapy for synthetic lethality in tumor treatment.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Adam, S., Rossi, S. E., Moatti, N., Zompit, M. D., Xue, Y. B., Ng, T. F., Durocher, D., et al. (2021). The CIP2A-TOPBP1 axis safeguards chromosome stability and is a synthetic lethal target for BRCA-mutated cancer. Nature Cancer. https://doi.org/10.1038/s43018-021-00266-w
Ashwell, S., Janetka, J. W., & Zabludoff, S. (2008). Keeping checkpoint kinases in line: New selective inhibitors in clinical trials. Expert Opinion on Investigational Drugs, 17(9), 1331–1340. https://doi.org/10.1517/13543784.17.9.1331
Blackford, A. N., Nieminuszczy, J., Schwab, R. A., Galanty, Y., Jackson, S. P., & Niedzwiedz, W. (2015). TopBP1 interacts with BLM to maintain genome stability but is dispensable for preventing BLM degradation. Molecular Cell, 57(6), 1133–1141. https://doi.org/10.1016/j.molcel.2015.02.012
Burrows, A. E., & Elledge, S. J. (2008). How ATR turns on: TopBP1 goes on ATRIP with ATR. Genes & Development, 22(11), 1416–1421. https://doi.org/10.1101/gad.1685108
Cescutti, R., Negrini, S., Kohzaki, M., & Halazonetis, T. D. (2010). TopBP1 functions with 53BP1 in the G1 DNA damage checkpoint. Embo Journal, 29(21), 3723–3732. https://doi.org/10.1038/emboj.2010.238
Choi, J. H., Lindsey-Boltz, L. A., & Sancar, A. (2009). Cooperative activation of the ATRcheckpoint kinase by TopBP1 and damaged DNA. Nucleic Acids Research, 37(5), 1501–1509. https://doi.org/10.1093/nar/gkn1075
Choi, S. H., Yang, H., Lee, S. H., Ki, J. H., Nam, D. H., & Yoo, H. Y. (2014). TopBP1 and Claspin contribute to the radioresistance of lung cancer brain metastases. Molecular Cancer. https://doi.org/10.1186/1476-4598-13-211
Chowdhury, P., Lin, G. E., Liu, K., Song, Y. C., Lin, F. T., & Lin, W. C. (2014). Targeting TopBP1 at a convergent point of multiple oncogenic pathways for cancer therapy. Nature Communications. https://doi.org/10.1038/Ncomms6476
Ciccia, A., & Elledge, S. J. (2010). The DNA damage response: Making it safe to play with knives. Molecular Cell, 40(2), 179–204. https://doi.org/10.1016/j.molcel.2010.09.019
Cortez, D., Wang, Y., Qin, J., & Elledge, S. J. (1999). Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science, 286(5442), 1162–1166. https://doi.org/10.1126/science.286.5442.1162
Day, M., Rappas, M., Ptasinska, K., Boos, D., Oliver, A. W., & Pearl, L. H. (2018). BRCT domains of the DNA damage checkpoint proteins TOPBP1/Rad4 display distinct specificities for phosphopeptide ligands. eLife. https://doi.org/10.7554/eLife.39979
Delacroix, S., Wagner, J. M., Kobayashi, M., Yamamoto, K., & Karnitz, L. M. (2007). The Rad9-Hus1-Rad1 (9–1-1) clamp activates checkpoint signaling via TopBP1. Genes & Development, 21(12), 1472–1477. https://doi.org/10.1101/gad.1547007
Feng, H., Lu, J., Song, X., Thongkum, A., Zhang, F., Lou, L., Gong, Z., et al. (2020). CK2 kinase-mediated PHF8 phosphorylation controls TopBP1 stability to regulate DNA replication. Nucleic Acids Research, 48(19), 10940–10952. https://doi.org/10.1093/nar/gkaa756
Forma, E., Brzezianska, E., Krzeslak, A., Chwatko, G., Jozwiak, P., Szymczyk, A., Brys, M., et al. (2013). Association between the c.*229C > T polymorphism of the topoisomerase II beta binding protein 1 (TopBP1) gene and breast cancer. Molecular Biology Reports, 40(5), 3493–3502. https://doi.org/10.1007/s11033-012-2424-z
Forma, E., Krzeslak, A., Bernaciak, M., Romanowicz-Makowska, H., & Brys, M. (2012). Expression of TopBP1 in hereditary breast cancer. Molecular Biology Reports, 39(7), 7795–7804. https://doi.org/10.1007/s11033-012-1622-z
Going, J. J., Nixon, C., Dornan, E. S., Boner, W., Donaldson, M. M., & Morgan, I. M. (2007). Aberrant expression of TopBP1 in breast cancer. Histopathology, 50(4), 418–424. https://doi.org/10.1111/j.1365-2559.2007.02622.x
Golan, T., Hammel, P., Reni, M., Van Cutsem, E., Macarulla, T., Hall, M. J., Kindler, H. L., et al. (2019). Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. New England Journal of Medicine, 381(4), 317–327. https://doi.org/10.1056/Nejmoa1903387
Gong, Z., Kim, J. E., Leung, C. C., Glover, J. N., & Chen, J. (2010). BACH1/FANCJ acts with TopBP1 and participates early in DNA replication checkpoint control. Molecular Cell, 37(3), 438–446. https://doi.org/10.1016/j.molcel.2010.01.002
Harper, J. W., & Elledge, S. J. (2007). The DNA damage response: Ten years after. Molecular Cell, 28(5), 739–745. https://doi.org/10.1016/j.molcel.2007.11.015
Huang, R. X., & Zhou, P. K. (2020). DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduction and Targeted Therapy, 5(1), 60. https://doi.org/10.1038/s41392-020-0150-x
Jackson, S. P., & Bartek, J. (2009). The DNA-damage response in human biology and disease. Nature, 461(7267), 1071–1078. https://doi.org/10.1038/nature08467
Jeon, Y., Ko, E., Lee, K. Y., Ko, M. J., Park, S. Y., Kang, J., Hwang, D. S., et al. (2011). TopBP1 deficiency causes an early embryonic lethality and induces cellular senescence in primary cells. Journal of Biological Chemistry, 286(7), 5414–5422. https://doi.org/10.1074/jbc.M110.189704
Karppinen, S. M., Erkko, H., Reini, K., Pospiech, H., Heikkinen, K., Rapakko, K., Winqvist, R., et al. (2006). Identification of a common polymorphism in the TopBP1 gene associated with hereditary susceptibility to breast and ovarian cancer. European Journal of Cancer, 42(15), 2647–2652. https://doi.org/10.1016/j.ejca.2006.05.030
Kumagai, A., Lee, J., Yoo, H. Y., & Dunphy, W. G. (2006). TopBP1 activates the ATR-ATRIP complex. Cell, 124(5), 943–955. https://doi.org/10.1016/j.cell.2005.12.041
Laine, A., Nagelli, S. G., Farrington, C., Butt, U., Cvrljevic, A. N., Vainonen, J. P., Westermarck, J., et al. (2021). CIP2A interacts with TopBP1 and drives basal-like breast cancer tumorigenesis. Cancer Research, 81(16), 4319–4331. https://doi.org/10.1158/0008-5472.CAN-20-3651
Leimbacher, P. A., Jones, S. E., Shorrocks, A. M. K., Zompit, M. D., Day, M., Blaauwendraad, J., Stucki, M., et al. (2019). MDC1 interacts with TOPBP1 to maintain chromosomal stability during mitosis. Molecular Cell, 74(3), 571. https://doi.org/10.1016/j.molcel.2019.02.014
Leung, C. C. Y., & Glover, J. N. M. (2011). BRCT domains easy as one, two, three. Cell Cycle, 10(15), 2461–2470. https://doi.org/10.4161/cc.10.15.16312
Leung, C. C., Gong, Z., Chen, J., & Glover, J. N. (2011). Molecular basis of BACH1/FANCJ recognition by TopBP1 in DNA replication checkpoint control. Journal of Biological Chemistry, 286(6), 4292–4301. https://doi.org/10.1074/jbc.M110.189555
Leung, C. C., Kellogg, E., Kuhnert, A., Hanel, F., Baker, D., & Glover, J. N. (2010). Insights from the crystal structure of the sixth BRCT domain of topoisomerase IIbeta binding protein 1. Protein Science, 19(1), 162–167. https://doi.org/10.1002/pro.290
Leung, C. C. Y., Sun, L. X., Gong, Z. H., Burkat, M., Edwards, R., Assmus, M., Glover, J. N. M., et al. (2013). Structural insights into recognition of MDC1 by TopBP1 in DNA replication checkpoint control. Structure, 21(8), 1450–1459. https://doi.org/10.1016/j.str.2013.06.015
Li, M. L., & Greenberg, R. A. (2012). Links between genome integrity and BRCA1 tumor suppression. Trends in Biochemical Sciences, 37(10), 418–424. https://doi.org/10.1016/j.tibs.2012.06.007
Li, T., & Chen, Z. J. (2018). The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. Journal of Experimental Medicine, 215(5), 1287–1299. https://doi.org/10.1084/jem.20180139
Liu, K., Bellam, N., Lin, H. Y., Wang, B., Stockard, C. R., Grizzle, W. E., & Lin, W. C. (2009). Regulation of p53 by TopBP1: A potential mechanism for p53 inactivation in cancer. Molecular and Cellular Biology, 29(10), 2673–2693. https://doi.org/10.1128/MCB.01140-08
Lord, C. J., & Ashworth, A. (2012). The DNA damage response and cancer therapy. Nature, 481(7381), 287–294. https://doi.org/10.1038/nature10760
Magda, A. B., Bogdanova, N. V., Bremer, M., Karstens, J. H., Hillemanns, P., & Dörk, T. (2010). TOPBP1 missense variant Arg309Cys and breast cancer in a German hospital-based case-control study. Journal of Negative Results in BioMedicine, 9, 1–4.
Montales, K., Kim, A., Ruis, K., & Michael, W. M. (2021). Structure-function analysis of TOPBP1’s role in ATR signaling using the DSB-mediated ATR activation in Xenopus egg extracts (DMAX) system. Science and Reports, 11(1), 467. https://doi.org/10.1038/s41598-020-80626-1
Mooser, C., Symeonidou, I. E., Leimbacher, P. A., Ribeiro, A., Shorrocks, A. K., Jungmichel, S., Stucki, M., et al. (2020). Treacle controls the nucleolar response to rDNA breaks via TOPBP1 recruitment and ATR activation. Nature Communications, 11(1), 123. https://doi.org/10.1038/s41467-019-13981-x
Moudry, P., Watanabe, K., Wolanin, K. M., Bartkova, J., Wassing, I. E., Watanabe, S., Bartek, J., et al. (2016). TOPBP1 regulates RAD51 phosphorylation and chromatin loading and determines PARP inhibitor sensitivity. Journal of Cell Biology, 212(3), 281–288. https://doi.org/10.1083/jcb.201507042
Nguyen, L. S., Wilkinson, M. F., & Gecz, J. (2014). Nonsense-mediated mRNA decay: Inter-individual variability and human disease. Neuroscience and Biobehavioral Reviews, 46(Pt 2), 175–186. https://doi.org/10.1016/j.neubiorev.2013.10.016
Paulsen, R. D., Soni, D. V., Wollman, R., Hahn, A. T., Yee, M. C., Guan, A., Cimprich, K. A., et al. (2009). A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Molecular Cell, 35(2), 228–239. https://doi.org/10.1016/j.molcel.2009.06.021
Rappas, M., Oliver, A. W., & Pearl, L. H. (2011). Structure and function of the Rad9-binding region of the DNA-damage checkpoint adaptor TopBP1. Nucleic Acids Research, 39(1), 313–324. https://doi.org/10.1093/nar/gkq743
Rebbeck, T. R., Mitra, N., Domchek, S. M., Wan, F., Chuai, S., Friebel, T. M., kConFab, et al. (2009). Modification of ovarian cancer risk by BRCA1/2-interacting genes in a multicenter cohort of BRCA1/2 mutation carriers. Cancer Research, 69(14), 5801–5810. https://doi.org/10.1158/0008-5472.CAN-09-0625
Santos-Pereira, J. M., & Aguilera, A. (2015). R loops: New modulators of genome dynamics and function. Nature Reviews Genetics, 16(10), 583–597. https://doi.org/10.1038/nrg3961
Sokka, M., Parkkinen, S., Pospiech, H., & Syvaoja, J. E. (2010). Function of TopBP1 in genome stability. SubCellular Biochemistry, 50, 119–141. https://doi.org/10.1007/978-90-481-3471-7_7
Sun, L. X., Huang, Y. H., Edwards, R. A., Yang, S. M., Blackford, A. N., Niedzwiedz, W., & Glover, J. N. M. (2017). Structural insight into BLM recognition by TopBP1. Structure, 25(10), 1582. https://doi.org/10.1016/j.str.2017.08.005
Wang, J., Gong, Z., & Chen, J. (2011). MDC1 collaborates with TopBP1 in DNA replication checkpoint control. Journal of Cell Biology, 193(2), 267–273. https://doi.org/10.1083/jcb.201010026
Wardlaw, C. P., Carr, A. M., & Oliyer, A. W. (2014). TopBP1: A BRCT-scaffold protein functioning in multiple cellular pathways. DNA Repair (amst), 22, 165–174. https://doi.org/10.1016/j.dnarep.2014.06.004
Yamane, K., Kawabata, M., & Tsuruo, T. (1997). A DNA- topoisomerase-11-binding protein with eight repeating regions similar to DNA-repair enzymes and to a cell-cycle regulator. European Journal of Biochemistry, 250(3), 794–799.
Yamane, K., Wu, X., & Chen, J. (2002). A DNA damage-regulated BRCT-containing protein, TopBP1, is required for cell survival. Molecular and Cellular Biology, 22(2), 555–566. https://doi.org/10.1128/mcb.22.2.555-566.2002
Zhang, J., Yan, Z., Wang, Y., Wang, Y., Guo, X., Jing, J., Wu, C., et al. (2020). Cancer-associated 53BP1 mutations induce DNA damage repair defects. Cancer Letters, 501, 43–54. https://doi.org/10.1016/j.canlet.2020.12.033
Zhou, Z. W., Liu, C., Li, T. L., Bruhn, C., Krueger, A., Min, W., Carr, A. M., et al. (2013). an essential function for the ATR-activation-domain (AAD) of TopBP1 in mouse development and cellular senescence. Plos Genetics. https://doi.org/10.1371/journal.pgen.1003702
Funding
This study was funded by the National Natural Science Foundation of China (No. 82002594, No. 32071277, No. 32171295, No. 81874160), the Hebei Natural Science Foundation for Outstanding Young Scholars (No. H2020201017), High-level Talents Research Start-up Project of Hebei University (No. 521000981352), Natural Science Foundation of Hebei province (No. C2021201012), S&T Program of Hebei (No. 216Z2602G) and Interdisciplinary Research Program of Natural Science of Hebei University (Nos. DXK202006, DXK202007).
Author information
Zhenzhen Yan and Xiuling Ge contributed equally to this work.
Authors and Affiliations
School of Life Sciences, Institute of Life Sciences and Green Development, Hebei University, Baoding, 071002, Hebei, China
Zhenzhen Yan, Xiuling Ge, Jiaxu Wang, Mengyao Li, Xiuhua Liu & Chen Wu
Westlake Laboratory of Life Sciences and Biomedicine, Hangzhou, Zhejiang, China
Xiaochun Yu
School of Life Sciences, Westlake University, Hangzhou, Zhejiang, China
Xiaochun Yu
Institute of Biology, Westlake Institute for Advanced Study, Hangzhou, Zhejiang, China
Xiaochun Yu
Department of Basic Medicine, Xinzhou Vocational and Technical College, Xinzhou, Shanxi, China
Xiuling Ge
Hebei Zhitong Bio-Pharmaceutical Co., Ltd, Hebei, China
Feng Xu
Contributions
CW, XY and XL conceived and designed the experiments; ZY and XG performed the experiments, analyzed the data and made figures in the manuscript; JW and XL made structure modeling; FX and ML did part of experiments; CW and XY wrote and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Correspondence to Xiuhua Liu, Xiaochun Yu or Chen Wu.
Ethics declarations
Conflict of interest
The authors declare no completing financial interests.
Consent for publication
All authors have approved the final submission and consented to publication.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file1 (DOCX 729 KB)
Supplementary file2 (DOCX 431 KB)
Rights and permissions
About this article
Cite this article
Yan, Z., Ge, X., Wang, J. et al. The impact of TOPBP1 mutations in human cancers on the DNA damage response. GENOME INSTAB. DIS. 3, 144–162 (2022). https://doi.org/10.1007/s42764-022-00072-3
Received05 January 2022
Revised26 February 2022
Accepted12 March 2022
Published27 April 2022
Issue DateJune 2022
DOIhttps://doi.org/10.1007/s42764-022-00072-3
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
TOPBP1
DNA damage repair
Checkpoint
Cancer-associated mutations
用户登录
还没有账号?
立即注册