






Histone methyltransferase GLP epigenetically activates GPCPD1 to sustain cancer cell metastasis and invasion
Original Research Paper
Open Access
He Wen, Minghui Shu, Jia-Yi Chen, Xiaofan Li, Qian Zhu, Jun Zhang, Yuan Tian, Xiaopeng Lu & Wei-Guo Zhu
Genome Instability & Disease (2022)
Abstract
Disordered choline metabolism is associated with tumor progression. Glycerophosphocholine phosphodiesterase 1 (GPCPD1) is critical for cleaving glycerophosphocholine (GPC) to produce choline. However, whether and how GPCPD1 is epigenetically regulated remains largely unknown. In the current study, we report that histone H3 lysine 9 (H3K9) methyltransferase GLP (G9a-like Protein) is essential for transcriptional activation of GPCPD1 through H3K9me1 to promote tumor cell migration and invasion. Knocking down GLP or inhibiting its methyltransferase activity impaired GPCPD1 expression and decreased the choline levels. Importantly, we confirmed that both GPCPD1 and choline levels are positively correlated with cancer cell migration. The reduced migration and invasion of GPCPD1-knockdown cells were rescued by choline treatment. Interestingly, GPCPD1 gene expression was found regulated by transcription factor Krüppel-like Factor 5 (KLF5). KLF5 recruitment was GLP-dependent and was indispensable for GPC-induced GPCPD1 expression. These data suggest that GLP promotes tumor cell migration and invasion by transcriptionally activating GPCPD1. GLP and KLF5 are potential therapeutic targets in future cancer treatment.
Introduction
Metabolic reprogramming is a predominant hallmark of cancer and an attractive process that can be targeted in cancer treatment (Pavlova & Thompson, 2016). The metabolism of choline is considered a critical aspect of the cancer metabolome and has thus been intensively investigated in the context of tumor progression (Aboagye & Bhujwalla, 1999; Ackerstaff et al., 2003; Glunde et al., 2006, 2011; Iorio et al., 2005). Cells undergoing malignant transformation produce high levels of total choline (tCho) and phosphocholine (PC), which is increasingly used in clinical diagnostics (Gillies & Morse, 2005; Glunde et al., 2010; Iorio et al., 2010; Podo, 1999). For example, increased tCho levels are associated with aggressiveness in both breast (Chen et al., 2011) and prostate cancers (Scheenen et al., 2011). In addition, increased PC accompanied by decreased glycerophosphocholine (GPC) is also found in breast and ovarian carcinomas, which causes a lower GPC/PC ratio in cancer samples as compared to the normal tissues (Aboagye & Bhujwalla, 1999; Iorio et al., 2005).
Glycerophosphocholine phosphodiesterase 1 (GPCPD1, also known as EDI3 or GDE5), is an essential enzyme for choline generation. Mechanistically, GPC could be cleaved by GPCPD1, resulting in generation of choline, which is possibly related to cancer cell migration (Lesjak et al., 2014). Unsurprisingly, GPCPD1 expression was also found higher in primary tumors of patients with ovarian and endometrial cancers, which is positively correlated with developed metastasis and poor survival outcomes (Stewart et al., 2012). Although GPDPD1 has been suggested to be an important factor in cancer development, the epigenetic regulation mechanism of GPCPD1 in cancer is poorly understood.
Histone lysine methylations have pivotal roles in various biological processes including the regulation of gene transcription. Histone lysine methylations are reversible and modulated by two groups of enzymes, namely histone lysine methyltransferases (KMTs) and lysine demethylases (KDMs). Importantly, the catalyzation conducted by KMTs or KDMs requires certain metabolic coenzymes, such as S-adenosylmethionine (SAM), α-ketoglutarate (α-KG) and flavin adenine dinucleotide (FAD). During cancer development, the cellular metabolites contents are always changed, possibly leading to altered behaviors of KMTs or KDMs to reprogram gene expression and promote the malignancy of tumor. In this scenario, the metabolic genes can also be targeted. It raises an exciting theory that the metabolites-regulated KMTs or KDMs participate in cancer metabolic reprogramming through transcriptional regulation of metabolic genes, which may serve as possible therapeutic targets in cancer treatment (Teperino et al., 2010).
G9a-like protein (GLP), also known as EHMT1, belongs to KMTs and plays a critical function in catalyzation of histone H3K9 mono-methylation (H3K9me1) and di-methylation (H3K9me2) in mammals. Histone lysine methyltransferases GLP and G9a usually function as heterodimers that are responsible for most of the H3K9me1 and H3K9me2 in the genome. GLP and G9a are important regulators of various cellular processes, including embryo development (Choi et al., 2018), immune response (Shinkai & Tachibana, 2011), and DNA damage repair (Lu et al., 2019; Yang et al., 2017). In the human genome, H3K9me1 is suggested to potentially associate with transcriptional activation, whereas H3K9me2 is usually involved in transcriptional repression (Barski et al., 2007). For example, G9a accumulation induced by hypoxia has been reported to induce increased levels of H3K9me2 and suppression of the targeted genes expression to promote cell survival (Chen et al., 2006). Interestingly, G9a can also activate the serine–glycine biosynthesis pathway to maintain cancer cell proliferation and survival in an epigenetic manner by catalyzing H3K9me1 (Ding et al., 2013). Although GLP is overexpressed in many cancers and correlates with survival (Huang et al., 2010; Shinkai & Tachibana, 2011), how GLP mediates cancer metastasis and invasion is not well understood.
Krüpple-like factor 5 (KLF5) is a transcription factor related to cell proliferation, migration and invasion. Mechanistically, KLF5 interacts with the GC-rich DNA region using its zinc-finger domain (Kojima et al., 1997) and regulates transcription of several downstream target genes, including Survivin (Zhu et al., 2006), Cyclin D1 (Bateman et al., 2004), and P21 (He et al., 2009). In cancer development, KLF5 is reported to enhance hepatocellular carcinoma (HCC) invasion and migration via inducing epithelial-mesenchymal transition (EMT) and stimulating the gene expression of matrix metalloproteinase 2 and 9 (MMP2 and MMP9) (An et al., 2019). In addition, cooperative binding of KLF5 with histone deacetylase 1 (HDAC1) on the insulin like growth factor 1 (IGF1) promoter can repress IGF1 expression to regulate the metastasis of prostate cancer (Ma et al., 2020). It is still unknown whether KLF5 is associated with the choline metabolism to regulate the cancer cells invasion and migration.
Here, we confirmed for the first time that histone methyltransferase GLP is epigenetically involved in choline metabolism and GPCPD1-related cancer cell migration and invasion. H3K9me1 levels were greatly increased at the GPCPD1 promoter by GPC treatment, together with enhanced GPCPD1 expression. Mechanistically, the enriched GLP at GPCPD1 promoter could recruit the transcription factor KLF5 and cooperate with KLF5 to regulate the expression of GPCPD1. Overall, this study uncovered a novel function of GLP in choline metabolism to promote tumor cell migration and invasion, implying that GLP and the regulated choline pathway are potential therapeutic targets in cancer therapy.
Materials and methods
Cell lines, antibodies, and reagents
HeLa and SW620 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (vol/vol) fetal bovine serum (FBS) (Gibco, NY, USA), 100 U/ml penicillin, and 100 μg/ml streptomycin. McCoy's 5a medium was used for HCT116 and HT29 cells culture and RPMI-1640 medium was used for DLD-1 cells culture, supplemented with the same amount of FBS, penicillin, and streptomycin. GLP knockdown HeLa cells and G9a KO HCT116 cells were described before (Lu et al., 2019; Yang et al., 2017). All cells were maintained in a 37 ℃ incubator (Panasonic, Osaka, Japan) with a 5% CO2 and humidified atmosphere. The antibodies used in this study included: anti-GPCPD1 (HPA039556) from Sigma; anti-H3K9me1 (ab8896), anti-H3K9me2 (ab1220), anti-EHMT1 (GLP, ab41969), anti-H3K4me2 (ab11946), anti-H3K4me3 (ab8580), anti-H3K36me3 (ab9050), anti-H3K79me3 (ab2621), anti-histone H3 (ab176842), anti-KLF5 (ab137676) from Abcam; anti-EHMT2 (G9a, 3306S) from Cell Signaling Technology; Anti-H3K27me3 (07449) from Millipore; anti-β-actin from Santa Cruz. The primary antibodies used 1:1,000 working diluent. Dipyridamole (S1895), BIX01294 (S8006), ML264 (S8196), and UNC0638 (S8071) were obtained from Selleck (Houston, TX, USA). We dissolved each inhibitor in dimethyl sulfoxide (DMSO) at a final concentration of 10 mM. Glycerophosphocholine (C824554) and Choline chloride (C805029) were purchased from Macklin. Each compound was dissolved in PBS at a final concentration of 100 mM.
RNA interference
All the siRNAs used in this research were synthesized by Genepharma (Suzhou, China) and transfected into cells using Lipofectamine 2000 (Invitrogen, CA, USA), according to the manufacturer’s procedure. The siRNAs sequences were as follows:
GPCPD1: 5’-GCAGAUUGUGUUAACUGUAACUU-3’;
EHMT1(GLP): 5’-GAUGCCAGCAGUCAUGCAAUU-3’;
EHMT1(GLP) #2: 5’-GGAUUCAGAUGUCACCUUA-3’;
KLF5: 5’-GAUGUAAAUAGAUGACAAACGUU-3’
KLF5 #2: 5’-GCUACAUAUAUGCAAUAUAAA-3’;
GATA2: 5’-GGAUUAGUGAACUAAGUUAUU-3’
HOXA5: 5’-CAUAAGUCAUGACAACAUAGG-3’
CREB1: 5’-CAAUGUGUUCAUUAUUAAAUA-3’
RHOXF1: 5’-GCACUUAUGUUAUCAAUAAAG-3’
MZF1: 5’-GGCGAUUACAUAAGUAUAAGC-3’
TFCP2: 5’-GGGAAUUGUGUGAUGUUUAAC-3’
POU3F4: 5’-CAAUUCAAACAAAGAAGAAUC-3’
GATA3: 5’-GAACUGUUGUAUAAAUUUAUU-3’
FOXO3: 5’-CCAAUGUGUUUCAACUUUAAA-3’
MEIS1: 5’-GGUGAUAGACGAUAGAGAAGG-3’
A pGPU6/Hygro vector was used for cloning the GLP siRNA sequence. shGLP constructs were transfected into HeLa cells for generation of stable GLP knockdown cells. The hygromycin was used for selection of stable transfected cells.
Reverse-transcription PCR
Isolation of total RNA was used TRIzol (Invitrogen) and chloroform. The extracts were precipitated in isopropanol, then the pellets were washed with 75% (vol/vol) ethanol, dried in air for 10 min, and dissolved in DNAase/RNAase free water (TIANGEN, Beijing, China). Two microgram RNA was used for synthesizing the cDNA with a cDNA Synthesis Kit (ReverTra Ace, TOYOBO, Osaka, Japan). The relative gene expressions of GPCPD1 and GAPDH were measured by performing polymerase chain reaction (PCR) and gel electrophoresis. The following primers were used: GPCPD1, 5’-GATCTGCCAGCAAAGGGATG-3’ and 5’-CTTTTGCCGAACCATTGTGC-3’; GAPDH, 5’-GAGTCAACGGATTTGGTCGT-3’, 5’-TTGATTTTGGAGGGATACTCG-3’.
Western blotting
Total proteins were extracted, quantified, and then analyzed by performing the SDS-PAGE and immunoblotting as we previously described (Zhu et al., 2004). Equal amounts of proteins (20–60 µg) from each sample were subjected to SDS-PAGE, and then transferred to nitrocellulose membranes. The membrane was incubated with the indicated antibodies, and then treated with anti-rabbit or anti-mouse (1:10,000) secondary antibodies conjugated with HRP, and visualized with ECL solution (GE Healthcare).
Chromatin immunoprecipitation (ChIP)-qPCR and ChIP-Re-ChIP
The ChIP assays were performed as previously described (Cao et al., 2016a, 2016b). In brief, after the treatment, cells from each sample were fixed with 1% paraformaldehyde and lysed in ChIP lysis buffer (50 mM Tris·HCl pH 8.0, 5 mM EDTA, 1% SDS with phosphatases and protease inhibitors) on ice for 10 min. The lysates were then sonicated on ice, 5 s on 5 s off for 2 min, at 30% intensity. The sonicated samples were centrifuged at 12,000 rpm (13,523 × g) for 10 min at 4 ℃. The supernatants were collected and precleared in ChIP dilution buffer (150 mM NaCl, 20 mM Tris·HCl pH 8.0, 2 mM EDTA, 1% Triton X-100) with protein G or A Sepharose beads and salmon DNA (Sigma, D1626) for over 2 h at 4 ℃ on a rotator. After centrifugation, the supernatants were moved to new tubes and 5% of the samples was taken as ChIP input, stored at – 20 ℃. The left pre-cleared samples were divided and immunoprecipitated with the indicated antibodies at 4℃ overnight on a rotator. Sepharose beads (G or A) were then added and incubated for another 3 h at 4 ℃ with rotation. The beads were washed as previously described (Li et al., 2018). Then, the precipitated samples were eluted from the beads by two times of 15 min incubation with 250 μL of ChIP elution buffer (0.1 M NaHCO3, 1% SDS). In ChIP-Re-ChIP experiments, the first elution was conduct by using 10 mM DTT in TE buffer (10 mM Tris–HCl pH 8.0, 1 mM EDTA pH 8.0.) and then diluted 10 times with ChIP dilution buffer (150 mM NaCl, 20 mM Tris·HCl pH 8.0, 2 mM EDTA, 1% Triton X-100) and subjected again to the above ChIP processes from antibody incubation. Crosslinking was inverted by incubating samples at 65 ℃ overnight. The DNA was purified by using a Cleaning Kit (Macherey–Nagel, 740609), according to the manufacturer’s instructions. Real-time PCR was carried out with an Analytikjena sequence detection system (qTOWER3G), using a thunderbird SYBR Green qPCR mix (TOYOBO, QPC-201). Primers used for PCR were from the GPCPD1 promoter sequence: 5’-AGCGTGCAACTCGAGCTATT-3’ and 5’-AGCGCCTGTAATTCCAGCTA-3’. Three repeats of PCR were performed for each sample. Then, we calculated the percentage of IP/input (2−△Ct) using the average of Ct values obtained from three PCR replications. The differences between two groups were assessed by unpaired two-tailed Student’s t tests using IP/input percentage values obtained from three independent experiments. All ChIP-qPCR data represent the means ± S.D. of three independent experiments to show the enrichment.
Co-immunoprecipitation
After treatment of GPC, cells were collected, washed with PBS and lysed in NP-40 lysis buffer (20 mM Tris–HCl pH 8.0, 137 mM NaCl, 10% glycerol, 1% Nonidet P-40, protease inhibitors) for 30 min on ice. After centrifugation at 13,000 rpm (15,871 × g) for 15 min at 4 ℃, the supernatant was moved in to new tube for immunoprecipitation. After taking the input, antibodies (each for 2 µg) were added for an overnight incubation on a rotator at 4 ℃. The washed Sepharose beads (G or A) were incubated for another 2 h at 4 ℃ with rotation. The beads were washed for over three times with the lysis buffer, and the samples were eluted with 2X SDS loading buffer and analyzed by western blotting.
Peptide pull-down assays
For GST-tagged protein purification, GST-GLP or GST-KLF5 vectors were individually transformed into BL21 cells. After induction using IPTG at 25 ℃ overnight, the expressed proteins were purified with glutathione-sepharose 4B beads (GE Healthcare, NY, USA). A molecular sieve was used for further purification. All the Biotin-labeled peptides were synthesized by Taopu Biotech (Shanghai, China). The sequences of the peptides used were as follows: Biotin-H3K9me0: MARTKQTARKSTGGKAPRKQLAGGK-Biotin; Biotin-H3K9me1: MARTKQTAR[K(me)]STGGKAPRKQLAGGK-Biotin. Biotin-tagged peptides were first incubated with streptavidin slurry from New England Biolabs (MA, USA) in binding buffer (150 mM NaCl, 50 mM Tris–HCl pH 7.4, 10% Glycerol, 1 mM EDTA, 2 mM MgCl2, 0.5% NP-40) at 4 °C overnight. After magnetic adsorption, the unbound peptides were discarded, and the peptide-bound slurries was re-suspended with binding buffer and incubated with 2 μg purified GST-GLP or GST-KLF5 proteins for 1 h at 4 °C. After magnetic adsorption, the slurries were re-suspended with washing buffer (300 mM NaCl, 50 mM Tris–HCl pH 7.4, 10% Glycerol, 1 mM EDTA, 2 mM MgCl2, 0.5% NP-40) and washed for three times. After that, the slurry-bound proteins were dissolved in 2X SDS loading buffer and boiled for analyzation by western blotting.
Cell migration assay
After the seed cells were grown to confluency in a 6-well plate, the scratches were made with pipet tips. The old culture medium was discarded to remove cells scratched off from dishes and changed with new culture medium that contained reduced serum (1% FBS) to decrease cell proliferation. Before scratching, the cells were transfected with siRNAs or vectors for 48 h, if necessary, then cells were scratched with a pipet tip. After scratching, the media were added the small molecular compounds, if necessary, such as choline (1 mM) and GPC (1 mM) were incubated for 48 h and dipyridamole (10 µM), UNC0638 (1 µM) and BIX01294 (5 µM) were incubated for 24 h, then the culture media was freshly changed. Images were captured under a microscopy at 0 h and at 48 h of incubation after the scratching. Microscope settings were kept the same for each measurement.
Invasion assay
Cell invasion assays were carried out in 24-well transwell plates with the inserts dividing upper and lower chambers. In brief, the cells were suspended using serum-free media and seeded on the upper chamber. Medium containing 10% FBS were added into the lower chamber. The transwell plates were cultured in a 37 °C incubator with 5% CO2 for 48 h. After transfection of HeLa and HCT116 cells with siRNA for 48 h, the cells were seeded. During the incubation, small molecular compounds were added to the media, if necessary, such as choline (1 mM) and GPC (1 mM) were incubated for 48 h and dipyridamole (10 µM), UNC0638 (1 µM) and BIX01294 (5 µM) were incubated for 24 h, then the culture media was freshly changed. After incubation, cells attached on the upper chamber were removed, whereas the invasive cells on the lower side of the insert were fixed with 4% paraformaldehyde (PFA) for 15 min and stained with 0.05% crystal violet for 20 min. The quantification of invaded cells were used Image J (National Institutes of Health).
Metabolite extraction for NMR spectroscopy
Metabolite extraction was performed on counted cells (4 × 106 cells) per sample. After transfection of HeLa and HCT116 cells with siRNA for 48 h or treatment of dipyridamole (10 µM), UNC0638 (1 µM) and BIX01294 (5 µM) for 24 h, the cells were collected. The collected cell were re-suspended with 200 µL MAD buffer (Methanol, Acetonitrile, Distilled water at the ratio 5: 3: 2). After 20 min extraction on ice, the samples were centrifuged at 15,000 × g for 20 min at 4 °C. After drying the supernatants using a vacuum centrifugator (Eppendorf, Concentrator plus), the metabolite pellets were dissolved with 500 µL NMR buffer (2 mM Na2HPO4 and 5 mM NaH2PO4 in D2O) with 0.025% trimethylsilyl propionic acid sodium salt-d4 (TSP, an internal standard).
NMR measurement
One-dimensional classical NMR spectrum was performed on a 600 MHz Bruker ASCEND NMR spectrometer (Bruker, Fallanden, Switzerland) prepared with a cryogenic triple resonance probe. The detection parameters were essentially the same as those previously reported (Wen et al., 2015). Chenomx database (Edmonton, Alberta, Canada) was used for identification of the metabolites, such as GPC and choline. Standard compounds were also used for comparison of experimental spectra by fitting the chemical shifts and J-coupling.
Statistical analysis
Gene expression profiling and interactive analyses (GEPIA, http://gepia.cancer-pku.cn/) is a newly developed webserver platform, which can be applied for analyzation of clinical data of tumor patients from TCGA and the GTEx databases (Tang et al., 2017). Based on GEPIA, we investigated the overall survival of GPCPD1 expression and correlation analysis between GPCPD1 and EHMT1 both in CESC and COAD tumor samples. Cutoff-high (75%) and cutoff-low (25%) for CESC or cutoff-low (35%) for COAD were applied for the thresholds of different GPCPD1 expression, respectively. Patient samples were separated into high and low groups based on GPCPD1 expression and analyzed using the risk ratio (95% confidence interval) and log rank test. Correlation coefficients were evaluated by Pearson correlation analysis. For comparison metastasis with non-metastasis colon adenocarcinomas, the clinical data were obtained from the TCGA database (https://www.cancer.gov/tcga) using the Sangerbox tools, an online platform for data analysis and dataset download (http://www.sangerbox.com/tool). Based on clinical information, we sorted metastatic and non-metastatic samples, meanwhile excluded the samples without obvious information. Then, we read out the values of GPCPD1 (ENSG00000125772) expression according to TCGA sample name and performed statistical analysis using the values. Unpaired two-tailed Student’s t-test was employed. P < 0.05 was considered to be significant (*P < 0.05; **P < 0.01; ***P < 0.001; NS, no significance). The data represent the means ± SD of three independent experiments.
Results
GPCPD1 is associated with cervical squamous cell carcinoma and colon adenocarcinoma metastasis
To examine whether GPCPD1 is clinically associated with cancer metastasis, we first compared GPCPD1 expression levels between non-metastasizing and metastasizing samples with mRNA-Seq data of colon adenocarcinomas (COAD) from the TCGA database. As shown in Fig. 1a, we found that GPCPD1 expression is significantly enhanced in metastasizing samples as compared with the non-metastasizing samples. We also obtained the clinical data of cervical squamous cell carcinoma (CESC) and COAD from TCGA database and performed a Kaplan–Meier survival analysis using GEPIA (http://gepia.cancer-pku.cn/). As expected, high GPCPD1 expression was positively correlated to poor survival in both cancer samples (Fig. 1b). Next, we detected GPCPD1 protein levels and its downstream metabolite choline levels in various colon cancer cell lines. We saw that GPCPD1 (Fig. 1c) and choline (Fig. 1d) levels were relatively lower in HT29 cells, higher in DLD-1 and HCT116 cells, and higher still in the SW620 cell line. By performing a scratch assay in these cell lines, we also found that the migration abilities gradually increased from HT29 to SW620 (Fig. 1e). We also found a positive correlation between the metastatic capacities of these cells and the expression levels of GPCPD1 (Fig. 1f, left) and the choline levels (Fig. 1f, right). These data suggest an appealing theory that cancer cell metastatic abilities can be regulated by GPCPD1 expression or the choline levels. Indeed, when we knocked down GPCPD1 by siRNA in HeLa and HCT116 cells (Fig. 1g), the choline levels were markedly decreased (Fig. 1h) and the migration and invasion capabilities of these cells were significantly reduced (Fig. 1i and j). In addition, the migration and invasion abilities were rescued when we transfected with flag-GPCPD1 in these cells (Fig. S1). These results suggest that GPCPD1 protein expression and choline levels are potential regulators of cancer cell metastasis.
Fig. 1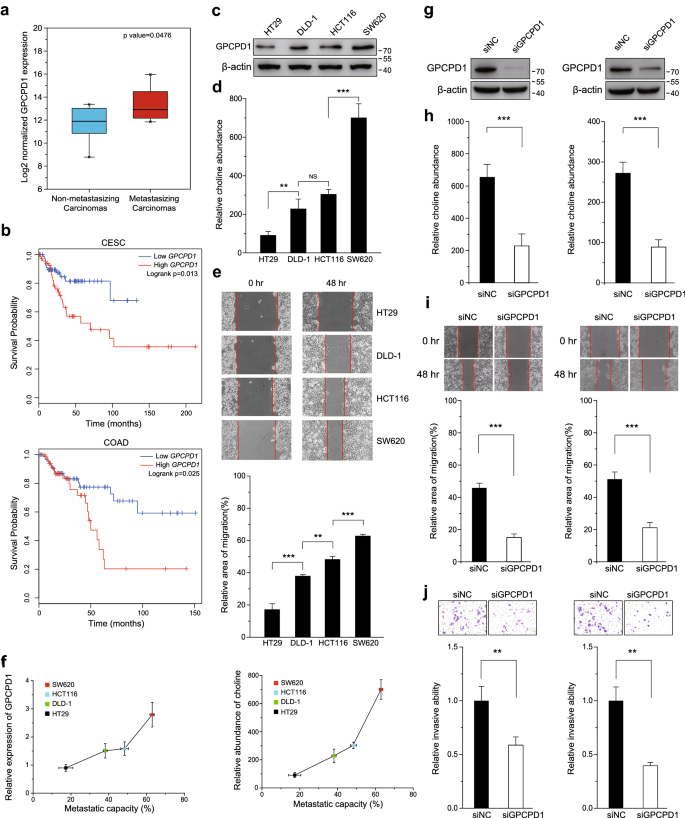
High GPCPD1 expression is associated with a poor prognosis in colon and cervical squamous cancer. a GPCPD1 mRNA expression analysis in metastasizing and non-metastasizing colon adenocarcinomas. The top of the box represents the 75th percentile, the bottom represents the 25th percentile, and the middle line represents the 50th percentile. The whiskers represent the highest and lowest values that are not outliers or extreme values. b Kaplan–Meier survival analysis of CESC and COAD patient samples based on GPCPD1 expression via GEPIA (log-rank test, p = 0.013, n(high) = 73, n(low) = 73, HR(high) = 2.4, p(HR) = 0.017 for CESC and p = 0.025, n(high) = 68, n(low) = 95, HR(high) = 2.1, p(HR) = 0.028 for COAD). c GPCPD1 was detected by western blotting in SW620, HCT116, DLD-1, and HT29 cells. Actin served as the loading control. d The relative amounts of choline detected by 1H-NMR spectroscopy in SW620, HCT116, DLD-1, and HT29 cells (n = 3 for each cell lines). e Cell migration assays were carried out in variety colon cancer cells, such as SW620, HCT116, DLD-1, and HT29 cells. The wound closure rate was determined as a percentage of the original wound area (down). f The correlation between metastatic capacities (data from Fig. 1e) and expression levels of GPCPD1 (left, data from Fig. 1c) or choline (right, data from Fig. 1d) in colon cancer cell lines. g GPCPD1 knockdown was detected by western blotting in HeLa (left) and HCT116 (right) cells transfected with non-specific siRNA negative control (siNC) or GPCPD1 siRNA. Actin served as the loading control. h The relative amounts of choline detected by 1H-NMR spectroscopy in HeLa (left, n = 5) and HCT116 (right, n = 5) cells transfected with non-specific siRNA negative control (siNC) or GPCPD1 siRNA. i, j Cell migration (i) and invasion (j) assays in HeLa (left) or HCT116 (right) cells transfected with non-specific siRNA negative control (siNC) or GPCPD1 siRNA. i The wound closure rate was determined as a percentage of the original wound area (down). j The relative invasive ability was normalized to siNC (down). All data represent the means ± S.D. of three independent experiments (**p < 0.01; ***p < 0.001; NS, no significance)
Full size imageGPCPD1 downstream metabolite choline sustains cancer cell migration and invasion
We next assessed whether the effect of GPCPD1 on cancer cell migration and invasion depends on its choline-release activity. To do so, we performed the scratch and transwell assays with HeLa and HCT116 cells, this time in the presence of dipyridamole (DIP), a phosphodiesterase inhibitor. As shown in Fig. S2 and Fig. 2a, DIP treatment had no effect on GPCPD1 expression (Fig. S2), but the choline levels were significantly decreased (Fig. 2a). In addition, migration and invasion abilities of these cells were also inhibited (Fig. 2b and c). We thus hypothesized that this reduction of migration and invasion abilities following GPCPD1 pharmacological inhibition might result from choline deprivation. To confirm our hypothesis, HeLa and HCT116 cells were treated with choline. We found that choline treatment induced the increment of migration and invasion capacities of these cells but did not affect GPCPD1 expression levels (Fig. 2d–f). Meanwhile, choline treatment also rescued the reduced migration and invasion phenotypes of both HeLa and HCT116 cells transfected with GPCPD1 siRNA (Fig. 2g and h). These findings imply that choline, the downstream metabolite of GPCPD1, promotes the migration and invasion capacity of cancer cells.
Fig. 2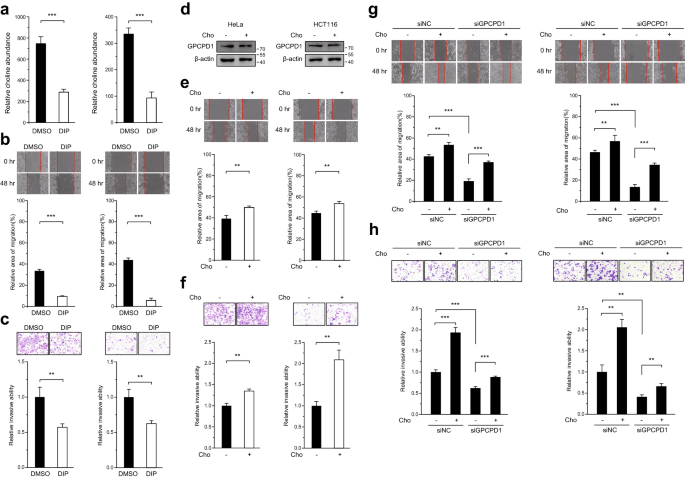
Choline promotes cell migration and invasion. a The relative amounts of choline detected by 1H-NMR spectroscopy in HeLa (left, n = 5) or HCT116 (right, n = 5) cells incubated with DMSO or 10 µM dipyridamole (DIP) for 24 h. b, c Cell migration (b) and invasion (c) assays in HeLa (left) or HCT116 (right) cells incubated with DMSO or DIP (10 µM). b The wound closure rate was determined as a percentage of the original wound area (down). c The relative invasive ability was normalized to DMSO (down). (d) GPCPD1 expression was detected by western blotting in HeLa (left) or HCT116 (right) cells incubated with or without 1 mM choline (Cho) for 24 h. Actin served as the loading control. e, f Cell migration (e) and invasion (f) assays in HeLa (left) or HCT116 (right) cells with or without 1 mM choline (Cho). e The wound closure rate was determined as a percentage of the original wound area (down). f The relative invasive ability was normalized to negative control (down). g, h Cell migration (g) and invasion (h) assays in HeLa (left) or HCT116 (right) cells transfected with non-specific siRNA negative control (siNC) or GPCPD1 siRNA for 48 h, and treated with or without 1 mM Choline (Cho). g The wound closure rate was determined as a percentage of the original wound area (down). h The relative invasive ability was normalized to siNC without choline (down). All data represent the means ± S.D. of three independent experiments (**p < 0.01; ***p < 0.001)
Full size imageGPC modulates GPCPD1 expression in vitro via GLP through H3K9 mono-methylation
We next considered whether the upstream metabolite of GPCPD1, namely GPC, can promote the migration and invasion of cancer cells. We treated HeLa and HCT116 cells with 1 mM GPC. We found that GPC treatment not only markedly enhanced tumor cell migration and invasion capacities as compared to untreated cells, but also enhanced the expression levels of GPCPD1 in these cells (Fig. 3a–c). In addition, we obtained the consistent results in HepG2 and 293 T cells (Fig. S3a–c). To study whether the higher expression of GPCPD1 upon GPC treatment depends on histone modification, we carried out a chromatin immunoprecipitation (ChIP) assay using several transcriptional regulation-related histone modification antibodies, including: H3K9me1, H3K9me2, H3K4me2, H3K4me3, H3K27me3, H3K36me3, and H3K79me3. We then performed qPCR to determine their relative enrichment around the GPCPD1 promoter (Fig. 3d). We found that H3K9me1 levels were markedly enriched at the promoter region of GPCPD1 compared with the other histone methylation sites tested upon GPC treatment (Fig. 3d). As is well known, H3K9me1 is usually modulated by the methyltransferases G9a and GLP. To study whether GLP and/or G9a triggers GPCPD1 promoter activity, we performed a chromatin immunoprecipitation assay using anti-GLP or anti-G9a antibodies. Following GPC treatment, we saw that GLP levels were significantly enhanced at regions around the GPCPD1 promoter. However, we did not see an obvious enrichment of G9a at the GPCPD1 promoter even under GPC treatment (Fig. 3e). In addition, GPCPD1 mRNA expression levels were not markedly changed when G9a was knocked out in HCT116 cells (Fig. S4a and 4b). These results indicate that GLP, but not G9a, is positively associated with GPCPD1 gene expression through H3K9me1 upon GPC treatment.
Fig. 3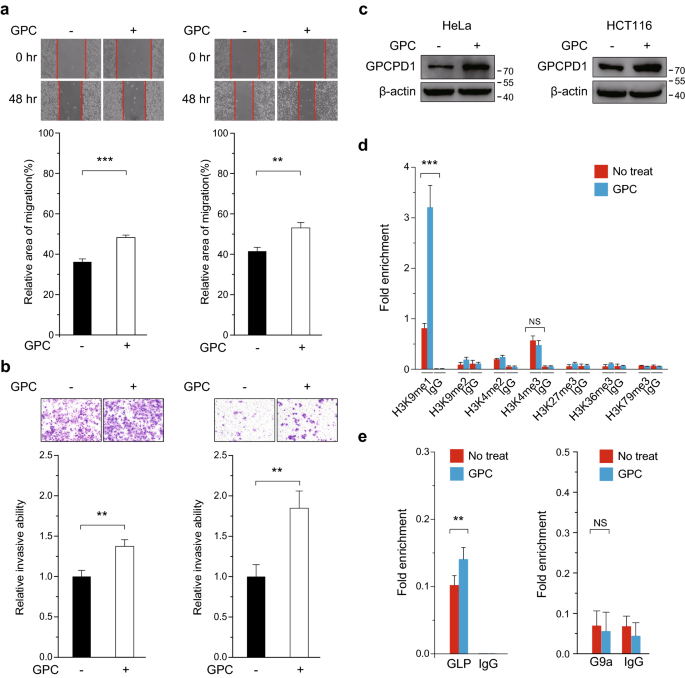
GPC activates GPCPD1’s expression by GLP through H3K9me1. a, b Cell migration (a) and invasion (b) assays in HeLa (left) or HCT116 (right) cells incubated with or without 1 mM GPC. a The wound closure rate was determined as a percentage of the original wound area (down). b The relative invasive ability was normalized to negative control (down). c GPCPD1 expression was detected by western blotting in HeLa (left) or HCT116 (right) cells incubated with or without 1 mM GPC for 24 h. Actin served as the loading control. d ChIP-qPCR was performed in HeLa cells incubated with (blue) or without (red) 1 mM GPC for 24 h and using indicated antibodies. IgG indicates the relative IgG control. e ChIP-qPCR was performed in HeLa cells incubated with (blue) or without (red) 1 mM GPC for 24 h using anti-GLP (left) or anti-G9a (right) antibodies. IgG indicates the relative IgG control. All data represent the means ± S.D. of three independent experiments (**p < 0.01; ***p < 0.001; NS, no significance)
Full size imageGLP silencing reduces GPCPD1 expression and cancer cell migration and invasion
To investigate whether GLP depletion reduces H3K9me1 levels at the promoter region of the GPCPD1 gene, we repeated our ChIP assay this time using anti-GLP and anti-H3K9me1 antibodies (Fig. 4a and b). In HeLa cells stably expressing GLP shRNA (shGLP cells; Fig. S5), GLP levels were markedly decreased at the promoter region of GPCPD1 as compared to those in control cells (Fig. 4a). Similarly, the H3K9me1 levels were also significantly reduced at the GPCPD1 promoter in these cells (Fig. 4b). Consistent with our earlier results, we did not detect any changes in the enrichment of G9a or H3K9me2 at the same region (Fig. S6). In addition, GLP silencing reduced the migration and invasion capacities of HeLa and HCT116 cells in conjunction with reduced GPCPD1 expression (Figs. 4c–e, S7). We also obtained the consistent results in HepG2 and 293 T cells (Fig. S3d–f). Furthermore, we found that the resulting reduction in GPCPD1 expression led to a greater accumulation of GPC and consequently a reduction in choline levels compared to control cells (Fig. 4f). As a confirmation, pharmacological inhibition of GLP activity using UNC0638 or BIX01294 also decreased GPCPD1 expression (Fig. 4g) and choline levels (Fig. 4h), and subsequently inhibited tumor migration and invasion (Fig. 4i and j). All these results suggest that GLP can promote cancer cell migration and invasion by regulating GPCPD1 expression.
Fig. 4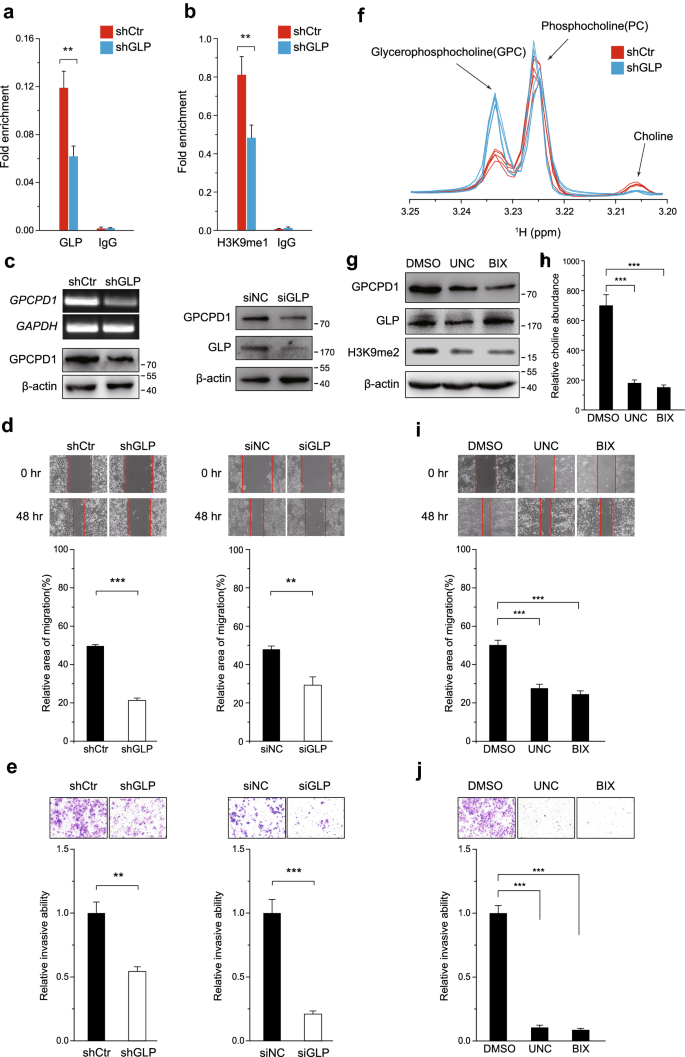
GLP knockdown or enzyme inactivation reduce GPCPD1 expression levels and cancer cell migration and invasion capacity. a, b ChIP-qPCR was performed in HeLa cells stably transfected with shRNA control (shCtr) or shRNA targeting GLP (shGLP) using anti-GLP (a) and anti-H3K9me1 (b). IgG indicates the relative IgG control. c GPCPD1 mRNA and protein expression levels were detected in HeLa cells stably transfected with control (shCtr) or GLP shRNA (left panel). HCT116 cells transfected with non-specific siRNA negative control (siNC) or GLP siRNA for 48 h (right panel) and whole cell extracts were analyzed by immunoblotting. GAPDH and actin served as the loading controls. d, e Cell migration (d) and invasion (e) assays in HeLa cells stably transfected with control (shCtr) or GLP shRNA (left panel) or HCT116 cells transfected with non-specific siRNA negative control (siNC) or GLP siRNA (right panel). d The wound closure rate was determined as a percentage of the original wound area (down). e The relative invasive ability was normalized to shCtr for HeLa or siNC for HCT116 cells (down). f The relative amounts of choline, GPC and PC detected by 1H-NMR spectroscopy in HeLa cells stably transfected control (shCtr) or GLP shRNA. Each group contains six duplicates. g, h GPCPD1 (g) and choline (h) levels in HeLa cells treated with DMSO, UNC0638 (1 µM) or BIX01294 (5 µM) for 24 h, respectively. g The whole cell extracts were analyzed by immunoblotting with the indicated antibodies. Actin served as the loading control. h The relative amounts of choline detected by 1H-NMR spectroscopy. Each group contains three duplicates. i, j Cell migration (i) and invasion (j) assays in HeLa cells treated with DMSO, UNC0638 (1 µM) or BIX01294 (5 µM) for 24 h. i The wound closure rate was determined as a percentage of the original wound area (down). j The relative invasive ability was normalized to DMSO (down). All data represent the means ± S.D. of three independent experiments (**p < 0.01; ***p < 0.001)
Full size imageCholine treatment rescued the reduced metastatic and invading phenotypes of GLP deficiency
We next checked whether the reduced metastatic and invading phenotypes account form GLP silencing in HeLa and HCT116 cells could be rescued by choline treatment. To do so, we performed scratch and transwell assays and found that choline could indeed enhance the migration and invasion capacities of GLP-silenced HeLa and HCT116 cells (Fig. 5a and b). GPCPD1 expression, however, did not increase upon GPC treatment in these cells (Fig. 5c). We also checked GLP and H3K9me1 enrichment at the GPCPD1 promoter in shGLP cells after GPC treatment and found no enhancement of H3K9me1 or GLP as compared to that in shGLP cells without GPC (Fig. 5d), suggesting that the increased H3K9me1 at GPCPD1 promoter was dependent on GLP. In addition, both G9a and H3K9me2 were not enriched at the GPCPD1 promoter and showed no obvious change between control and shGLP HeLa cells (Fig. S8). Conversely, when overexpressing WT-GLP in the shGLP cells, we observed that cell migration and GPCPD1 levels were significantly improved and GPC treatment even promoted a further increment (Fig. 5e and f). By contrast, we saw no enhancement of migration or GPCPD1 levels in shGLP cells overexpressing an enzymatically inactive GLP mutant (MT-GLP; C1203A) even under GPC treatment (Fig. 5e and f). In addition, MT-GLP overexpression was unable to induce an increase in H3K9me1 levels on the promoter region of GPCPD1 as compared with cells overexpressing WT-GLP (Fig. 5g). We also evaluated the correlation between GLP and GPCPD1 in cancer samples from CESC and COAD patients. GPCPD1 levels were positively correlated with expression levels of GLP (EHMT1) in CESC (n = 304, r = 0.2, p = 0.00049) and COAD (n = 261, r = 0.14, p = 0.023) tissues (Fig. 5h). Overall, these data prove that GLP has a key role in modulating GPCPD1-mediated cell migration and invasion.
Fig. 5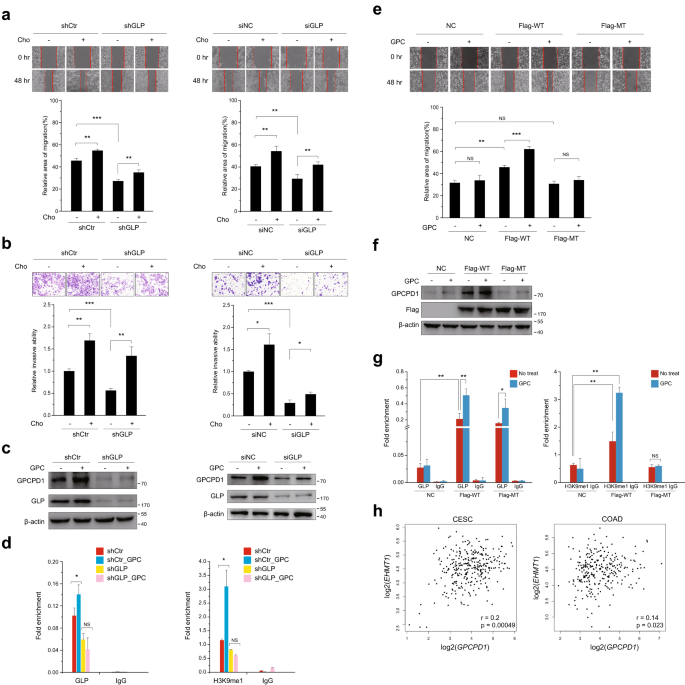
Supplemental choline rescues GLP silencing-induced cell phenotypes. a, b Cell migration (a) and invasion (b) assays were carried out in HeLa cells stably transfected with control (shCtr) or GLP shRNA (left panel) or HCT116 cells transfected with non-specific siRNA negative control (siNC) or GLP siRNA (right panel), each group with or without 1 mM choline (Cho). a The wound closure rate was determined as a percentage of the original wound area (down). b The relative invasive ability was normalized to shCtr for HeLa or siNC for HCT116 without choline, respectively (down). c GPCPD1 and GLP expression levels were detected by western blot analysis in HeLa cells stably transfected with control (shCtr) or GLP shRNA (left panel) or HCT116 cells transfected with non-specific siRNA negative control (siNC) or GLP siRNA (right panel), each group with or without 1 mM GPC for 24 h. Actin served as the loading control. d After treating GPC for 24 h, ChIP-qPCR was performed in HeLa cells stably transfected with control (shCtr) or GLP shRNA using anti-GLP (left panel) or anti-H3K9me1 (right panel) antibodies. IgG indicates the relative IgG control. e–g Cell migration (e), western blot (f), and ChIP-qPCR (g) assays were carried out with or without 1 mM GPC in HeLa stably expressing GLP shRNA and rescued with wild-type GLP (Flag-WT) or enzymatically dead GLP (Flag-MT). NC, negative control. e The wound closure rate was determined as a percentage of the original wound area (down). f Actin served as the loading control. Flag was used to indicate the transfection efficacy. g IgG indicates the relative IgG control. h Gene expression correlation analysis between GLP (EHMT1) and GPCPD1 in tumor samples of CESC (left) and COAD (right). R and p value indicated at bottom of the figures. All data represent the means ± S.D. of three independent experiments (*p < 0.05; **p < 0.01; ***p < 0.001; NS, no significance)
Full size imageTranscription factor KLF5 activates GPCPD1 expression in response to GPC treatment
GLP typically cooperates with other co-modulators to activate the expression of target genes (Levy et al., 2011). In our final analyses, we therefore investigated whether GLP requires an additional transcription factor to co-regulate GPCPD1 expression. We started by searching the JASPAR database (http://jaspar.genereg.net/) and found several candidates that might modulate GPCPD1’s expression. We then investigated the expression levels of GPCPD1 in HeLa cells individually transfected with the siRNA of each candidate (Fig. S9). We found that knocking down the transcription factor Krüppel-like Factor 5 (KLF5) in HeLa cells reduced GPCPD1 expression levels compared with that of the control. Intriguingly, data from endogenous co-immunoprecipitation (co-IP) assays revealed a physiological interaction between GLP and KLF5 when using either anti-KLF5 or anti-GLP antibodies. In addition, this endogenous interaction between GLP and KLF5 was significantly enhanced after GPC treatment (Fig. 6a and b). Interestingly, we did not observe a direct interaction between H3K9me1 and KLF5 in vitro, whereas the interaction between H3K9me1 and GLP (positive control) was successfully detected (Fig. S10). Next, we detected KLF5 enrichment at the GPCPD1 promoter upon GPC treatment in HeLa cells. As shown in Fig. 6c, in control cells, GPC treatment induced an increased enrichment of KLF5 at the GPCPD1 promoter, however, such increased enrichment was not observed in shGLP cells (Fig. 6c). Moreover, overexpression of KLF5 in GLP-silenced HeLa cells was not enhanced GPCPD1 expression levels and the abilities of invasion compared with that of the control (Fig. S11). These results indicated that the recruitment of KLF5 onto GPCPD1 promoter was dependent on GLP. In addition, we carried out a ChIP-Re-ChIP assay and found that combinatorial recruitment of GLP and KLF5 to the GPCPD1 promoter was enhanced upon GPC treatment (Fig. 6d). Importantly, we found that the GPCPD1 expression levels were reduced in KLF5-silenced HeLa and HCT116 cells (Figs. 6e, S12a), even no increment upon GPC treatment in KLF5-silenced HeLa cells (Fig. 6e). At the phenotypic level, migration and invasion capacities of KLF5-silenced HeLa and HCT116 cells were decreased (Figs. 6f–g, S12b, c) and GPC treatment also failed to improve the migration and invasion capacity in KLF5-silenced HeLa cells (Fig. 6f and g). In addition, pharmacological inhibition of KLF expression using ML264 also increased GPC accumulation, decreased choline levels and KLF5 enrichment at the GPCPD1 promoter in HeLa cells, and reduced GPCPD1 expression and subsequently inhibited tumor migration and invasion in HeLa and HCT116 cells (Fig. S13). Moreover, combined treatment of ML264 and BIX01294 inhibited the capacities of migration and invasion more effectively than ML264 or BIX01294 single treatment in HeLa and HCT116 cells (Fig. S14). These results proved that GLP recruits and cooperates with transcription factor KLF5 to regulate the GPCPD1 gene expression in cancer cells.
Fig. 6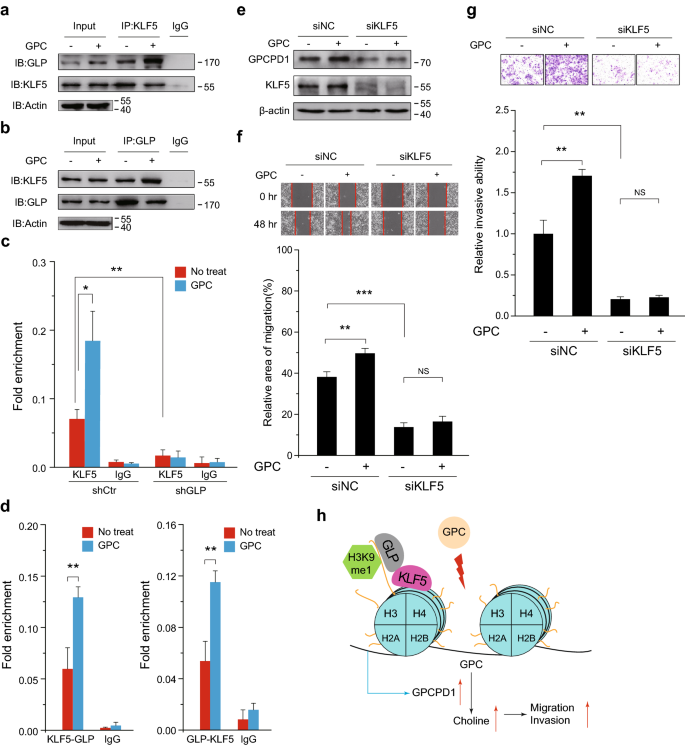
GLP recruits KLF5 to the GPCPD1 promoter. a, b HeLa cells were treated with or without 1 mM GPC for 24 h and the whole cell lysates were extracted for Co-IP using anti-KLF5 (a) or anti-GLP (b) antibodies, followed by western blotting using anti-GLP or anti-KLF5 antibodies to detect the endogenous interaction between KLF5 and GLP, respectively. Actin served as the loading control. c ChIP-qPCR was performed in HeLa cells stably transfected with control (shCtr) or GLP shRNA with (blue) or without (red) 1 mM GPC for 24 h, using an anti-KLF5 antibody. IgG indicates the relative IgG control. d ChIP-Re-ChIP was performed in HeLa cells with (blue) or without (red) 1 mM GPC for 24 h. The relative antibodies successively used for ChIP (left) and for Re-ChIP (right) were indicated at the bottom of graph. IgG indicates the relative IgG control. e GPCPD1 expression was detected by western blotting in HeLa cells transfected with non-specific siRNA negative control (siNC) or KLF5 siRNA, each group with or without 1 mM GPC for 24 h. Actin served as the loading control. f, g Cell migration (f) and invasion (g) assays were carried out in HeLa cells transfected with non-specific siRNA negative control (siNC) or KLF5 siRNA and treated with or without 1 mM GPC. f The wound closure rate was determined as a percentage of the original wound area (down). g The relative invasive ability was normalized to siNC without GPC (down). h A schematic showing a possible mechanism. After GPC treatment, GLP modifies H3K9 mono-methylation and recruits KLF5 to the GPCPD1 promoters to activate its transcription and permit choline biosynthesis, resulting in heightened migration and invasion capacity of cancer cells. All data represent the means ± S.D. of three independent experiments (*p < 0.05; **p < 0.01; ***p < 0.001; NS, no significance)
Full size imageDiscussion
In this research, we aimed to understand whether and how the expression of GPCPD1 is epigenetically regulated. By performing a series of cell-based analyses, we derived a new mechanism that GLP activates GPCPD1 gene expression by increasing H3K9me1 levels and recruiting KLF5 around GPCPD1 promoter to increase choline production, which in turn promotes cancer cell migration and invasion (Fig. 6h). Considering the association between enhanced PC levels and malignancy (M. D. Cao et al., 2016a, 2016b), inhibiting GPCPD1 activation can restrict GPC cleavage and choline production, which ultimately reduces cancer cell migration.
Outside of this study, little was known about the epigenetic regulation of GPCPD1. To gain an understanding of such regulation, we performed a series of ChIP assays using histone modification antibodies. We screened histone modifications known to transcriptionally modulate gene expression, namely: the methylation of H3K9 and H3K27 (that modifies transcriptional repression) and the methylation of H3K4, H3K36, and H3K79 (that modifies transcriptional activation), and found H3K9me1 specifically involved in GPCPD1 expression regulation. Different methylation status on the same histone lysine residues may have different roles in transcriptional regulation. For example, tri-methylations of H3K27 and H3K79 are always associated with gene suppression, whereas mono-methylations of H3K27 and H3K79 are linked with gene activation (Barski et al., 2007). In addition, both H3K9me2 and H3K9me3 are highly involved in gene silencing, whereas higher H3K9me1 levels are detected in more active promoters surrounding the transcription start sites (TSSs) (Barski et al., 2007). Moreover, serine-glycine biosynthetic enzyme genes have been shown transcriptionally activated by H3K9me1 (Black et al., 2012; Ding et al., 2013). In current study, we found that H3K9me1 positively correlates with GPCPD1 expression, supporting the association of H3K9me1 with active chromatin under certain circumstances.
G9a and GLP are H3K9 methyltransferases that primarily catalyze mono- and di-methylation of H3K9 (Tachibana et al., 2005). Consistently, we identified that GLP is recruited to the GPCPD1 promoter region upon GPC treatment and activates GPCPD1 expression through H3K9me1. However, G9a did not seem to have the same effect around the GPCPD1 promoter and we saw no obvious change in GPCPD1 expression in G9a knockout HCT116 cells. This result can be explained as GLP and G9a have been revealed to show different biological properties in some cases. For instance, it was reported that G9a and GLP have distinct H3K9 methylation binding preferences (Collins et al., 2008). In addition, during mice embryonic development, their H3K9 methylation-binding deficient mutations led to different phenotypes (Liu et al., 2015). Also, mutations of EHMT1 gene that is coding GLP are related with the rare Kleefstra syndrome; it is worth noting that the G9a gene is unrelated to the syndrome (Benevento et al., 2016). It has been shown that GLP (not G9a) influences chromatin states and myogenic differentiation through repression of methyl-CpG-binding protein 2 (MeCP2) (Choi et al., 2018). In terms of gene expression regulation, previous studies found that G9a is involved in the silencing of CASP1 (caspase 1) promoter by inducing H3K9me2 (Huang et al., 2017) and required for activating the serine biosynthesis pathway by catalyzing H3K9me1 (Ding et al., 2013). GLP is enriched at the promoters of RelA targeted genes to induce H3K9me2 for the regulation of cell proliferation and inflammation (Levy et al., 2011). In most of previous studies, only one of the two is investigated to show the functions of GLP or G9a in gene regulation. Based on our findings, we think GLP and G9a can individually regulate gene expression in some cases. Therefore, it is intriguing to determine whether G9a and GLP function together or they individually operate in certain gene regulation and other biological processes in the future.
Importantly, we showed that GLP cooperates with KLF5 to transcriptionally activate GPCPD1. This finding is not the first to implicate the role of KLF5 in cancer pathogenesis. Actually, KLF5 was already found highly expressed in high-grade breast cancers (Jia et al., 2019) and KLF5 high expression was also related with a poor prognosis in patients suffering from colorectal cancer (Takagi et al., 2020). So did the cellular metastasis in hepatocellular carcinoma (An et al., 2019) and thyroid cancer (Ma et al., 2018). Moreover, KLF5 can transcriptionally activate a large number of oncogenes (Frigo et al., 2009; Jia et al., 2016). Our data provide further evidence that KLF5 is a newly identified regulator of tumor choline metabolism that contributes to cancer cell migration and invasion. Overall, we revealed a novel mechanism by which GLP and KLF5 promote GPCPD1 expression to produce choline and promote cancer cell migration and invasion. We propose that GLP, KLF5 and the regulated choline metabolic pathway may serve as potential therapeutic targets for dealing with the migration and invasion of cancer.
Availability of data and materials
Not applicable.
Code availability
Not applicable.
References
Aboagye, E. O., & Bhujwalla, Z. M. (1999). Malignant transformation alters membrane choline phospholipid metabolism of human mammary epithelial cells. Cancer Research, 59(1), 80–84. http://www.ncbi.nlm.nih.gov/pubmed/9892190
Ackerstaff, E., Glunde, K., & Bhujwalla, Z. M. (2003). Choline phospholipid metabolism: A target in cancer cells? Journal of Cellular Biochemistry, 90(3), 525–533. https://doi.org/10.1002/jcb.10659
An, T., Dong, T., Zhou, H., Chen, Y., Zhang, J., Zhang, Y., Li, Z., & Yang, X. (2019). The transcription factor Kruppel-like factor 5 promotes cell growth and metastasis via activating PI3K/AKT/Snail signaling in hepatocellular carcinoma. Biochemical and Biophysical Research Communications, 508(1), 159–168. https://doi.org/10.1016/j.bbrc.2018.11.084
Barski, A., Cuddapah, S., Cui, K., Roh, T. Y., Schones, D. E., Wang, Z., Wei, G., Chepelev, I., & Zhao, K. (2007). High-resolution profiling of histone methylations in the human genome. Cell, 129(4), 823–837. https://doi.org/10.1016/j.cell.2007.05.009
Bateman, N. W., Tan, D., Pestell, R. G., Black, J. D., & Black, A. R. (2004). Intestinal tumor progression is associated with altered function of KLF5. Journal of Biological Chemistry, 279(13), 12093–12101. https://doi.org/10.1074/jbc.M311532200
Benevento, M., Iacono, G., Selten, M., Ba, W., Oudakker, A., Frega, M., Keller, J., Mancini, R., Lewerissa, E., Kleefstra, T., Stunnenberg, H. G., Zhou, H., van Bokhoven, H., & Nadif Kasri, N. (2016). Histone methylation by the kleefstra syndrome protein EHMT1 mediates homeostatic synaptic scaling. Neuron, 91(2), 341–355. https://doi.org/10.1016/j.neuron.2016.06.003
Black, J. C., Van Rechem, C., & Whetstine, J. R. (2012). Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Molecular Cell, 48(4), 491–507. https://doi.org/10.1016/j.molcel.2012.11.006
Cao, L. L., Wei, F., Du, Y., Song, B., Wang, D., Shen, C., Lu, X., Cao, Z., Yang, Q., Gao, Y., Wang, L., Zhao, Y., Wang, H., Yang, Y., & Zhu, W. G. (2016a). ATM-mediated KDM2A phosphorylation is required for the DNA damage repair. Oncogene, 35(3), 301–313. https://doi.org/10.1038/onc.2015.81
Cao, M. D., Cheng, M., Rizwan, A., Jiang, L., Krishnamachary, B., Bhujwalla, Z. M., Bathen, T. F., & Glunde, K. (2016b). Targeting choline phospholipid metabolism: GDPD5 and GDPD6 silencing decrease breast cancer cell proliferation, migration, and invasion. NMR in Biomedicine, 29(8), 1098–1107. https://doi.org/10.1002/nbm.3573
Chen, H., Yan, Y., Davidson, T. L., Shinkai, Y., & Costa, M. (2006). Hypoxic stress induces dimethylated histone H3 lysine 9 through histone methyltransferase G9a in mammalian cells. Cancer Research, 66(18), 9009–9016. https://doi.org/10.1158/0008-5472.CAN-06-0101
Chen, J. H., Mehta, R. S., Baek, H. M., Nie, K., Liu, H., Lin, M. Q., Yu, H. J., Nalcioglu, O., & Su, M. Y. (2011). Clinical characteristics and biomarkers of breast cancer associated with choline concentration measured by 1H MRS. NMR in Biomedicine, 24(3), 316–324. https://doi.org/10.1002/nbm.1595
Choi, M. H., Palanichamy Kala, M., Ow, J. R., Rao, V. K., Suriyamurthy, S., & Taneja, R. (2018). GLP inhibits heterochromatin clustering and myogenic differentiation by repressing MeCP2. Journal of Molecular Cell Biology, 10(2), 161–174. https://doi.org/10.1093/jmcb/mjx038
Collins, R. E., Northrop, J. P., Horton, J. R., Lee, D. Y., Zhang, X., Stallcup, M. R., & Cheng, X. (2008). The ankyrin repeats of G9a and GLP histone methyltransferases are mono- and dimethyllysine binding modules. Nature Structural & Molecular Biology, 15(3), 245–250. https://doi.org/10.1038/nsmb.1384
Ding, J., Li, T., Wang, X., Zhao, E., Choi, J. H., Yang, L., Zha, Y., Dong, Z., Huang, S., Asara, J. M., Cui, H., & Ding, H. F. (2013). The histone H3 methyltransferase G9A epigenetically activates the serine-glycine synthesis pathway to sustain cancer cell survival and proliferation. Cell Metabolism, 18(6), 896–907. https://doi.org/10.1016/j.cmet.2013.11.004
Frigo, D. E., Sherk, A. B., Wittmann, B. M., Norris, J. D., Wang, Q., Joseph, J. D., Toner, A. P., Brown, M., & McDonnell, D. P. (2009). Induction of Kruppel-like factor 5 expression by androgens results in increased CXCR4-dependent migration of prostate cancer cells in vitro. Molecular Endocrinology, 23(9), 1385–1396. https://doi.org/10.1210/me.2009-0010
Gillies, R. J., & Morse, D. L. (2005). In vivo magnetic resonance spectroscopy in cancer. Annual Review of Biomedical Engineering, 7, 287–326. https://doi.org/10.1146/annurev.bioeng.7.060804.100411
Glunde, K., Ackerstaff, E., Mori, N., Jacobs, M. A., & Bhujwalla, Z. M. (2006). Choline phospholipid metabolism in cancer: Consequences for molecular pharmaceutical interventions. Molecular Pharmaceutics, 3(5), 496–506. https://doi.org/10.1021/mp060067e
Glunde, K., Artemov, D., Penet, M. F., Jacobs, M. A., & Bhujwalla, Z. M. (2010). Magnetic resonance spectroscopy in metabolic and molecular imaging and diagnosis of cancer. Chemical Reviews, 110(5), 3043–3059. https://doi.org/10.1021/cr9004007
Glunde, K., Bhujwalla, Z. M., & Ronen, S. M. (2011). Choline metabolism in malignant transformation. Nature Reviews Cancer, 11(12), 835–848. https://doi.org/10.1038/nrc3162
He, M., Han, M., Zheng, B., Shu, Y. N., & Wen, J. K. (2009). Angiotensin II stimulates KLF5 phosphorylation and its interaction with c-Jun leading to suppression of p21 expression in vascular smooth muscle cells. Journal of Biochemistry, 146(5), 683–691. https://doi.org/10.1093/jb/mvp115
Huang, J., Dorsey, J., Chuikov, S., Perez-Burgos, L., Zhang, X., Jenuwein, T., Reinberg, D., & Berger, S. L. (2010). G9a and Glp methylate lysine 373 in the tumor suppressor p53. Journal of Biological Chemistry, 285(13), 9636–9641. https://doi.org/10.1074/jbc.M109.062588
Huang, T., Zhang, P., Li, W., Zhao, T., Zhang, Z., Chen, S., Yang, Y., Feng, Y., Li, F., Shirley Liu, X., Zhang, L., Jiang, G., & Zhang, F. (2017). G9A promotes tumor cell growth and invasion by silencing CASP1 in non-small-cell lung cancer cells. Cell Death & Disease, 8(4), e2726. https://doi.org/10.1038/cddis.2017.65
Iorio, E., Mezzanzanica, D., Alberti, P., Spadaro, F., Ramoni, C., D’Ascenzo, S., Millimaggi, D., Pavan, A., Dolo, V., Canevari, S., & Podo, F. (2005). Alterations of choline phospholipid metabolism in ovarian tumor progression. Cancer Research, 65(20), 9369–9376. https://doi.org/10.1158/0008-5472.CAN-05-1146
Iorio, E., Ricci, A., Bagnoli, M., Pisanu, M. E., Castellano, G., Di Vito, M., Venturini, E., Glunde, K., Bhujwalla, Z. M., Mezzanzanica, D., Canevari, S., & Podo, F. (2010). Activation of phosphatidylcholine cycle enzymes in human epithelial ovarian cancer cells. Cancer Research, 70(5), 2126–2135. https://doi.org/10.1158/0008-5472.CAN-09-3833
Jia, L., Zhou, Z., Liang, H., Wu, J., Shi, P., Li, F., Wang, Z., Wang, C., Chen, W., Zhang, H., Wang, Y., Liu, R., Feng, J., & Chen, C. (2016). KLF5 promotes breast cancer proliferation, migration and invasion in part by upregulating the transcription of TNFAIP2. Oncogene, 35(16), 2040–2051. https://doi.org/10.1038/onc.2015.263
Jia, X., Shi, L., Wang, X., Luo, L., Ling, L., Yin, J., Song, Y., Zhang, Z., Qiu, N., Liu, H., Deng, M., He, Z., Li, H., & Zheng, G. (2019). KLF5 regulated lncRNA RP1 promotes the growth and metastasis of breast cancer via repressing p27kip1 translation. Cell Death & Disease, 10(5), 373. https://doi.org/10.1038/s41419-019-1566-5
Kojima, S., Kobayashi, A., Gotoh, O., Ohkuma, Y., Fujii-Kuriyama, Y., & Sogawa, K. (1997). Transcriptional activation domain of human BTEB2, a GC box-binding factor. Journal of Biochemistry, 121(2), 389–396. https://doi.org/10.1093/oxfordjournals.jbchem.a021600
Lesjak, M. S., Marchan, R., Stewart, J. D., Rempel, E., Rahnenfuhrer, J., & Hengstler, J. G. (2014). EDI3 links choline metabolism to integrin expression, cell adhesion and spreading. Cell Adhesion & Migration, 8(5), 499–508. https://doi.org/10.4161/cam.29284
Levy, D., Kuo, A. J., Chang, Y., Schaefer, U., Kitson, C., Cheung, P., Espejo, A., Zee, B. M., Liu, C. L., Tangsombatvisit, S., Tennen, R. I., Kuo, A. Y., Tanjing, S., Cheung, R., Chua, K. F., Utz, P. J., Shi, X., Prinjha, R. K., Lee, K., … Gozani, O. (2011). Lysine methylation of the NF-kappaB subunit RelA by SETD6 couples activity of the histone methyltransferase GLP at chromatin to tonic repression of NF-kappaB signaling. Nature Immunology, 12(1), 29–36. https://doi.org/10.1038/ni.1968
Li, M., Hou, T., Gao, T., Lu, X., Yang, Q., Zhu, Q., Li, Z., Liu, C., Mu, G., Liu, G., Bao, Y., Wen, H., Wang, L., Wang, H., Zhao, Y., Gu, W., Yang, Y., & Zhu, W. G. (2018). p53 cooperates with SIRT6 to regulate cardiolipin de novo biosynthesis. Cell Death & Disease, 9(10), 941. https://doi.org/10.1038/s41419-018-0984-0
Liu, N., Zhang, Z., Wu, H., Jiang, Y., Meng, L., Xiong, J., Zhao, Z., Zhou, X., Li, J., Li, H., Zheng, Y., Chen, S., Cai, T., Gao, S., & Zhu, B. (2015). Recognition of H3K9 methylation by GLP is required for efficient establishment of H3K9 methylation, rapid target gene repression, and mouse viability. Genes & Development, 29(4), 379–393. https://doi.org/10.1101/gad.254425.114
Lu, X., Tang, M., Zhu, Q., Yang, Q., Li, Z., Bao, Y., Liu, G., Hou, T., Lv, Y., Zhao, Y., Wang, H., Yang, Y., Cheng, Z., Wen, H., Liu, B., Xu, X., Gu, L., & Zhu, W. G. (2019). GLP-catalyzed H4K16me1 promotes 53BP1 recruitment to permit DNA damage repair and cell survival. Nucleic Acids Research, 47(21), 10977–10993. https://doi.org/10.1093/nar/gkz897
Ma, J. B., Bai, J. Y., Zhang, H. B., Jia, J., Shi, Q., Yang, C., Wang, X., He, D., & Guo, P. (2020). KLF5 inhibits STAT3 activity and tumor metastasis in prostate cancer by suppressing IGF1 transcription cooperatively with HDAC1. Cell Death & Disease, 11(6), 466. https://doi.org/10.1038/s41419-020-2671-1
Ma, Y., Wang, Q., Liu, F., Ma, X., Wu, L., Guo, F., Zhao, S., Huang, F., & Qin, G. (2018). KLF5 promotes the tumorigenesis and metastatic potential of thyroid cancer cells through the NF-kappaB signaling pathway. Oncology Reports, 40(5), 2608–2618. https://doi.org/10.3892/or.2018.6687
Pavlova, N. N., & Thompson, C. B. (2016). The emerging hallmarks of cancer metabolism. Cell Metabolism, 23(1), 27–47. https://doi.org/10.1016/j.cmet.2015.12.006
Podo, F. (1999). Tumour phospholipid metabolism. NMR in Biomedicine, 12(7), 413–439. https://doi.org/10.1002/(sici)1099-1492(199911)12:7%3c413::aid-nbm587%3e3.0.co;2-u
Scheenen, T. W., Futterer, J., Weiland, E., van Hecke, P., Lemort, M., Zechmann, C., Schlemmer, H. P., Broome, D., Villeirs, G., Lu, J., Barentsz, J., Roell, S., & Heerschap, A. (2011). Discriminating cancer from noncancer tissue in the prostate by 3-dimensional proton magnetic resonance spectroscopic imaging: A prospective multicenter validation study. Investigative Radiology, 46(1), 25–33. https://doi.org/10.1097/rli.0b013e3181f54081
Shinkai, Y., & Tachibana, M. (2011). H3K9 methyltransferase G9a and the related molecule GLP. Genes & Development, 25(8), 781–788. https://doi.org/10.1101/gad.2027411
Stewart, J. D., Marchan, R., Lesjak, M. S., Lambert, J., Hergenroeder, R., Ellis, J. K., Lau, C. H., Keun, H. C., Schmitz, G., Schiller, J., Eibisch, M., Hedberg, C., Waldmann, H., Lausch, E., Tanner, B., Sehouli, J., Sagemueller, J., Staude, H., Steiner, E., & Hengstler, J. G. (2012). Choline-releasing glycerophosphodiesterase EDI3 drives tumor cell migration and metastasis. Proceedings of the National Academy of Sciences of the United States of America, 109(21), 8155–8160. https://doi.org/10.1073/pnas.1117654109
Tachibana, M., Ueda, J., Fukuda, M., Takeda, N., Ohta, T., Iwanari, H., Sakihama, T., Kodama, T., Hamakubo, T., & Shinkai, Y. (2005). Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3–K9. Genes & Development, 19(7), 815–826. https://doi.org/10.1101/gad.1284005
Takagi, Y., Sakai, N., Yoshitomi, H., Furukawa, K., Takayashiki, T., Kuboki, S., Takano, S., Suzuki, D., Kagawa, S., Mishima, T., Nakadai, E., Miyauchi, H., Matsubara, H., & Ohtsuka, M. (2020). High expression of Kruppel-like factor 5 is associated with poor prognosis in patients with colorectal cancer. Cancer Science, 111(6), 2078–2092. https://doi.org/10.1111/cas.14411
Tang, Z., Li, C., Kang, B., Gao, G., Li, C., & Zhang, Z. (2017). GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Research, 45(W1), W98–W102. https://doi.org/10.1093/nar/gkx247
Teperino, R., Schoonjans, K., & Auwerx, J. (2010). Histone methyl transferases and demethylases; can they link metabolism and transcription? Cell Metabolism, 12(4), 321–327. https://doi.org/10.1016/j.cmet.2010.09.004
Wen, H., Xu, W. J., Jin, X., Oh, S., Phan, C. H., Song, J., Lee, S. K., & Park, S. (2015). The roles of IP3 receptor in energy metabolic pathways and reactive oxygen species homeostasis revealed by metabolomic and biochemical studies. Biochimica et Biophysica Acta, 1853(11 Pt A), 2937–2944. https://doi.org/10.1016/j.bbamcr.2015.07.020
Yang, Q., Zhu, Q., Lu, X., Du, Y., Cao, L., Shen, C., Hou, T., Li, M., Li, Z., Liu, C., Wu, D., Xu, X., Wang, L., Wang, H., Zhao, Y., Yang, Y., & Zhu, W. G. (2017). G9a coordinates with the RPA complex to promote DNA damage repair and cell survival. Proceedings of the National Academy of Sciences of the United States of America, 114(30), E6054–E6063. https://doi.org/10.1073/pnas.1700694114
Zhu, N., Gu, L., Findley, H. W., Chen, C., Dong, J. T., Yang, L., & Zhou, M. (2006). KLF5 Interacts with p53 in regulating survivin expression in acute lymphoblastic leukemia. Journal of Biological Chemistry, 281(21), 14711–14718. https://doi.org/10.1074/jbc.M513810200
Zhu, W. G., Hileman, T., Ke, Y., Wang, P., Lu, S., Duan, W., Dai, Z., Tong, T., Villalona-Calero, M. A., Plass, C., & Otterson, G. A. (2004). 5-aza-2’-deoxycytidine activates the p53/p21Waf1/Cip1 pathway to inhibit cell proliferation. Journal of Biological Chemistry, 279(15), 15161–15166. https://doi.org/10.1074/jbc.M311703200
Funding
We would like to thank Dr. Jin-Tang Dong from Southern University of Science and Technology School of Medicine for kindly sharing the KLF5 vector. This study was supported by National Natural Science Foundation of China (32090030, 32090033,31701099, 81530074, 81720108027 and 82002988); National Key R&D Program of China (2017YFA0503900); Science and Technology Program of Guangdong Province in China (2017B030301016); Shenzhen Municipal Commission of Science and Technology Innovation (JCYJ20200109114214463); Shenzhen Bay Laboratory (SZBL2019062801011); Natural Science Foundation of Guangdong Province (2017A030310459).
Author information
He Wen and Minghui Shu contributed equally to this work.
Authors and Affiliations
Guangdong Key Laboratory for Genome Stability and Human Disease Prevention, Department of Biochemistry and Molecular Biology, Shenzhen University Health Science Center, Shenzhen, 518055, China
He Wen, Minghui Shu, Jia-Yi Chen, Qian Zhu, Jun Zhang, Yuan Tian, Xiaopeng Lu & Wei-Guo Zhu
Shenzhen Key Laboratory of Marine Bioresource and Eco-Environmental Science, Shenzhen Key Laboratory of Microbial Genetic Engineering,, College of Life Sciences and Oceanography, Shenzhen University, Shenzhen, 518055, China
Xiaofan Li
Shenzhen Bay Laboratory, Shenzhen University Health Science Center, Shenzhen, 518055, China
Wei-Guo Zhu
International Cancer Center, Shenzhen University Health Science Center, Shenzhen, 518055, China
Wei-Guo Zhu
Marshall Laboratory of Biomedical Engineering, Shenzhen University Health Science Center, Shenzhen, 518055, China
Wei-Guo Zhu
Contributions
HW and XL designed the study; HW and MS performed the experimental work; XL carried out the NMR measurement; HW, XL and WZ wrote the paper; JC, QZ, JZ, YT, and XL assisted in the experimental process and contributed analytic tools. All authors read and approved the final manuscript.
Corresponding authors
Correspondence to Xiaopeng Lu or Wei-Guo Zhu.
Ethics declarations
Conflict of interest
The authors declare no potential conflicts of interest. Wei-Guo Zhu is Editor-in-Chief of Genome Instability and Disease.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file1 (DOCX 12217 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article
Cite this article
Wen, H., Shu, M., Chen, JY. et al. Histone methyltransferase GLP epigenetically activates GPCPD1 to sustain cancer cell metastasis and invasion. GENOME INSTAB. DIS. (2022). https://doi.org/10.1007/s42764-022-00083-0
Received06 July 2022
Revised26 August 2022
Accepted07 September 2022
Published19 September 2022
DOIhttps://doi.org/10.1007/s42764-022-00083-0
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
G9a-like protein
Glycerophosphocholine phosphodiesterase 1
KLF5
H3K9me1
用户登录
还没有账号?
立即注册