






Exploring the DNA damage response pathway for synthetic lethality
Review Article
Genome Instability & Disease (2022)
Abstract
Synthetic lethality (SL) describes a situation in which the occurrence of one genetic event maintains cell viability, whereas the co-occurrence of two genetic events lead to cell death. DNA damage response (DDR) pathway represents the most attractive synthetic lethality targets, since genomic instability is a hallmark of cancers due to the accumulation of DNA mutation during the process of DNA damage response. The definition of synthetic lethality will help to target cancer cells precisely and tackle the undruggable targets. In recent years, the success of DNA damage response inhibitors in cancer treatment highlights the potential of this approach. In this review, we will highlight the concept of synthetic lethality, strategy as well as the most recent development in this area.
Introduction
DNA damage occurs constantly in cells undergoing exogenous and endogenous stress, such as reactive oxygen species (ROS) produced by cell metabolism, ionizing radiation (IR), genotoxic agents and so on (Matt & Hofmann, 2016). Several types ofDNA damage have been reported over past decades: single-strand breaks, double-strand breaks, base damage, DNA across-linking and clustered damage sites. Normal cells maintain genomic stability through an inter-related network of molecular pathways.
Cancer is considered a disease that accumulates driver mutations. As a hallmark of cancer, genomic instability often accumulate in cancers with deficiency in DNA damage response (DDR) pathway (Hanahan, 2022). DDR encompasses many interactive signal pathways and machineries mediating DNA damage sensing, DNA repair, cell cycle arrest and apoptosis (as shown in Fig. 1). The DNA repair mechanism is one of the most interesting targets for synthetic lethality as many cancers are genetically defective in one or more components of the DNA repair pathways, including homologous recombination (HR), Non-homologous end joining (NHEJ), microhomology-mediated end-joining (MMEJ), base excision repair (BER), mismatch repair (MMR),
nucleotide excision repair (BER), and translesion synthesis (TLS). The mutation of tumor-related DNA damage repair protein will force cells to utilize alternative ways to overcome DNA repair deficiency. The conceptual framework of synthetic lethality (SL) provides a paradigm to precisely target tumor cells while sparing normal cells. Most cancer cells are deficient in at least one or more DDRs, so these cancer cells become highly dependent on alternate pathways, and inhibition of the alternate pathways will kill these cancer cells, resulting in SL.
Fig. 1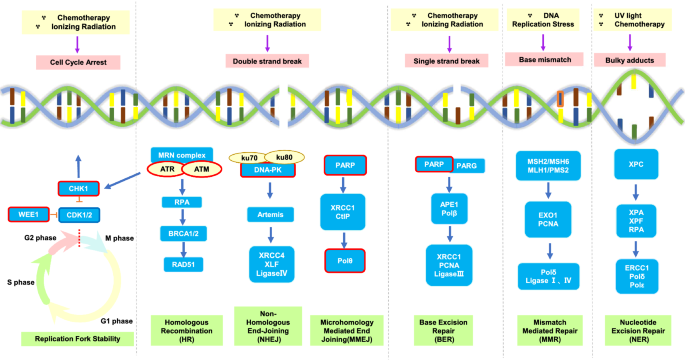
DNA damage response pathways and the related synthetic lethality targets. There are several types of DNA damage response (DDR) pathways including: double strand break (DSB), single strand break (SSB), base mismatch and bulky adducts due to replication. Cell cycle arrest is also involved in DNA damage response. These pathways lead to the activation of intricate signal cascades. DDR pathways mitigate DNA lesion and replication; therefore, the deficiency and/or inhibition of these pathways results in SSBs and DSBs accumulation. Red boxes highlight some important targets which undergoing clinical trials. Poly (ADP-ribose) polymerase (PARP) enzymes are crucial to initiate the downstream mechanism of DSB and SSB. DSB repair occurs mainly through non-homologous end joining (NHEJ) and homologous recombination (HR). Microhomology-mediated end joining (MMEJ) sometimes is active through polymerase Theta (Polθ) as an alternative end joining pathway. The kinases ATR and ATM have key roles in HR signaling and in maintaining replication fork stability via cell cycle regulation by targeting their downstream molecular, CHK1/2. The activity of DNA-dependent protein kinase (DNA-PK) is essential for NHEJ. WEE1 is a nuclear kinase that can modulate mitotic entry by inhibiting cyclin dependent kinase (CDK) 1/2. Drugs targeting these pivotal components of the DDR pathways are undergoing preclinical research and clinical testing. MRN, MRE11, RAD50 and NBS1 complex; RPA, replication protein A; BRCA, breast cancer susceptibility gene; XRCC, X-Ray repair cross complementing; XLF, XRCC4-like factor; CtIP, CtBP-interacting protein; PARG, poly(ADP-ribose) glycohydrolase; APE, apurinic/apyrimidinic endonuclease; PCNA, proliferating cell nuclear antigen;MSH, MutS homologs;MLH, MutL homologs; PMS, mismatch repair protein; EXO1, exonuclease 1; XPA, XPC, XPF, xeroderma pigmentosum A, C, F; ERCC, excision repair cross-complementing
Full size imageThe concept of SL was first described in Drosophila in 1922 (Bridges, 1922) and defined by authors in 1946 (Dobzhansky, 1946). At the end of the twentieth century, Hartwell and colleagues proposed applying the principle of SL to identify new lesions in cancer cells with known defects using yeast for the first time (Hartwell et al., 1997). However, the screens were limited in model organisms until the advent of RNA interference (RNAi) (Mullenders & Bernards, 2009). Application of the concept in human cell line systems was expanded rapidly. Until 2005, the first interaction between poly (ADP-ribose)-polymerase (PARP)1 and breast cancer susceptibility gene (BRCA) 2 was discovered based on the theory of SL: the loss of either gene is compatible with cell viability (normal cells), whereas the simultaneous mutations lead to cell death (when BRCA gene deficient tumor cells are exposed to PARP inhibitors) (Bryant et al., 2005; Farmer et al., 2005) (Fig. 2). These results opened the prelude to a novel targeted therapy in cancers (Brown et al., 2017).
Fig. 2
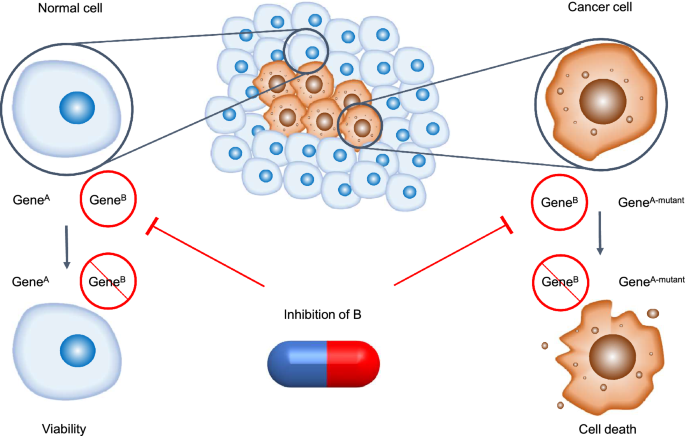
The conceptual framework of synthetic lethality. In normal cell, an individual genetic event is compatible with cell viability (left), whereas, in cancer cells with original mutations, the occurrence of multiple genetic events leads to cell death (right). The capsule represents corresponding inhibitor administration
In addition to BRCA1/2 mutations, there are many aberrant genetic or epigenetic changes in human tumors. Therapeutic approaches based on SL theory are promising: a large number of cancers related molecular targets have been screened, and some pharmacological inhibitors were developed based on these screenings. However, this situation is relative rare. In addition, some targets are loss of function of tumor suppressor genes which are undruggable.
Recent studies mainly target the machineries of the DDR pathways (shown in Fig. 1), which will be discussed later. In this review, we will discuss the development of SL screening strategy, some archetypal examples of genes based on the SL theory to present the development and state of the art status of research targeting SL related to DDR in cancer.
Screening strategy
The synthetic lethal screening is a method of isolating novel mutants whose survival is dependent on a gene of interest. Experimental determination of synthetic lethal interactions is based on identifying genes, mainly after inactivation, that display a lethal phenotype in a specific genotypic context. There are many ways to identify genotypes that can interfere with the expression of individual genes or can be used for large-scale screening. These include: yeast screening, small-molecule compound libraries screens, short interfering RNAs (siRNAs) libraries screening, short hairpin RNAs (shRNAs) libraries screening, and more recently guide RNAs (gRNAs) libraries for clustered regularly interspaced short palindromic repeats (CRISPR) screening as well comprehensive bioinformatics analysis based on the above experimental results.
Yeast screening
Yeast genome is evolutionarily conserved with human. Yeast was initially used to screen to determine the role of synthetic lethal and anticancer therapeutic targets (Hartwell et al., 1997; Kroll et al., 1996). Hartwell et al. (Hartwell et al., 1997) conducted a small-scale screening using 70 different syngeneic strains from budding yeast with deletion of DNA damage response genes corresponding to United States Food and Drug Administration (FDA)-approved chemotherapeutic drugs. 96-well plates are used to meet the transformations needs of a large number of different yeast strains or plasmids (Fleming & Gitler, 2011). Synthetic genetic arrays (SGA) were initially used to explore synthetic lethal interactions (Ooi et al., 2006) Over the past decade, SGA methods have enabled the explosive growth of yeast genetic analysis thanks to the ability to perform thousands of pairwise genetic crosses in parallel (Adames et al., 2019).
At present, the yeast screening alone for SL is mostly limited to the related mechanism research of some target proteins such as pol2 (Garg et al., 2020; Schwer et al., 2021) and ribosome biogenesis related proteins (Gómez-Herreros et al., 2017; Rodríguez-Galán et al., 2021). With the development of computer methodology, the homologous synthetic lethal interaction partners obtained in yeast screening and the molecular targets known in clinical tumors are usually screened jointly using machine learning algorithm.
Small molecule screening
The concept of SL was expanded from describing interactions between genetic events to also include a situation between a genetic event and a chemical compound treatment (Hartwell et al., 1997). Drug screening is primarily based on mutational data from large-scale cell lines and screening data from high-throughput drug libraries to identify susceptibility mutations that constitute synthetic lethal interactions (Liu et al., 2018).
The libraries are mainly based on FDA-approved drugs. Small molecular kinase compounds such as polo-like kinase 1 (PLK1) and RhoA/Rho kinase (ROCK) have been screened as therapeutic targets for KRAS-mutant cancers (Wang et al., 2016). Proteasome inhibitor bortezomib (BTZ) /the histone deacetylase (HDAC) inhibitor vorinostat (SAHA) was nominated as potential therapeutic targets for MYC-driven cancers (Wang et al., 2019). As for pancreatic ductal adenocarcinoma (PDAC) cells with low argininosuccinate synthetase 1 (ASS1) expression, HDAC inhibitors/Arg deprivation showed synthetical lethality (Kim et al., 2020).
The advantage of drug screening is that it can directly provide existing compounds as potential clinical translation targets, and it is easy to optimize and study drug resistance against known compounds. However, the effectiveness and specificity of drug screening is usually lower than that of gene knockout due to insufficient drug inhibition and related toxic side effects (Mullard, 2017). Furthermore, due to the theoretical basis of library screening of small-molecule compounds, this screening is mainly based on specific mutations, and subsequent studies are difficult to provide information about the genetic background of the disease and biologically relevant network signals.
RNA interference screening
The advent of libraries of siRNAs or shRNAs, as well as small molecule compounds libraries, has enabled the genome-wide study of specific mutations in mammalian cells (Chan & Giaccia, 2011). Since the inhibitors of PARP are highly lethal to cells with deficiencies in BRCA1/2, PARP inhibitor (PARPi) synthetic lethal siRNA screen was conducted to identify more kinases (Turner et al., 2008). Recently, the Cancer Cell Line Encyclopedia (Barretina et al., 2012) was exploited to create a list of essential genes and synthetic lethal interactions. Project DRIVE conducted shRNA screen using an average of 20 shRNAs per gene in 398 cancer cell lines to verify viability effect of 7837 genes (McDonald et al., 2017). Project Achilles developed DEMETER method which could leverage the observation of shRNA off-target effects in an analysis of 501 genome-scale loss-of-function screens (Tsherniak et al., 2017). Protein arginine methyltransferase 5 (PRMT5) was identified as a potential vulnerability in cells with loss of methylthioadenosine phosphorylase (MTAP) through shRNA screens (Kryukov, et al., 2016).
RNAi screening can accurately identify the target gene–gene interaction. On the other hand, it may be difficult to find a paired compound for the identified targets due to molecular weight or structure. In addition, since a shRNA and siRNA sequence can bind to and down-regulate mRNA unrelated to the target gene, it leads to multiple basically unavoidable false positives in genetic screening, leading to limited clinical applications (Champagne et al., 2020).
CRISPR screening
CRISPR genome-wide screening has revolutionized loss-of-function screening, with CRISPR technology improving transcriptional repression, reducing off-target efficiency in RNAi screening, and increasing sensitivity to essential gene scores (Evers et al., 2016; Morgens et al., 2016). Project Score introduces a genome-scale library which includes 941 CRISPR-Cas9 screens in 324 cell lines from 30 cancers (Behan et al., 2019). Werner syndrome ATP-dependent helicase (WRN) is an example of novel druggable target nominated by CRISPR–Cas9 screens (Behan et al., 2019; Lieb et al., 2019).
Based on CRISPR technology, paired gRNAs for paralog genetic interaction mapping (pgPEN) was developed in order to exclude false negatives caused by redundancy between related genes (Parrish et al., 2021). This approach is helpful in discovery of synthetic lethal paired targets. For instance, for validated targeted drugs such as PARPi, using CRISPR screening combined with olaparib treatment, a study found that the loss of amplification in liver cancer 1(ALC1) was synthetic lethal with homologous recombination deficiency (HRD), which attribute to sensitivity of PARPi (Hewitt et al., 2021).
In vitro, CRISPR screens in immortalized cell lines are generally not limited in cell number, while screening in vivo is often limited by cell number, so more screens targeting specific gene families, biochemical routes, or protein types are more likely to be suitable for well-designed pooled libraries. Combining or facilitating new analytical tools may gradually address existing limitations.
Bioinformatic screening
As mentioned above, various types of screening are combined with bioinformatics analysis, and new algorithms have been developed for more precise screening (Behan et al., 2019; Tsherniak et al., 2017). Conventional screening combined with computational methods is a very powerful technique for elucidating fundamental cancer genes (Iorio et al., 2018; Wang et al., 2015, 2019). CRISPR score (CS) and gene-trap score (GTS) were computed score from conventional screen of CRISPR-Cas9, the differences between the two scores are verified by functional profiling experiments conducted in yeast Saccharomyces cerevisiae. Gene-trap screens provide similar measurement accuracy of the cell-essentiality of human genes as CRISPR (Wang et al., 2015). PAn-canceR Inferred Synthetic lethalities (PARIS) was a machine learning approach to identify cancer vulnerabilities (Benfatto et al., 2021). 15 high confidence SL interactions within 549 DNA damage repair (DDR) genes were predicted using PARIS. Some of them had been experimental validated. For instance, various cancer cell lines with cyclin dependent kinase inhibitor 2A (CDKN2A) damaging mutations to show sensitivity to thymidylate synthase (TYMS) mutant. In addition, bioinformatics screens can also analyze the genomic sequencing results of biopsies from patients with different prognosis, revealing synthetic lethal relationships for treatment and new lesion sites that are beneficial for clinical treatment (Wheeler et al., 2021).
One of the challenges for bioinformatics analysis is deciding how to normalize data from different sources so that the functional genome of interest can be accurately applied to multiple cancer subgroups. In addition, the continuous evolution of drug resistance also requires more data mining for auxiliary screening, and bioinformatics screening is expected to provide a powerful approach (Huang et al., 2020).
Current target
PARP
BRCA1/2 are tumor suppressor genes that, when heterozygous mutated in the germ line, confer an elevated risk of several cancers including breast, ovarian, pancreatic, and prostate cancer. Functional BRCA1/2 proteins are important for double strand break (DSB) repair by HR (Lord & Ashworth, 2017). When HR, a conservative mechanism, becomes deficient, non-conservative forms of DSB repair predominate, such as NHEJ. These processes lead to DNA aberration accumulation, resulting in cancer initiation or progression. PARP1 and PARP2 enzymes are important sensors of DNA damage and signal transducers in DDR pathway (Satoh & Lindahl, 1992). PARP1 binds damaged DNA at the site of single strand DNA breaks (SSBs) and other DNA damage, which causes a series of allosteric changes in the structure of PARP1 that recruits catalytic PARylation-prone proteins. This regulates the stability of replication forks and post-translational modification that permits a DNA repair process. PARP1 eventually PARylates itself (autoPARylation), which causes its release from repaired DNA (Dawicki-McKenna et al., 2015; Eustermann et al., 2015; Vos et al., 2012). The inhibition of PARP can “trap” PARP1 on DNA, impeding subsequent events from happening and therefore interfering with the catalytic cycle of PARP1 (Murai et al., 2012, 2014). Normally, these errors are repaired by HR during S phase, otherwise resulting in cell death in HR-deficient tumor cells.
Two articles describing the relationship between PARP inhibition and BRCA1/2 mutation opened a new era of SL in 2005 (Bryant et al., 2005; Farmer et al., 2005). The PARPi were introduced into clinic as a combination treatment with temozolomide in patients with metastatic melanoma (Plummer et al., 2013). A phase I olaparib clinical trial, which involved carriers with BRCA1/2 mutations, showed that 12 of the 19 patients benefit from olaparib with less severe side effects than those receiving conventional chemotherapy (Fong et al., 2009). Subsequently, phase 2 and 3 clinical trials, which included various types of cancers, proved the clinical benefit of olaparib (Audeh et al., 2010; Kaufman et al., 2015; Mateo et al., 2020), thus providing sufficient evidence for FDA approval as a treatment for advanced ovarian cancer patients (Kim et al., 2015). Increasing types of refractory advanced solid tumors, such as high-risk breast cancer (Tutt et al., 2021), platinum-sensitive ovarian cancer with BRCA1/2 mutation (Banerjee et al., 2021; Poveda et al., 2021), platinum-sensitive pancreatic ductal adenocarcinoma cancer (Javle et al., 2021), and even primary triple negative breast cancer (TNBC) without previous chemotherapy (Eikesdal et al., 2021) benefit from Olaparib monotherapy. Currently, FDA has approved four PARPi for clinical utility, including olaparib, rucaparib (Coleman et al., 2017; Swisher et al., 2017), niraparib (González-Martín et al., 2019) and talazoparib (Litton et al., 2018), whereas several inhibitors are currently in clinical trials (Table 1). Their mechanisms of inhibition are similar, but chemical structures, pharmacodynamic, clinical doses and indications vary.






BRCAness is used to define the tumors that lack germline BRCA mutations but harbor some similar characteristics, particularly HR deficiency. BRCAness tumors present a phenocopies of BRCA-mutant tumors (Lord & Ashworth, 2016). This phenotype is secondary to defects in encoding genes, such as recombination protein A(RAD51), replication protein A (RPA)1, NBS1, ataxia telangiectasia-mutated gene (ATM), recombinant ataxia telangiectasia and rad3-related protein (ATR), partner and localizer of BRCA2 (PALB2), checkpoint kinases (CHK) 1/2 and Fanconi anemia complementation group (FANC) gene family, that are involved in DDR process, and were also shown to confer sensitivity to PARPi (Pilié et al., 2019). BRCAness provides a wider range of application for PARPi (Mateo et al., 2020).
The increasing use of PARPi in clinical practice to treat different tumors has raised the issue of acquired resistance, and the consequent problems of disease recurrence and dismal survival outcomes (Audeh et al., 2010). Several mechanisms of resistance have been investigated, and ongoing studies are focusing on strategies to address this challenge.
There are several reasons underlying PARPi resistance mechanism.
BRCA1/2-dependent HR restoration
BRCA1/2 protein can be re-expressed by reversing mutations or epigenetic changes. Re-expressed BRCA can re-recruit RAD51 to sites of DNA damage and reduce genomic instability (Edwards et al., 2008; Sakai et al., 2008). Preclinical studies have reported that patient- derived xenograft (PDX) models derived from patients with BRCA1 mutations and methylation are resistant to PARPi (Brugge, 2016; Kondrashova et al., 2018). This resistance is associated with mutation-induced restoration of the BRCA1 reading frame and methylation. Analysis of HR-related genes that contribute to resistance to PARPi or platinum-based chemotherapy identified mutational hotspots on BRCA1/2 (Pettitt et al., 2020). In addition, mutations in other HR-related genes, such as RAD51 and PALB2, affect drug resistance (Goodall et al., 2017; Kondrashova et al., 2017).
Independent of BRCA-related HR restoration
Restoration of HR can complete DNA damage repair through other compensatory pathways. Deletion of the NHEJ factor p53-binding protein 1(53BP1) reverses the balance of repair pathways toward HR (Bouwman et al., 2010; Bunting et al., 2010; Cao et al., 2009). Studies at both the cellular and animal levels show that deletion of 53BP1 rescues HR defects, resulting in resistance to PARPi. Other mutations have been shown to regulate DNA end-resection via downstream proteins of 53BP1, such as Rif1 (Virgilio et al., 2013), Rev7 (Xu et al., 2015) and shieldin complex (Callen et al., 2020; Dev et al., 2018), shifting repair from NHEJ to HR. Downstream of these proteins, there is also the CTC1-STN1-TEN1 (CST) complex, the loss of which leads to recovery of end excision, resulting in resistance to PARPi (Barazas et al., 2018; Mirman et al., 2018).
Replication forks stabilization
BRCA1 and BRCA2 are not only required for HR—these proteins also control the stability and protection of replication forks under replication stress. In the absence of BRCA1/2, MRE11 uncontrollably excises stalled forks, resulting in increased genomic instability (Schlacher et al., 2011). If MER11 recruitment is blocked via depletion of the MLL3/4 complex protein PTIP, the nucleosome remodeling factor CHD4, or chromatin remodeling complex SMARCAL1, resistance to PARPi can develop in BRCA1/2-deficient cells due to protection of the replication fork (Guillemette et al., 2015; Ray Chaudhuri et al., 2016; Taglialatela et al., 2017). RADX depletion in BRCA2-mutated cells also restores protection of replication forks (Dungrawala et al., 2017). Endonuclease homologous protein MUS81 functions similarly to MRE11 degrading stalled replication forks. Inhibition of the methyltransferase EZH2 affects the recruitment of MUS81, resulting in resistance to PARPi (Rondinelli et al., 2017). It is worth noting that WRN helicase inhibitors screened in recent years can also enhance the functions of MRE11 and MUS81, increase NHEJ and chromosomal instability. In BRCA-deficient cells, WRN helicase inhibitor markedly potentiated Olaparib cytotoxicity (Datta et al., 2021). More research is needed to determine whether it can reverse the resistance of PARPi.
Loss of cell cycle control
In response to genotoxic stressors that impede DNA replication, cells enter a condition of replication stress, which leads to activation of the DDR pathway and subsequent inhibition of cell cycle progression. The cell cycle regulates the selection of DSB repair pathways (Hustedt & Durocher, 2016). In G1 phase, 53BP1 and Rif1 localize to damage sites, promoting the NHEJ pathway. When DNA end resection occurs in S/G2 phase, they promote the HR pathway for repair (Symington & Gautier, 2011). Cyclin-dependent kinases (CDKs), as cell cycle checkpoints, mediate the phosphorylation of MRN (MRE11, RAD50 and NBS1) complexes and CtBP-interacting protein (CtIP) (Tomimatsu et al., 2014), thereby affecting the sensitivity to PARPi. Loss-of-function (LOF) mutations in CDK12 can affect sensitivity to PARPi such as olaparib and veliparib (Bajrami et al., 2014; Joshi et al., 2014). ATR, CHK1/2, and WEE1-like protein kinase (WEE1), are key kinases involved in replication stress response (RSR) through regulation of cell cycle arrest. The activated ATR-CHK1 pathway suppresses cell cycle progression by inactivating the cell division cycle 25 (CDC25), a CDK activator (Mailand et al., 2000). WEE1, activated by CHK1, are involved in CDK1/2 activity and abrogation of G2 cell-cycle checkpoint, and helps the HR-deficient cells step into mitosis (Colicchia et al., 2017). It has been reported that the combination of ATR-CHK1-WEE1 axis inhibitor and PARPi can reverse PARPi resistance (Chiappa, 2022; Kim et al., 2020).
DDR-independent way
PARPi bind the catalytic domain by competing with the cofactor NAD + . Therefore, mutations in PARP itself may result in a decrease in inhibitor affinity or preservation of the endogenous function of the enzyme (Pettitt, 2018). This mechanism will lead to the occurrence of drug resistance. Mutations in PARP1 that affect PARP1 trapping have been identified in ovarian tumors resistant to PARPi. PAR Glycohydrolase (PARG) is an enzyme that can remove PAR chains from proteins. In genetically engineered mouse models of BRCA1/2-deficient breast tumors, loss of PARG makes them resistant to PARPi. (Gogola et al., 2018) PARG-deficient cells exposed to PARPi still retain sufficient PARylation of target proteins to induce DNA damage signaling cascades.
Alternative DDR way
Microhomology-mediated end-joining (MMEJ) normally plays a minor role, but can be upregulated as a compensatory mechanism in the context of HR deficiency (Chang et al., 2017). DNA polymerase theta (Polθ) is a key factor driving this error-prone pathway. Polθ is based on short regions (> 2 bp) of sequence homology that join two broken DNA strands (Nik-Zainal et al., 2012). Synthetic lethal interactions between Polθ depletion and HR-related proteins were demonstrated in 2015 (Ceccaldi et al., 2015; Mateos-Gomez et al., 2015). DNA-repair processes involving regions of microhomology may be able to repair DSBs through MMEJ. The antibiotic novobiocin (NVB) inhibits Polθ activity; inhibition of Polθ selectively kills HR-deficient tumors. NVB still delayed tumor growth in a PDX model with loss of BRCA1 and 53BP1 function, suggesting that Polθ inhibition may also be a viable option for tumors that have acquired resistance to PARPi due to loss of DNA end protection (Zhou et al., 2021).
ATR
ATR and ATM work through overlapping but distinct pathways to initiate DDR. Upon detection of DSBs, NBS1 in the MRN complex recruits and activates ATM, which responds to and facilitates repair of DSBs produced throughout the cell cycle (Ha et al., 2019). The ATM gene is non-essential, despite germline mutations leading to a disorder characterized by extreme radiation sensitivity and increased risk of developing certain cancers (Lavin, 2008). On the other hand, the ATR gene is essential, and biallelic loss of gene function leads to early embryonic lethality (Klein et al., 2000). During the initial phase of HR, replication fork stalling by DNA end resection activates ATR through RPA bound ssDNA (Menolfi et al., 2018). Activation of oncogenes and loss of control of the G1 checkpoint leads to frenzied replication of tumor cells. This makes tumor cells dependent on the S and G2/M phase checkpoints, and the replication pressure increases sharply when tumor cells enter S phase (Bradbury et al., 2020). ATR is important for cell cycle checkpoints in S and G2/M phases and inhibits CDK activity through the phosphorylation of CDC25A via CHK1 (Stracker et al., 2009). ATR inhibitors can lead to tumor cell death by selectively targeting tumor cells that are out of control of the G1 cycle checkpoint (Kumar et al., 2014). In addition, the association with various proteins involved in HR, such as BRCA (Yazinski et al., 2017), PALB2, RAD51 (Ahlskog et al., 2016), and the RecQ helicases (Datta et al., 2021), suggest a potential of combining other DDR inhibitors. ATR inhibitors undergoing clinical trials are listed (Table 1).
Currently, ATR inhibitor development lags behind other DDR proteins, including PARP and CHK1, the downstream target of ATR itself. This may be due to the difficulty in establishing high-throughput drug screening in vitro due to the molecular size of ATR. Berzosertib, an ATR inhibitor, has been tested in multiple studies; berzosertib monotherapy was well tolerated and no dose-limiting toxicities were observed (Yap et al., 2020). Recently reported studies have focused on the combination of berzosertib with gemcitabine or cisplatin in high-risk ovarian cancer (Gorecki et al., 2020; Konstantinopoulos et al., 2020, 2021; Pal et al., 2021; Thomas et al., 2018; Yap et al., 2020). Berzosertib's indications are also expanding, with 36% (9/25) objective response rates in patients with small cell neuroendocrine cancers (SCNCs) receiving berzosertib in combination with topotecan, and encouraging results were observed in patients with platinum-resistant SCNCs durable tumor regression (Thomas et al., 2021). Similarly, ceralasertib has already passed Phase I clinical trials (Dillon et al., 2018). Ceralasertib has been combined with different chemotherapy regimens, such as topoisomerase inhibitors paclitaxel and carboplatin (Kim et al., 2021; Yap et al., 2021) and extended the potential indication to colorectal cancer (Inoue et al., 2021) and triple negative breast cancer (Wilson et al., 2022) in preclinical and early clinical studies. Phase 2 trial in combination with immune checkpoint inhibitor in advanced melanoma offers more possibilities for patients who have failed immune checkpoint therapy (Kim et al., 2022). Clinical trials of other ATR inhibitors are still in phase I (Table 1).
So far, clinical studies on ATR inhibitor resistance have not been reported. With the perspective of cell cycle regulation, the findings from a genome-wide CRISPR Screen showed that CDC25A determines sensitivity to ATR inhibitors (Ruiz et al., 2016). Cells deficient in the CDC25A gene do not enter mitosis prematurely, which leads to resistance to high-dose ATR inhibitors. However, WEE1 inhibitors could force mitosis entry of CDC25A-deficient cells and restore the cytotoxicity of ATR inhibitors. Several preclinical studies have identified mechanisms of ATR inhibitor resistance in vitro (Henssen, 2017). Endogenous piggyBac transposable element derived 5 (PGBD5) deletion is associated with the resistance of human tumor cells to AZD6738 (ceralasertib) (Henssen, 2017). Ceralasertib causes unrepaired DNA damage to accumulate in PGBD5-expressing cells, leading to apoptosis in G1 phase and dividing cells. Treatment toxicity also needs attention, although hematologic toxicity as an adverse effect is dose limited. The optimal scheduling and sequencing of these drugs with their respective partners is unclear, and the full results of these trials are anticipated with great interest.
DNA-PK
DNA-dependent protein kinase (DNA-PK) which is a critical enzyme involved in the NHEJ pathway of DNA repair, is a member of the PI3K–mTOR enzyme family (Brown et al., 2017). DNA-PK is composed of Ku with a ~ 460 kDa catalytic subunit (DNA-PKcs), which can be recruited with X-Ray repair cross complementing (XRCC4), DNA ligase IV (LIG4), XRCC4-like factor (XLF). Autophosphorylation at DSB ends by a Ku80-Ku70 heterodimer induces a conformational change to facilitate repair, which plays a pivotal role in the NHEJ DSB repair pathway (Gell & Jackson, 1999; Singleton et al., 1999). It has been reported that DNA-PK inhibits tumor growth by regulating tumor-associated transcriptional pathways (Goodwin & Knudsen, 2014). In addition to being important for the repair of exogenous DNA DSBs, canonical NHEJ also plays a critical role in repairing endogenous DSBs generated during physiological processes such as V(D)J and class-switching recombination (Blunt et al., 1995).
Thus, DNA-PK inhibitors lead to stabilization of DNA ends, hinder NHEJ and may interfere with other repair processes, including HR resection. Monotherapy had little effect on cancer cell or tumor viability, because most endogenous DSBs appear in the context of DNA replication where the preferred repair pathway is HR. Most clinical trials test the combination treatment with radio-chemotherapy. CC-115, a small-molecule mTOR and DNA-PK inhibitor (Mortensen et al., 2015), has completed phase I clinical trial which included 44 advanced solid or hematologic malignancy participants receiving CC-115 monotherapy. In addition, highly selective DNA-PK inhibitor, AZD7648, is an efficient sensitizer of radiation- and doxorubicin-induced DNA damage, inducing sustained tumor regressions in preclinical studies (Fok et al., 2019; Nakamura et al., 2021). Based on the AZD7648 preclinical data, phase 1/2a clinical trial consisting of 234 patients receiving AZD7648 alone and in combination with olaparib has recently been initiated (Goldberg et al., 2020). Furthermore, a novel inhibitor named peposertib (M3814) is currently in phase 1 clinical trials under monotherapy and in combination with chemo-radiotherapies for advanced solid tumor patients (Bussel et al., 2021).
DNA-PK inhibitor resistance has also been reported in clinical trials. CC-115, a dual mammalian target of rapamycin (mTOR)/DNA-PK inhibitor, was reported as a substrate of ATP-binding cassette G2 (ABCG2), a member of the ATP-binding cassette transporter superfamily, the overexpression of ABCG2 significantly increased the resistance to CC-115. Use of small molecular inhibitors to inhibit ABCG2 function to make cancer cells sensitive to CC-115 (Beebe & Zhang, 2019). Cryo-electron microscopy (cryo-EM) structural findings about human DNA-PKcs in complex with four inhibitors should greatly assist future development in drug design targeting DNA-PKcs (Liang et al., 2022).
CHK1
CHK1/2 are also cell cycle checkpoint kinases that target ATR/ATM, respectively, and are involved in preventing cell cycle progression upon DNA damage and repair (Stracker et al., 2009). The selective inhibition of CHK1 has been predicted to be particularly sensitive in those p53-defective cells because the loss of both pathways (CHK1 and/or CHK2–p53 pathway) is lethal (Kaelin, 2005; Levesque & Eastman, 2007) As mentioned earlier, a variety of inhibitors can regulate the cell cycle by interacting with CHK1, especially ATR inhibitors. This means that a series of cell cycle checkpoints have potential synthetic lethal targets targeted by CHK1 inhibitors. Synthetic lethal-based markers for indication of CHK1 inhibitors are expanding. Autophagic tumor-suppressor protein AMBRA1 is an upstream master regulator of the transition from G1 to S phase and thereby mitigates replication stress. This helps to maintain genomic integrity during DNA replication. CHK1 kinase is a potential therapeutic target in tumors harboring AMBRA1 deficiency (Maiani et al., 2021).
The current clinical trials for CHK1-selective inhibitors mainly includes SRA737 and prexasertib. SRA737 is an oral CHK1 inhibitor being tested as a monotherapy and in combination with gemcitabine for non-Hodgkin’s lymphoma and advanced malignancies (Sen et al., 2019). Prexasertib (LY2606368) has completed the phase I clinical trials in advanced tumors (Hong et al., 2016, 2018; Lee et al., 2018). A comparative study focused on the activity and off-target effects of 3 CHK1 inhibitors tested in clinical trials. Among MK-8776, SRA737, and LY2606368, LY2606368 is suggested to be a prospective inhibitor with most selectivity to CHK1 (Ditano & Eastman, 2021). Based on the high synergy with other genes involved in replication-dependent DNA damage, combination therapy with such drugs is focused during clinical trials. Prexasertib was tested in various combination with different drugs in the respective phase I trials. Prexasertib can be combined with standard-of-care agents (cisplatin, cetuximab, and 5-fluorouracil) (Moore et al., 2021), cetuximab-radiotherapy (Yang et al., 2021) and olaparib (Do et al., 2021).
While effective CHK1 inhibitors are advancing in the clinic, resistance is also rapidly emerging. The decrease in endogenous and exogenous events-induced replication stress will reduce the stress generated by CHK1 inhibitors. It has been reported that the acquired resistance of CHK1 inhibitor is associated with innate immunity (Lowery et al., 2019). The expression of immune-related E2F/ G2M/SAC genes contributes to prexasertib resistance. The increased E2F/G2M/SAC expression mitigate replication stress and is highly associated with resistance to prexasertib in lung cancer cells (Blosser et al., 2020). Li et al. (Li et al., 2020) reported that a little-known gene FAM122A/PABIR1 confers cellular resistance to CHK1 inhibitors and ATR inhibitors via regulation of WEE1. Thus, a combination of a CHK1 inhibitor plus a WEE1 inhibitor can overcome CHK1 inhibitor resistance of these tumor cells, thereby enhancing efficacy. Furthermore, tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein zeta (YWHAE) was found to be a key determinant of sensitivity to CHK1 inhibition along with the discovery of other resistance markers (Wang et al., 2021). With the development of high-throughput CRISPR and RNAi screening technologies, researchers can obtain more interaction targets, and comprehensive mechanisms are being explored.
WEE1
WEE1 is a serine-threonine kinase that triggers G2/M arrest by inhibiting phosphorylation on Tyr15 of CDK1, thereby preventing cells from entering mitosis. Wee1 inhibition may lead to elevated CDK1 activity and failure of cells to adequately repair DNA damage through the G2/M checkpoint, leading to mitotic catastrophe and cell death. Inhibition of WEE1 also catalyzes the phosphorylation of CDK2, resulting in abnormal DNA replication and DSB (Matheson et al., 2016; Geenen JJJ & Schellens JHM, 2017; Moiseeva et al., 2019). In addition, tumor suppressor genes such as p53 can regulate the G1 cell cycle checkpoint, and in p53-depleted tumor cells, the G2 cell cycle checkpoint occupies a more important position. Thus, inhibition of WEE1 mediates mitotic death in p53-deficient cells, making them more sensitive to genotoxic stress (Diab et al., 2019; Liang et al., 2020).
Adavosertib is a promising WEE1 inhibitor that is undergoing clinical trials with safety result. In phase I clinical trial, 2 of 25 refractory solid tumor patients carrying BRCA-mutated tumor reported partial response to adavosertib monotherapy (Do et al., 2015). In a Phase 2 clinical trial in Recurrent Uterine Serous Carcinoma tumors, 10 of 34 patients receiving monotherapy were complete responders, for an objective response rate of 29.4% (95% CI, 15.1 to 47.5) (Liu et al., 2021). However, no clear molecular biomarker can be exploited in this study. A sensitizing effect of adavosertib on chemoradiotherapy is also expected after a favorable outcome inquired from phase I clinical trial (Leijen et al., 2016). Both single and combined use of adavosertib have high frequency of adverse events, mainly gastrointestinal related adverse events such as diarrhea, vomiting and anemia, which cast a shadow on the further expansion of adavosertib (Liu et al., 2021). Recently, a novel WEE1 inhibitor, ZN-c3, has demonstrated better selectivity and higher solubility, which can reduce the differences in drug exposure between patients and limit the toxicity observed with adavosertib in clinical trials. (Huang et al., 2021).
Although WEE1 inhibitors have shown promising results in the clinical trials, drug resistance is unpreventable. Through a high-throughput unbiased proteomic profiling (RPPA), Sen et al. demonstrated that small cell lung cancer (SCLC) models with primary resistance to adavosertib harbor a higher expression level of AXL and phosphorylated ribosomal S6 and that WEE1 inhibitors combined with AXL or mTOR inhibitors could overcome resistance (Sen et al., 2017). Lewis et al. (Lewis et al., 2019) found that cells with acquired resistance to adavosertib have higher expression levels of Wee1-related kinase Myt1 than sensitive cells. The suppression of MYT1 promoted CDK1 activity and helped overcome adavosertib resistance. Furthermore, Li et al. (Li et al., 2020) reconfirmed that complementary activated mTOR pathway contributes to Wee1 inhibitor resistance. Therefore, the dual inhibition of WEE1 and mTOR will induce immense DNA replication stress, resulting in replication fork collapse. The discovery of corresponding resistance markers will help to overcome and reverse this dilemma, which needs more research into the mechanism.
Novel targets
As mentioned above, inspiration from SL expands clinical therapeutic approaches targeting DNA damage and facilitates research progress on various DDR inhibitors. The results of PARPi are encouraging, and increasing research has focused on targeting other DDR pathway components that could serve as synthetic lethal therapies. In addition to mentioned inhibitors, although the corresponding research and development is still in the preclinical stage, the following molecules are also expected to achieve excellent clinical translation.
Polθ
Cells maintain a balance of DSB repair pathway choices to avoid deleterious events. HR and C-NHEJ, as the predominant mechanisms of DSB repair in mammalian cells, cooperate to maximize genomic stability. Polθ assumes a critical role for repair of DSB repair once either the HR or NHEJ apparatuses are attenuated (Ceccaldi et al., 2015; Mateos-Gomez et al., 2015; Wyatt et al., 2016). There is an alternative repair pathway referred to as MMEJ (or alternative EJ, al-EJ) to describe the repair signature left at sites of DNA repair. Polθ is involved in this error prone form of DSB repair, which is suppressed and largely inactive in cells with functional HR pathways. Thus, the survival of BRCA1/2-deficient cancer cells, in which HR pathways are compromised, seem to be dependent on MMEJ. Inhibition of Polθ induces SL which could also reverse the resistance of PARPi.
Substantial progress has been made validating Polθ as an ideal drug target. Silencing Polθ could enhance the effects of PARP inhibition agents in HR-deficient ovarian cancer cells (Ceccaldi et al., 2015; Mateos-Gomez et al., 2015). The preclinical studies focus on Polθ inhibitors are emerging. Through high-throughput small molecule screen, a known antibiotic NVB was identified as a Polθ inhibitors that kills HR-deficient tumor cell in vitro and in vivo by increasing DSB end resection (Zhou et al., 2021). It was reported that only after the onset of the mitosis, Polθ-mediated end joining (TMEJ) repairs DSBs initiated during the S phase in BRCA2-deficient cells. During this process, RAD52 plays an important role as well as BRCA2. The proteins of those two genes lead to stalling of the polymerase function of Polθ (Llorens-Agost et al., 2021). Furthermore, Polθ inhibitors, ART558, was discovered which elicits DNA damage and SL in BRCA-deficient tumor cells and strengthen the efficacy of PARPi. In addition, tumors with defects of 53BP1/Shieldin complex shows a sensitivity to ART558 (Zatreanu et al., 2021). The exact molecular mechanism by which TMEJ transfers genomic instability under different context of DSB remains uncertain. Using single-molecule imaging, Schaub et al. (Schaub et al., 2022) revealed Polθ-helicase (Polθ-h) is a highly processive single-stranded DNA (ssDNA) motor protein. Polθ-h can strip Replication Protein A (RPA) from ssDNA, also has a limited capacity for disassembling RAD51 filaments. Polθ-h can build bridges between two non-complementary DNA strands in trans, which can be resolved by PARylation of Polθ-h by PARP-1.
Polθ inhibitor could elicit BRCA-gene SL and PARPi synergy, as well as targeting PARPi-resistance. The most appropriate clinical background for the application, especially the schedule of combination with inhibitors target HR pathways remains to be determined.
FEN1 and APEX2
In addition to Polθ, flap endonuclease 1 (FEN1) and APEX2 were also identified as BRCA synthetic lethal genes using shRNA and CRISPR screens (Mengwasser et al., 2019). BRCA-mutant cells require the 5′ flap endonuclease activity of Fen1 and the PCNA-binding domain of APE2 (a paralog of the apurinic/apyrimidinic endonuclease APE1). FEN1 was first validated as a drug target in BRCA-deficient tumors by increase DSB load or replication stress during MMEJ. Similar SL relationships identified in Saccharomyces cerevisiae (Guo et al., 2020). N-hydroxy pyrimidinedione-based FEN1 inhibitors were synthesized to determine the effect at both the cellular and animal level. APEX2-BRCA SL was related to 3′-blocking lesions induced by topoisomerase I (TOP1) processing of genomic ribonucleotides (Álvarez-Quilón et al., 2020).
Werner syndrome ATP-dependent helicase (WRN)
Recent study based on CRISPR/Cas9-mediated knockout and RNAi screens verified that a RecQ DNA helicase, WRN, is selectively essential in microsatellite instability (MSI) models but not microsatellite stable (MSS) (Behan et al., 2019). MSI is a phenotype that occurs during replication of microsatellite repeat regions due to impaired mismatch repair (MMR) (Kunkel & Erie, 2015). Inactivating mutations or epigenetic silencing of MMR pathway induce to MSI which occurs in many types of human cancers in colon, stomach, rectal, esophageal and ovarian carcinomas (Cortes-Ciriano et al., 2017).
WRN, as one of five human RecQ-like helicases (Hickson, 2003), plays a major role in HR-mediated replication fork restart and preventing fork stalling. It was discovered that WRN depletion leads to cell cycle arrest, DNA damage accumulation and apoptosis specifically in MSI cells. WRN depletion also significantly reduced growth of tumor with MSI in the preclinical study (Chan et al., 2019). Other RecQ genes (RecQL1, BLM, and RecQL5) were not necessary for MSI cell lines, this suggests a WRN-specific dependency. WRN dependency was significantly associated with hypermethylation of MLH1 promoter as well as mutations in MMR gene MSH6 and histone methyltransferase gene MLL2 (Behan et al., 2019). MSI cancer cells need WRN helicase activity to relieve harmful DNA damage caused by MMR defects. Contemporaneous studies also confirmed these findings (Kategaya et al., 2019; Lieb, et al,. 2019). Recent report present that 55 of these 60 (92%) preclinical MSI colorectal cancer models were found to be dependent on WRN for survival (Picco et al., 2021). In view of this, WRN inhibitors can be expected to be used to develop precise therapeutic approaches of MSI cancer.
Combination strategies and new treatment
The development of synthetic lethal drugs has several limitations. Cancers usually do not rely on a single DNA repair pathway to survive. DNA repair pathways in the cells may overlap or be distinct, resulting in drug resistance to synthetic lethal drugs. DNA repair pathways might share some DNA repair proteins. Therefore, monotherapy of lethal drugs may have off-target side effects-no response for tumors and/or intolerable injuries in normal tissue that possess important protein shared domain of the targeted repair component. Increasing studies put the spotlight on combination therapy to overcome these limitations and find new inspiration from it.
The main role of chemoradiotherapy is to mediate DNA damage, and the activated DDR signaling contributes to chemoradiotherapy resistant. Therefore, combining DDR inhibitors with chemoradiotherapy may alleviate this resistance. Furthermore, targeting the compensatory DDR pathway may sensitize multiple DDR signaling-deficient cancers to chemoradiotherapy. Various studies have reported that PARPi can alleviate drug resistance by altering the immune microenvironment and inducing cross-presentation, thereby enhancing the efficacy of immune checkpoint inhibitors (ICIs) (Li et al., 2019). DNA damage induced by PARPi enhances immune priming through various molecular mechanisms and upregulates the expression of programmed death ligand 1 (PD-L1) (Färkkilä et al., 2020). Combinations of PARPi and immune checkpoint inhibitors have shown promising results.
In a large phase 3, double-blind, randomized trial, patients with high-risk breast cancer who had received local treatment and neoadjuvant or adjuvant chemotherapy were assigned to Olaparib or placebo (Tutt et al., 2021). The 3-year invasive disease-free survival (85.9% vs 77.1%, P < 0.001) and 3-year distant disease-free survival (87.5% vs 80.4%, P < 0.001)) are improved significantly. In preclinical study, AZD7648 potentiated the efficacy of olaparib in BRCA-deficient ovarian cancer (Anastasia et al., 2022). Type I IFN signaling was enhanced in mice treated with the combination when compared with radiation therapy treatment alone. AZD6738 has been demonstrated to enhance the anti-tumor activity of chemo-radiotherapy, Olaparib and ICI treatment (Nakamura et al., 2021; Sheng, 2020). The combination of inhibitors trigger dual SL is emerging due to the evolution of screening technology. Simultaneous targeting of PARP1 and RAD52 with inhibitors result in synergistic accumulation of DSBs and eradication of BRCA-deficient but not BRCA-proficient tumor cells (Sullivan-Reed et al., 2018).
The differential efficacy of combination therapy suggests that a comprehensive understanding of the individual DDR pathway inhibition and different DNA damage of by radiochemical approaches is necessary. Optimizing drug arrangements could exploit potential differences in repair kinetics between normal and cancer cells, thus potentially increasing tumor damage while sparing normal tissue. The order of combination administration also needs to be carefully considered to optimize synergistic effects. Likewise, selecting patients with tumors with specific genes or phenotypes may provide a therapeutic window for this combination.
In recent years, there have been many attempts to clinically apply the proteolysis-targeting chimeras (PROTAC) method in cancer treatment. To develop PARP-targeted PROTACs, PARP inhibitors were linked to E3 ubiquitin ligase ligands via flexible linkers. This bivalent molecule tightly links the E3 ligase and PARPs, where PARPs is ubiquitinated and subsequently degraded by the proteasomal pathway. PROTACs using olaparib, rucaparib, and niraparib derivatives were developed and tested in different cells (Cao et al., 2020; Zhang et al., 2020; Zhao et al., 2019). Compared with traditional PARPi, they show better cytotoxicity and have broader utility beyond BRCA-mutated cancers. PROTACs can also increase the specificity of some inhibitors. Potent PROTAC degrader PP-C8 based on the noncovalent dual inhibitors of CDK12/13 was synthesized to target CDK12 more specifically than to CDK13 (Niu et al., 2022). Importantly, PP-C8 and PARP inhibitors show a synergistic antiproliferative effect in TNBC.
Applying new combination strategies and new technologies continues to mature and optimize, exciting results are expected in the near future. If innovative exploration and accumulation are continued, powerful treatment with new mechanisms are expected to be developed and translate to clinical application.
Conclusion
Synthetic lethal genes with oncogenic driver mutations are not necessarily mutated in cancer. Therefore, the development of synthetic lethal interactions in tumor cells may significantly increase the number of tumor drug targets. Cells with a tumor suppressor gene deletion may develop a greater dependence on another gene that may not be an oncogene, a phenomenon known as non-oncogene dependence (Hahn et al., 2021). DNA damage response, frequently occurring in genetical unstable tumors, provides numerous targets to promote or inhibit tumor development (Huang & Zhou, 2021). When used in combination with a second drug that is synthetically lethal from the first drug, its clinical effect can be greatly enhanced as compared to a single drug.
Characterization of cancer genomes provides insight into altered genes across tumor cells, transforming our understanding of cancer biology, and enabling the customization of therapeutic strategies. Experimental determination of lethal interactions is based on identifying genes that, after inactivation, display a lethal phenotype in a specific genotypic context. There are many ways to identify genotypes that can interfere with the expression of individual genes or can be used for large-scale screening. These include: a library of siRNAs, a library of shRNAs, and, more recently, a large library of gRNAs for Crispr/Cas9 genome editing (Behan et al., 2019; Bolck, 2022). All these techniques can be applied in high-throughput screening, either analyzing each gene or reagent in a single pool, or in a mixed format, where thousands of vectors are combined in a pool.
With the advancement of screening technology and drug development, SL will open new avenues for the development of cancer therapy by targeting cancers with specific mutations and finding precisely targeted targets. Novel powerful screening tools and in-depth understanding of this concept will lead to new drug targets for different genetic backgrounds, providing opportunities for expanding application to patients and breaking through treatment bottlenecks. Despite the great potential of this approach, many barriers to clinical translation remain. The implementation of SL in anti-cancer strategies is still in its infancy. In the future, more efforts will be needed to clarify detailed mechanisms between SL targets and to introduce new powerful and compelling targets and hypotheses.
Data availability
The data supporting the findings of this study are available within the submitted article.
References
Adames, N. R., Gallegos, J. E., & Peccoud, J. (2019). Yeast genetic interaction screens in the age of CRISPR/Cas. Current Genetics, 65(2), 307–327.
Ahlskog, J. K., Larsen, B. D., Achanta, K., & Sørensen, C. S. (2016). ATM/ATR-mediated phosphorylation of PALB2 promotes RAD51 function. EMBO Reports, 17(5), 671–681.
Álvarez-Quilón, A., et al. (2020). Endogenous DNA 3’ Blocks Are Vulnerabilities for BRCA1 and BRCA2 deficiency and are reversed by the APE2 nuclease. Molecular Cell, 78(6), 1152-1165.e1158.
Anastasia, A., et al. (2022). The DNA-PK Inhibitor AZD7648 Sensitizes patient-derived ovarian cancer xenografts to pegylated liposomal doxorubicin and olaparib preventing abdominal metastases. Molecular Cancer Therapeutics, 21(4), 555–567.
Audeh, M. W., et al. (2010). Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet (london, England), 376(9737), 245–251.
Bajrami, I., et al. (2014). Genome-wide profiling of genetic synthetic lethality identifies CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity. Cancer Research, 74(1), 287–297.
Banerjee, S., et al. (2021). Maintenance olaparib for patients with newly diagnosed advanced ovarian cancer and a BRCA mutation (SOLO1/GOG 3004): 5-year follow-up of a randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet Oncology, 22(12), 1721–1731.
Barazas, M., et al. (2018). The CST complex mediates end protection at double-strand breaks and promotes PARP inhibitor sensitivity in BRCA1-Deficient cells. Cell Reports, 23(7), 2107–2118.
Barretina, J., et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature, 483(7391), 603–607.
Beebe, J., & Zhang, J. T. (2019). CC-115, a dual mammalian target of Rapamycin/DNA-Dependent Protein kinase inhibitor in clinical trial, is a substrate of ATP-Binding Cassette G2, a risk factor for CC-115 resistance. The Journal of Pharmacology and Experimental Therapeutics, 371(2), 320–326.
Behan, F. M., et al. (2019). Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature, 568(7753), 511–516.
Benfatto, S., et al. (2021). Uncovering cancer vulnerabilities by machine learning prediction of synthetic lethality. Molecular Cancer, 20(1), 111.
Blosser, W. D., et al. (2020). A pan-cancer transcriptome analysis identifies replication fork and innate immunity genes as modifiers of response to the CHK1 inhibitor prexasertib. Oncotarget, 11(3), 216–236.
Blunt, T., et al. (1995). Defective DNA-dependent protein kinase activity is linked to V(D)J recombination and DNA repair defects associated with the murine scid mutation. Cell, 80(5), 813–823.
Bolck, H. A., et al. (2022). RNAi screening uncovers a synthetic sick interaction between CtIP and the BARD1 tumor suppressor. Cells, 11(4), 643.
Bouwman, P., et al. (2010). 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nature Structural & Molecular Biology, 17(6), 688–695.
Bradbury, A., Hall, S., Curtin, N., & Drew, Y. (2020). Targeting ATR as cancer therapy: A new era for synthetic lethality and synergistic combinations? Pharmacology & Therapeutics, 207, 107450.
Bridges, C. B. (1922). The origin of variations ins sexual and sex-limited characters. The American Naturalist, 56, 51–63.
Brown, J. S., O’Carrigan, B., Jackson, S. P., & Yap, T. A. (2017). Targeting DNA repair in cancer: Beyond PARP inhibitors. Cancer Discovery, 7(1), 20–37.
Bryant, H. E., et al. (2005). Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature, 434(7035), 913–917.
Bunting, S. F., et al. (2010). 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell, 141(2), 243–254.
Callen, E., et al. (2020). 53BP1 Enforces Distinct Pre- and Post-resection blocks on homologous recombination. Molecular Cell, 77(1), 26-38.e27.
Cao, C., et al. (2020). Discovery of SK-575 as a highly potent and efficacious proteolysis-targeting chimera Degrader of PARP1 for treating cancers. Journal of Medicinal Chemistry, 63(19), 11012–11033.
Cao, L., et al. (2009). A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Molecular Cell, 35(4), 534–541.
Ceccaldi, R., et al. (2015). Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature, 518(7538), 258–262.
Champagne, J., et al. (2020). TAG-RNAi overcomes off-target effects in cancer models. Oncogene, 39(4), 935–945.
Chan, D. A., & Giaccia, A. J. (2011). Harnessing synthetic lethal interactions in anticancer drug discovery. Nature Reviews Drug Discovery, 10(5), 351–364.
Chan, E. M., et al. (2019). WRN helicase is a synthetic lethal target in microsatellite unstable cancers. Nature, 568(7753), 551–556.
Chang, H. H. Y., Pannunzio, N. R., Adachi, N., & Lieber, M. R. (2017). Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nature Reviews. Molecular Cell Biology, 18(8), 495–506.
Chiappa, M., et al. (2022). Combinations of ATR, Chk1 and Wee1 inhibitors with olaparib are active in olaparib resistant Brca1 proficient and deficient murine ovarian cells. Cancers, 14(7), 1807.
Coleman, R. L., et al. (2017). Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet (london, England), 390(10106), 1949–1961.
Colicchia, V., et al. (2017). PARP inhibitors enhance replication stress and cause mitotic catastrophe in MYCN-dependent neuroblastoma. Oncogene, 36(33), 4682–4691.
Cortes-Ciriano, I., Lee, S., Park, W. Y., Kim, T. M., & Park, P. J. (2017). A molecular portrait of microsatellite instability across multiple cancers. Nature Communications, 8, 15180.
Datta, A., et al. (2021a). WRN helicase safeguards deprotected replication forks in BRCA2-mutated cancer cells. Nature Communications, 12(1), 6561.
Datta, A., Dhar, S., Awate, S., & Brosh, R. M., Jr. (2021b). Synthetic lethal interactions of RECQ helicases. Trends in Cancer, 7(2), 146–161.
Dawicki-McKenna, J. M., et al. (2015). PARP-1 activation requires local unfolding of an autoinhibitory domain. Molecular Cell, 60(5), 755–768.
de Klein, A., et al. (2000). Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Current Biology : CB, 10(8), 479–482.
De Vos, M., Schreiber, V., & Dantzer, F. (2012). The diverse roles and clinical relevance of PARPs in DNA damage repair: Current state of the art. Biochemical Pharmacology, 84(2), 137–146.
Dev, H., et al. (2018). Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nature Cell Biology, 20(8), 954–965.
Di Virgilio, M., et al. (2013). Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science (new York, n.y.), 339(6120), 711–715.
Diab, A., et al. (2019). Multiple Defects Sensitize p53-deficient head and neck cancer cells to the WEE1 kinase inhibition. Molecular Cancer Research : MCR, 17(5), 1115–1128.
Dillon, M. T., et al. (2018). PATRIOT: A phase I study to assess the tolerability, safety and biological effects of a specific ataxia telangiectasia and Rad3-related (ATR) inhibitor (AZD6738) as a single agent and in combination with palliative radiation therapy in patients with solid tumours. Clinical and Translational Radiation Oncology, 12, 16–20.
Ditano, J. P., & Eastman, A. (2021). Comparative Activity and Off-Target Effects in Cells of the CHK1 Inhibitors MK-8776, SRA737, and LY2606368. ACS Pharmacology & Translational Science, 4(2), 730–743.
Do, K., et al. (2015). Phase I Study of Single-Agent AZD1775 (MK-1775), a Wee1 kinase inhibitor, in patients with refractory solid tumors. Journal of Clinical Oncology : Official Journal of the American Society of Clinical Oncology, 33(30), 3409–3415.
Do, K. T., et al. (2021). Phase 1 Combination Study of the CHK1 Inhibitor prexasertib and the PARP Inhibitor olaparib in high-grade serous ovarian cancer and other solid tumors. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research, 27(17), 4710–4716.
Dobzhansky, T. (1946). Genetics of natural populations; recombination and variability in populations of Drosophila pseudoobscura. Genetics, 31(3), 269–290.
Dungrawala, H., et al. (2017). RADX Promotes genome stability and modulates chemosensitivity by regulating RAD51 at replication forks. Molecular Cell, 67(3), 374-386.e375.
Edwards, S. L., et al. (2008). Resistance to therapy caused by intragenic deletion in BRCA2. Nature, 451(7182), 1111–1115.
Eikesdal, H. P., et al. (2021). Olaparib monotherapy as primary treatment in unselected triple negative breast cancer. Annals of Oncology : Official Journal of the European Society for Medical Oncology, 32(2), 240–249.
Eustermann, S., et al. (2015). Structural basis of detection and signaling of DNA single-strand breaks by human PARP-1. Molecular Cell, 60(5), 742–754.
Evers, B., et al. (2016). CRISPR knockout screening outperforms shRNA and CRISPRi in identifying essential genes. Nature Biotechnology, 34(6), 631–633.
Färkkilä, A., et al. (2020). Immunogenomic profiling determines responses to combined PARP and PD-1 inhibition in ovarian cancer. Nature Communications, 11(1), 1459.
Farmer, H., et al. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature, 434(7035), 917–921.
Fleming, M. S., & Gitler, A. D. (2011). High-throughput yeast plasmid overexpression screen. Journal of Visualized Experiments: Jove. https://doi.org/10.3791/2836
Fok, J. H. L., et al. (2019). AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nature Communications, 10(1), 5065.
Fong, P. C., et al. (2009). Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. The New England Journal of Medicine, 361(2), 123–134.
Garg, A., Shuman, S., & Schwer, B. (2020). A genetic screen for suppressors of hyper-repression of the fission yeast PHO regulon by Pol2 CTD mutation T4A implicates inositol 1-pyrophosphates as agonists of precocious lncRNA transcription termination. Nucleic Acids Research, 48(19), 10739–10752.
Geenen, J. J. J., & Schellens, J. H. M. (2017). Molecular pathways: targeting the protein kinase wee1 in cancer. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research, 23(16), 4540–4544.
Gell, D., & Jackson, S. P. (1999). Mapping of protein-protein interactions within the DNA-dependent protein kinase complex. Nucleic Acids Research, 27(17), 3494–3502.
Gogola, E., et al. (2018). Selective Loss of PARG Restores PARylation and counteracts PARP inhibitor-mediated synthetic lethality. Cancer Cell, 33(6), 1078-1093.e1012.
Goldberg, F. W., et al. (2020). The Discovery of 7-Methyl-2-[(7-methyl[1,2,4]triazolo[1,5-a]pyridin-6-yl)amino]-9-(tetrahydro-2H-pyran-4-yl)-7,9-dihydro-8H-purin-8-one (AZD7648), a Potent and Selective DNA-Dependent Protein Kinase (DNA-PK) Inhibitor. Journal of Medicinal Chemistry, 63(7), 3461–3471.
Gómez-Herreros, F., et al. (2017). The ribosome assembly gene network is controlled by the feedback regulation of transcription elongation. Nucleic Acids Research, 45(16), 9302–9318.
González-Martín, A., et al. (2019). Niraparib in patients with newly diagnosed advanced ovarian cancer. The New England Journal of Medicine, 381(25), 2391–2402.
Goodall, J., et al. (2017). Circulating Cell-Free DNA to guide prostate cancer treatment with PARP inhibition. Cancer Discovery, 7(9), 1006–1017.
Goodwin, J. F., & Knudsen, K. E. (2014). Beyond DNA repair: DNA-PK function in cancer. Cancer Discovery, 4(10), 1126–1139.
Gorecki, L., Andrs, M., Rezacova, M., & Korabecny, J. (2020). Discovery of ATR kinase inhibitor berzosertib (VX-970, M6620): Clinical candidate for cancer therapy. Pharmacology & Therapeutics, 210, 107518.
Guillemette, S., et al. (2015). Resistance to therapy in BRCA2 mutant cells due to loss of the nucleosome remodeling factor CHD4. Genes & Development, 29(5), 489–494.
Guo, E., et al. (2020). FEN1 endonuclease as a therapeutic target for human cancers with defects in homologous recombination. Proceedings of the National Academy of Sciences of the United States of America, 117(32), 19415–19424.
Ha, G. H., et al. (2019). Pellino1 regulates reversible ATM activation via NBS1 ubiquitination at DNA double-strand breaks. Nature Communications, 10(1), 1577.
Hahn, W. C., et al. (2021). An expanded universe of cancer targets. Cell, 184(5), 1142–1155.
Hanahan, D. (2022). Hallmarks of cancer: New dimensions. Cancer Discovery, 12(1), 31–46.
Hartwell, L. H., Szankasi, P., Roberts, C. J., Murray, A. W., & Friend, S. H. (1997). Integrating genetic approaches into the discovery of anticancer drugs. Science New York, n.y., 278(5340), 1064–1068.
Henssen, A. G., et al. (2017). Therapeutic targeting of PGBD5-induced DNA repair dependency in pediatric solid tumors. Science Translational Medicine., 9(414), eaam078.
Hewitt, G., et al. (2021). Defective ALC1 nucleosome remodeling confers PARPi sensitization and synthetic lethality with HRD. Molecular Cell, 81(4), 767-783.e711.
Hickson, I. D. (2003). RecQ helicases: Caretakers of the genome. Nature Reviews. Cancer, 3(3), 169–178.
Hong, D., et al. (2016). Phase I Study of LY2606368, a checkpoint kinase 1 Inhibitor, in patients with advanced cancer. Journal of Clinical Oncology : Official Journal of the American Society of Clinical Oncology, 34(15), 1764–1771.
Hong, D. S., et al. (2018). Evaluation of Prexasertib, a checkpoint kinase 1 inhibitor, in a phase Ib study of patients with squamous cell carcinoma. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research, 24(14), 3263–3272.
Huang, A., Garraway, L. A., Ashworth, A., & Weber, B. (2020). Synthetic lethality as an engine for cancer drug target discovery. Nature Reviews Drug Discovery, 19(1), 23–38.
Huang, P. Q., et al. (2021). Discovery of ZN-c3, a Highly Potent and Selective Wee1 inhibitor undergoing evaluation in clinical trials for the treatment of cancer. Journal of Medicinal Chemistry, 64(17), 13004–13024.
Huang, R., & Zhou, P. K. (2021). DNA damage repair: Historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal Transduction and Targeted Therapy, 6(1), 254.
Hustedt, N., & Durocher, D. (2016). The control of DNA repair by the cell cycle. Nature Cell Biology, 19(1), 1–9.
Inoue, A., et al. (2021). Sequential Administration of XPO1 and ATR inhibitors enhances therapeutic response in TP53-mutated colorectal cancer. Gastroenterology, 161(1), 196–210.
Iorio, F., et al. (2018). Unsupervised correction of gene-independent cell responses to CRISPR-Cas9 targeting. BMC Genomics, 19(1), 604.
Javle, M., et al. (2021). Olaparib monotherapy for previously treated pancreatic cancer with DNA damage repair genetic alterations other than germline BRCA variants: findings from 2 phase 2 nonrandomized clinical trials. JAMA Oncology, 7(5), 693–699.
Joshi, P. M., Sutor, S. L., Huntoon, C. J., & Karnitz, L. M. (2014). Ovarian cancer-associated mutations disable catalytic activity of CDK12, a kinase that promotes homologous recombination repair and resistance to cisplatin and poly(ADP-ribose) polymerase inhibitors. The Journal of Biological Chemistry, 289(13), 9247–9253.
Kaelin, W. G., Jr. (2005). The concept of synthetic lethality in the context of anticancer therapy. Nature Reviews. Cancer, 5(9), 689–698.
Kategaya, L., Perumal, S. K., Hager, J. H., & Belmont, L. D. (2019). Werner syndrome helicase is required for the survival of cancer cells with microsatellite instability. iScience, 13, 488–497.
Kaufman, B., et al. (2015). Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. Journal of Clinical Oncology : Official Journal of the American Society of Clinical Oncology, 33(3), 244–250.
Kim, G., et al. (2015). FDA approval summary: Olaparib monotherapy in patients with deleterious germline BRCA-Mutated advanced ovarian cancer treated with three or more lines of chemotherapy. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research, 21(19), 4257–4261.
Kim, H., et al. (2020b). Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nature Communications, 11(1), 3726.
Kim, R., et al. (2022). Phase II study of ceralasertib (AZD6738) in combination with durvalumab in patients with advanced/metastatic melanoma who have failed prior anti-PD-1 therapy. Annals of Oncology : Official Journal of the European Society for Medical Oncology, 33(2), 193–203.
Kim, S. S., et al. (2020). Histone deacetylase inhibition is synthetically lethal with arginine deprivation in pancreatic cancers with low argininosuccinate synthetase 1 expression. Theranostics, 10(2), 829–840.
Kim, S. T., et al. (2021). Phase I Study of Ceralasertib (AZD6738), a Novel DNA damage repair agent, in combination with weekly paclitaxel in refractory cancer. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research, 27(17), 4700–4709.
Kondrashova, O., et al. (2017). Secondary somatic mutations restoring RAD51C and RAD51D associated with acquired resistance to the PARP inhibitor rucaparib in high-grade ovarian carcinoma. Cancer Discovery, 7(9), 984–998.
Kondrashova, O., et al. (2018). Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nature Communications, 9(1), 3970.
Konstantinopoulos, P. A., et al. (2020). Berzosertib plus gemcitabine versus gemcitabine alone in platinum-resistant high-grade serous ovarian cancer: A multicentre, open-label, randomised, phase 2 trial. The Lancet. Oncology, 21(7), 957–968.
Konstantinopoulos, P. A., et al. (2021). A Replication stress biomarker is associated with response to gemcitabine versus combined gemcitabine and ATR inhibitor therapy in ovarian cancer. Nature Communications, 12(1), 5574.
Kroll, E. S., Hyland, K. M., Hieter, P., & Li, J. J. (1996). Establishing genetic interactions by a synthetic dosage lethality phenotype. Genetics, 143(1), 95–102.
Kryukov, G. V., et al. (2016). MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science(new York, n.y.), 351(6278), 1214–1218.
Kumar, A., et al. (2014). ATR mediates a checkpoint at the nuclear envelope in response to mechanical stress. Cell, 158(3), 633–646.
Kunkel, T. A., & Erie, D. A. (2015). Eukaryotic mismatch repair in relation to DNA replication. Annual Review of Genetics, 49, 291–313.
Lavin, M. F. (2008). Ataxia-telangiectasia: From a rare disorder to a paradigm for cell signalling and cancer. Nature Reviews. Molecular Cell Biology, 9(10), 759–769.
Lee, J. M., et al. (2018). Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: A first-in-class proof-of-concept phase 2 study. The Lancet. Oncology, 19(2), 207–215.
Leijen, S., et al. (2016). Phase I Study Evaluating WEE1 Inhibitor AZD1775 as monotherapy and in combination with gemcitabine, cisplatin, or carboplatin in patients with advanced solid tumors. Journal of Clinical Oncology : Official Journal of the American Society of Clinical Oncology, 34(36), 4371–4380.
Levesque, A. A., & Eastman, A. (2007). p53-based cancer therapies: Is defective p53 the Achilles heel of the tumor? Carcinogenesis, 28(1), 13–20.
Lewis, C. W., et al. (2019). Upregulation of Myt1 promotes acquired resistance of cancer cells to Wee1 inhibition. Cancer Research, 79(23), 5971–5985.
Li, A., et al. (2019). Prospects for combining immune checkpoint blockade with PARP inhibition. Journal of Hematology & Oncology, 12(1), 98.
Li, F., et al. (2020a). CHK1 Inhibitor Blocks Phosphorylation of FAM122A and promotes replication stress. Molecular Cell, 80(3), 410-422.e416.
Li, F., et al. (2020b). mTOR inhibition overcomes primary and acquired resistance to Wee1 inhibition by augmenting replication stress in epithelial ovarian cancers. American Journal of Cancer Research, 10(3), 908–924.
Liang, J., et al. (2020). Genome-Wide CRISPR-Cas9 screen reveals selective vulnerability of ATRX-Mutant Cancers to WEE1 inhibition. Cancer Research, 80(3), 510–523.
Liang, S., et al. (2022). Structural insights into inhibitor regulation of the DNA repair protein DNA-PKcs. Nature, 601(7894), 643–648.
Lieb, S., et al. (2019). Werner syndrome helicase is a selective vulnerability of microsatellite instability-high tumor cells. eLife, 8, e43333.
Litton, J. K., et al. (2018). Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. The New England Journal of Medicine, 379(8), 753–763.
Liu, J. F., et al. (2021). Phase II study of the WEE1 inhibitor adavosertib in recurrent uterine serous carcinoma. Journal of Clinical Oncology : Official Journal of the American Society of Clinical Oncology, 39(14), 1531–1539.
Liu, L., et al. (2018). Synthetic lethality-based identification of targets for anticancer drugs in the human signaling network. Science Reports, 8(1), 8440.
Llorens-Agost, M., et al. (2021). POLθ-mediated end joining is restricted by RAD52 and BRCA2 until the onset of mitosis. Nature Cell Biology, 23(10), 1095–1104.
Lord, C. J., & Ashworth, A. (2016). BRCAness revisited. Nature Reviews Cancer, 16(2), 110–120.
Lord, C. J., & Ashworth, A. (2017). PARP inhibitors: Synthetic lethality in the clinic. Science (new York, n.y.), 355(6330), 1152–1158.
Lowery, C. D., et al. (2019). Broad Spectrum Activity of the Checkpoint Kinase 1 Inhibitor Prexasertib as a single agent or chemopotentiator across a range of preclinical pediatric tumor models. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research, 25(7), 2278–2289.
Maiani, E., et al. (2021). AMBRA1 regulates cyclin D to guard S-phase entry and genomic integrity. Nature, 592(7856), 799–803.
Mailand, N., et al. (2000). Rapid destruction of human Cdc25A in response to DNA damage. Science (new York, n.y.), 288(5470), 1425–1429.
Mateo, J., et al. (2020). Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): A multicentre, open-label, randomised, phase 2 trial. The Lancet Oncology, 21(1), 162–174.
Mateos-Gomez, P. A., et al. (2015). Mammalian polymerase θ promotes alternative NHEJ and suppresses recombination. Nature, 518(7538), 254–257.
Matheson, C. J., Backos, D. S., & Reigan, P. (2016). Targeting WEE1 kinase in cancer. Trends in Pharmacological Sciences, 37(10), 872–881.
Matt, S., & Hofmann, T. G. (2016). The DNA damage-induced cell death response: A roadmap to kill cancer cells. Cellular and Molecular Life Sciences: CMLS, 73(15), 2829–2850.
McDonald, E. R., 3rd., et al. (2017). Project DRIVE: A compendium of cancer dependencies and synthetic lethal relationships uncovered by large-scale Deep RNAi Screening. Cell, 170(3), 577-592.e510.
Mengwasser, K. E., et al. (2019). Genetic Screens Reveal FEN1 and APEX2 as BRCA2 synthetic lethal targets. Molecular Cell, 73(5), 885-899.e886.
Menolfi, D., et al. (2018). Kinase-dead ATR differs from ATR loss by limiting the dynamic exchange of ATR and RPA. Nature Communications, 9(1), 5351.
Mirman, Z., et al. (2018). 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polα-dependent fill-in. Nature, 560(7716), 112–116.
Moiseeva, T. N., Qian, C., Sugitani, N., Osmanbeyoglu, H. U., & Bakkenist, C. J. (2019). WEE1 kinase inhibitor AZD1775 induces CDK1 kinase-dependent origin firing in unperturbed G1- and S-phase cells. Proceedings of the National Academy of Sciences of the United States of America, 116(48), 23891–23893.
Moore, K. N., et al. (2021). A Phase 1b Trial of Prexasertib in combination with standard-of-care agents in advanced or metastatic cancer. Targeted Oncology, 16(5), 569–589.
Morgens, D. W., Deans, R. M., Li, A., & Bassik, M. C. (2016). Systematic comparison of CRISPR/Cas9 and RNAi screens for essential genes. Nature Biotechnology, 34(6), 634–636.
Mortensen, D. S., et al. (2015). Optimization of a Series of Triazole Containing Mammalian Target of Rapamycin (mTOR) kinase inhibitors and the discovery of CC-115. Journal of Medicinal Chemistry, 58(14), 5599–5608.
Mullard, A. (2017). Synthetic lethality screens point the way to new cancer drug targets. Nature Reviews Drug Discovery, 16(9), 589–591.
Mullenders, J., & Bernards, R. (2009). Loss-of-function genetic screens as a tool to improve the diagnosis and treatment of cancer. Oncogene, 28(50), 4409–4420.
Murai, J., et al. (2012). Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Research, 72(21), 5588–5599.
Murai, J., et al. (2014). Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Molecular Cancer Therapeutics, 13(2), 433–443.
Nakamura, K., et al. (2021). Inhibition of DNA-PK with AZD7648 sensitizes tumor cells to radiotherapy and induces type I IFN-dependent durable tumor control. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research, 27(15), 4353–4366.
Nik-Zainal, S., et al. (2012). Mutational processes molding the genomes of 21 breast cancers. Cell, 149(5), 979–993.
Niu, T., et al. (2022). Noncovalent CDK12/13 dual inhibitors-based PROTACs degrade CDK12-Cyclin K complex and induce synthetic lethality with PARP inhibitor. European Journal of Medicinal Chemistry, 228, 114012.
Ooi, S. L., et al. (2006). Global synthetic-lethality analysis and yeast functional profiling. Trends in Genetics : TIG, 22(1), 56–63.
Pal, S. K., et al. (2021). Effect of Cisplatin and gemcitabine with or without berzosertib in patients with Advanced urothelial carcinoma: a phase 2 randomized clinical trial. JAMA Oncology, 7(10), 1536–1543.
Parrish, P. C. R., et al. (2021). Discovery of synthetic lethal and tumor suppressor paralog pairs in the human genome. Cell Reports, 36(9), 109597.
Pettitt, S. J., et al. (2018). Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nature Communications., 9(1), 1849.
Pettitt, S. J., et al. (2020). Clinical BRCA1/2 reversion analysis identifies hotspot mutations and predicted neoantigens associated with therapy resistance. Cancer Discovery, 10(10), 1475–1488.
Picco, G., et al. (2021). Werner helicase is a synthetic-lethal vulnerability in mismatch repair-deficient colorectal cancer refractory to targeted therapies, chemotherapy, and immunotherapy. Cancer Discovery, 11(8), 1923–1937.
Pilié, P. G., Gay, C. M., Byers, L. A., O’Connor, M. J., & Yap, T. A. (2019). PARP inhibitors: Extending benefit beyond BRCA-Mutant cancers. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research, 25(13), 3759–3771.
Plummer, R., et al. (2013). A phase II study of the potent PARP inhibitor, Rucaparib (PF-01367338, AG014699), with temozolomide in patients with metastatic melanoma demonstrating evidence of chemopotentiation. Cancer Chemotherapy and Pharmacology, 71(5), 1191–1199.
Poveda, A., et al. (2021). Olaparib tablets as maintenance therapy in patients with platinum-sensitive relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A final analysis of a double-blind, randomised, placebo-controlled, phase 3 trial. The Lancet Oncology, 22(5), 620–631.
Ray Chaudhuri, A., et al. (2016). Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature, 535(7612), 382–387.
Rodríguez-Galán, O., et al. (2021). A functional connection between translation elongation and protein folding at the ribosome exit tunnel in Saccharomyces cerevisiae. Nucleic Acids Research, 49(1), 206–220.
Rondinelli, B., et al. (2017). EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nature Cell Biology, 19(11), 1371–1378.
Ruiz, S., et al. (2016). A Genome-wide CRISPR Screen Identifies CDC25A as a determinant of sensitivity to ATR inhibitors. Molecular Cell, 62(2), 307–313.
Sakai, W., et al. (2008). Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature, 451(7182), 1116–1120.
Satoh, M. S., & Lindahl, T. (1992). Role of poly(ADP-ribose) formation in DNA repair. Nature, 356(6367), 356–358.
Schaub, J. M., Soniat, M. M., & Finkelstein, I. J. (2022). Polymerase theta-helicase promotes end joining by stripping single-stranded DNA-binding proteins and bridging DNA ends. Nucleic Acids Research, 50(7), 3911–3921.
Schlacher, K., et al. (2011). Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell, 145(4), 529–542.
Schwer, B., Garg, A., Jacewicz, A., & Shuman, S. (2021). Genetic screen for suppression of transcriptional interference identifies a gain-of-function mutation in Pol2 termination factor Seb1. Proceedings of the National Academy of Sciences of the United States of America, 118(33), e2108105118.
Sen, T., et al. (2017). Targeting AXL and mTOR pathway overcomes primary and acquired resistance to WEE1 inhibition in small-cell lung cancer. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research, 23(20), 6239–6253.
Sen, T., et al. (2019). Combination Treatment of the Oral CHK1 Inhibitor, SRA737, and low-dose gemcitabine enhances the effect of programmed death ligand 1 blockade by modulating the immune microenvironment in SCLC. Journal of Thoracic Oncology : Official Publication of the International Association for the Study of Lung Cancer, 14(12), 2152–2163.
Sheng, H., et al. (2020). ATR inhibitor AZD6738 enhances the antitumor activity of radiotherapy and immune checkpoint inhibitors by potentiating the tumor immune microenvironment in hepatocellular carcinoma. Journal for Immunotherapy of Cancer, 8(1), e000340.
Singleton, B. K., Torres-Arzayus, M. I., Rottinghaus, S. T., Taccioli, G. E., & Jeggo, P. A. (1999). The C terminus of Ku80 activates the DNA-dependent protein kinase catalytic subunit. Molecular and Cellular Biology, 19(5), 3267–3277.
Stracker, T. H., Usui, T., & Petrini, J. H. (2009). Taking the time to make important decisions: The checkpoint effector kinases Chk1 and Chk2 and the DNA damage response. DNA Repair, 8(9), 1047–1054.
Sullivan-Reed, K., et al. (2018). Simultaneous targeting of PARP1 and RAD52 triggers dual synthetic lethality in BRCA-deficient tumor cells. Cell Reports, 23(11), 3127–3136.
Swisher, E. M., et al. (2017). Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. The Lancet Oncology, 18(1), 75–87.
Symington, L. S., & Gautier, J. (2011). Double-strand break end resection and repair pathway choice. Annual Review of Genetics, 45, 247–271.
Taglialatela, A., et al. (2017). Restoration of replication fork stability in BRCA1- and BRCA2-deficient cells by inactivation of SNF2-Family fork remodelers. Molecular Cell, 68(2), 414-430.e418.
Ter Brugge, P., et al. (2016). Mechanisms of therapy resistance in patient-derived xenograft Models of BRCA1-Deficient breast cancer. Journal of the National Cancer Institute. https://doi.org/10.1093/jnci/djw148
Thomas, A., et al. (2018). Phase I Study of ATR Inhibitor M6620 in combination with topotecan in patients with advanced solid tumors. Journal of Clinical Oncology : Official Journal of the American Society of Clinical Oncology, 36(16), 1594–1602.
Thomas, A., et al. (2021). Therapeutic targeting of ATR yields durable regressions in small cell lung cancers with high replication stress. Cancer Cell, 39(4), 566-579.e567.
Tomimatsu, N., et al. (2014). Phosphorylation of EXO1 by CDKs 1 and 2 regulates DNA end resection and repair pathway choice. Nature Communications, 5, 3561.
Tsherniak, A., et al. (2017). Defining a cancer dependency map. Cell, 170(3), 564-576.e516.
Turner, N. C., et al. (2008). A synthetic lethal siRNA screen identifying genes mediating sensitivity to a PARP inhibitor. The EMBO Journal, 27(9), 1368–1377.
Tutt, A. N. J., et al. (2021). Adjuvant olaparib for patients with BRCA1- or BRCA2-mutated breast cancer. The New England Journal of Medicine, 384(25), 2394–2405.
van Bussel, M. T. J., et al. (2021). A first-in-man phase 1 study of the DNA-dependent protein kinase inhibitor peposertib (formerly M3814) in patients with advanced solid tumours. British Journal of Cancer, 124(4), 728–735.
Wang, C., et al. (2021). Genetic vulnerabilities upon inhibition of DNA damage response. Nucleic Acids Research, 49(14), 8214–8231.
Wang, J., et al. (2016). Suppression of KRas-mutant cancer through the combined inhibition of KRAS with PLK1 and ROCK. Nature Communications, 7, 11363.
Wang, J., et al. (2019). FDA-approved drug screen identifies proteasome as a synthetic lethal target in MYC-driven neuroblastoma. Oncogene, 38(41), 6737–6751.
Wang, T., et al. (2015). Identification and characterization of essential genes in the human genome. Science (new York, n.y.), 350(6264), 1096–1101.
Wang, W., et al. (2019). Combined gene essentiality scoring improves the prediction of cancer dependency maps. eBioMedicine, 50, 67–80.
Wheeler, D. A., et al. (2021). Molecular features of cancers exhibiting exceptional responses to treatment. Cancer Cell, 39(1), 38-53.e37.
Wilson, Z., et al. (2022). ATR Inhibitor AZD6738 (Ceralasertib) exerts antitumor activity as a monotherapy and in combination with chemotherapy and the PARP inhibitor olaparib. Cancer Research, 82(6), 1140–1152.
Wyatt, D. W., et al. (2016). Essential Roles for Polymerase θ-mediated end joining in the repair of chromosome breaks. Molecular Cell, 63(4), 662–673.
Xu, G., et al. (2015). REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature, 521(7553), 541–544.
Yang, E. S., et al. (2021). A Phase 1b trial of prexasertib in combination with chemoradiation in patients with locally advanced head and neck squamous cell carcinoma. Radiotherapy and Oncology : Journal of the European Society for Therapeutic Radiology and Oncology, 157, 203–209.
Yap, T. A., et al. (2020). Phase I Trial of First-in-Class ATR Inhibitor M6620 (VX-970) as monotherapy or in combination with carboplatin in patients with advanced solid tumors. Journal of Clinical Oncology : Official Journal of the American Society of Clinical Oncology, 38(27), 3195–3204.
Yap, T. A., et al. (2021). Ceralasertib (AZD6738), an Oral ATR kinase inhibitor, in combination with carboplatin in patients with advanced solid tumors: A Phase I Study. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research, 27(19), 5213–5224.
Yazinski, S. A., et al. (2017). ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes & Development, 31(3), 318–332.
Zatreanu, D., et al. (2021). Polθ inhibitors elicit BRCA-gene synthetic lethality and target PARP inhibitor resistance. Nature Communications, 12(1), 3636.
Zhang, Z., et al. (2020). Identification of probe-quality degraders for Poly(ADP-ribose) polymerase-1 (PARP-1). Journal of Enzyme Inhibition and Medicinal Chemistry, 35(1), 1606–1615.
Zhao, Q., Lan, T., Su, S., & Rao, Y. (2019). Induction of apoptosis in MDA-MB-231 breast cancer cells by a PARP1-targeting PROTAC small molecule. Chemical Communications (cambridge, England), 55(3), 369–372.
Zhou, J., et al. (2021). A first-in-class Polymerase theta inhibitor selectively targets homologous-recombination-deficient tumors. Nature Cancer, 2(6), 598–610.
Funding
This study was supported by the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences to M.D (2021-RC310-013), Beijing Hope Run Special Fund of Cancer Foundation of China to M.D (LC2021R02) and the CAMS Innovation Fund for Medical Sciences to M.D (2021-I2M-1-067).
Author information
Authors and Affiliations
Department of Radiation Oncology, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100021, China
Xin Xu
Department of Dermatology, University of California San Diego, San Diego, CA, USA
Somaira Nowsheen
State Key Laboratory of Molecular Oncology and Department of Radiation Oncology, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100021, China
Min Deng
Contributions
XX: data curation, writing—original draft. SN: writing—review and editing. MD: conceptualization, funding acquisition, resources, supervision, writing—review and editing.
Corresponding author
Correspondence to Min Deng.
Ethics declarations
Conflict of interests
The authors declare that they have no competing financial interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article
Cite this article
Xu, X., Nowsheen, S. & Deng, M. Exploring the DNA damage response pathway for synthetic lethality. GENOME INSTAB. DIS. (2022). https://doi.org/10.1007/s42764-022-00087-w
Received01 July 2022
Revised30 September 2022
Accepted07 October 2022
Published02 November 2022
DOIhttps://doi.org/10.1007/s42764-022-00087-w
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
Synthetic lethality
DNA damage response
DNA repair pathway
用户登录
还没有账号?
立即注册