






MiRNA let-7i promotes radiation-induced pulmonary epithelial-mesenchymal transition by targeting IL-10
Original Research Paper
Shenghui Zhou, Xin Liang, Zewen Sun, Xueping Li, Jiaojiao Zhu, Zhihua Yang, Xiujie Pan, Yilong Wang, Yongqing Gu & Maoxiang Zhu
Genome Instability & Disease 3, 271–284 (2022)
Abstract
Radiation-induced pulmonary fibrosis (RIPF) is a common complication of thoracic tumors, lacking targeted therapeutic strategies, which seriously affects the quality of life. Numerous studies have confirmed that epithelial to mesenchymal transition (EMT) plays an important role in RIPF, but the specific mechanism remains unclear. We verified by mice lung tissue miRNA microarray screen combined with alveolar type II epithelial cells (AECII) and found that miRNA-let-7i was significantly highly expressed after ionizing radiation (IR). It was also confirmed that miRNA-let-7i inhibited its expression by targeted binding to the 3ʹUTR of IL-10, promoting the phosphorylation activation of AKT, and subsequently inducing the EMT of AECII. In conclusion, our study confirmed the role and specific mechanism of miRNA-let-7i promoting AECII cells EMT to participate in RIPF and provided a new target for the prevention and treatment of RIPF.
Graphical Abstract

Introduction
Radiation-induced pulmonary fibrosis (RIPF) is a frequently occurring complication from radiotherapy of thoracic tumors, as many as 35% of patients with lung cancer or breast cancer receiving thoracic radiotherapy developed secondary radiation pneumonitis and a high risk of RIPF (Carver et al., 2007). The characteristics of RIPF include alveolar epithelial cell injury, excessive proliferation of fibroblasts and significant deposition of interstitial extracellular matrix (ECM), ultimately leading to disruption of normal lung architecture and respiratory failure (Chen et al., 2019). Accumulative evidence suggests that epithelial to mesenchymal transition (EMT) occurred in the alveolar epithelial cells was critical cellular mechanism in the pathogenesis of fibrosis (Zhang et al., 2019; Phan et al., 2021; X. Liang et al., 2021; Liu et al., 2019). However, the molecular mechanisms underlying RIPF are still unclear.
EMT was a set of multiple and dynamic transitional states between the epithelial and mesenchymal phenotypes, not involved a single binary decision. EMT and its intermediate states have been identified as crucial drivers of organ fibrosis which modifies the adhesion molecules expressed by epithelial cells, allowing them to adopt a migratory and invasive behavior (Nieto et al., 2016; Thiery et al., 2009). The research shows that lung epithelial cells undergo EMT and contribute substantially to the lung fibroblast population during experimentally induced lung fibrosis (Andugulapati et al., 2020). EMT is one of the several proposed mechanisms through which fibroblasts and myofibroblasts are generated. The main features of EMT in epithelial cell shape were driven by the complementary modulation of cell to another cell adhesion and cell-substrate interaction, mediating by N-cadherin and E-cadherin, as well as fibronectin and integrin receptors, respectively (Dongre & Weinberg, 2019). The significance of EMT to the fibrogenic process may go beyond direct generation of fibroblasts and may include indirect effects related to release of cytokines by injured epithelial cells, thereby creating a pro-fibrogenic environment (Mittal, 2018). Ample evidence gained with in vitro studies, animal models or tissue samples of human fibrotic lungs has established that EMT occurs in lung fibrosis, in which lung epithelial cells acquire mesenchymal phenotypes after injury (Hewlett et al., 2018; Kong et al., 2020). Therefore, EMT reportedly plays a critical role in the pathogenesis of pulmonary fibrosis.
During the process of EMT, many pathways regulate EMT. The phosphatidylinositol-3-kinase (PI3K)/Akt signaling pathway plays a key role in regulating cell proliferation and EMT (Xu et al., 2015). Phosphatidylinositol-3-kinase (PI3K) as a lipid kinase, generates PIP3 that regulates the translocation of major downstream effector Akt to plasma membrane (PM) as a second messenger. Then, Akt is activated by phosphorylation modification of threonine 308 and serine 473 (Shorning et al., 2020). Radiation-damaged AT2 cells have the PI3K/Akt pathway activated, which controls EMT and other important physiologic processes (Yeon et al., 2017).
MicroRNAs (miRNAs) are endogenously encoded, a class of evolution conserved (19–22 nt in length) non-coding small RNAs, which regulate gene expression by binding to the 3′-UTR of targets and inhibiting the translation or degradation of target mRNAs. It is presumed that about one-third of human genes are regulated by miRNAs (Lu & Rothenberg, 2018). Increasing evidence suggests that dysregulated miRNAs play essential roles in the development of lung injury and fibrosis (Kadota et al., 2021). Recently, a number of studies have proved that miRNAs targeting EMT are also involved in the initial stage and progression of pulmonary fibrosis and may be new therapeutic targets for fibrogenic lung diseases (Liu et al. 2019). In our previous research, we found that miRNA-155-5p inhibits EMT by targeting GSK-3β during pulmonary fibrosis caused by radiation (D. Wang et al., 2021). Inhibition of miRNA-486-3p induces EMT during RIPF by targeting Snail (Yan et al., 2022).
However, the role of miRNAs let-7i in RIPF has been poorly studied. In the current study, we aim to investigate the role of miRNAs let-7i and its target IL-10 in mediating radiation-induced EMT via the PI3K / AKT pathway induced EMT using a previously established mouse model of RIPF (Xiong et al., 2015) and to search for new molecular therapy targets for RIPF.
Materials and methods
Animals
SPF male C57BL/6 wild-type (WT) mice, 6–8 weeks old, were purchased from Vital River Laboratory Animal Technology Co., Ltd (Beijing, China) and raised in Institute of Radiation Medicine, Institute of Military Medical Research, Laboratory Animals Center (Beijing, China).
miRNA microarray analysis
Mice were anesthetized with 1% sodium pentobarbital (anesthetic dose of 50 mg/kg) and fixed on an irradiation device. The 10 cm-thick lead bricks were shielded from the rest of the chest and 60CO γ rays 20 Gy were used to shot full chest. The mice were sacrificed after 14 days of irradiation and the lung tissues of the control and irradiated mice were used for miRNA microarray analysis. Alveolar septal thickening and increased collagen deposition could be observed at/after 14 days post-irradiation in our previous study (Xiong et al., 2015). Three samples for each group. A leaf lung tissue each sample was used to detect, and then the rest of tissues was temporarily placed at−80 °C. MiRNA microarray analysis was performed by CapitalBio Technology Co (Beijing, China).
Cell culture, irradiation and transfection
The human lung adenocarcinoma A549 cells were saved by this laboratory. A549 cells were cultured in Dulbecco’s Modified Eagle Media (DMEM) medium (Hyclone, USA) supplemented with 10% fetal bovine serum (FBS). All cells were cultured in media containing 100 U/ml penicillin and 100 μg/ml streptomycin and 5% CO2, 95% air at 37 °C.
Radiation-induced A549 cells epithelial-mesenchymal transition model was conducted by 60Co γ-ray 6 Gy and cultured in an incubator for 48 h. Cells equilibrated overnight in medium without FBS were used before irradiation.
Transfection of 100 nM let-7i inhibitor (GenePharma, Suzhou, China) and IL-10 siRNA (GenePharma, Suzhou, China) were performed 48 h using lipofectamine 2000 (Invitrogen, USA) before irradiation and transfection of 50 nM let-7i mimic (GenePharma, Suzhou, China) following the manufacturer’s protocol.
Western blotting
Proteins were extracted from A549 cells. The load samples were prepared. Each well was equally loaded and electrophoresed on SDS-PAGE gel at 80 V. After 30 min, it was changed to 120 V for 50 min. Then, transferred to a PVDF membrane at 100 V for 2 h. The membrane was blocked at room temperature for 1 h in 5% milk prepared in TBST and the primary antibodies E-cadherin (Abeam, USA; ab76055, 1:1,000), N-cadherin (Abeam, USA; ab18203, 1:1,000), Twist (Abeam, USA; ab175430, 1:1,000), GAPDH (Santa Cruz, USA; sc-25778, 1:1,000), IL-10 (1:1000 dilution), AKT (Abcam, ab179463, 1:1000 dilution), p-AKT (Abcam, USA; ab81283, phospho S473, 1:1000 dilution) were incubated overnight at 4 °C. After that, the membranes were incubated with HRP-conjugated IgG (1:4000 dilution) at room temperature for 1 h.
Flow cytometry
After transfected with let-7i mimic for 48 h, A549 cells were digested with 0.25% trypsin to prepare single cell suspensions. Then, stained cells with FITC-conjugated anti-Zo-1 antibody (Abcam, 61357, 1:100 dilution), PE-conjugated anti- E-cadherin antibody (BD Pharmingen, 562526, 1:100 dilution) and APC-conjugated anti- N-cadherin antibody (eBioscience, 17–3259, 1:100 dilution) respectively and examined on a FACS Calibur cytometer (BD Biosciences, USA). The resulting populations were analyzed with FlowJo software (Treestar, Ashland, OR).
Quantitative real-time -PCR (qRT-PCR)
The total RNA of the cells and the lung tissues were extracted using Trizol Reagent (TIANGEN, DP421). The quality and the concentration of the extracted RNA were detected by Thermo Nanodrop 2000. RNA was reversely transcribed according to miRcute Plus miRNA First-Strand cDNA Synthesis Kit instruction (TIANGEN, KR211). The reaction was under the condition of 42 °C for 60 min, 95 °C for 3 min. The primer sequences were shown in Table 1. Then qRT-PCR reaction was conducted using miRcute Plus miRNA qPCR Detection Kit (SYBR Green) (TIANGEN, FR411) on a Bio-Rad Real-Time PCR System (California, USA) under the condition of pre-denaturalizing at 95 °C for 15 min, denaturalizing at 94 °C for 20 s, annealing and extending at 60 °C for 34 s, which were cycled for 45 times. The expression levels of let-7i were normalized to U6 and that of other mRNAs were β-actin. The results were calculated via 2−ΔΔCt method.
Table 1 Primer sequence for qRT-PCR assay
Full size table
Immunofluorescence staining
To detect the intracellular localization of and changes in E-cadherin and N-cadherin, A549 cells were transfected with let-7i inhibitor and negative control, then irradiated after 48 h. The samples were treated as follows: fixed with 4% polyformaldehyde, permeabilized with 0.25% Triton X-100 and blocked with 3% BSA, each step at room temperature for 30 min. Then samples were incubated in primary antibody E-cadherin (Abeam, USA; ab76055, 1: 200 dilution) and N-cadherin (Abeam, USA; ab18203, 1: 200 dilution) overnight at 4 °C. After that, the samples were incubated in goat anti-rat IgG (FITC, 1: 400 dilution) and mouse anti-rabbit IgG (APC, 1: 400 dilution) for 1 h in dark. Nuclei were counterstained with DAPI in PBS (1: 500 dilution) for 20 min. Rinsed with pre-chilled PBS after each step. Then observed under fluorescence microscope and took pictures with NIS-ELIMENTS software.
Dual luciferase assay
TargetScan (www.targetscan.org/vert_71/), miRanda (www.microrna.org) and Pictar (http://pictar.mdc-berlin.de/) database was used for predicting the target genes for miR-let-7i. MicroRNA biological prediction website was taken for target gene analysis of let-7i. Binding sequence of the IL-10 3ʹUTR and let-7i was obtained, then wild sequence and mutant sequence were synthesized and connected onto the pmirGLO Dual-Luciferase miRNA Target Expression Vector by GenePharma company (Suzhou, China). A549 cells were inoculated in a 96-well plate and then transfected with let-7i negative control (NC)/mimic and IL-10 wild/mutant plasmid after cell adherence. The cells were collected after 48 h. The effect on the luciferase activity of 3’UTR in IL-10 was detected according to Dual-Glo® Luciferase Assay System (Promega, USA) instruction and tested using Biochemistry luminometer (TurnerBioSystems, TD 20/20).
Statistical analysis
SPSS 16.0 statistical software was used to analyze all experimental data. The results were expressed as mean ± standard deviation (`x ± SD). Independent sample t-test was used. P < 0.05 indicated that the difference was statistically significant.
Results
Radiation induces let-7i highly expression
To investigate the effect of radiation on miRNA expression in lung tissues, C57BL/6 mice were irradiated via 60Co γ ray 20 Gy and euthanized at day 14 (14 days post radiation is a key time point for RIPF), and then lung tissues were taken for miRNA microarray analysis. Comparing the miRNAs that were differentially expressed between the control and irradiated groups. (Fig. 1a) The results indicated that miRNA let-7i was significantly upregulated in lung tissues. (Fig. 1b). After validation by qRT-PCR, the expression of let-7i increased 1.8 folds, which was consistent with the results of the microarray (Fig. 1c). To further verify the expression of let-7i by IR, we irradiated A549 cells with 60Co γ ray 6 Gy and found that let-7i level was persistently upregulated after 0.5 h post-irradiation. Interestingly, this increased expression trend continued for 48 h. The fold increase was 2.6 times (Fig. 1d). Based on these results, higher miRNA let-7i expression possibly was a crucial factor in RIPF and potentially represents a prognostic indicator for patients with pulmonary fibrosis in the clinic.
Fig. 1
Radiation Induces upregulation of let-7i in lung tissues of C57BL/6 mice and A549 cells. C57BL/6 mice were irradiated via.60Co γ-ray 20 Gy and sacrificed at day 14, then lung tissues were taken for miRNA microarray analysis. a. The heat map hierarchal clustering dendrogram analysis of miRNA expression in control and irradiation groups; b. Changes in let-7i levels of miRNA microarray; c. Expression of let-7i in lungs of mice detected by RT-qPCR; d. Radiation induces upregulation of let-7i in A549 cells as times go. (n = 3, *p < 0.05 versus control)
Let-7i overexpression augments EMT of AECII
Our previous experimental results have shown that radiation-induced EMT plays an important role in RIPF and the expression of let-7i was upregulated in lung tissues and A549 cells by IR (Xiong et al., 2015). Hence, we investigated the role of let-7i in EMT of A549 cells. We first transfected the let-7i mimics into A549 cells and detected the transfection effect by qRT-PCR. The results showed that the transfected group expressed 4.5 times more than the control group (Fig. 2a). We assessed the expression levels of EMT markers in let-7i overexpression A549 cells to elucidate the relationship between let-7i and the EMT. Notably, let-7i overexpression markedly decreased the protein expression of the epithelial markers E-cadherin, whereas the expression of the mesenchymal indicators N-cadherin, Vimentin and Twist were increased (Fig. 2b). In addition, EMT represents a continuum with epithelial/ mesenchymal hybrid and fully transitioned mesenchymal-like states. These results thus are associated with both hybrid (N-cadherin+ZO-1+) and further differentiated (N-cadherin+ZO-1−) phenotypes. Proportions of N-cadherin+ZO-1+ cells and N-cadherin+ZO-1− cells in group transfected with let-7i mimic were considerably larger than that in control (Fig. 2c, d). Taken together, these data indicated that let-7i induces EMT of AECII.
Fig. 2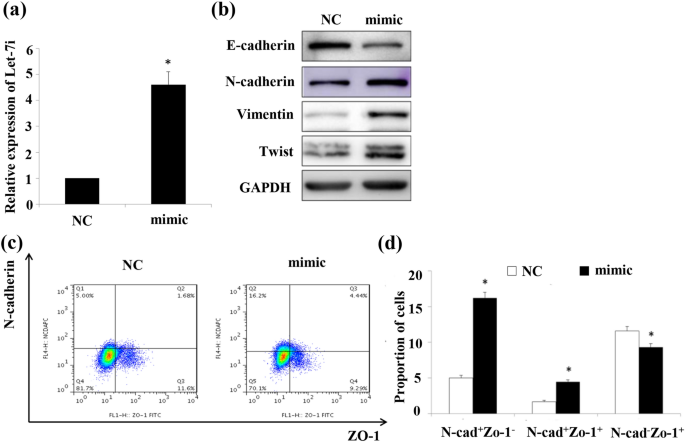
Let-7i overexpression induces EMT of A549 cells. A549 cells were transfected with let-7i mimic or negative controls (NC). After 48 h, expression level of let-7i was measured by qRT-PCR and that of relative EMT proteins were WB and Flow cytometry. a. Expression level of let-7i; b. Expression levels of relative EMT proteins detected by WB; c. Expression levels of relative EMT proteins detected by Flow cytometry; d. Bar graphs depicting changes in relative density according to c. (n = 3, *p < 0.05 versus negative control)
Let-7i deficiency prevents radiation-induced EMT of AECII
To further investigate the role of let-7i in radiation-induced EMT, we used let-7i inhibitor to suppress let-7i in A549 cells. The effect of inhibition was examined by qRT-PCR. As shown in Fig. 3a, the expression of let-7i in suppression group was lower expression about 0.5 times than that in control. After irradiated for 48 h, the protein expression of E-cadherin, N-cadherin, Vimentin, and Twist were measured by Western blot. The results showed that radiation-induced downregulated E-cadherin levels and upregulated N-cadherin, Vimentin, and Twist levels. However, inhibition of let-7i blocked the changes caused by radiation (Fig. 3b). Furthermore, it was also proved that let-7i inhibition increased expression of E-cadherin but reduced N-cadherin expression using immunofluorescence staining with IR (Fig. 3c) collectively, our results indicated that inhibited let-7i alleviates the process of EMT induced by radiation.
Fig. 3
Let-7i Inhibition Prevents Radiation-induced EMT of A549 cells. A549 cells were transfected with let-7i inhibitor or negative controls (NC). After 48 h, expression level of let-7i was measured by qRT-PCR and that of relative EMT proteins were WB. a. Expression level of let-7i; b. Expression levels of relative EMT proteins detected by WB; c. Bar graphs depicting changes in relative density of E-cadherin, N-cadherin, Vimentin, and Twist. The density values of blots were normalized to the GAPDH; d. E-cadherin and N-cadherin were detected by immunofluorescence Staining
Let-7i directly targeted inhibits IL-10 expression in AECII
From the study above, we can conclude that let-7i plays a pivotal role in the process of EMT. However, the molecular mechanisms of let-7i mediating radiation-induced EMT are still unclear. Therefore, in light of the reported methods (Gu et al., 2020; Wang et al., 2019; Zhou et al., 2021), Targetscan, miRanda and Pictar were used to predict the target downstream of let-7i. The results have shown the 3ʹ-UTR sequence of IL-10 mRNA combined with let-7i in whole three websites, which indicated that IL-10 is high possible to be a target gene. Therefore, we then detected the mRNA and protein levels of IL-10 in mouse lung tissues by qRT-PCR and Western blot. As shown in (Fig. 4a, b), the expression of IL-10 mRNA and protein were both decreased in mouse lung tissues after irradiation. Subsequently, we detected IL-10 expression in let-7i overexpression and knockdown A549 cells to elucidate the relationship between let-7i and IL-10. The results revealed that overexpression of let-7i could significantly downregulate the expression levels of IL-10 mRNA and protein (Fig. 4c, d). Next, we found that expression of IL-10 was upregulated in A549 cells following let-7i inhibition. (Fig. 4e, f) To validate this potential association whether let-7i is capable of regulating IL-10 via the binding sites in its 3ʹ-UTR, we constructed the 3ʹ-UTR containing the predicted let-7i binding site (wild-type group, WT) or mutant (mutant group, MT) downstream of the firefly luciferase coding region in the pmirGLO Dual-Luciferase vector. The luciferase vectors together with let-7i mimic or mimic control were transfected into A549 cells. The results revealed that the luciferase activity in the wild group was significantly reduced, but there was no significant change in the mutant group (Fig. 4g, h). These data demonstrated that let-7i directly targets 3’-UTR of IL-10 and inhibits its expression.
Fig. 4
Let-7i directly targets and downregulates expression of IL-10. A549 cells were transfected with let-7i mimic or inhibitor. After 48 h, mRNA and protein levels of IL-10 and GAPDH were measured by Western blot and qRT-PCR. a. Relative expression of let-7i and IL-10 mRNA in lung tissues of C57BL/6 mice after irradiation compared with control measured by qRT-PCR. b. Relative expression of let-7i and IL-10 mRNA in lung tissues of C57BL/6 mice after irradiation compared with control measured by Western blot. c. Let-7i overexpression on IL-10 mRNA expression measured by qRT-PCR. d. Let-7i overexpression on IL-10 protein expression measured by Western blot. e. Let-7i suppression on IL-10 mRNA expression measured by qRT-PCR. f. Let-7i suppression on IL-10 mRNA expression measured by Western blot. g. Sequences of let-7i and the putative target sequence in the IL-10 mRNA (wild-type, WT) or an engineered mutant of this sequence (mutant, MT) for the luciferase activity assay. h. The activity assay of dual luciferase reporter gene, activity assay of luciferase correlated with let-7i mimic and IL-10 3ʹUTR WT/MT in A549 cells, let-7i inhibited the luciferase activity of WT plasmid but it did not influence that of MT plasmid, compared with the control group, *P < 0.05)
High expression of let-7i promote EMT though inhibits IL-10
Recent studies indicate that alteration in the level of myokine IL-10 might exert a direct or indirect inhibitory effect on EMT (Kim et al., 2021). Furthermore, we investigated whether knockdown IL-10 could promote the progression of EMT mediated by let-7i. For this experiment, we transfected A549 cells with siRNAs targeting IL-10, and the resulting exhibited dramatically decreased IL-10 expression by Western blot. To detect the protein levels of E-cadherin, N-cadherin, Vimentin, and Twist, as shown in Fig. 5a. These results indicated that downregulation of IL-10 could promote EMT. In addition, knockdown of IL-10 and inhibited let-7i induced a decrease in E-cadherin and an increase in N-cadherin, Vimentin, and Twist (Fig. 5b) Therefore, we found that IL-10 knockdown promoted EMT although it also reversed the effects of the let-7i inhibitor. These data provide evidence that let-7i/ IL-10 promotes EMT in vitro.
Fig. 5
Knockdown of IL-10 reversed EMT attenuated by low expression of let-7i. A549 cells were both treated with IL-10 siRNA and negative control (NC), relative expression levels of EMT proteins were detected by Western blot after 48 h. a. Relative expression levels of proteins in A549 cells. b. A549 cells were transfected with let-7i inhibitor and IL-10 siRNA, then irradiated after transfection for 48 h, relative expression levels of EMT proteins were detected by Western blot after 48 h
Let-7i activated the PI3K/AKT signaling pathway by targeting IL-10 in A549 cells in vitro
Considering that PI3K/Akt pathway is the pivotal pathway to regulating process of EMT and involved in the RIPF.(Xu et al., 2021; Yeon et al., 2017) Here, we measured levels of proteins involved in the PI3K/AKT pathway to determine whether let-7i could target IL-10 regulates radiation-induced EMT through PI3K/AKT pathway. We found that inhibition of IL-10 and overexpression of let-7i were both associated with an increase of p-AKT at Ser473, but there was no significant change in the expression of total AKT (Fig. 6a, b). We then assessed the effects of let-7i inhibition on radiation-induced p-AKT at Ser473. The results showed that let-7i inhibition could reduce p-AKT at Ser473 increased by IR (Fig. 6c). Moreover, let-7i and IL-10 inhibition together reversed let-7i inhibition–induced decreases in p-AKT at Ser473 levels in A549 cells (Fig. 6d). Taken together, these results indicated that let-7i could regulate the PI3K/AKT pathway through IL-10 and further regulate radiation-induced EMT.
Fig. 6
Let-7i Targets IL-10 to mediate radiation-induced EMT via the PI3K / AKT pathway. A549 cells were transfected with let-7i mimic, let-7i inhibitor and IL-10 siRNA or negative control for 48 h. Cells were then treated with or without irradiation for 48 h, and protein levels of p-AKT at Ser473, AKT and GAPDH were measured by Western blot. a. Relative expression of AKT and p-AKT at Ser473 after IL-10 inhibition; b. Relative expression of AKT and p-AKT at Ser473 after let-7i expression; c. Effects of let-7i inhibition on PI3K/AKT pathway-related proteins expression in A549 cells induced by radiation; d. Effects of inhibiting let-7i and IL-10 simultaneously on PI3K/AKT pathway-related protein expression in A549 cells induced by radiation. e. A schematic model for ionizing radiation upregulates miR-let-7i in alveolar epithelial cells that induce EMT contributing to RIPF through IL-10/PI3K/AKT pathway
Discussion
RIPF is a frequently occurred complication from radiotherapy threatening the health and life of thoracic malignancies patients (Turrisi et al., 1999). Despite increasing evidence, the detailed molecular mechanism of RIPF remains to be elucidated. The role of type 2 EMT in the pathogenesis of pulmonary fibrosis has recently received a great deal of attention (Chang et al., 2021). Our previous study revealed that Tregs participate in EMT regulation in RIPF (Xiong et al., 2015). There are extensive studies that show that miRNAs regulate lung fibrosis by inducing EMT in epithelial cells by modulating the expression of specific ligands, receptors, and signaling pathways account for their effects on pulmonary fibrosis (Liang et al., 2021; Su et al., 2020). For instance, miR-26a alleviated TGF-β1 and BLM-induced EMT in A549 cells and mice by regulating HMGA2 in idiopathic pulmonary fibrosis, whereas miR-26a inhibition resulted in lung epithelial cells transforming into myofibroblasts in vitro and in vivo (Liang et al., 2014). Lentivirus-mediated over-expression of miR-338* can reduce the EMT procedure by target smoothened (SMO) and thus alleviate lung fibrosis induced by bleomycin in mice (Zhuang et al., 2016). MiR-503 was shown to be downregulated in pulmonary fibrosis mice induced by silica, whereas upregulation of miR-503 increased the protein expression level of E-cadherin and decreased the expression levels of vimentin and α-SMA via targeting PI3K p85, thus alleviating the process of EMT in vivo (Yan et al., 2017). MiRNA-targeted EMT-based therapy for RIPF may be an effective therapeutic tool to improve current treatment challenges. In this study, the profiles of miRNAs in the lungs of C57/BL6 mice with 20 Gy-irradiated were identified as compared with those non-irradiated. Our data demonstrated that miRNA let-7i was significantly upregulated by 60Co γ-rays irradiation. Therefore, we assumed that the upregulation of let-7i was associated with radiation-induced EMT and RIPF.
MiRNA let-7i, located on chromosome 12, belongs to the let-7 family. Let-7 family is composed of thirteen mature let-7 miRNAs included let-7a-1/a-2/a-3/b/c/d/e/f-1/f-2/g/i, mir-98, and miR-202, which is one of the first miRNAs to be discovered (Lee et al., 2016; Roush & Slack, 2008). Previous studies focus on the role of let-7 in regulation of tumors (Li & Liao, 2021; Pandolfini et al., 2019). Recently, several studies have demonstrated that let-7 plays a role in fibrosis disease. For example, let-7d was decreased in the lungs of IPF patients and transcriptionally repressed by TGF-β1. Transfection of let-7d into primary fibroblasts decreased the expression of the mesenchymal markers α-smooth muscle actin, N-cadherin, fibroblast-specific protein-1, and fibronectin, as well as an increase in the epithelial markers tight junction protein-1 and keratin 19 (Huleihel et al., 2014). What’s more, let-7c and let-7 g were reported to be increased in endothelial-to-mesenchymal transition (Yazarlou et al., 2021). To demonstrate our hypothesis, we first paid special attention to let-7i after further validation of its increased expression in the A549 cells after irradiation with RT-qPCR analysis. These results suggested that overexpression of let-7i induced EMT, while inhibition of let-7i prevented radiation-induced EMT in A549 cells. However, there are limitations as we did not use cell-type specific experimental strategies in this article due to technical limitations.
Ling Xie and co-workers also found that let-7i was upregulated in lungs of rats with lung injury induced by radiation, which is consistent with our findings (Xie et al., 2014). However, the effect of let-7i on EMT of RIPF has not been studied. In the current study, we have identified that let-7i overexpression significantly promoted EMT in A549 cells, which may cause pulmonary fibrosis, while knockdown of let-7i alleviated EMT induced by irradiation in mice. Whereas Wang et al. reported that let-7i was downregulated in cardiac inflammation and fibrosis induced by angiotensin II (Wang et al., 2015). Meanwhile, cardiac inflammation and fibrosis was inhibited in let-7i-infused mouse in a dose-dependent manner. This phenomenon may occur due to differences in tissue microenvironment and cell types in different organs resulting in discrepancies in the role of miRNAs.
It is well known that miRNAs play biological role mainly by interacting with target gene 3’-UTR. Here, we predicted the target genes via Targetscan, miRBase and Pictar websites and found that IL-10 is a target of let-7i. Recently, IL-10 came into focus as fibrosis-related immunosuppressive cytokine. K Nakagome demonstrated that delivery of IL-10 in vivo suppresses the development of bleomycin-induced pulmonary fibrosis (Nakagome et al., 2006). Moreover, IL-10 suppresses the production and activation of TGF-β which is a key growth factor that directly promotes the synthesis of collagen in the lung. In myofibroblasts, IL-10 may inhibit the proliferation and collagen synthesis as well (Lee & Jang, 2018; Sziksz et al., 2015). IL-10 is also essential for the maintenance of epithelial layer integrity. Y. Jin demonstrated that the expression of fibronectin, α-SMA, fibroblast-specific protein-1, vimentin, and MMP-2 were increased in renal fibrosis of mice lacking IL-10 (Jin et al., 2013). However, few studies have directly looked into the role of IL-10 in EMT. In this study, we investigated the role of IL-10 in EMT and found that suppresses of IL-10 could cause decrease in E-cadherin and increased in N-cadherin, vimentin, and twist. Moreover, knockdown of IL-10 could recover radiation-induced EMT lessened by let-7i suppression. These results suggested that let-7i could mediate radiation-induced EMT via IL-10.
Although the mechanism of EMT is a complex network including multiple signaling pathways, such as TGF-β/Smad2/Smad3, PI3K/AKT and et al., are proven to regulate EMT process. An interesting finding in our study is that let-7i mediated EMT and RPF via PI3K/Akt signaling pathway. We found that inhibition of IL-10 and overexpression of let-7i were both associated with an increase of p-AKT. Moreover, let-7i and IL-10 inhibition together reversed let-7i inhibition–induced decreases in p-AKT levels. Previous studies have revealed that PI3K/Akt signaling pathway plays a critical role in EMT and organ fibrosis (Wanas et al., 2022; Chen et al., 2020). For example, microRNA-29b could prevent tubulointerstitial injury by attenuating renal tubular epithelial cell-mesenchymal transition and repressing PI3K/AKT signaling pathway (Hu et al., 2021). miRNA-34c-5p could target Fra-1 to inhibit pulmonary fibrosis induced by silica through p53 and chromosome 10/phosphatidylinositol-4,5-bisphosphate3-kinase/protein kinase B (PTEN/PI3K/AKT) signaling pathway (Pang et al., 2022).
To the best of our knowledge, this is the first report showed let-7i functions in pulmonary fibrosis in mice. We confirmed the important role of let-7i in regulating EMT of PI3K/AKT signaling pathway by targeting IL-10 expression in RIPF. In brief, these results suggest that let-7i contributes to EMT, thereby mediating pulmonary fibrosis. The results have significant implications in understanding the molecular mechanisms underlying the RIPF. Although our study results are not fully comprehensive, they may also provide research strategies for the molecular therapeutic target of RIPF to some extent. We hope that more research in clinical applications for RIPF patients should also be investigated further.
Data availability
The datasets analyzed during the current study are available from the corresponding author on reasonable request.
References
Andugulapati, S. B., Gourishetti, K., Tirunavalli, S. K., Shaikh, T. B., & Sistla, R. (2020). Biochanin-A ameliorates pulmonary fibrosis by suppressing the TGF-β mediated EMT, myofibroblasts differentiation and collagen deposition in in vitro and in vivo systems. Phytomedicine: International Journal of Phytotherapy and Phytopharmacology, 78, 153298. https://doi.org/10.1016/j.phymed.2020.153298
Carver, Joseph R., Shapiro, Charles L., Ng, Andrea, Jacobs, Linda, Schwartz, Cindy, Virgo, Katherine S., Hagerty, Karen L., Somerfield, Mark R., Vaughn, David J., ASCO Cancer Survivorship Expert Panel. (2007). American society of clinical oncology clinical evidence review on the ongoing care of adult cancer survivors: cardiac and pulmonary late effects. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology, 25(25), 3991–4008. https://doi.org/10.1200/JCO.2007.10.9777
Chang, K.-W., Zhang, X., Lin, S.-C., Lin, Y.-C., Li, C.-H., Akhrymuk, I., Lin, S.-H., & Lin, C.-C. (2021). Atractylodin suppresses TGF-β-mediated epithelial-mesenchymal transition in alveolar epithelial cells and attenuates bleomycin-induced pulmonary fibrosis in mice. International Journal of Molecular Sciences, 22(20), 11152. https://doi.org/10.3390/ijms222011152
Chen, H., Chen, Na., Li, F., Sun, L., Jicong, Du., Chen, Y., Cheng, F., et al. (2020). Repeated radon exposure induced lung injury and epithelial-mesenchymal transition through the PI3K/AKT/MTOR pathway in human bronchial epithelial cells and mice. Toxicology Letters, 334(November), 4–13. https://doi.org/10.1016/j.toxlet.2020.09.008
Chen, Z., Zhiqiang, Wu., & Ning, W. (2019). Advances in molecular mechanisms and treatment of radiation-induced pulmonary fibrosis. Translational Oncology, 12(1), 162–169. https://doi.org/10.1016/j.tranon.2018.09.009
Dongre, A., & Weinberg, R. A. (2019). New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nature Reviews. Molecular Cell Biology, 20(2), 69–84. https://doi.org/10.1038/s41580-018-0080-4
Gu, H., Shi, Si., Xiao, F., Huang, Z., Jun, Xu., Chen, G., Zhou, K., Lixia, Lu., & Yin, X. (2020). MiR-1-3p regulates the differentiation of mesenchymal stem cells to prevent osteoporosis by targeting secreted frizzled-related protein 1. Bone, 137(August), 115444. https://doi.org/10.1016/j.bone.2020.115444
Hewlett, J. C., Kropski, J. A., & Blackwell, T. S. (2018). Idiopathic pulmonary fibrosis: epithelial-mesenchymal interactions and emerging therapeutic targets. Matrix Biology: Journal of the International Society for Matrix Biology, 71–72(October), 112–127. https://doi.org/10.1016/j.matbio.2018.03.021
Hu, S., Hongtao, Hu., Wang, R., He, H., & Shui, H. (2021). MicroRNA-29b prevents renal fibrosis by attenuating renal tubular epithelial cell-mesenchymal transition through targeting the PI3K/AKT pathway. International Urology and Nephrology, 53(9), 1941–1950. https://doi.org/10.1007/s11255-021-02836-4
Huleihel, Luai, Ben-Yehudah, Ahmi, Milosevic, Jadranka, Guoying, Yu., Pandit, Kusum, Sakamoto, Koji, Yousef, Hanadie, et al. (2014). Let-7d MicroRNA affects mesenchymal phenotypic properties of lung fibroblasts. American Journal of Physiology. Lung Cellular and Molecular Physiology, 306(6), L534-542. https://doi.org/10.1152/ajplung.00149.2013
Jin, Yuanmeng, Liu, Ruijie, Xie, Jingyuan, Xiong, Huabao, He, John Cijiang, & Chen, Nan. (2013). Interleukin-10 deficiency aggravates kidney inflammation and fibrosis in the unilateral ureteral obstruction mouse model. Laboratory Investigation; a Journal of Technical Methods and Pathology, 93(7), 801–11. https://doi.org/10.1038/labinvest.2013.64
Kadota, T., Fujita, Yu., Araya, J., Watanabe, N., Fujimoto, S., Kawamoto, H., Minagawa, S., et al. (2021). Human bronchial epithelial cell-derived extracellular vesicle therapy for pulmonary fibrosis via inhibition of TGF-β-WNT crosstalk. Journal of Extracellular Vesicles, 10(10), e12124. https://doi.org/10.1002/jev2.12124
Kim, J.-S., Galvão, D. A., Newton, R. U., Gray, E., & Taaffe, D. R. (2021). Exercise-induced myokines and their effect on prostate cancer. Nature Reviews. Urology, 18(9), 519–542. https://doi.org/10.1038/s41585-021-00476-y
Kong, D., Zhang, Z., Chen, L., Huang, W., Zhang, F., Ling Wang, Yu., Wang, P. C., & Zheng, S. (2020). Curcumin blunts epithelial-mesenchymal transition of hepatocytes to alleviate hepatic fibrosis through regulating oxidative stress and autophagy. Redox Biology, 36(September), 101600. https://doi.org/10.1016/j.redox.2020.101600
Lee, H., Han, S., Kwon, C. S., & Lee, D. (2016). Biogenesis and regulation of the Let-7 MiRNAs and their functional implications. Protein & Cell, 7(2), 100–113. https://doi.org/10.1007/s13238-015-0212-y
Lee, H. J., & Jang, Y. J. (2018). Recent understandings of biology, prophylaxis and treatment strategies for hypertrophic scars and keloids. International Journal of Molecular Sciences, 19(3), E711. https://doi.org/10.3390/ijms19030711
Li, C.-H., & Liao, C.-C. (2021). The metabolism reprogramming of MicroRNA Let-7-mediated glycolysis contributes to autophagy and tumor progression. International Journal of Molecular Sciences, 23(1), 113. https://doi.org/10.3390/ijms23010113
Liang, H., Gu, Y., Li, T., Zhang, Y., Huangfu, L., Hu, M., Zhao, D., et al. (2014). Integrated analyses identify the involvement of MicroRNA-26a in epithelial-mesenchymal transition during idiopathic pulmonary fibrosis. Cell Death & Disease, 5(May), e1238. https://doi.org/10.1038/cddis.2014.207
Liang, X., Yan, Z., Wang, P., Liu, Y., Ao, X., Liu, Z., Wang, D., et al. (2021). Irradiation activates MZF1 to inhibit MiR-541-5p expression and promote epithelial-mesenchymal transition (EMT) in radiation-induced pulmonary fibrosis (RIPF) by upregulating slug. International Journal of Molecular Sciences, 22(21), 11309. https://doi.org/10.3390/ijms222111309
Liu, Z., Liang, X., Li, X., Liu, X., Zhu, M., Yongqing, Gu., & Zhou, P. (2019). MiRNA-21 functions in ionizing radiation-induced epithelium-to-mesenchymal transition (EMT) by downregulating PTEN. Toxicology Research, 8(3), 328–340. https://doi.org/10.1039/c9tx00019d
Lu, T. X., & Rothenberg, M. E. (2018). MicroRNA. The Journal of Allergy and Clinical Immunology, 141(4), 1202–1207. https://doi.org/10.1016/j.jaci.2017.08.034
Mittal, V. (2018). Epithelial Mesenchymal Transition in Tumor Metastasis. Annual Review of Pathology, 13(January), 395–412. https://doi.org/10.1146/annurev-pathol-020117-043854
Nakagome, K., Dohi, M., Okunishi, K., Tanaka, R., Miyazaki, J., & Yamamoto, K. (2006). In vivo IL-10 gene delivery attenuates bleomycin induced pulmonary fibrosis by inhibiting the production and activation of TGF-beta in the lung. Thorax, 61(10), 886–894. https://doi.org/10.1136/thx.2005.056317
Nieto, M. A., Huang, R.-J., Jackson, R. A., & Thiery, J. P. (2016). EMT: 2016. Cell, 166(1), 21–45. https://doi.org/10.1016/j.cell.2016.06.028
Pandolfini, L., Barbieri, I., Bannister, A. J., Hendrick, A., Andrews, B., Webster, N., Murat, P., et al. (2019). METTL1 promotes Let-7 MicroRNA processing via M7G methylation. Molecular Cell, 74(6), 1278-1290.e9. https://doi.org/10.1016/j.molcel.2019.03.040
Pang, X., Shi, H., Chen, X., Li, C., Shi, B., Yeo, A. J., Lavin, M. F., et al. (2022). MiRNA-34c-5p targets Fra-1 to inhibit pulmonary fibrosis induced by silica through P53 and PTEN/PI3K/Akt signaling pathway. Environmental Toxicology, 37(8), 2019–2032. https://doi.org/10.1002/tox.23547
Phan, T. H., Giang, P. P., Nasrallah, G. K., Giordo, R., Eid, A. H., Fois, A. G., Zinellu, A., Mangoni, A. A., & Pintus, G. (2021). Emerging cellular and molecular determinants of idiopathic pulmonary fibrosis. Cellular and Molecular Life Sciences: CMLS, 78(5), 2031–2057. https://doi.org/10.1007/s00018-020-03693-7
Roush, S., & Slack, F. J. (2008). The Let-7 family of MicroRNAs. Trends in Cell Biology, 18(10), 505–516. https://doi.org/10.1016/j.tcb.2008.07.007
Shorning, B. Y., Dass, M. S., Smalley, M. J., & Pearson, H. B. (2020). The PI3K-AKT-MTOR pathway and prostate cancer: at the crossroads of AR, MAPK, and WNT signaling. International Journal of Molecular Sciences, 21(12), E4507. https://doi.org/10.3390/ijms21124507
Su, J., Morgani, S. M., David, C. J., Wang, Q., Er, E. E., Huang, Y.-H., Basnet, H., et al. (2020). TGF-β orchestrates fibrogenic and developmental EMTs via the RAS effector RREB1. Nature, 577(7791), 566–571. https://doi.org/10.1038/s41586-019-1897-5
Sziksz, E., Pap, D., Lippai, R., Béres, N. J., Fekete, A., Szabó, A. J., & Vannay, Á. (2015). Fibrosis related inflammatory mediators: role of the il-10 cytokine family. Mediators of Inflammation, 2015, 764641. https://doi.org/10.1155/2015/764641
Thiery, J. P., Acloque, H., Huang, R. Y. J., & Angela Nieto, M. (2009). Epithelial-mesenchymal transitions in development and disease. Cell, 139(5), 871–890. https://doi.org/10.1016/j.cell.2009.11.007
Turrisi, A. T., Kim, K., Blum, R., Sause, W. T., Livingston, R. B., Komaki, R., Wagner, H., Aisner, S., & Johnson, D. H. (1999). Twice-daily compared with once-daily thoracic radiotherapy in limited small-cell lung cancer treated concurrently with cisplatin and etoposide. The New England Journal of Medicine, 340(4), 265–271. https://doi.org/10.1056/NEJM199901283400403
Wanas, H., El Shereef, Z., Rashed, L., & Aboulhoda, B. E. (2022). Ticagrelor ameliorates bleomycin-induced pulmonary fibrosis in rats by the inhibition of TGF-Β1/Smad3 and PI3K/AKT/MTOR pathways. Current Molecular Pharmacology, 15(1), 227–238. https://doi.org/10.2174/1874467214666210204212533
Wang, D., Liu, Z., Yan, Z., Liang, X., Liu, X., Liu, Y., Wang, P., Bai, C., Yongqing, Gu., & Zhou, P.-K. (2021). MiRNA-155-5p inhibits epithelium-to-mesenchymal transition (EMT) by targeting GSK-3β during radiation-induced pulmonary fibrosis. Archives of Biochemistry and Biophysics, 697(January), 108699. https://doi.org/10.1016/j.abb.2020.108699
Wang, H., Tan, Z., Hui, Hu., Liu, H., Tangwei, Wu., Zheng, C., Wang, X., et al. (2019). MicroRNA-21 promotes breast cancer proliferation and metastasis by targeting LZTFL1. BMC Cancer, 19(1), 738. https://doi.org/10.1186/s12885-019-5951-3
Wang, X., Wang, Hong-Xia., Li, Yu-Lin., Zhang, Cong-Cong., Zhou, Chun-Yu., Wang, Lei, Xia, Yun-Long., Jie, Du., & Li, Hui-Hua. (2015). MicroRNA Let-7i negatively regulates cardiac inflammation and fibrosis. Hypertension (Dallas, Tex.: 1979), 66(4), 776–85. https://doi.org/10.1161/HYPERTENSIONAHA.115.05548
Xie, L., Zhou, J., Zhang, S., Chen, Q., Lai, R., Ding, W., Song, ChuanJun, Meng, XingJun, & Jinchang, Wu. (2014). Integrating MicroRNA and MRNA expression profiles in response to radiation-induced injury in rat lung. Radiation Oncology (london, England), 9(May), 111. https://doi.org/10.1186/1748-717X-9-111
Xiong, S., Pan, X., Long, Xu., Yang, Z., Guo, R., Yongqing, Gu., Li, R., et al. (2015). Regulatory T cells promote β-catenin–mediated epithelium-to-mesenchyme transition during radiation-induced pulmonary fibrosis. International Journal of Radiation Oncology, Biology, Physics, 93(2), 425–435. https://doi.org/10.1016/j.ijrobp.2015.05.043
Xu, W., Yang, Z., & Nonghua, Lu. (2015). A new role for the PI3K/Akt signaling pathway in the epithelial-mesenchymal transition. Cell Adhesion & Migration, 9(4), 317–324. https://doi.org/10.1080/19336918.2015.1016686
Xu, Z., Jia, K., Wang, H., Gao, F., Zhao, S., Li, F., & Hao, J. (2021). METTL14-Regulated PI3K/Akt signaling pathway via PTEN affects HDAC5-mediated epithelial-mesenchymal transition of renal tubular cells in diabetic kidney disease. Cell Death & Disease, 12(1), 32. https://doi.org/10.1038/s41419-020-03312-0
Yan, W., Qiuyun, Wu., Yao, W., Li, Y., Liu, Yi., Yuan, J., Han, R., Yang, J., Ji, X., & Ni, C. (2017). MiR-503 modulates epithelial-mesenchymal transition in silica-induced pulmonary fibrosis by targeting PI3K P85 and is sponged by LncRNA MALAT1. Scientific Reports, 7(1), 11313. https://doi.org/10.1038/s41598-017-11904-8
Yan, Z., Ao, X., Liang, X., Chen, Z., Liu, Y., Wang, P., Wang, D., et al. (2022). Transcriptional inhibition of MiR-486-3p by BCL6 upregulates snail and induces epithelial-mesenchymal transition during radiation-induced pulmonary fibrosis. Respiratory Research, 23(1), 104. https://doi.org/10.1186/s12931-022-02024-7
Yazarlou, Fatemeh, Kadkhoda, Sepideh, & Ghafouri-Fard, Soudeh. (2021). Emerging role of Let-7 family in the pathogenesis of hematological malignancies. Biomedicine & Pharmacotherapy = Biomedecine & Pharmacotherapie., 14, 112334. https://doi.org/10.1016/j.biopha.2021.112334
Yeon, L. S., Jeong, E. K., Min Kyung, Ju., Jeon, H. M., Kim, M. Y., Kim, C. H., Park, H. G., Han, S. I., & Kang, H. S. (2017). Induction of metastasis, cancer stem cell phenotype, and oncogenic metabolism in cancer cells by ionizing radiation. Molecular Cancer, 16(1), 10. https://doi.org/10.1186/s12943-016-0577-4
Zhang, C., Zhu, X., Hua, Y., Zhao, Q., Wang, K., Zhen, L., Wang, G., et al. (2019). YY1 mediates TGF-Β1-induced EMT and pro-Fibrogenesis in alveolar epithelial cells. Respiratory Research, 20(1), 249. https://doi.org/10.1186/s12931-019-1223-7
Zhou, W., Ye, S., & Wang, W. (2021). MiR-217 alleviates high-glucose-induced vascular smooth muscle cell dysfunction via regulating ROCK1. Journal of Biochemical and Molecular Toxicology, 35(3), e22668. https://doi.org/10.1002/jbt.22668
Zhuang, Yi., Dai, J., Wang, Y., Zhang, H., Li, X., Wang, C., Cao, M., et al. (2016). MiR-338* targeting smoothened to inhibit pulmonary fibrosis by epithelial-mesenchymal transition. American Journal of Translational Research, 8(7), 3206–3213.
Acknowledgements
We are grateful to Ping Ge and Xiaodan Liu for their laboratory assistance.
Funding
This research was funded by grants from the National Natural Science Foundation of China (82073488, 81773359, 81472910 and 31470827).
Author information
Shenghui Zhou and Xin Liang have contributed equally to this work.
Authors and Affiliations
Hengyang Medical College, University of South China, Hengyang, 421001, Hunan, China
Shenghui Zhou & Yongqing Gu
Beijing Key Laboratory for Radiobiology, Beijing Institute of Radiation Medicine, AMMS, Beijing, 100850, China
Shenghui Zhou, Xin Liang, Zewen Sun, Xueping Li, Jiaojiao Zhu, Zhihua Yang, Xiujie Pan, Yilong Wang, Yongqing Gu & Maoxiang Zhu
School of Public Health, Tianjin Medical University, Tianjin, 300070, China
Xin Liang
School of Life Science, Shihezi University, Shihezi, 832003, Xinjiang, China
Xueping Li & Yongqing Gu
Contributions
SZ and XL: drafted the manuscript, analyzed the data and interpreted the data, conducted experiments; ZS, XL and JZ: analyzed the data and interpreted the data, conducted some experiments; ZY, XP and YW: analyzed the data and interpreted the data, helped with some experiments; YG: conceived and designed the study; revised the manuscript; project administration; MZ: revised the manuscript critically for important intellectual content; project administration.
Corresponding authors
Correspondence to Yongqing Gu or Maoxiang Zhu.
Ethics declarations
Conflict of interest
The authors declare non-financial interests that are directly or indirectly related to the work submitted for publication.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article
Cite this article
Zhou, S., Liang, X., Sun, Z. et al. MiRNA let-7i promotes radiation-induced pulmonary epithelial-mesenchymal transition by targeting IL-10. GENOME INSTAB. DIS. 3, 271–284 (2022). https://doi.org/10.1007/s42764-022-00089-8
Received27 August 2022
Revised09 October 2022
Accepted12 October 2022
Published19 October 2022
Issue DateDecember 2022
DOIhttps://doi.org/10.1007/s42764-022-00089-8
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
MiRNA-let-7i
Alveolar type II epithelial cells
Onizing radiation
Epithelial to mesenchymal transition
Radiation-induced pulmonary fibrosis
用户登录
还没有账号?
立即注册