






PRMT1 and PRMT5: on the road of homologous recombination and non-homologous end joining
Review Article
Genome Instability & Disease (2022)
Abstract
DNA double-strand breaks (DSBs) are widely accepted to be the most deleterious form of DNA lesions that pose a severe threat to genome integrity. Two predominant pathways are responsible for repair of DSBs, homologous recombination (HR) and non-homologous end-joining (NHEJ). HR relies on a template to faithfully repair breaks, while NHEJ is a template-independent and error-prone repair mechanism. Multiple layers of regulation have been documented to dictate the balance between HR and NHEJ, such as cell cycle and post-translational modifications (PTMs). Arginine methylation is one of the most common PTMs, which is catalyzed by protein arginine methyltransferases (PRMTs). PRMT1 and PRMT5 are the predominate PRMTs that promote asymmetric dimethylarginine and symmetric dimethylarginine, respectively. They have emerged to be crucial regulators of DNA damage repair. In this review, we summarize current understanding and unaddressed questions of PRMT1 and PRMT5 in regulation of HR and NHEJ, providing insights into their roles in DSB repair pathway choice and the potential of targeting them for cancer therapy.
Introduction
DNA double-strand breaks (DSBs) are the most cytotoxic and complex DNA lesions, which are potentially induced by a range of exogenous and endogenous sources, such as chemotoxic agents, γ-irradiation, metabolic alterations, and transcription-replication collisions (Mehta & Haber, 2014). Improper repair of DSBs results in genomic instability, a hallmark of many human diseases, particularly cancer (Jackson & Bartek, 2009; Khanna & Jackson, 2001). To maintain genome integrity in response to various DNA damage challenges, cells have evolved multiple mechanisms to repair DSBs, with homologous recombination (HR) and non-homologous end-joining (NHEJ) being the two major repair pathways (Haber, 2000; Harper & Elledge, 2007; Lieber, 2008; San Filippo et al., 2008). Multiple mechanisms have been reported to modulate DSB repair by HR and NHEJ pathways (Scully et al., 2019; Shrivastav et al., 2008), including posttranslational modifications (PTMs) (Huen & Chen, 2008; Polo & Jackson, 2011).
HR
HR is commonly considered as an error-free repair mechanism because it copies a homologous sequence from a template (San Filippo et al., 2008; Wright et al., 2018). HR is a multiple-steps process (Fig. 1, left). It is initiated by 5’-3’ end resection, a step executed by the MRE11-Rad50-NBS1 (MRN) complex and CtIP (Takeda et al., 2007). MRN complex has multiple roles in this process: acting as the DSB sensor to recognize and bind broken DNA ends (Carney et al., 1998; Usui et al., 1998), serving as a platform to recruit CtIP and ataxia-telangiectasia-mutant (ATM) protein kinase to the damage sites (Lee & Paull, 2005; Wang et al., 2013), releasing Ku from DNA ends (Foster et al., 2011; Jensen & Russell, 2016; Langerak et al., 2011; Mimitou & Symington, 2010), and generating a short single-stranded DNA (ssDNA) (Buis et al., 2008; Hopkins & Paull, 2008; Trujillo et al., 1998). CtIP promotes the endonuclease activity of the MRN complex to facilitate the initiation of end resection (Sartori et al., 2007). The short ssDNA is then extended by exonuclease 1 (EXO1), DNA replication helicase/nuclease 2 (DNA2), and Bloom syndrome protein (BLM) (Cannavo et al., 2013; Cejka et al., 2010; Gravel et al., 2008; Nimonkar et al., 2011; Niu et al., 2010; Shim et al., 2010). Upon exposure, ssDNA is immediately bound by replication protein A (RPA), which is then replaced by RAD51 to form a nucleofilament for homology search and strand invasion (Sigurdsson et al., 2001; Sugiyama & Kowalczykowski, 2002; Sung, 1997; Wang & Haber, 2004; Wold, 1997). During the HR process, many mediators and effectors have also been identified and play critical roles (Sung & Klein, 2006). Notably, BRCA1 is a versatile mediator in HR: displacing p53 binding protein 1 (53BP1) to allow resection (Bouwman et al., 2010; Bunting et al., 2010), interacting with CtIP to promote resection (Wang et al., 2000; Zhong et al., 1999), forming a complex with PALB2 and BRCA2 to replace RPA with RAD51 for filament formation (Prakash et al., 2015), and binding BARD1 to enhance the recombinase activity of RAD51 (Zhao et al., 2017).
Fig. 1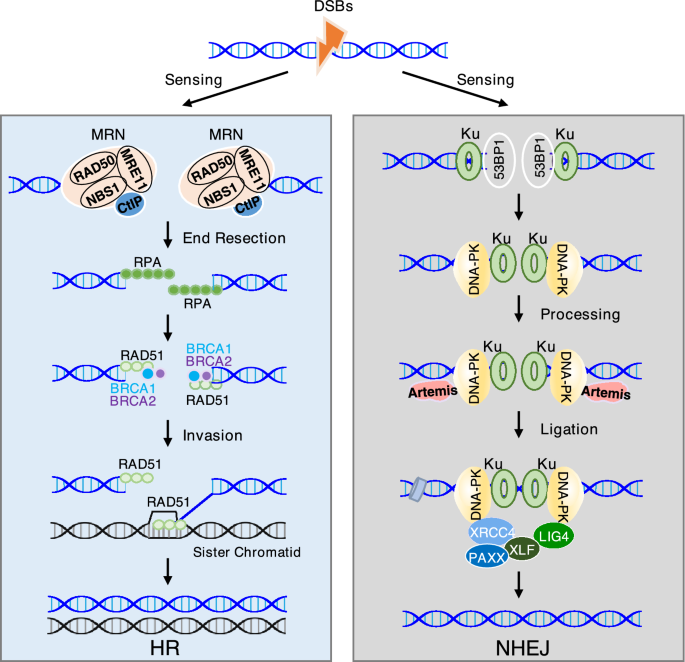
The process and major components of HR and NHEJ. (Left) HR is initiated by sensing the DNA double-strand breaks (DSBs) via the MRN complex, followed by DNA end resection and loading of repair factors, including CtIP, RPA, RAD51, BRCA1/2, and others to complete the DSB repair. (Right) DSBs also can be recognized by KU complex, which serves as a platform to recruit 53BP1, DNA-PK, and ligation complex (XRCC4, XLF, PAXX, and LIG4) for NHEJ repair
Full size imageNHEJ
NHEJ is widely accepted as an error-prone repair pathway, which directly rejoins DNA ends throughout the cell cycle with minimal resection and independent of a template (Ciccia & Elledge, 2010; Mahaney et al., 2009; Scully et al., 2019). This pathway consists of four major components, including the DNA protein kinase (DNA-PK) complex, nucleases, DNA polymerases, and the DNA ligase complex (Fig. 1, right). The DNA-PK complex is composed of KU70, KU80, and DNA-PK catalytic subunit (DNA-PKcs) (Jin & Weaver, 1997; Singleton et al., 1999; Spagnolo et al., 2006). KU70 and KU80 form a heterodimer (KU) to recognize and bind the DSB ends, serving as a toolbelt for docking of other repair proteins (Featherstone & Jackson, 1999; Mari et al., 2006; Uematsu et al., 2007). The KU heterodimer recruits DNA-PKcs to the DNA ends and subsequently activates downstream repair signaling (Gottlieb & Jackson, 1993; Uematsu et al., 2007). Prior to ligation, the broken DNA ends generally requires processing by nucleases and DNA polymerases. DNA-PKcs phosphorylates and activates the Artemis nuclease to resect incompatible DNA ends (Goodarzi et al., 2006). Another nuclease Aprataxin and PNKP-like factor (APLF) is a 3′ exonuclease and contributes to the DNA-end configuration (Kanno et al., 2007). KU also interacts with two DNA polymerases, Pol μ and Pol λ, to modify the DNA ends (Lee et al., 2004; Ma et al., 2004; Mahajan et al., 2002). Finally, the processed DNA ends are ligated by the DNA ligase complex that consists of DNA ligase IV (LIG4), X-ray repair cross-complementing protein 4 (XRCC4) (Costantini et al., 2007; Mari et al., 2006; Nick McElhinny et al., 2000), XRCC4-like factor (XLF) (Yano et al., 2008), and paralogue of XRCC4 and XLF (PAXX) (Ochi et al., 2015; Xing et al., 2015). In addition to the core components, 53BP1 is a key determinant of NHEJ (Mirman & de Lange, 2020; Panier & Boulton, 2014). It interacts with Rap1-interacting factor 1 (RIF1) and Pax transactivation domain-interacting protein (PTIP) to antagonize DNA end resection (Callen et al., 2013; Chapman et al., 2013; Zimmermann et al., 2013). Its recruitment to broken DNA ends also increases the local chromatin mobility for synapsis of DNA breaks (Dimitrova et al., 2008; Lottersberger et al., 2015), thereby promoting NHEJ and suppressing HR.
Arginine methylation
Protein arginine methylation is one of common posttranslational modifications. It is carried out by protein arginine methyltransferases (PRMTs), which transfer a methyl group from S-adenosyl methionine (SAM) to the nitrogen of arginine residues (Tewary et al., 2019). In mammals, nine known members of the PRMT family can be categorized into three types based on the structurally defined formations of methylated arginine as follows: Type I (PRMT1, PRMT2, PRMT3, PRMT4, PRMT6, and PRMT8) and Type II PRMTs (PRMT5 and PRMT9) catalyze the formation of ω-NG-monomethyl arginine (MMA) and then further catalyze the formation of ω-NG,NG-asymmetric dimethyl arginine (ADMA) and ω-NG,N′G-symmetric dimethyl arginine (SDMA), respectively, while PRMT7 is the sole member of Type III enzyme responsible for generation of MMA only (Fig. 2) (Bedford & Clarke, 2009; Blanc & Richard, 2017). PRMT1 is the predominate type I methyltransferase responsible for as much as 85% of total methylated arginine residues (Tang et al., 2000) and PRMT5 is the main type II methyltransferase (Karkhanis et al., 2011). PRMTs commonly target the RGG/RG motifs (arginine and glycine rich motifs) for methylation (Thandapani et al., 2013). However, CARM1/PRMT4 has a preference for arginine in PGM-rich motif (proline, glycine, and methionine rich motif) (Cheng et al., 2007; Shishkova et al., 2017), while PRMT7 preferentially methylates RXR motifs surrounded by multiple lysines (Feng et al., 2013). PRMT1 and PRMT5 are frequently overexpressed in various human cancers, such as breast cancer, colorectal cancer, lung cancer, and leukemia (Yang & Bedford, 2013). Moreover, their high expression is correlated with poor prognosis and survival of cancer patients (Lattouf et al., 2019; Mathioudaki et al., 2008; Powers et al., 2011). Thus, both PRMT1 and PRMT5 are potential therapeutic targets. Multiple inhibitors of PRMT1/5 have been developed and several PRMT5 inhibitors are currently being evaluated in clinical trials (Hwang et al., 2021; Jarrold & Davies, 2019; Li et al., 2019).
Fig. 2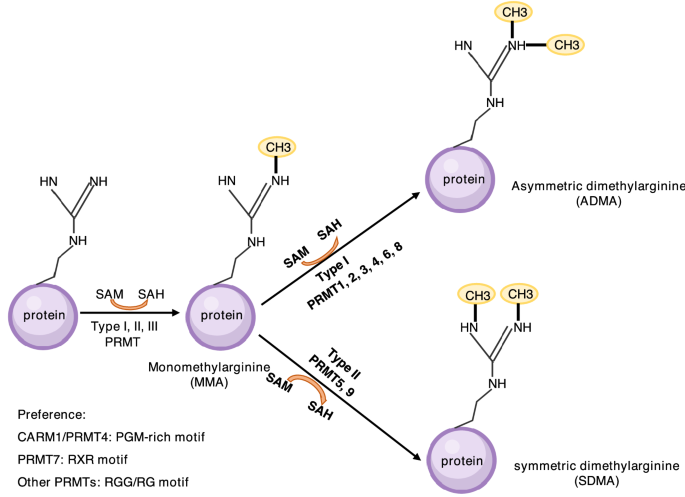
Arginine methylation. There are three types of methylarginine, including ADMA, SDMA, and MMA, which are catalyzed by PRMTs of type I (PRMT1, 2, 3, 4, 6, 8), type II (PRMT5, 9), and type III (PRMT7), respectively. RGG/RG motif is favored by most of PRMTs. CARM1/PRMT4 and PRMT7 prefer to methylate PGM-rich motif and RXR motif, respectively
Full size imageNumerous substrates of PRMT1 and PRMT5, including histones and non-histone substrates, have been identified (Bedford, 2007; Kim & Ronai, 2020; Thiebaut et al., 2021), through which they play crucial roles in many fundamental cellular processes, including transcription (Lee & Stallcup, 2009; Lee et al., 2005), RNA splicing and translation (Boisvert et al., 2002; Fong et al., 2019; Li et al., 2021; Lorton & Shechter, 2019), cell signaling (Albrecht et al., 2018; Feng et al., 2019; Geoghegan et al., 2015; Liao et al., 2015; Yin et al., 2021), and DNA repair (Auclair & Richard, 2013; Brobbey et al., 2022). This review summarizes current understanding of PRMT1 and PRMT5 in regulation of HR and NHEJ and discusses unaddressed questions, providing insights into their roles in DSB repair pathway choice.
Loss of PRMT1 and PRMT5 causes genomic instability
PRMT1 is required for cell proliferation, survival, and tumor growth (Giuliani et al., 2021; Hsu et al., 2017; Hua et al., 2020; Yamagata et al., 2008). PRMT1 knockout in mice is embryonic lethality. Loss of PRMT1 in mouse embryonic fibroblasts (MEFs) leads to several defects, including spontaneous DNA damage, chromosomal rearrangements, cell cycle arrest, and growth inhibition (Yu et al., 2009), suggesting a critical role of PRMT1 in maintenance of genome stability. PRMT1-deficient cells are hypersensitive to DNA damaging agents, such as etoposide, cisplatin, 5-fluorouracil and temozolomide (He et al., 2020; Musiani et al., 2020; Yu et al., 2009).
PRMT5 also plays a critical role in cell growth and survival (Chiang et al., 2017; Jing et al., 2018; Sheng & Wang, 2016; Zencheck et al., 2012), knockout of which abolishes early mouse development and leads to embryonic lethality (Tee et al., 2010). Loss of PRMT5 triggers DNA damage response (DDR) and elevates transposable elements in primordial germ cells (PGCs) (Kim et al., 2014). In contrast, depletion of PRMT5 in glioblastoma cells promotes the degradation of γH2AX by SMURF2 and consequently impairs DDR (Du et al., 2019). Thus, PRMT5 may employ different mechanisms to protect genome integrity in normal and cancer cells. Like PRMT1, PRMT5 inhibition sensitizes cancer cells to DNA damaging agents, including etoposide, ionizing irradiation, hydroxyurea, and camptothecin (He et al., 2011; Hu et al., 2015; Owens et al., 2020; Rehman et al., 2018).
Two major mechanisms have been documented by which PRMT1 and PRMT5 regulate DNA repair. They directly methylate DNA repair proteins to control their stability, enzymatic activity, and recruitment to DNA damage sites (Auclair & Richard, 2013; Brobbey et al., 2022). PRMT1 and PRMT5 also functions as epigenetic activators to promote transcription of DNA repair regulators (Giuliani et al., 2021; Hamard et al., 2018; Owens et al., 2020; Tan et al., 2019).
PRMT1 and PRMT5 modulate HR and NHEJ pathway choice
PRMT1 promotes HR by methylating MRE11, BRCA1, and hnRNPUL1
MRE11, the key component of the MRN complex, acts as a primary sensor to recognize broken DNA ends and initiate resection for the HR (Stracker & Petrini, 2011). MRE11 contains a conserved glycine-arginine-rich (GAR) motif, which is often a target for PRMTs (Najbauer et al., 1993). Indeed, MRE11 is methylated at the GAR motif by PRMT1. This methylation is necessary for its 3′ to 5′ exonuclease activity and recruitment to DNA damage sites but has no impact on its interaction with RAD50 and NBS1 to form MRN complex (Boisvert et al., 2005a). Mutating the arginine residues in the GAR motif to lysine residues (MRE11RK/RK) impairs the recruitment of RPA and RAD51 to the DNA damage sites and ATR activation. As a result, mice harboring the MRE11RK/RK mutation exhibits chromosome instability and are hypersensitive to γ-irradiation (Yu et al., 2012). These studies demonstrate that PRMT1-mediated methylation of MRE11 is critical for DNA end resection (Fig. 3a). Future studies are needed to demonstrate how this methylation event promotes MRE11 activity.
Fig. 3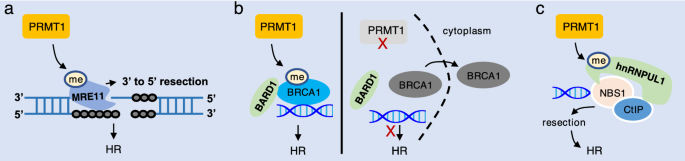
PRMT1 promotes HR by methylating HR proteins. a PRMT1 methylates MRE11 to promote its exonuclease activity for resection, thereby promoting HR. b PRMT1 methylates BRCA1 to enhance its interaction with BARD1 and DNA for HR repair. Deficiency in PRMT1 disrupts these interactions and translocates BRCA1 to cytoplasm. c PRMT1 catalyzes methylation of hnRNPIL1 to promote resection for HR
Full size imageBRCA1 is a well-known tumor suppressor, which is frequently mutated in hereditary breast and ovarian cancers (Mersch et al., 2015). BRCA1 is a key player of HR pathway by promoting DNA end resection and homologous template search and pairing (Prakash et al., 2015). Several PTMs have been identified for BRCA1 in response to DNA damage, including phosphorylation (Ouchi, 2006), ubiquitylation (Choudhury et al., 2004), SUMOylation (Morris et al., 2009), and arginine methylation (Guendel et al., 2010; Montenegro et al., 2020). PRMT1 associates with and methylates BRCA1 at the fragment of 504–802 amino acids with R610 being the predicted methylation site. Arginine methylation of BRCA1 alerts its DNA binding and interaction with its partners under normal condition (Guendel et al., 2010). Later study showed that in response to irradiation, the S-adenosylmethionine (SAM) levels are increased, which subsequently activates PRMT1 to promote BRCA1 arginine methylation. Depletion of PRMT1 promotes BRCA1 translocation from nucleus to cytoplasm and prevents BRCA1 and BARD1 interaction and foci formation after irradiation, leading to impairment of DSB repair by HR (Fig. 3b) (Montenegro et al., 2020). However, several following questions still need to be addressed: What are the methylated residues? Whether arginine methylation of BRCA1 affects its interaction with other partners, such as CtIP, PALB2, and BRCA2, in response irradiation and other DNA damaging agents? Whether arginine methylation interplays with other PTMs to control BRCA1 localization and function in HR repair? How arginine methylation cooperates with BRCA1 mutations to promote cancer progression?
hnRNPUL1 is an RNA binding protein that plays a critical in RNA metabolism (Geuens et al., 2016). Interestingly, hnRNPUL1 interacts with NBS1 and CtIP and accumulates on the DNA damage sites to promote DSB resection and HR (Polo et al., 2012). PRMT1 methylates hnRNPUL1 at R612, R618, R620, R639, R645, R656, and R661 within an RGG/RG motif, which is required for its ability to interaction with NBS1 and recruitment to DNA damage sites (Gurunathan et al., 2015). These findings suggest that PRMT1 may promote HR by methylating hnRNPUL1 (Fig. 3c). It will be interesting to know whether PRMT1-mediated methylation of hnRNPUL1 regulates its functions on RNA metabolism and consequently controls the expression of DNA repair genes.
PRMT5 promotes HR by methylating KLF4
Kruppel-like factor 4 (KLF4) is a multifunctional transcription factor involved in stem cell renewal (Jiang et al., 2008), cell cycle control (Chen et al., 2003), and genome stability regulation (El-Karim et al., 2013). PRMT5 was identified as a KLF4 interacting protein by mass spectrometric analyses and methylated KLF4 at three arginine residues (R374, R376, and R378), which was enhanced by irradiation. This methylation event stabilizes KLF4 by preventing its ubiquitination and degradation by E3 ligase VHL. Mutating the three arginine residues to lysine residues (KLF43K) increased foci formation of 53BP1 at DNA damage sites (Fig. 4), suggesting a defect in HR and enhanced NHEJ. As a result, chromosomal instability was significantly increased in KLF43K expressing cells upon irradiation (Hu et al., 2015). Further study showed that either knockdown of PRMT5 or KLF43K mutant impairs DNA resection and BRCA1 foci formation, thereby suppressing HR pathway (Checa-Rodriguez et al., 2020). Therefore, PRMT5-mediated methylation of KLF4 facilitates DSB repair by HR. Future studies are warranted to elucidate the mechanisms by which KLF4 arginine methylation affect DNA resection. Does it regulate MRN complex recruitment and activity? Whether it interplays with PRMT1-mediated methylation of MRE11?
Fig. 4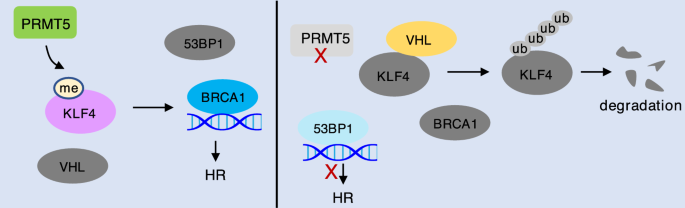
PRMT5 promotes HR by methylating KLF4. PRMT5 mediated arginine methylation of KLF4 impairs KLF4 interaction with VHL and consequently stabilizes KLF4 to promote DNA resection and BRCA1 recruitment for HR repair
Full size imagePRMT1 and PRMT5 regulate NHEJ by methylating 53BP1
53BP1, a protein interacting with tumor suppressor p53 (Iwabuchi et al., 1994), is an early participant translocating to DNA damage sites and plays a determinant role in HR and NHEJ pathway choice (Mirman & de Lange, 2020; Panier & Boulton, 2014; Schultz et al., 2000). Binding of 53BP1 at the DSBs protects DNA ends from resection to limit ssDNA formation, consequently promoting NHEJ (Bunting et al., 2010; Chapman et al., 2013; Paiano et al., 2021). 53BP1 contains a GAR motif, which is in the region responsible for 53BP1 binding to ssDNA and double-stranded DNA (dsDNA) (Iwabuchi et al., 2003). Interestingly, the GAR motif of 53BP1 is methylated by both PRMT1 and PRMT5 (Fig. 5) (Boisvert et al., 2005b; Hwang et al., 2020). PRMT1 catalyzes ADMA on GAR motif at three major arginine residues, R1400, R1401, and R1403. Mutation of these arginine residues markedly reduced 53BP1 binding to both ssDNA and dsDNA in vitro. However, PRMT1-mediated GAR methylation is neither enhanced nor dispensable for 53BP1 oligomerization and foci formation in response to DNA damage by treatment with etoposide (Adams et al., 2005; Boisvert et al., 2005b). A recent study showed that PRMT5 competes with PRMT1 to symmetrically dimethylate the GAR motif of 53BP1 at five arginine residues, including R1396, R1398, R1400, R1401, and R1403. Mutating these arginine residues or knockdown of PRMT5 destabilizes 53BP1 protein and decreases the intensity of 53BP1 foci upon treatment of etoposide. However, it is unknown whether the SDMA of 53BP1 is regulated by DNA damage. As both the GAR-containing DNA binding domain and the Tudor domain (methylation reader) are necessary and sufficient for 53BP1 foci formation, it will be critical to investigate whether arginine methylation of 53BP1 by PRMT1 and PRMT5 interplays with the Tudor domain to control 53BP1 loading on DSBs for regulation of NHEJ.
Fig. 5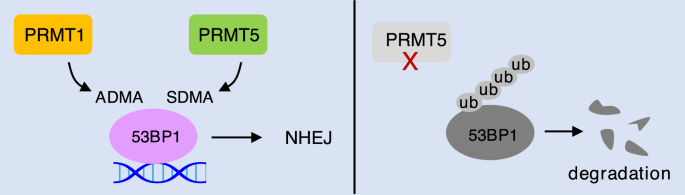
PRMT1 and PRMT5 compete for 53BP1 methylation to regulate NHEJ. The GAR motif of 53BP1 can be methylated by both PRMT1 and PRMT5. Deficiency in PRMT5 leads to 53BP1 degradation
Full size imagePRMT1 and PRMT5 epigenetically regulate HR and NHEJ
A recent study found that in response to replication stress by treatment of cisplatin, DNA-PK interacts with PRMT1 to mediate PRMT1 recruitment to chromatin, leading to increase of H4R3 methylation (H4R3me2a) and induction of the senescence-associated secretory phenotype (SASP)-transcriptional program in ovarian cancer cells (Fig. 6a). However, analysis of Gene Ontology (GO) terms for the genes downregulated by cisplatin revealed no enrichment of any categories (Musiani et al., 2020). Thus, it is unknown whether recruitment of PRMT1 to chromatin plays a role in HR and NHEJ. Another study found that pharmacological PRMT1 inhibition in pancreatic cancer cells significantly downregulates pathways involved in DNA repair and HR pathway. Both mRNA and protein levels of several key HR genes, including BRCA1, BRCA2, and RAD51, are markedly decreased upon PRMT1 inhibition (Fig. 6b) (Giuliani et al., 2021). Moreover, PRMT1 promotes methylation of plakophilin 2 (PKP2) at R101 to enhance its interaction with β-catenin, leading to stabilization of β-catenin to increase the expression of LIG4 in lung cancer cells (Fig. 6c). Overexpression of PKP2 wildtype, but not the R101A or R101K, restores LIG4 expression and radioresistance in PKP2-depleted cells, indicating that arginine methylation of PKP2 is involved NHEJ-mediated DNA repair (Cheng et al., 2021).
Fig. 6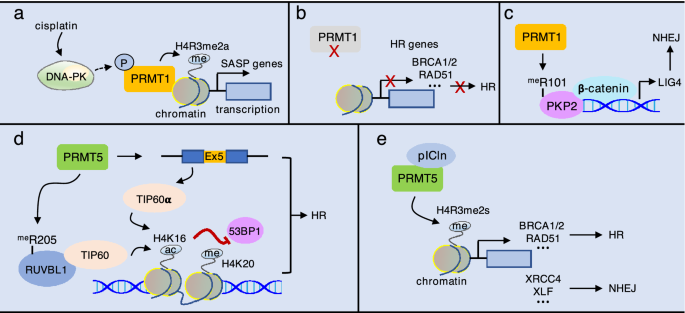
Epigenetic role of PRMT1 and PRMT5 in HR and NHEJ. a In response to cisplatin treatment, DNA-PK likely phosphorylates PRMT1 to recruit PRMT1 to chromatin for H4R3 methylation (H4R3me2a), which promotes expression of SASP gens. b Inhibition of PRMT1 suppresses the expression of HR gens, including BRCA1/2, RAD51, and others. c PRMT1-mediated methylation of PKP2 at R101 enhances its interaction with β-catenin, leading to increase of LIG4 expression. d PRMT5 methylates the activator RUVBL1 at R205 and promotes TIP60α expression to increase acetylation of H4K16, which antagonizes 53BP1 binding to methylated H4K20. e PRMT5 interacts with pICln to promote H4R3me2s and the expression of both HR and NHEJ genes
Full size imagePRMT5 also regulates DSB repair choice through epigenetic mechanisms. Using a approach of immunoprecipitation in combination with mass spectrometry, the AAA+ ATPase RUVBL1 was identified as a PRMT5-interacting protein, which is a component of TIP60, SWR1, and INO80 complexes involved in DNA repair (Dong et al., 2014; Ikura et al., 2000; Jha & Dutta, 2009; Wu et al., 2007). This interaction led to methylation of RUVB1 at R205, deficiency in which increases 53BP1 foci and conversely decreases RPA and RAD51 foci in response to irradiation. Mechanically, the methyl-deficient RUVBL1 mutant (RUVBL1R205K) reduces TIP60 histone acetyltransferase (HAT) activity toward H4K16 acetylation, resulting in enhanced 53BP1 binding to methylated H4K20 and consequent impairment of DNA end resection and defect in HR (Clarke et al., 2017). In addition, TIP60 has two alternative transcripts, TIP60α (full-length) and TIP60β (exclusion of exon 5) with TIP60α being more active (McAllister et al., 2002; Ran & Pereira-Smith, 2000). PRMT5 knockout significantly reduces TIP60α expression and H4 acetylation, leading to similar DNA repair defects as RUVBL1R205K in response to ionizing radiation (Hamard et al., 2018). These studies demonstrate that PRMT5 promotes TIP60 activity by enhancing its interaction with a RUVBL1 and increasing TIP60α expression, promoting HR (Fig. 6d).
Differently, Owens et al. authors reported that knockdown of PRMT5 in prostate cancer cells impairs Ku70 and RAD51 foci formation likely by decreasing the expression of DNA repair gens in both HR and NHEJ pathways, including RAD51, RAD51D, RAD51AP1, BRCA1, BRCA2, DNAPKcs, XRCC4, and XLF. They further found that PRMT5 cooperates with pICln, but not MEP50 or RUVBL1/TIP60, to promote H4R3me2s for the activation of these DNA repair genes in response to DNA damage (Fig. 6e) (Owens et al., 2020). However, it is unclear whether PRMT5 regulates the expression of these genes simultaneously or in a cell-cycle-dependent manner, which would be an important question for future studies.
In summary, PRMT1 and PRMT5 may balance HR and NHEJ by directly methylating their components or epigenetically regulating the expression of DNA repair genes (Table 1).
Table 1 Mechanisms and substrates of PRMT1 and PRMT5 in HR and NHEJFull size table

Targeting PRMT1 and PRMT5 for cancer therapy
As a better understanding of arginine methylation and the development of multiple PRMT inhibitors (Table 2), PRMTs have been emerged as an ideal therapeutic target for cancers (Hwang et al., 2021; Jarrold & Davies, 2019; Li et al., 2019). GSK3368715 is the only Type I PRMT inhibitor that has been evaluated in clinical trial for Diffuse Large B-cell Lymphoma (DLBCL) and solid tumors with MTAP deficiency (NCT03666988). However, this clinical trial has been terminated due to unsatisfied overall benefit-risk. Several PRMT5 inhibitors are also under clinical studies. GSK3326595 (NCT04676516, Phase II) and PF-06939999 (NCT03854227, Phase I) have been tested for early stage breast cancer and advanced/metastatic solid tumors, respectively. SCR-6920 is in Phase I for patients with advanced malignant tumors (NCT05528055). PRT811 is in Phase I for patients with advanced solid tumors and recurrent glioma (NCT04089449). TNG908 (NCT05275478) and MRTX1719 (NCT05245500) are in Phase I/II for advanced or metastatic solid tumors with MTAP deficiency. JNJ-64619178 is tested for patients with advanced solid tumor, NHL, or lower risk MDS (NCT03573310). PRT543 is evaluated for patients with advanced solid tumors and hematologic malignancies (NCT03886831). In addition, AMG 193 in combination with Docetaxel is in phase I/II for MTAP-null solid tumor (NCT05094336), while GSK3326595 in combination with Pembrolizumab is in phase I for solid tumors and NHL (NCT02783300).
Table 2 PRMT Inhibitors in clinicFull size table
Given that inhibition of PRMT1 and PRMT5 result in DNA repair defects, a combination PRMT inhibitors and DDR-related drugs could be a potential strategy to improve cancer therapy (Table 3). PRMT1 inhibitor MS023 synergizes with PARP inhibitor BMN-673 in non-small cell lung cancer (NSCLC) (Dominici et al., 2021)and enhances sensitivity to cisplatin in SK-OV3 ovarian cancer cells (Musiani et al., 2020). PRMT5 inhibitor GSK3186000A and PARP inhibitors Olaparib also have a synergistic effect on acute myeloid leukemia (AML) cells (Hamard et al., 2018). Furthermore, radiotherapy is one of the most common treatment for more than 50% human cancers but is limited by adverse toxicity and resistance. Targeting PRMT1 and PRMT5 may be ideal for radiosensitization to benefit cancer patients.
Table 3 Combination of PRMT inhibitors and DNA damaging agentsFull size table

Data availability statement
No research data is included in this review article. Please consult the authors of the individual studies for access to research data and materials.
References
Adams, M. M., Wang, B., Xia, Z., Morales, J. C., Lu, X., Donehower, L. A., et al. (2005). 53BP1 oligomerization is independent of its methylation by PRMT1. Cell Cycle, 4(12), 1854–1861. https://doi.org/10.4161/cc.4.12.2282
Albrecht, L. V., Ploper, D., Tejeda-Munoz, N., & De Robertis, E. M. (2018). Arginine methylation is required for canonical Wnt signaling and endolysosomal trafficking. Proceedings of the National Academy of Sciences of the United States of America, 115(23), E5317–E5325. https://doi.org/10.1073/pnas.1804091115
Auclair, Y., & Richard, S. (2013). The role of arginine methylation in the DNA damage response. DNA Repair (Amst), 12(7), 459–465. https://doi.org/10.1016/j.dnarep.2013.04.006
Bedford, M. T. (2007). Arginine methylation at a glance. Journal of Cell Science, 120(Pt 24), 4243–4246. https://doi.org/10.1242/jcs.019885
Bedford, M. T., & Clarke, S. G. (2009). Protein arginine methylation in mammals: Who, what, and why. Molecular Cell, 33(1), 1–13. https://doi.org/10.1016/j.molcel.2008.12.013
Blanc, R. S., & Richard, S. (2017). Arginine methylation: The coming of age. Molecular Cell, 65(1), 8–24. https://doi.org/10.1016/j.molcel.2016.11.003
Boisvert, F. M., Cote, J., Boulanger, M. C., Cleroux, P., Bachand, F., Autexier, C., et al. (2002). Symmetrical dimethylarginine methylation is required for the localization of SMN in Cajal bodies and pre-mRNA splicing. Journal of Cell Biology, 159(6), 957–969. https://doi.org/10.1083/jcb.200207028
Boisvert, F. M., Dery, U., Masson, J. Y., & Richard, S. (2005a). Arginine methylation of MRE11 by PRMT1 is required for DNA damage checkpoint control. Genes & Development, 19(6), 671–676. https://doi.org/10.1101/gad.1279805
Boisvert, F. M., Rhie, A., Richard, S., & Doherty, A. J. (2005b). The GAR motif of 53BP1 is arginine methylated by PRMT1 and is necessary for 53BP1 DNA binding activity. Cell Cycle, 4(12), 1834–1841. https://doi.org/10.4161/cc.4.12.2250
Bouwman, P., Aly, A., Escandell, J. M., Pieterse, M., Bartkova, J., van der Gulden, H., et al. (2010). 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nature Structural & Molecular Biology, 17(6), 688–695. https://doi.org/10.1038/nsmb.1831
Brobbey, C., Liu, L., Yin, S., & Gan, W. (2022). The role of protein arginine methyltransferases in DNA damage response. International Journal of Molecular Science. https://doi.org/10.3390/ijms23179780
Buis, J., Wu, Y., Deng, Y., Leddon, J., Westfield, G., Eckersdorff, M., et al. (2008). Mre11 nuclease activity has essential roles in DNA repair and genomic stability distinct from ATM activation. Cell, 135(1), 85–96. https://doi.org/10.1016/j.cell.2008.08.015
Bunting, S. F., Callen, E., Wong, N., Chen, H. T., Polato, F., Gunn, A., et al. (2010). 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell, 141(2), 243–254. https://doi.org/10.1016/j.cell.2010.03.012
Callen, E., Di Virgilio, M., Kruhlak, M. J., Nieto-Soler, M., Wong, N., Chen, H. T., et al. (2013). 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell, 153(6), 1266–1280. https://doi.org/10.1016/j.cell.2013.05.023
Cannavo, E., Cejka, P., & Kowalczykowski, S. C. (2013). Relationship of DNA degradation by Saccharomyces cerevisiae exonuclease 1 and its stimulation by RPA and Mre11-Rad50-Xrs2 to DNA end resection. Proceedings of the National Academy of Sciences of the United States of America, 110(18), E1661-1668. https://doi.org/10.1073/pnas.1305166110
Carney, J. P., Maser, R. S., Olivares, H., Davis, E. M., Le Beau, M., Yates, J. R., 3rd., et al. (1998). The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: Linkage of double-strand break repair to the cellular DNA damage response. Cell, 93(3), 477–486. https://doi.org/10.1016/s0092-8674(00)81175-7
Cejka, P., Cannavo, E., Polaczek, P., Masuda-Sasa, T., Pokharel, S., Campbell, J. L., et al. (2010). DNA end resection by Dna2-Sgs1-RPA and its stimulation by Top3-Rmi1 and Mre11-Rad50-Xrs2. Nature, 467(7311), 112–116. https://doi.org/10.1038/nature09355
Chapman, J. R., Barral, P., Vannier, J. B., Borel, V., Steger, M., Tomas-Loba, A., et al. (2013). RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Molecular Cell, 49(5), 858–871. https://doi.org/10.1016/j.molcel.2013.01.002
Checa-Rodriguez, C., Cepeda-Garcia, C., Ramon, J., Lopez-Saavedra, A., Balestra, F. R., Dominguez-Sanchez, M. S., et al. (2020). Methylation of the central transcriptional regulator KLF4 by PRMT5 is required for DNA end resection and recombination. DNA Repair (Amst). https://doi.org/10.1016/j.dnarep.2020.102902
Chen, X., Whitney, E. M., Gao, S. Y., & Yang, V. W. (2003). Transcriptional profiling of Kruppel-like factor 4 reveals a function in cell cycle regulation and epithelial differentiation. Journal of Molecular Biology, 326(3), 665–677. https://doi.org/10.1016/s0022-2836(02)01449-3
Cheng, C., Pei, X., Li, S. W., Yang, J., Li, C., Tang, J., et al. (2021). CRISPR/Cas9 library screening uncovered methylated PKP2 as a critical driver of lung cancer radioresistance by stabilizing beta-catenin. Oncogene, 40(16), 2842–2857. https://doi.org/10.1038/s41388-021-01692-x
Cheng, D., Cote, J., Shaaban, S., & Bedford, M. T. (2007). The arginine methyltransferase CARM1 regulates the coupling of transcription and mRNA processing. Molecular Cell, 25(1), 71–83. https://doi.org/10.1016/j.molcel.2006.11.019
Chiang, K., Zielinska, A. E., Shaaban, A. M., Sanchez-Bailon, M. P., Jarrold, J., Clarke, T. L., et al. (2017). PRMT5 is a critical regulator of breast cancer stem cell function via histone methylation and FOXP1 expression. Cell Reports, 21(12), 3498–3513. https://doi.org/10.1016/j.celrep.2017.11.096
Choudhury, A. D., Xu, H., & Baer, R. (2004). Ubiquitination and proteasomal degradation of the BRCA1 tumor suppressor is regulated during cell cycle progression. Journal of Biological Chemistry, 279(32), 33909–33918. https://doi.org/10.1074/jbc.M403646200
Ciccia, A., & Elledge, S. J. (2010). The DNA damage response: Making it safe to play with knives. Molecular Cell, 40(2), 179–204. https://doi.org/10.1016/j.molcel.2010.09.019
Clarke, T. L., Sanchez-Bailon, M. P., Chiang, K., Reynolds, J. J., Herrero-Ruiz, J., Bandeiras, T. M., et al. (2017). PRMT5-dependent methylation of the TIP60 coactivator RUVBL1 is a key regulator of homologous recombination. Molecular Cell, 65(5), 900-916 e907. https://doi.org/10.1016/j.molcel.2017.01.019
Costantini, S., Woodbine, L., Andreoli, L., Jeggo, P. A., & Vindigni, A. (2007). Interaction of the Ku heterodimer with the DNA ligase IV/Xrcc4 complex and its regulation by DNA-PK. DNA Repair (Amst), 6(6), 712–722. https://doi.org/10.1016/j.dnarep.2006.12.007
Dimitrova, N., Chen, Y. C., Spector, D. L., & de Lange, T. (2008). 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature, 456(7221), 524–528. https://doi.org/10.1038/nature07433
Dominici, C., Sgarioto, N., Yu, Z., Sesma-Sanz, L., Masson, J. Y., Richard, S., et al. (2021). Synergistic effects of type I PRMT and PARP inhibitors against non-small cell lung cancer cells. Clinical Epigenetics, 13(1), 54. https://doi.org/10.1186/s13148-021-01037-1
Dong, S., Han, J., Chen, H., Liu, T., Huen, M. S. Y., Yang, Y., et al. (2014). The human SRCAP chromatin remodeling complex promotes DNA-end resection. Current Biology, 24(18), 2097–2110. https://doi.org/10.1016/j.cub.2014.07.081
Du, C., Hansen, L. J., Singh, S. X., Wang, F., Sun, R., Moure, C. J., et al. (2019). A PRMT5-RNF168-SMURF2 axis controls H2AX proteostasis. Cell Reports, 28(12), 3199–3211. https://doi.org/10.1016/j.celrep.2019.08.031
El-Karim, E. A., Hagos, E. G., Ghaleb, A. M., Yu, B., & Yang, V. W. (2013). Kruppel-like factor 4 regulates genetic stability in mouse embryonic fibroblasts. Molecular Cancer. https://doi.org/10.1186/1476-4598-12-89
Featherstone, C., & Jackson, S. P. (1999). Ku, a DNA repair protein with multiple cellular functions? Mutation Research, 434(1), 3–15. https://doi.org/10.1016/s0921-8777(99)00006-3
Feng, J., Dang, Y., Zhang, W., Zhao, X., Zhang, C., Hou, Z., et al. (2019). PTEN arginine methylation by PRMT6 suppresses PI3K-AKT signaling and modulates pre-mRNA splicing. Proceedings of the National Academy of Sciences of the United States of America, 116(14), 6868–6877. https://doi.org/10.1073/pnas.1811028116
Feng, Y., Maity, R., Whitelegge, J. P., Hadjikyriacou, A., Li, Z., Zurita-Lopez, C., et al. (2013). Mammalian protein arginine methyltransferase 7 (PRMT7) specifically targets RXR sites in lysine- and arginine-rich regions. Journal of Biological Chemistry, 288(52), 37010–37025. https://doi.org/10.1074/jbc.M113.525345
Fong, J. Y., Pignata, L., Goy, P. A., Kawabata, K. C., Lee, S. C., Koh, C. M., et al. (2019). Therapeutic targeting of RNA splicing catalysis through inhibition of protein arginine methylation. Cancer Cell, 36(2), 194-209 e199. https://doi.org/10.1016/j.ccell.2019.07.003
Foster, S. S., Balestrini, A., & Petrini, J. H. (2011). Functional interplay of the Mre11 nuclease and Ku in the response to replication-associated DNA damage. Molecular and Cellular Biology, 31(21), 4379–4389. https://doi.org/10.1128/MCB.05854-11
Geoghegan, V., Guo, A., Trudgian, D., Thomas, B., & Acuto, O. (2015). Comprehensive identification of arginine methylation in primary T cells reveals regulatory roles in cell signalling. Nature Communications. https://doi.org/10.1038/ncomms7758
Geuens, T., Bouhy, D., & Timmerman, V. (2016). The hnRNP family: Insights into their role in health and disease. Human Genetics, 135(8), 851–867. https://doi.org/10.1007/s00439-016-1683-5
Giuliani, V., Miller, M. A., Liu, C. Y., Hartono, S. R., Class, C. A., Bristow, C. A., et al. (2021). PRMT1-dependent regulation of RNA metabolism and DNA damage response sustains pancreatic ductal adenocarcinoma. Nature Communications, 12(1), 4626. https://doi.org/10.1038/s41467-021-24798-y
Goodarzi, A. A., Yu, Y., Riballo, E., Douglas, P., Walker, S. A., Ye, R., et al. (2006). DNA-PK autophosphorylation facilitates Artemis endonuclease activity. EMBO Journal, 25(16), 3880–3889. https://doi.org/10.1038/sj.emboj.7601255
Gottlieb, T. M., & Jackson, S. P. (1993). The DNA-dependent protein kinase: Requirement for DNA ends and association with Ku antigen. Cell, 72(1), 131–142. https://doi.org/10.1016/0092-8674(93)90057-w
Gravel, S., Chapman, J. R., Magill, C., & Jackson, S. P. (2008). DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes & Development, 22(20), 2767–2772. https://doi.org/10.1101/gad.503108
Guendel, I., Carpio, L., Pedati, C., Schwartz, A., Teal, C., Kashanchi, F., et al. (2010). Methylation of the tumor suppressor protein, BRCA1, influences its transcriptional cofactor function. PLoS ONE, 5(6), e11379. https://doi.org/10.1371/journal.pone.0011379
Gurunathan, G., Yu, Z., Coulombe, Y., Masson, J. Y., & Richard, S. (2015). Arginine methylation of hnRNPUL1 regulates interaction with NBS1 and recruitment to sites of DNA damage. Science and Reports. https://doi.org/10.1038/srep10475
Haber, J. E. (2000). Partners and pathwaysrepairing a double-strand break. Trends in Genetics, 16(6), 259–264. https://doi.org/10.1016/s0168-9525(00)02022-9
Hamard, P. J., Santiago, G. E., Liu, F., Karl, D. L., Martinez, C., Man, N., et al. (2018). PRMT5 regulates DNA repair by controlling the alternative splicing of histone-modifying enzymes. Cell Reports, 24(10), 2643–2657. https://doi.org/10.1016/j.celrep.2018.08.002
Harper, J. W., & Elledge, S. J. (2007). The DNA damage response: Ten years after. Molecular Cell, 28(5), 739–745. https://doi.org/10.1016/j.molcel.2007.11.015
He, L., Hu, Z., Sun, Y., Zhang, M., Zhu, H., Jiang, L., et al. (2020). PRMT1 is critical to FEN1 expression and drug resistance in lung cancer cells. DNA Repair (Amst). https://doi.org/10.1016/j.dnarep.2020.102953
He, W., Ma, X., Yang, X., Zhao, Y., Qiu, J., & Hang, H. (2011). A role for the arginine methylation of Rad9 in checkpoint control and cellular sensitivity to DNA damage. Nucleic Acids Research, 39(11), 4719–4727. https://doi.org/10.1093/nar/gkq1264
Hopkins, B. B., & Paull, T. T. (2008). The P. furiosus mre11/rad50 complex promotes 5′ strand resection at a DNA double-strand break. Cell, 135(2), 250–260. https://doi.org/10.1016/j.cell.2008.09.054
Hsu, J. H., Hubbell-Engler, B., Adelmant, G., Huang, J., Joyce, C. E., Vazquez, F., et al. (2017). PRMT1-mediated translation regulation is a crucial vulnerability of cancer. Cancer Research, 77(17), 4613–4625. https://doi.org/10.1158/0008-5472.CAN-17-0216
Hu, D., Gur, M., Zhou, Z., Gamper, A., Hung, M. C., Fujita, N., et al. (2015). Interplay between arginine methylation and ubiquitylation regulates KLF4-mediated genome stability and carcinogenesis. Nature Communications. https://doi.org/10.1038/ncomms9419
Hua, Z. Y., Hansen, J. N., He, M., Dai, S. K., Choi, Y., Fulton, M. D., et al. (2020). PRMT1 promotes neuroblastoma cell survival through ATF5. Oncogenesis, 9(5), 50. https://doi.org/10.1038/s41389-020-0237-9
Huen, M. S., & Chen, J. (2008). The DNA damage response pathways: At the crossroad of protein modifications. Cell Research, 18(1), 8–16. https://doi.org/10.1038/cr.2007.109
Hwang, J. W., Cho, Y., Bae, G. U., Kim, S. N., & Kim, Y. K. (2021). Protein arginine methyltransferases: Promising targets for cancer therapy. Experimental & Molecular Medicine, 53(5), 788–808. https://doi.org/10.1038/s12276-021-00613-y
Hwang, J. W., Kim, S. N., Myung, N., Song, D., Han, G., Bae, G. U., et al. (2020). PRMT5 promotes DNA repair through methylation of 53BP1 and is regulated by Src-mediated phosphorylation. Commun Biol, 3(1), 428. https://doi.org/10.1038/s42003-020-01157-z
Ikura, T., Ogryzko, V. V., Grigoriev, M., Groisman, R., Wang, J., Horikoshi, M., et al. (2000). Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell, 102(4), 463–473. https://doi.org/10.1016/s0092-8674(00)00051-9
Iwabuchi, K., Bartel, P. L., Li, B., Marraccino, R., & Fields, S. (1994). Two cellular proteins that bind to wild-type but not mutant p53. Proceedings of the National Academy of Sciences of the United States of America, 91(13), 6098–6102. https://doi.org/10.1073/pnas.91.13.6098
Iwabuchi, K., Basu, B. P., Kysela, B., Kurihara, T., Shibata, M., Guan, D., et al. (2003). Potential role for 53BP1 in DNA end-joining repair through direct interaction with DNA. Journal of Biological Chemistry, 278(38), 36487–36495. https://doi.org/10.1074/jbc.M304066200
Jackson, S. P., & Bartek, J. (2009). The DNA-damage response in human biology and disease. Nature, 461(7267), 1071–1078. https://doi.org/10.1038/nature08467
Jarrold, J., & Davies, C. C. (2019). PRMTs and arginine methylation: Cancer’s best-kept secret? Trends in Molecular Medicine, 25(11), 993–1009. https://doi.org/10.1016/j.molmed.2019.05.007
Jensen, K. L., & Russell, P. (2016). Ctp1-dependent clipping and resection of DNA double-strand breaks by Mre11 endonuclease complex are not genetically separable. Nucleic Acids Research, 44(17), 8241–8249. https://doi.org/10.1093/nar/gkw557
Jha, S., & Dutta, A. (2009). RVB1/RVB2: Running rings around molecular biology. Molecular Cell, 34(5), 521–533. https://doi.org/10.1016/j.molcel.2009.05.016
Jiang, J., Chan, Y. S., Loh, Y. H., Cai, J., Tong, G. Q., Lim, C. A., et al. (2008). A core Klf circuitry regulates self-renewal of embryonic stem cells. Nature Cell Biology, 10(3), 353–360. https://doi.org/10.1038/ncb1698
Jin, S., & Weaver, D. T. (1997). Double-strand break repair by Ku70 requires heterodimerization with Ku80 and DNA binding functions. EMBO Journal, 16(22), 6874–6885. https://doi.org/10.1093/emboj/16.22.6874
Jing, P., Zhao, N., Ye, M., Zhang, Y., Zhang, Z., Sun, J., et al. (2018). Protein arginine methyltransferase 5 promotes lung cancer metastasis via the epigenetic regulation of miR-99 family/FGFR3 signaling. Cancer Letters. https://doi.org/10.1016/j.canlet.2018.04.019
Kanno, S., Kuzuoka, H., Sasao, S., Hong, Z., Lan, L., Nakajima, S., et al. (2007). A novel human AP endonuclease with conserved zinc-finger-like motifs involved in DNA strand break responses. EMBO Journal, 26(8), 2094–2103. https://doi.org/10.1038/sj.emboj.7601663
Karkhanis, V., Hu, Y. J., Baiocchi, R. A., Imbalzano, A. N., & Sif, S. (2011). Versatility of PRMT5-induced methylation in growth control and development. Trends in Biochemical Sciences, 36(12), 633–641. https://doi.org/10.1016/j.tibs.2011.09.001
Khanna, K. K., & Jackson, S. P. (2001). DNA double-strand breaks: Signaling, repair and the cancer connection. Nature Genetics, 27(3), 247–254. https://doi.org/10.1038/85798
Kim, H., & Ronai, Z. A. (2020). PRMT5 function and targeting in cancer. Cell Stress, 4(8), 199–215. https://doi.org/10.15698/cst2020.08.228
Kim, S., Gunesdogan, U., Zylicz, J. J., Hackett, J. A., Cougot, D., Bao, S., et al. (2014). PRMT5 protects genomic integrity during global DNA demethylation in primordial germ cells and preimplantation embryos. Molecular Cell, 56(4), 564–579. https://doi.org/10.1016/j.molcel.2014.10.003
Langerak, P., Mejia-Ramirez, E., Limbo, O., & Russell, P. (2011). Release of Ku and MRN from DNA ends by Mre11 nuclease activity and Ctp1 is required for homologous recombination repair of double-strand breaks. PLoS Genetics, 7(9), e1002271. https://doi.org/10.1371/journal.pgen.1002271
Lattouf, H., Poulard, C., & Le Romancer, M. (2019). PRMT5 prognostic value in cancer. Oncotarget, 10(34), 3151–3153. https://doi.org/10.18632/oncotarget.26883
Lee, D. Y., Teyssier, C., Strahl, B. D., & Stallcup, M. R. (2005). Role of protein methylation in regulation of transcription. Endocrine Reviews, 26(2), 147–170. https://doi.org/10.1210/er.2004-0008
Lee, J. H., & Paull, T. T. (2005). ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science, 308(5721), 551–554. https://doi.org/10.1126/science.1108297
Lee, J. W., Blanco, L., Zhou, T., Garcia-Diaz, M., Bebenek, K., Kunkel, T. A., et al. (2004). Implication of DNA polymerase lambda in alignment-based gap filling for nonhomologous DNA end joining in human nuclear extracts. Journal of Biological Chemistry, 279(1), 805–811. https://doi.org/10.1074/jbc.M307913200
Lee, Y. H., & Stallcup, M. R. (2009). Minireview: Protein arginine methylation of nonhistone proteins in transcriptional regulation. Molecular Endocrinology, 23(4), 425–433. https://doi.org/10.1210/me.2008-0380
Li, W. J., He, Y. H., Yang, J. J., Hu, G. S., Lin, Y. A., Ran, T., et al. (2021). Profiling PRMT methylome reveals roles of hnRNPA1 arginine methylation in RNA splicing and cell growth. Nature Communications, 12(1), 1946. https://doi.org/10.1038/s41467-021-21963-1
Li, X., Wang, C., Jiang, H., & Luo, C. (2019). A patent review of arginine methyltransferase inhibitors (2010–2018). Expert Opinion on Therapeutic Patents, 29(2), 97–114. https://doi.org/10.1080/13543776.2019.1567711
Liao, H. W., Hsu, J. M., Xia, W., Wang, H. L., Wang, Y. N., Chang, W. C., et al. (2015). PRMT1-mediated methylation of the EGF receptor regulates signaling and cetuximab response. The Journal of Clinical Investigation, 125(12), 4529–4543. https://doi.org/10.1172/JCI82826
Lieber, M. R. (2008). The mechanism of human nonhomologous DNA end joining. Journal of Biological Chemistry, 283(1), 1–5. https://doi.org/10.1074/jbc.R700039200
Lorton, B. M., & Shechter, D. (2019). Cellular consequences of arginine methylation. Cellular and Molecular Life Sciences, 76(15), 2933–2956. https://doi.org/10.1007/s00018-019-03140-2
Lottersberger, F., Karssemeijer, R. A., Dimitrova, N., & de Lange, T. (2015). 53BP1 and the LINC complex promote microtubule-dependent DSB mobility and DNA repair. Cell, 163(4), 880–893. https://doi.org/10.1016/j.cell.2015.09.057
Ma, Y., Lu, H., Tippin, B., Goodman, M. F., Shimazaki, N., Koiwai, O., et al. (2004). A biochemically defined system for mammalian nonhomologous DNA end joining. Molecular Cell, 16(5), 701–713. https://doi.org/10.1016/j.molcel.2004.11.017
Mahajan, K. N., Nick McElhinny, S. A., Mitchell, B. S., & Ramsden, D. A. (2002). Association of DNA polymerase mu (pol mu) with Ku and ligase IV: Role for pol mu in end-joining double-strand break repair. Molecular and Cellular Biology, 22(14), 5194–5202. https://doi.org/10.1128/MCB.22.14.5194-5202.2002
Mahaney, B. L., Meek, K., & Lees-Miller, S. P. (2009). Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. The Biochemical Journal, 417(3), 639–650. https://doi.org/10.1042/BJ20080413
Mari, P. O., Florea, B. I., Persengiev, S. P., Verkaik, N. S., Bruggenwirth, H. T., Modesti, M., et al. (2006). Dynamic assembly of end-joining complexes requires interaction between Ku70/80 and XRCC4. Proceedings of the National Academy of Sciences of the United States of America, 103(49), 18597–18602. https://doi.org/10.1073/pnas.0609061103
Mathioudaki, K., Papadokostopoulou, A., Scorilas, A., Xynopoulos, D., Agnanti, N., & Talieri, M. (2008). The PRMT1 gene expression pattern in colon cancer. British Journal of Cancer, 99(12), 2094–2099. https://doi.org/10.1038/sj.bjc.6604807
McAllister, D., Merlo, X., & Lough, J. (2002). Characterization and expression of the mouse tat interactive protein 60 kD (TIP60) gene. Gene, 289(1–2), 169–176. https://doi.org/10.1016/s0378-1119(02)00546-2
Mehta, A., & Haber, J. E. (2014). Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harbor Perspectives in Biology, 6(9), a016428. https://doi.org/10.1101/cshperspect.a016428
Mersch, J., Jackson, M. A., Park, M., Nebgen, D., Peterson, S. K., Singletary, C., et al. (2015). Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer, 121(2), 269–275. https://doi.org/10.1002/cncr.29041
Mimitou, E. P., & Symington, L. S. (2010). Ku prevents Exo1 and Sgs1-dependent resection of DNA ends in the absence of a functional MRX complex or Sae2. EMBO Journal, 29(19), 3358–3369. https://doi.org/10.1038/emboj.2010.193
Mirman, Z., & de Lange, T. (2020). 53BP1: A DSB escort. Genes & Development, 34(1–2), 7–23. https://doi.org/10.1101/gad.333237.119
Montenegro, M. F., Gonzalez-Guerrero, R., Sanchez-Del-Campo, L., Pinero-Madrona, A., Cabezas-Herrera, J., & Rodriguez-Lopez, J. N. (2020). PRMT1-dependent methylation of BRCA1 contributes to the epigenetic defense of breast cancer cells against ionizing radiation. Science and Reports, 10(1), 13275. https://doi.org/10.1038/s41598-020-70289-3
Morris, J. R., Boutell, C., Keppler, M., Densham, R., Weekes, D., Alamshah, A., et al. (2009). The SUMO modification pathway is involved in the BRCA1 response to genotoxic stress. Nature, 462(7275), 886–890. https://doi.org/10.1038/nature08593
Musiani, D., Giambruno, R., Massignani, E., Ippolito, M. R., Maniaci, M., Jammula, S., et al. (2020). PRMT1 is recruited via DNA-PK to chromatin where it sustains the senescence-associated secretory phenotype in response to cisplatin. Cell Reports, 30(4), 1208-1222 e1209. https://doi.org/10.1016/j.celrep.2019.12.061
Najbauer, J., Johnson, B. A., Young, A. L., & Aswad, D. W. (1993). Peptides with sequences similar to glycine, arginine-rich motifs in proteins interacting with RNA are efficiently recognized by methyltransferase(s) modifying arginine in numerous proteins. Journal of Biological Chemistry, 268(14), 10501–10509.
Nick McElhinny, S. A., Snowden, C. M., McCarville, J., & Ramsden, D. A. (2000). Ku recruits the XRCC4-ligase IV complex to DNA ends. Molecular and Cellular Biology, 20(9), 2996–3003. https://doi.org/10.1128/MCB.20.9.2996-3003.2000
Nimonkar, A. V., Genschel, J., Kinoshita, E., Polaczek, P., Campbell, J. L., Wyman, C., et al. (2011). BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes & Development, 25(4), 350–362. https://doi.org/10.1101/gad.2003811
Niu, H., Chung, W. H., Zhu, Z., Kwon, Y., Zhao, W., Chi, P., et al. (2010). Mechanism of the ATP-dependent DNA end-resection machinery from Saccharomyces cerevisiae. Nature, 467(7311), 108–111. https://doi.org/10.1038/nature09318
Ochi, T., Blackford, A. N., Coates, J., Jhujh, S., Mehmood, S., Tamura, N., et al. (2015). DNA repair. PAXX, a paralog of XRCC4 and XLF, interacts with Ku to promote DNA double-strand break repair. Science, 347(6218), 185–188. https://doi.org/10.1126/science.1261971
Ouchi, T. (2006). BRCA1 phosphorylation: Biological consequences. Cancer Biology & Therapy, 5(5), 470–475. https://doi.org/10.4161/cbt.5.5.2845
Owens, J. L., Beketova, E., Liu, S., Tinsley, S. L., Asberry, A. M., Deng, X., et al. (2020). PRMT5 cooperates with pICln to function as a master epigenetic activator of DNA double-strand break repair genes. iScience, 23(1), 100750. https://doi.org/10.1016/j.isci.2019.100750
Paiano, J., Zolnerowich, N., Wu, W., Pavani, R., Wang, C., Li, H., et al. (2021). Role of 53BP1 in end protection and DNA synthesis at DNA breaks. Genes & Development, 35(19–20), 1356–1367. https://doi.org/10.1101/gad.348667.121
Panier, S., & Boulton, S. J. (2014). Double-strand break repair: 53BP1 comes into focus. Nature Reviews Molecular Cell Biology, 15(1), 7–18. https://doi.org/10.1038/nrm3719
Polo, S. E., Blackford, A. N., Chapman, J. R., Baskcomb, L., Gravel, S., Rusch, A., et al. (2012). Regulation of DNA-end resection by hnRNPU-like proteins promotes DNA double-strand break signaling and repair. Molecular Cell, 45(4), 505–516. https://doi.org/10.1016/j.molcel.2011.12.035
Polo, S. E., & Jackson, S. P. (2011). Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes & Development, 25(5), 409–433. https://doi.org/10.1101/gad.2021311
Powers, M. A., Fay, M. M., Factor, R. E., Welm, A. L., & Ullman, K. S. (2011). Protein arginine methyltransferase 5 accelerates tumor growth by arginine methylation of the tumor suppressor programmed cell death 4. Cancer Research, 71(16), 5579–5587. https://doi.org/10.1158/0008-5472.CAN-11-0458
Prakash, R., Zhang, Y., Feng, W., & Jasin, M. (2015). Homologous recombination and human health: The roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harbor Perspectives in Biology, 7(4), a016600. https://doi.org/10.1101/cshperspect.a016600
Ran, Q., & Pereira-Smith, O. M. (2000). Identification of an alternatively spliced form of the Tat interactive protein (Tip60), Tip60(beta). Gene, 258(1–2), 141–146. https://doi.org/10.1016/s0378-1119(00)00410-8
Rehman, I., Basu, S. M., Das, S. K., Bhattacharjee, S., Ghosh, A., Pommier, Y., et al. (2018). PRMT5-mediated arginine methylation of TDP1 for the repair of topoisomerase I covalent complexes. Nucleic Acids Research, 46(11), 5601–5617. https://doi.org/10.1093/nar/gky291
San Filippo, J., Sung, P., & Klein, H. (2008). Mechanism of eukaryotic homologous recombination. Annual Review of Biochemistry. https://doi.org/10.1146/annurev.biochem.77.061306.125255
Sartori, A. A., Lukas, C., Coates, J., Mistrik, M., Fu, S., Bartek, J., et al. (2007). Human CtIP promotes DNA end resection. Nature, 450(7169), 509–514. https://doi.org/10.1038/nature06337
Schultz, L. B., Chehab, N. H., Malikzay, A., & Halazonetis, T. D. (2000). p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. Journal of Cell Biology, 151(7), 1381–1390. https://doi.org/10.1083/jcb.151.7.1381
Scully, R., Panday, A., Elango, R., & Willis, N. A. (2019). DNA double-strand break repair-pathway choice in somatic mammalian cells. Nature Reviews Molecular Cell Biology, 20(11), 698–714. https://doi.org/10.1038/s41580-019-0152-0
Sheng, X., & Wang, Z. (2016). Protein arginine methyltransferase 5 regulates multiple signaling pathways to promote lung cancer cell proliferation. BMC Cancer. https://doi.org/10.1186/s12885-016-2632-3
Shim, E. Y., Chung, W. H., Nicolette, M. L., Zhang, Y., Davis, M., Zhu, Z., et al. (2010). Saccharomyces cerevisiae Mre11/Rad50/Xrs2 and Ku proteins regulate association of Exo1 and Dna2 with DNA breaks. EMBO Journal, 29(19), 3370–3380. https://doi.org/10.1038/emboj.2010.219
Shishkova, E., Zeng, H., Liu, F., Kwiecien, N. W., Hebert, A. S., Coon, J. J., et al. (2017). Global mapping of CARM1 substrates defines enzyme specificity and substrate recognition. Nature Communications. https://doi.org/10.1038/ncomms15571
Shrivastav, M., De Haro, L. P., & Nickoloff, J. A. (2008). Regulation of DNA double-strand break repair pathway choice. Cell Research, 18(1), 134–147. https://doi.org/10.1038/cr.2007.111
Sigurdsson, S., Van Komen, S., Bussen, W., Schild, D., Albala, J. S., & Sung, P. (2001). Mediator function of the human Rad51B-Rad51C complex in Rad51/RPA-catalyzed DNA strand exchange. Genes & Development, 15(24), 3308–3318. https://doi.org/10.1101/gad.935501
Singleton, B. K., Torres-Arzayus, M. I., Rottinghaus, S. T., Taccioli, G. E., & Jeggo, P. A. (1999). The C terminus of Ku80 activates the DNA-dependent protein kinase catalytic subunit. Molecular and Cellular Biology, 19(5), 3267–3277. https://doi.org/10.1128/MCB.19.5.3267
Spagnolo, L., Rivera-Calzada, A., Pearl, L. H., & Llorca, O. (2006). Three-dimensional structure of the human DNA-PKcs/Ku70/Ku80 complex assembled on DNA and its implications for DNA DSB repair. Molecular Cell, 22(4), 511–519. https://doi.org/10.1016/j.molcel.2006.04.013
Stracker, T. H., & Petrini, J. H. (2011). The MRE11 complex: Starting from the ends. Nature Reviews Molecular Cell Biology, 12(2), 90–103. https://doi.org/10.1038/nrm3047
Sugiyama, T., & Kowalczykowski, S. C. (2002). Rad52 protein associates with replication protein A (RPA)-single-stranded DNA to accelerate Rad51-mediated displacement of RPA and presynaptic complex formation. Journal of Biological Chemistry, 277(35), 31663–31672. https://doi.org/10.1074/jbc.M203494200
Sung, P. (1997). Function of yeast Rad52 protein as a mediator between replication protein A and the Rad51 recombinase. Journal of Biological Chemistry, 272(45), 28194–28197. https://doi.org/10.1074/jbc.272.45.28194
Sung, P., & Klein, H. (2006). Mechanism of homologous recombination: Mediators and helicases take on regulatory functions. Nature Reviews Molecular Cell Biology, 7(10), 739–750. https://doi.org/10.1038/nrm2008
Takeda, S., Nakamura, K., Taniguchi, Y., & Paull, T. T. (2007). Ctp1/CtIP and the MRN complex collaborate in the initial steps of homologous recombination. Molecular Cell, 28(3), 351–352. https://doi.org/10.1016/j.molcel.2007.10.016
Tan, D. Q., Li, Y., Yang, C., Li, J., Tan, S. H., Chin, D. W. L., et al. (2019). PRMT5 modulates splicing for genome integrity and preserves proteostasis of hematopoietic stem cells. Cell Reports, 26(9), 2316-2328 e2316. https://doi.org/10.1016/j.celrep.2019.02.001
Tang, J., Frankel, A., Cook, R. J., Kim, S., Paik, W. K., Williams, K. R., et al. (2000). PRMT1 is the predominant type I protein arginine methyltransferase in mammalian cells. Journal of Biological Chemistry, 275(11), 7723–7730. https://doi.org/10.1074/jbc.275.11.7723
Tee, W. W., Pardo, M., Theunissen, T. W., Yu, L., Choudhary, J. S., Hajkova, P., et al. (2010). Prmt5 is essential for early mouse development and acts in the cytoplasm to maintain ES cell pluripotency. Genes & Development, 24(24), 2772–2777. https://doi.org/10.1101/gad.606110
Tewary, S. K., Zheng, Y. G., & Ho, M. C. (2019). Protein arginine methyltransferases: Insights into the enzyme structure and mechanism at the atomic level. Cellular and Molecular Life Sciences, 76(15), 2917–2932. https://doi.org/10.1007/s00018-019-03145-x
Thandapani, P., O’Connor, T. R., Bailey, T. L., & Richard, S. (2013). Defining the RGG/RG motif. Molecular Cell, 50(5), 613–623. https://doi.org/10.1016/j.molcel.2013.05.021
Thiebaut, C., Eve, L., Poulard, C., & Le Romancer, M. (2021). Structure, activity, and function of PRMT1. Life (basel). https://doi.org/10.3390/life11111147
Trujillo, K. M., Yuan, S. S., Lee, E. Y., & Sung, P. (1998). Nuclease activities in a complex of human recombination and DNA repair factors Rad50, Mre11, and p95. Journal of Biological Chemistry, 273(34), 21447–21450. https://doi.org/10.1074/jbc.273.34.21447
Uematsu, N., Weterings, E., Yano, K., Morotomi-Yano, K., Jakob, B., Taucher-Scholz, G., et al. (2007). Autophosphorylation of DNA-PKCS regulates its dynamics at DNA double-strand breaks. Journal of Cell Biology, 177(2), 219–229. https://doi.org/10.1083/jcb.200608077
Usui, T., Ohta, T., Oshiumi, H., Tomizawa, J., Ogawa, H., & Ogawa, T. (1998). Complex formation and functional versatility of Mre11 of budding yeast in recombination. Cell, 95(5), 705–716. https://doi.org/10.1016/s0092-8674(00)81640-2
Wang, H., Shi, L. Z., Wong, C. C., Han, X., Hwang, P. Y., Truong, L. N., et al. (2013). The interaction of CtIP and Nbs1 connects CDK and ATM to regulate HR-mediated double-strand break repair. PLoS Genetics, 9(2), e1003277. https://doi.org/10.1371/journal.pgen.1003277
Wang, X., & Haber, J. E. (2004). Role of Saccharomyces single-stranded DNA-binding protein RPA in the strand invasion step of double-strand break repair. PLoS Biology, 2(1), E21. https://doi.org/10.1371/journal.pbio.0020021
Wang, Y., Cortez, D., Yazdi, P., Neff, N., Elledge, S. J., & Qin, J. (2000). BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes & Development, 14(8), 927–939.
Wold, M. S. (1997). Replication protein A: A heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annual Review of Biochemistry. https://doi.org/10.1146/annurev.biochem.66.1.61
Wright, W. D., Shah, S. S., & Heyer, W. D. (2018). Homologous recombination and the repair of DNA double-strand breaks. Journal of Biological Chemistry, 293(27), 10524–10535. https://doi.org/10.1074/jbc.TM118.000372
Wu, S., Shi, Y., Mulligan, P., Gay, F., Landry, J., Liu, H., et al. (2007). A YY1-INO80 complex regulates genomic stability through homologous recombination-based repair. Nature Structural & Molecular Biology, 14(12), 1165–1172. https://doi.org/10.1038/nsmb1332
Xing, M., Yang, M., Huo, W., Feng, F., Wei, L., Jiang, W., et al. (2015). Interactome analysis identifies a new paralogue of XRCC4 in non-homologous end joining DNA repair pathway. Nature Communications. https://doi.org/10.1038/ncomms7233
Yamagata, K., Daitoku, H., Takahashi, Y., Namiki, K., Hisatake, K., Kako, K., et al. (2008). Arginine methylation of FOXO transcription factors inhibits their phosphorylation by Akt. Molecular Cell, 32(2), 221–231. https://doi.org/10.1016/j.molcel.2008.09.013
Yang, Y., & Bedford, M. T. (2013). Protein arginine methyltransferases and cancer. Nature Reviews Cancer, 13(1), 37–50. https://doi.org/10.1038/nrc3409
Yano, K., Morotomi-Yano, K., Wang, S. Y., Uematsu, N., Lee, K. J., Asaithamby, A., et al. (2008). Ku recruits XLF to DNA double-strand breaks. EMBO Reports, 9(1), 91–96. https://doi.org/10.1038/sj.embor.7401137
Yin, S., Liu, L., Brobbey, C., Palanisamy, V., Ball, L. E., Olsen, S. K., et al. (2021). PRMT5-mediated arginine methylation activates AKT kinase to govern tumorigenesis. Nature Communications, 12(1), 3444. https://doi.org/10.1038/s41467-021-23833-2
Yu, Z., Chen, T., Hebert, J., Li, E., & Richard, S. (2009). A mouse PRMT1 null allele defines an essential role for arginine methylation in genome maintenance and cell proliferation. Molecular and Cellular Biology, 29(11), 2982–2996. https://doi.org/10.1128/MCB.00042-09
Yu, Z., Vogel, G., Coulombe, Y., Dubeau, D., Spehalski, E., Hebert, J., et al. (2012). The MRE11 GAR motif regulates DNA double-strand break processing and ATR activation. Cell Research, 22(2), 305–320. https://doi.org/10.1038/cr.2011.128
Zencheck, W. D., Xiao, H., & Weiss, L. M. (2012). Lysine post-translational modifications and the cytoskeleton. Essays in Biochemistry. https://doi.org/10.1042/bse0520135
Zhao, W., Steinfeld, J. B., Liang, F., Chen, X., Maranon, D. G., Jian Ma, C., et al. (2017). BRCA1-BARD1 promotes RAD51-mediated homologous DNA pairing. Nature, 550(7676), 360–365. https://doi.org/10.1038/nature24060
Zhong, Q., Chen, C. F., Li, S., Chen, Y., Wang, C. C., Xiao, J., et al. (1999). Association of BRCA1 with the hRad50-hMre11-p95 complex and the DNA damage response. Science, 285(5428), 747–750. https://doi.org/10.1126/science.285.5428.747
Zimmermann, M., Lottersberger, F., Buonomo, S. B., Sfeir, A., & de Lange, T. (2013). 53BP1 regulates DSB repair using Rif1 to control 5’ end resection. Science, 339(6120), 700–704. https://doi.org/10.1126/science.1231573
Acknowledgements
We apologize for original papers we were not able to cite but were covered by reviews instead. W.G. and S.Y. were supported by NIH grant (R35GM146749) and American Cancer Society (RSG-22-068-01-TBE). L.L were supported by the Hollings Cancer Center Abney Postdoctoral Fellowship.
Author information
Authors and Affiliations
Department of Biochemistry and Molecular Biology, Hollings Cancer Center, Medical University of South Carolina, Charleston, SC, 29425, USA
Shasha Yin, Liu Liu & Wenjian Gan
Corresponding author
Correspondence to Wenjian Gan.
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article
Cite this article
Yin, S., Liu, L. & Gan, W. PRMT1 and PRMT5: on the road of homologous recombination and non-homologous end joining. GENOME INSTAB. DIS. (2022). https://doi.org/10.1007/s42764-022-00095-w
Received29 October 2022
Accepted28 November 2022
Published07 December 2022
DOIhttps://doi.org/10.1007/s42764-022-00095-w
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
PRMT1
PRMT5
Homologous recombination
Non-homologous end joining
DNA repair pathway choice
用户登录
还没有账号?
立即注册