






Synthetic lethality in personalized cancer therapy
Review Article
Ying Pang, Meng Cheng, Mu Chen, Kaijun Zhao, Yuanyuan Yin, Min Liu, Jing Zhang & Chunlong Zhong
Genome Instability & Disease 4, 121–135 (2023)
Abstract
Genomic instability is one of the hallmarks of tumors that contributes to tumor heterogeneity and drug resistance. Genetically targeted cancer therapy that focused on genome instability has been a direction for tumor treatment of great interest. Synthetic lethality provides a new approach for the treatment of tumor suppressor gene especially DNA damage response pathway mutation-caused cancers that were not considered as a target in traditional genetic treatment previously. Here, we summarize the systematic classification of synthetic lethality and its mechanism which was divided into gene level, including cellular signaling pathway, cell cycle regulation, metabolism, and epigenetic regulation. The goal of this review was to help deepen the understanding of the mechanism of synthetic lethality and guide the direction for exploring the new synthetic lethal relationships.
Introduction
Cancer is characterized by genome instability and mutation, multiple genetics, cancer metabolism, and tumor immune microenvironment, which results in self-sufficiency in growth signals, resisting cell death, avoiding immune destruction, tissue invasion and metastasis, and drug resistance (Hanahan & Weinberg, 2011; Macheret & Halazonetis, 2015; Pavlova & Thompson, 2016). For cancer treatment, chemotherapeutic agents and radiation therapy are mainly focused on inducing DNA damage. However, toxic effects and chemo-resistance often result in clinical treatment failure. One of the characteristics of cancer cells is the abnormalities in DNA damage response (DDR), comprising DNA repair and cell-cycle checkpoint pathways, which is an attractive target for cancer therapy (Hu & Guo, 2020). On the one hand, the defects in DDR promote the clonal evolution of cancer cells through mutation accumulation. On the other hand, the same defects also create genomic instabilities in cancer cells, which increase the sensitivity to DNA damage agents, thereby improving the clinical efficacy of anticancer treatments and patient outcomes (Pilié et al., 2019). In recent studies, synthetic lethality has been indicated as a potential clinical treatment strategy that could improve the clinical prognosis of patients (Brown et al., 2017; Hu & Guo, 2020; Lord & Ashworth, 2017).
Synthetic lethality was originally developed from genetic studies in drosophila and yeast model systems. The concept emphasizes that mutations of two separate genes result in cell death while either of them leads to cell survival (Bender & Pringle, 1991; Hennessy et al., 1991; Kaiser & Schekman, 1990; Lucchesi, 1968; Wright & Dobzhansky, 1946). This provides a novel therapeutic strategy in which inhibition of synthetic lethal partners in tumor cells can kill cancers without harming normal cells of the body. Unlike traditional therapy focusing on “oncogene addiction”, in which a particular oncogene causes a subtype of cancer and inhibition of this oncogene with small molecules or antibodies can specifically kill cancer cells without affecting normal cells, synthetic lethal therapies rely on the synthetic lethal relationship and involve genes from oncogenes to tumor suppressors, the DDR pathway, cell cycle regulation, cancer metabolism, and epigenetic regulation (Myers et al., 2020; O’Neil et al., 2017). With the discovery of new synthetic lethal interactions, cancers driven by tumor suppressor genes mutated will be druggable.
Synthetic lethal therapy has been gradually applied in clinical cancer treatment in recent years. Some small molecular inhibitors focused on the DNA repair pathway have been put into clinical trials and demonstrated to improve the clinical prognosis of patients. Such as Poly (ADP-ribose) polymerase (PARP) inhibitors apply in breast cancer, ovarian cancer, pancreatic cancer, and prostate cancer (Bryant et al., 2005; Farmer et al., 2005; George et al., 2017; Hu & Guo, 2020; Robson et al., 2017; Sandhu et al., 2013). With the development of bioinformatics and novel experimental approaches, more and more new synthetic lethal interactions have been identified. The underlying synthetic lethal interactions will promote novel and effective synthetic lethality strategies to improve tumor progression and patient outcomes. This review will describe the classification of synthetic lethality and the mechanisms behind it, and summarize the clinical application of small-molecule inhibitors that focus on the synthetic lethal relationship.
Synthetic lethality classification
Synthetic lethality is a phenomenon that occurs between two genes. Either gene mutated or overexpressed alone is viable, but both genes inactivate simultaneously resulting in cell death (O’Neil et al., 2017). Synthetic lethality can be classified into nonconditional synthetic lethality and conditional synthetic lethality according to whether the synthetic lethal interaction depends on certain conditions. In addition, nonconditional synthetic lethality is further divided into classic synthetic lethality and synthetic dosage lethality (Fig. 1; O’Neil et al., 2017).
Fig.1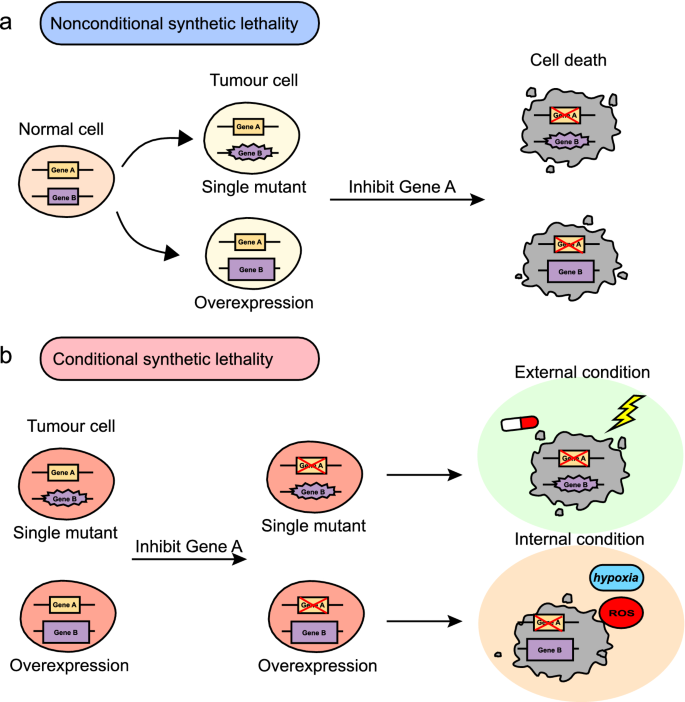
Classification of synthetic lethality. Synthetic lethality is divided into nonconditional synthetic lethality and conditional synthetic lethality. a Nonconditional synthetic lethality. Single mutation or overexpression of gene B alone is viable in tumor cells. Inhibition of gene A in cells with a mutation or overexpression of gene B results in synthetic lethality. b Conditional synthetic lethality. Several synthetic lethal interactions may be dependent on certain internal conditions, such as genetic background, hypoxia, high ROS, etc., or external conditions, such as DNA-damaging agents and radiation. Without these conditions, inhibition of gene A in cells with a mutation or overexpression of gene B could still survive
Classic synthetic lethality is that mutation of either gene in a pair does not affect cell viability, while the inactivation of both genes results in cell death. PARP and BRCA1/2 are synthetic lethal partners belonging to classic synthetic lethality. Both of them play a key role in DNA double-strand breaks (DSBs) repair. PARP inhibitors are proved effective for BRCA1/2-mutated ovarian cancer and breast cancer (George et al., 2017; Robson et al., 2017). PI5P4Ks (phosphatidylinositol-5-phosphate 4-kinases) is essential for growth in the absence of p53 in breast cancers. Inhibitors of PI5PK4s could be effective in preventing or treating cancers with mutations in TP53 (Emerling et al., 2013).
Synthetic dosage lethality is that overexpression of one gene combined with the inactivation of another gene results in cell death, such as Mad2 (mitotic arrest deficiency 2) and PP2A (protein phosphatase 2). Mad2 is a critical component of the spindle checkpoint, which is overexpressed in many cancer cells. PP2A inhibitors could inhibit the growth of Mad2-overexpressed cancer cells, such as liver cancer, lung cancer, and malignant lymphoma (Bian et al., 2014). TDP1 (tyrosyl-DNA-phosphdiesterase1) encodes the protein that repairs stalled topoisomerase I-DNA complexes and repairs free-radical mediated DSBs. Inhibition of its synthetic lethal partner RPD3, a histone deacetylase, could kill cancer cells overexpressing TDP1 in rhabdomyosarcoma (Duffy et al., 2016).
Conditional synthetic lethal interactions are dependent on certain internal conditions, such as high ROS (reactive oxygen species), hypoxia, etc., or external conditions, such as radiation and drugs. Without these conditions, the synthetic lethal relationship does not hold. PARP inhibition itself did not affect homologous recombination (HR) expression or function. But PRAP inhibition increases clonogenic killing in HR-deficient hypoxic cells. In glioblastoma cell lines that contain mutations in the cohesin component STAG2 (stromal antigen 2), PARP inhibition resulted in synthetic cytotoxicity in response to the DNA-damaging chemotherapeutic temozolomide (Bailey et al., 2014; Chan et al., 2010).
Synthetic lethality mechanism
Multiple factors, such as the genetic background, tumor immune microenvironment, tumor metabolism, and the DDR pathway, affect the mechanism of synthetic lethality. Here we divide the mechanism of synthetic lethality into the DNA repair level, cellular signaling pathway level, cell cycle regulation level, metabolism level, and epigenetic regulation level.
Synthetic lethality at the DNA repair level
The DDR is a complex network of signaling pathways that cells develop to respond to the genetic alterations induced by exogenous or endogenous lesions inflicted on their DNA. Two genes that are important for genomic stability maintenance are usually synthetic lethal. Several synthetic lethal interactions are based on DDR. PARP and BRCA1/2 are synthetic lethal partners which have been used for synthetic lethality therapeutic strategies in clinics. PARP can recognize DNA single-strand breaks (SSBs), which is a common cell damage. BRCA2 is an important tumor suppressor which promotes the formation of RAD51 filaments at DNA breaks in HR-mediated DNA damage repair (Davies et al., 2001). Inhibition of PARP converts SSBs into DSBs during replication, which can be repaired by BRCA-dependent HR. But in the BRCA1/2-deficit cells, accumulation of the irreparable toxic DSBs will result in replication fork collapse, genomic instability, and cell death (Helleday, 2011; Fig. 2). Four-stranded G-quadruplex (G4) structures of DNA increase the genomic instability, leading to more DNA damage which also relies on the HR pathway to repair (Varshney et al., 2020). CX-5461 and its related drug CX-3543 are G4 stabilizers, with specific toxicity to HR and non-homologous end joining (NHEJ) deficient cancer cells (Xu et al., 2017). BRCA1/2 inactivation not only creates vulnerability to PARP inhibitors and G4 ligands but also has a synthetic lethal relationship with APEX2 (apurinic/apyrimidinic endodeoxyribonuclease 2) and FEN1 (flap structure-specific endonuclease 1). Chemically inhibiting APEX2 and FEN1 leads to BRCA-deficient cell death (Mengwasser et al., 2019).
Fig.2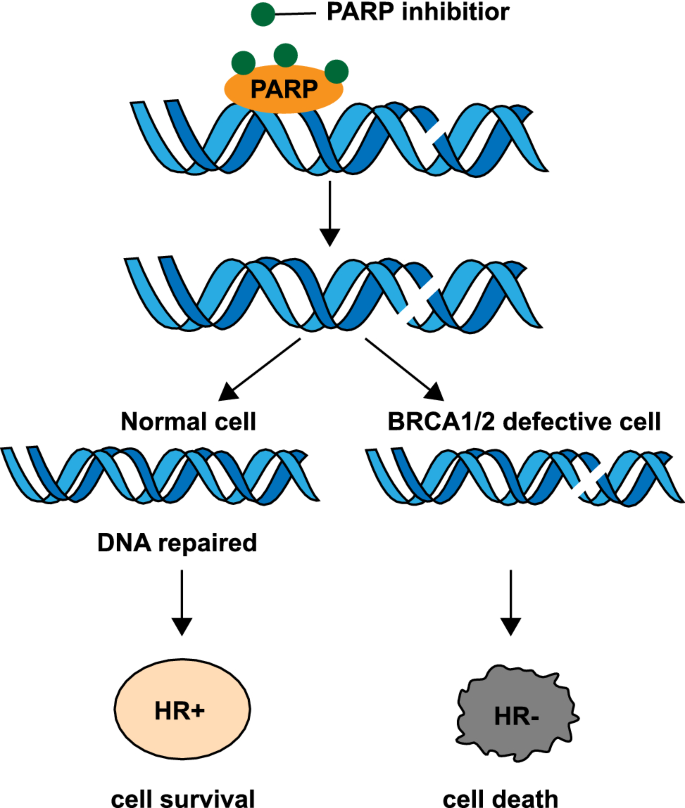
PARP inhibitors and BRCA1/2 defection. PARP inhibitors trap the PARP proteins on the DNA repair intermediates, causing BRCA1/2-defect cells to accumulate more toxic DSBs, thus leading to cell death
DNA polymerase θ (Polθ also known as POLQ, encoded by POLQ) plays an important role in the NHEJ pathway. Polθ-mediated repair pathway and the HR pathway form a synthetic lethal partner in epithelial ovarian cancers (EOCs; Ceccaldi et al., 2015). As the Polθ-mediated NHEJ pathway has a complementary relationship with the HR pathway, the knockdown of Polθ upregulates HR activity in normal cells while enhancing cell death in HR-deficient EOCs. Consistent with these results, genetic inactivation of an HR gene (Fancd2) and POLQ in mice results in embryonic lethality. This is a representative example of synthetic lethality at the gene level.
Defects in AT-rich interactive domain 1A (ARID1A), a component of the SWI/SNF chromatin remodeling complex, sensitize tumor cells to clinical inhibitors of ATR (Ataxia-Telangiectasia Mutated (ATM) and Rad3-related protein kinase), a critical component of the cellular DDR. Mechanistically, inhibition of ATR triggers apoptosis in ARID1A-deficient cells as ARID1A deficiency results in topoisomerase 2A and cell cycle defects, which cause an increased reliance on ATR checkpoint activity (Williamson et al., 2016). Another example is that ARID1A deficiency impairs DSBs end resection and thereby impairs DSBs-induced ATR signaling, which sensitizes cancer cells to PARP inhibitors in vivo and in vitro (Shen et al., 2015).
Microsatellite instability (MSI) is a form of genetic hypermutability that arises from impaired mismatch repair. There is a synthetic lethal relationship between the RecQ DNA helicase, Werner syndrome protein (WRN), and MSI (Behan et al., 2019; Chan et al., 2016; Kategaya et al., 2019). Recently found that TA-dinucleotide repeats undergo large-scale expansions and form non-B DNA secondary structures in MSI cells. These structures stall replication forks, activate the ATR checkpoint kinase, and require unwinding by the WRN helicase. In the absence of WRN, the expanded TA-dinucleotide repeats are susceptible to cleavage by the MUS81 nuclease, leading to massive chromosome shattering. This provides a new synthetic lethal therapeutic that targets WRN for MSI-associated cancers (van Wietmarschen et al., 2020).
Synthetic lethality at the cellular signaling pathway level
Establishing synthetic lethal interactions at the pathway level is more significant, as proteins are the gene expression products that play a direct role in cell life activities. After identifying the synthetic lethal interaction of genes, the mechanisms involved in these crucial pathways will be further studied at the protein level by researchers. This section will discuss the synthetic lethality mechanism at the signaling pathway level.
ARID1A is a tumor suppressor with a high frequency of inactivating mutations in many cancers, such as ovarian clear cell carcinoma (Wiegand et al., 2010), gastric cancer (Wang et al., 2011), and colorectal cancer (Muzny et al., 2012). The enhancer of zeste homolog 2 (EZH2), a histone methyltransferase and catalytic component of PRC2 (polycomb repressive complex 2), generates the lysine 27 trimethylation mark on histone H3 (H3K27me3) by its catalytic SET domain to silence gene expression (Cao & Zhang, 2004). ARID1A, which is mutated in over 50% of ovarian clear carcinomas, has a synthetic lethal interaction with EZH2. Benjamin G. Bitler et al. demonstrated that this synthetic lethal interaction is based on PI3K/AKT signaling (Bitler et al., 2015). Inhibition of EZH2 causes ARID1A-mutated ovarian cancer cell death by inhibiting PI3K/AKT signaling (Fig. 3). Moreover, EZH2 inhibitors also decreased the viability of ARID1A-deficient cells in a dose-dependent manner by PI3K/AKT signaling in gastric cancer (Yamada et al., 2021).
Fig.3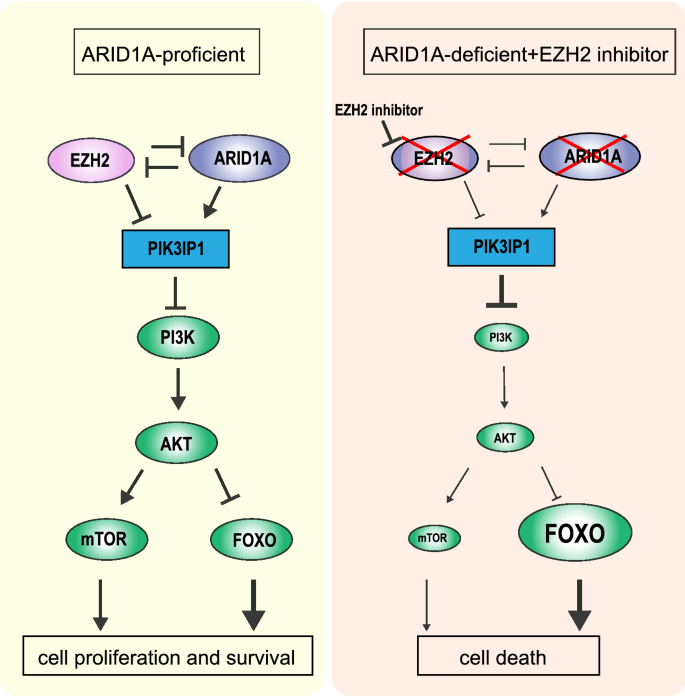
Synthetic lethality between ARID1A and EZH2. In ARID1A-deficient cells, inhibition of EZH2 leads to inhibition of mTOR and overexpression of FOXO, causing cell death
Mutations in the Ras family of GTPases are associated with poor clinical prognoses, which are commonly found in acute myeloid leukemia (AML) and many other human cancers (Cox et al., 2014). Wang et al., (2017) found that all Ras-driven cancers require PAK activity to fully activate MAPK signaling, with each cancer subtype activating the PAKs via distinct mechanisms. The enzymes catalyzing the latter two steps of the Ras processing pathway, Rce1, and Icmt, do display synthetic lethality with oncogenic Ras. It might be a new synthetic lethal therapeutic target.
MyD88 loss and mutations in the Wnt/β-catenin pathway constitute a pair of synthetic lethality partners in intestinal tumor epithelial cells (Kajino-Sakamoto et al., 2021). MyD88 is an adaptor protein of the TLR (toll-like receptor)/IL-1β (interleukin-1β) signaling that has been implicated as a signal transducer in the integrity of the intestines as well as in their tumorigenesis (Takeda et al., 2003). MyD88 deficiency resulted in the inactivation of the JNK-mTORC1, NF-κB, and Wnt/β-catenin pathways in tumor cells, all of which play important roles in the carcinogenesis and maintenance of colorectal cancer (CRC; Fig. 4).
Fig.4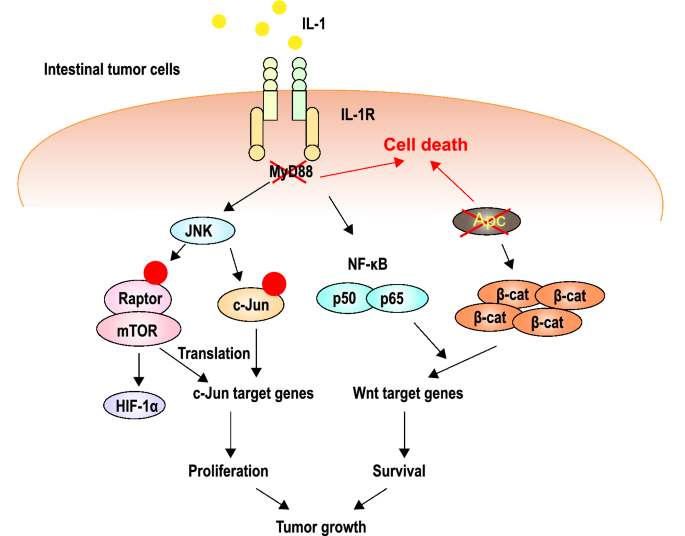
APC inactivation results in constitutive activation of Wnt/β-catenin signaling, which is required for the maintenance of advanced CRC. Inhibition of MyD88 causes inactivation of the tumor cell proliferation signaling pathways JNK-mTORC1, NF-κB, and Wnt/β-catenin. The combination of the two causes cell death
Synthetic lethality in cell cycle regulation
Cancer is characterized by disruption of cell cycle checkpoints, which evades growth suppressors and sustains cell proliferation. Dysregulation of the cyclin D-cyclin-dependent kinase 4/6 (CDK4/6) pathway mediated G1-S transition provides a new therapeutic strategy of synthetic lethality (Walker et al., 1998). B-rapidly accelerated fibrosarcoma (BRAF) is a serine/threonine kinase that is mutated in more than 60% of malignant melanomas and causes over-proliferation of melanoma cells (Millington, 2013). Checkpoint kinase 1 (CHK1) is a serine/threonine-specific protein kinase that coordinates the DDR and cell cycle checkpoint response at several cellular stages. Melanoma cells use a feedback loop between BRAF and CHK1 to manage replication stress, minimize DNA damage, and sustain cell proliferation. Inhibition of both of these proteins leads to impeded cell cycle arrest, aberrant mitosis, and apoptosis (Barnaba & LaRocque, 2021). AMBRA1 (activating molecule in beclin-1-regulated autophagy), a scaffold factor that regulates cyclin D to guard S-phase entry and genomic integrity, has a synthetic lethal relationship with CHK1. AMBRA1 loss leads to replication stress, resulting in DNA damage. Through CHK1 signaling, cells can tolerate replication stress. AMBRA1 deficiency increases the sensitivity to CHK1 inhibition, leading to synthetic lethality that could be exploited in cancer treatments (Maiani et al., 2021).
WEE1 is a nuclear serine/threonine-protein kinase that directly phosphorylates tyrosine residues on CDK1, thereby inactivating it and effectively inducing cell cycle arrest at the G2-M transition (McGowan & Russell, 1993). Similar to CHK1, this protein is essential for tumor cells to induce cell cycle arrest when presented with DNA damage. Combined treatment of CHK1 and WEE1 inhibitors has synergistic antitumor effects in head and neck cancer (van Harten et al., 2019), ovarian cancer (Carrassa et al., 2012), and neuroblastoma (Russell et al., 2013). Low-dose CHK1 inhibition and gemcitabine combined with high-dose WEE1 inhibition have also been shown to synergistically suppress pancreatic adenocarcinoma growth while maintaining tolerability by limiting toxic adverse effects (Koh et al., 2018).
The prototypical tumor suppressor genes, retinoblastoma or RB1, which are mutated in some of the most aggressive and hard-to-treat malignancies such as small-cell lung cancer (SCLC) and triple-negative breast cancer, have a synthetic lethal relationship with aurora kinase A (AURKA; Burkhart & Sage, 2008). The function of the RB1 product, RB1, in controlling the G1-S transition in the cell cycle is well understood (Sherr & McCormick, 2002). Multiple groups have reported that loss of RB1 leads to a hyperactivated or “primed” spindle-assembly checkpoint (SAC; Manning & Dyson, 2012). SAC is essential for AURKA inhibitor cytotoxicity in RB1-deficient cancers. Gong et al. uncover that AURKA inhibitor leads to durable regression of RB1mut tumor xenografts at exposures that are well tolerated in rodents (Gong et al., 2019). ARID1A has a synthetic lethal interaction with AURKA in CRC cells. Inhibiting AURKA activity in ARID1A-deficient cells significantly increases G2/M arrest and induces cellular multinucleation and apoptosis (Wu et al., 2018).
Synthetic lethality in cell metabolism
Metabolic reprogramming is a hallmark of cancer and plays an important role in maintaining the tumor microenvironment. Some synthetic lethality strategies focus on cell metabolism. Glutamate-cysteine ligase synthetase catalytic subunit (GCLC) is a rate-limiting enzyme for antioxidant glutathione (GSH) synthesis. Hideaki Ogiwara et al. found that ARID1A-deficient cancer cells are specifically vulnerable to inhibition of GSH and GCLC because inhibition of GCLC markedly decreased GSH in ARID1A-deficient cancer cells, leading to apoptotic cell death triggered by excessive amounts of ROS (Ogiwara et al., 2019). Further studies demonstrate that the vulnerability of ARID1A-deficient cancer cells to GSH inhibition is caused by decreased GSH synthesis due to impaired SLC7A11 expression, which is regulated by ARID1A-mediated chromatin remodeling. Thus, the synthetic lethality interaction between ARID1A and SLC7A11 is through cell metabolism.
KRAS is a major driver in lung adenocarcinoma. The SLC7A11/glutathione axis displays metabolic synthetic lethality with oncogenic KRAS (Hu et al., 2020). Mutant KRAS drives the reprogramming of glutathione metabolism, resulting in increased intracellular cystine levels and glutathione biosynthesis (Cox et al., 2014; Lv et al., 2016). SLC7A11 (solute carrier family 7 member 11) is the functional light chain subunit of the cystine/glutamate antiporter system xc− and confers specificity for cystine uptake, a major rate-limiting factor in glutathione biosynthesis (Koppula et al., 2018). Silencing SLC7A11 by genetic depletion or inhibiting by SLC7A11 inhibitor, HG106, resulted in selective killing across a panel of KRAS mutant cancer cells in vitro and tumor growth inhibition in vivo. Furthermore, HG106 increased oxidative stress- and ER stress-mediated cell apoptosis in KRAS-mutant cells (Hu et al., 2020).
IDH1 (isocitrate dehydrogenase) and Nrf2 (nuclear factor-erythroid-derived 2-like 2) are other synthetic lethal partners based on GSH metabolism. IDH1 mutation is a common genetic abnormality in human malignancies such as gliomas (Yan et al., 2009). IDH1-mutated cancer cells exhibit addiction to Nrf2-governed antioxidative pathways. Blockade of Nrf2 transcriptional activity by triptolide compromised the expression of GCLC, GCLM, and SLC7A11, which are essential regulatory genes for glutathione metabolism, leading to apoptotic changes in IDH1-mutated cells via redox catastrophe (Yu et al., 2020; Fig. 5).
Fig.5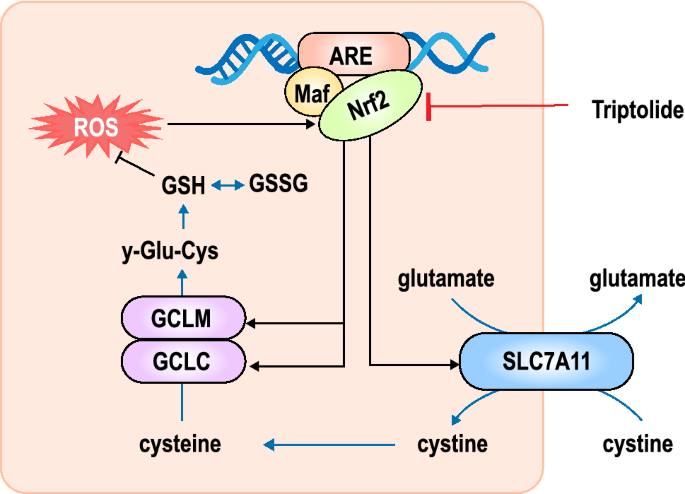
Inhibition of the transcriptional activity of Nrf2 by triptolide leads to down-regulation of the expression of GCLC, GCLM, and SLC7A11, resulting in the disruption of the glutathione metabolism and increasing intracellular oxidative damage, leading to cell apoptosis in IDH1 mutant glioma cells
SMARCA4 (SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily A, member 4) is one of the components of the SWI/SNF chromatin remodeling complex, which has a synthetic lethal interaction with oxidative phosphorylation (OXPHOS). SMARCA4 acts as a bona fide tumor suppressor and cooperates with p53 loss and Kras activation in lung cancer. Gene expression analyses revealed the signature of enhanced OXPHOS in SMARCA4 mutant tumors. Thus, inhibition of OXPHOS by small molecular inhibitors creates a new energy-driving synthetic lethality therapeutic strategy by limiting oxygen consumption and decreasing respiratory capacity (Lissanu Deribe et al., 2018).
Synthetic lethality in epigenetic regulation
Epigenetic modifications have been found to play a more and more important role in the onset and development of malignant tumors. Many epigenetic-related synthetic lethal targets have been identified and small molecule inhibitors have been developed. Histone acetyl-transferase gene CREBBP and its paralogue EP300 were found to have a synthetic lethal relationship (Morel et al., 2017; Ogiwara et al., 2016). CREBBP and EP300 encode mammalian enzymes which are expressed ubiquitously. These enzymes interact with more than 400 transcription factors and catalyze the modification of lysines on both histone and non-histone proteins, playing roles as global transcriptional co-activators. In diffuse large B cell lymphoma (DLBCL) and follicular lymphoma (FL), these two enzymes share common as well as distinct transcriptional targets in sub-compartments of the germinal center (GC) reaction. Research shows that CREBBP-DLBCL might depend on the residual EP300 activity. Therefore, inhibition of CREBBP and EP300 was preferentially toxic to CREBBP-mutated DLBCL (Meyer et al., 2019).
H3K27M and isocitrate-dehydrogenase 1 (IDH1) form a synthetic lethal partner. Because they hijack a conserved and critical metabolic pathway in opposing ways to maintain their preferred epigenetic state, H3.3K27M and IDH1 mutation (mIDH1) are mutually exclusive (Chung et al., 2020). The tricarboxylic acid (TCA) cycle intermediate alpha-ketoglutarate (α-KG), which serves as a critical co-factor for H3K27M demethylases. Inhibition of enzymes related to α-KG generation, such as IDH1, resulted in global H3K27me3, which contributed to shifting chromatin accessibility to mainly closed states, thus suppressing tumor cell proliferation. During the further in vivo experiment, inhibition of IDH1 activity prolonged overall survival in diffuse intrinsic pontine gliomas (DIPG) animal models (Fig. 6). Therefore, inhibition of IDH is a new synthetic lethal therapeutic which shows maximal beneficial effects.
Fig.6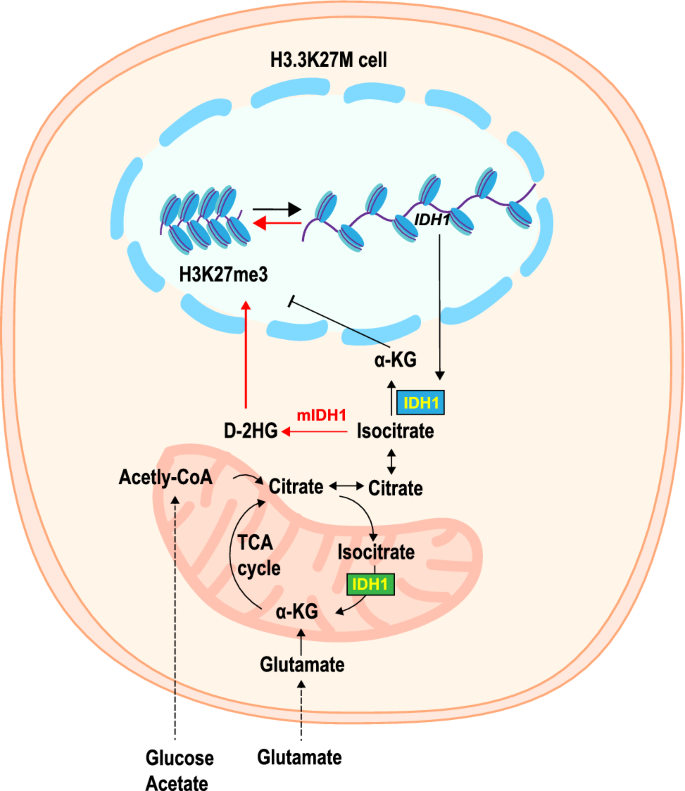
Glycolysis, glutaminolysis, and TCA cycle metabolism were enhanced with high α-KG production to maintain low H3K27me3 in H3.3K27M cells. Inhibition of key enzymes in glycolysis or glutaminolysis increased H3K27me3, altering chromatin accessibility
In PTEN-deficient tumors, the chromatin helicase DNA-binding factor CHD1 has been identified as a possible synthetic essential gene (Zhao et al., 2017). CHD1 depletion profoundly and specifically suppressed cell proliferation, survival, and tumorigenic potential in PTEN-deleted prostate and breast cancers. Mechanistically, PTEN mediates the degradation of CHD1 via the ubiquitination-proteasome process by the AKT/GSK3β/β-TrCP pathway. But in cancer, PTEN deficiency results in CHD1 protein stabilization, which engages and maintains the H3K4me3 mark to activate cancer promoting gene expression, including the TNFα/NF-κB network, which is known to promote prostate cancer (Huang et al., 2001; Jin et al., 2015; Karin & Greten, 2005). Far more research has found that deletion of CHD1 in PTEN-null models of prostate cancer profoundly impairs tumor progression in conjunction with dramatic remodeling of the TME towards an anti-tumor immune profile (Zhao et al., 2020). The mutually exclusive deletion pattern of PTEN and CHD1 is also seen in breast and colorectal adenocarcinoma, in addition to prostate cancer (Zhao et al., 2017).
Clinical development of synthetic lethality
With the mechanism behind synthetic lethality being unveiled, more and more preclinical studies have targeted inhibiting synthetic lethal partners. In 2005, Helleday and Ashworth discovered that PARP and HR-related gene BRCA1/2 were the classical synthetic lethal partners (Bryant et al., 2005; Farmer et al., 2005). BRCA1/2 mutant cells are more sensitive to PARP inhibition than normal cells. After that, the PARP inhibitors developed rapidly, and several PARP inhibitors have been applied in clinical practice for BRCA1/2-mutant tumors. In addition to PARP inhibitors, drugs based on other synthetic lethal relationships, including both oncogenes and tumor-suppressor genes such as TP53, KRAS, and MYC also have been tested in clinical trials (Table 1).


Table 1 Clinical trials on DDR targets based on synthetic lethality
ATR inhibition is synthetically lethal to TP53- or ATM-defective chronic lymphocytic leukemia (CLL) cells (Kwok et al., 2015, 2016). ATR, a serine/threonine protein kinase, regulates replication initiation, preventing aberrant and excessive replication origin firing, and also mediates the response to replication stress, stabilizing and repairing DNA replication forks (Couch et al., 2013; Toledo et al., 2013; Zeman & Cimprich, 2014). ATR inhibition makes functional ATM and p53 necessary for maintaining genome integrity, with ATM being necessary for homologous recombination repair (HRR) to restore replication fork structure and both ATM and p53 for arresting cell cycle advancement to permit repair (Bieging et al., 2014; Shiloh & Ziv, 2013). Previously, ATR deletion in p53-deficient mice and ATR inhibition in tumor cell lines have demonstrated this synthetically lethal interaction between ATR and p53 or ATM deficiency (Reaper et al., 2011; Ruzankina et al., 2009; Sangster-Guity et al., 2011). AZD6738, a novel ATR inhibitor, has been demonstrated in-vitro and in-vivo efficacy in a novel therapeutic approach that specifically targets p53-null or ATM-null CLL cells (Kwok et al., 2015).In addition, non-small cell lung cancer cells defective in both ATM and p53 are particularly sensitive to ATR inhibition in vitro (Weber et al., 2013).
Conclusion
Synthetic lethality is merely a simple concept in genetics, but it has great potential for cancer treatment. The concept of synthetic lethality provides a new path for the exploration of cancer treatment by targeting the synthetic lethal targets that make sense for the treatment of cancer caused by the inactivation of tumor suppressor genes. In this review, we propose a systematic classification of synthetic lethality and summarize the synthetic lethal mechanism. The mechanism behind synthetic lethality has been gradually revealed and has been used for clinical treatment guidelines, and multiple inhibitors based on synthetic lethal pairs have been entered into clinical trials. Targeting synthetic lethal partners could expand the effects of cancer therapy and reduce the side effects while increasing the therapeutic window and lowering the effective drug doses. Yeast screens, drug screens, RNA interference (RNAi) screens, clustered regularly interspaced short palindromic repeats (CRISPR) screens, and bioinformatics screens have been used to help identify and validate potential synthetic lethal partner genes. With more synthetic lethal partners being identified, changes move from novel target discovery to clinically effective medicines. Although multiple DDR inhibitors, such as PARP inhibitors, have been developed and tested in various stages and some progress has been made, the emergence of drug resistance to PARP inhibitors raises a new challenge. According to the concept of conditional synthetic lethality, the internal and external settings also needed to be taken into consideration while exploring new synthetic lethal relationships. Furthermore, synthetic lethal effects in multiple pathways is an important future direction with promising prospects for resolving the issue of synthetic lethal effects resistance. Drug combinations are more effective at combatting cancer than a single agent, such as PARP inhibitors can exacerbate levels of replication stress, micronuclei, and R-loops in PBRM1(Polybromo 1)-defective cells. Combining with ATR inhibitors can induce cyclin GMP-AMP synthase/stimulator of interferon genes (cGAS/STING) innate immune signaling, which provides possible methods for patients with PBRM1-defective clear cell renal cell carcinomas based on synthetic lethality (Chabanon et al., 2021). This therapeutic strategy of combining two DDR inhibitors provides a novel direction for synthetic lethal treatment. We are certain that synthetic lethality merits additional investigation and implementation in the realm of cancer therapy in the future.
Data availability
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
References
Bailey, M. L., O’Neil, N. J., van Pel, D. M., Solomon, D. A., Waldman, T., & Hieter, P. (2014). Glioblastoma cells containing mutations in the cohesin component STAG2 are sensitive to PARP inhibition. Molecular Cancer Therapeutics, 13, 724–732. https://doi.org/10.1158/1535-7163.MCT-13-0749
Barnaba, N., & LaRocque, J. R. (2021). Targeting cell cycle regulation via the G2-M checkpoint for synthetic lethality in melanoma. Cell Cycle, 20, 1041–1051. https://doi.org/10.1080/15384101.2021.1922806
Behan, F. M., Iorio, F., Picco, G., Gonçalves, E., Beaver, C. M., Migliardi, G., Santos, R., Rao, Y., Sassi, F., Pinnelli, M., et al. (2019). Prioritization of cancer therapeutic targets using CRISPR–Cas9 screens. Nature, 568, 511–516. https://doi.org/10.1038/s41586-019-1103-9
Bender, A., & Pringle, J. R. (1991). Use of a screen for synthetic lethal and multicopy suppressee mutants to identify two new genes involved in morphogenesis in Saccharomyces cerevisiae. Molecular and Cellular Biology, 11, 1295–1305. https://doi.org/10.1128/MCB.11.3.1295-1305.1991
Bian, Y., Kitagawa, R., Bansal, P. K., Fujii, Y., Stepanov, A., & Kitagawa, K. (2014). Synthetic genetic array screen identifies PP2A as a therapeutic target in Mad2-overexpressing tumors. Proceedings of the National Academy of Sciences, 111, 1628–1633. https://doi.org/10.1073/PNAS.1315588111
Bieging, K. T., Mello, S. S., & Attardi, L. D. (2014). Unravelling mechanisms of p53-mediated tumour suppression. Nature Reviews Cancer, 14, 359–370. https://doi.org/10.1038/NRC3711
Bitler, B. G., Aird, K. M., Garipov, A., Li, H., Amatangelo, M., Kossenkov, A. V., Schultz, D. C., Liu, Q., Shih, I. M., Conejo-Garcia, J. R., et al. (2015). Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nature Medicine, 21, 231–238. https://doi.org/10.1038/nm.3799
Brown, J. S., O’Carrigan, B., Jackson, S. P., & Yap, T. A. (2017). Targeting DNA repair in cancer: Beyond PARP inhibitors. Cancer Discovery, 7, 20–37. https://doi.org/10.1158/2159-8290.CD-16-0860
Bryant, H. E., Schultz, N., Thomas, H. D., Parker, K. M., Flower, D., Lopez, E., Kyle, S., Meuth, M., Curtin, N. J., & Helleday, T. (2005). Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature, 434, 913–917. https://doi.org/10.1038/NATURE03443
Burkhart, D. L., & Sage, J. (2008). Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nature Reviews Cancer, 8, 671–682. https://doi.org/10.1038/NRC2399
Cao, R., & Zhang, Y. (2004). The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Current Opinion in Genetics & Development, 14, 155–164. https://doi.org/10.1016/J.GDE.2004.02.001
Carrassa, L., Chilà, R., Lupi, M., Ricci, F., Celenza, C., Mazzoletti, M., Broggini, M., & Damia, G. (2012). Combined inhibition of Chk1 and Wee1: In vitro synergistic effect translates to tumor growth inhibition in vivo. Cell Cycle, 11, 2507–2517. https://doi.org/10.4161/CC.20899
Ceccaldi, R., Liu, J. C., Amunugama, R., Hajdu, I., Primack, B., Petalcorin, M. I. R., O’Connor, K. W., Konstantinopoulos, P. A., Elledge, S. J., Boulton, S. J., et al. (2015). Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature, 518, 258–262. https://doi.org/10.1038/nature14184
Chabanon, R. M., Morel, D., Eychenne, T., Colmet-Daage, L., Bajrami, I., Dorvault, N., Garrido, M., Meisenberg, C., Lamb, A., Ngo, C., et al. (2021). PBRM1 deficiency confers synthetic lethality to DNA repair inhibitors in cancer. Cancer Research, 81, 2888–2902. https://doi.org/10.1158/0008-5472.CAN-21-0628
Chan, E. M., Tsukasa, S., McFarland, J. M., Gaeta, B., Dumont, N., Gonzalez, A., McPartlan, J. S., Li, T., Zhang, Y., Liu, J. B., et al. (2016). WRN helicase is a synthetic lethal target in microsatellite unstable cancers Edmond. Physiology & Behavior, 176, 139–148. https://doi.org/10.1038/s41586-019-1102-x.WRN
Chan, N., Pires, I. M., Bencokova, Z., Coackley, C., Luoto, K. R., Bhogal, N., Lakshman, M., Gottipati, P., Oliver, F. J., Helleday, T., et al. (2010). Contextual synthetic lethality of cancer cell kill based on the tumor microenvironment. Cancer Research, 70, 8045–8054. https://doi.org/10.1158/0008-5472.CAN-10-2352
Chung, C., Sweha, S. R., Pratt, D., Tamrazi, B., Panwalkar, P., Banda, A., Bayliss, J., Hawes, D., Yang, F., Lee, H. J., et al. (2020). Integrated metabolic and epigenomic reprograming by H3K27M mutations in diffuse intrinsic pontine gliomas. Cancer Cell, 38, 334-349.e9. https://doi.org/10.1016/J.CCELL.2020.07.008
Couch, F. B., Bansbach, C. E., Driscoll, R., Luzwick, J. W., Glick, G. G., Bétous, R., Carroll, C. M., Jung, S. Y., Qin, J., Cimprich, K. A., et al. (2013). ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes & Development, 27, 1610–1623. https://doi.org/10.1101/GAD.214080.113
Cox, A. D., Fesik, S. W., Kimmelman, A. C., Luo, J., & Der, C. J. (2014). Drugging the undruggable RAS: Mission possible? Nature Reviews Drug Discovery, 13, 828–851. https://doi.org/10.1038/nrd4389
Davies, A. A., Masson, J. Y., McIlwraith, M. J., Stasiak, A. Z., Stasiak, A., Venkitaraman, A. R., & West, S. C. (2001). Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Molecular Cell, 7, 273–282. https://doi.org/10.1016/S1097-2765(01)00175-7
Duffy, S., Fam, H. K., Wang, Y. K., Styles, E. B., Kim, J. H., Ang, J. S., Singh, T., Larionov, V., Shah, S. P., Andrews, B., et al. (2016). Overexpression screens identify conserved dosage chromosome instability genes in yeast and human cancer. Proceedings of the National Academy of Sciences, 113, 9967–9976. https://doi.org/10.1073/PNAS.1611839113
Emerling, B. M., Hurov, J. B., Poulogiannis, G., Tsukazawa, K. S., Choo-Wing, R., Wulf, G. M., Bell, E. L., Shim, H. S., Lamia, K. A., Rameh, L. E., et al. (2013). Depletion of a putatively druggable class of phosphatidylinositol kinases inhibits growth of p53-null tumors. Cell, 155, 844. https://doi.org/10.1016/J.CELL.2013.09.057
Farmer, H., McCabe, H., Lord, C. J., Tutt, A. H. J., Johnson, D. A., Richardson, T. B., Santarosa, M., Dillon, K. J., Hickson, I., Knights, C., et al. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature, 434, 917–921. https://doi.org/10.1038/NATURE03445
George, A., Kaye, S., & Banerjee, S. (2017). Delivering widespread BRCA testing and PARP inhibition to patients with ovarian cancer. Nature Reviews Clinical Oncology, 14, 284–296. https://doi.org/10.1038/nrclinonc.2016.191
Gong, X., Du, J., Parsons, S. H., Merzoug, F. F., Webster, Y., Iversen, P. W., Chio, L. C., Van Horn, R. D., Lin, X., Blosser, W., et al. (2019). Aurora A kinase inhibition is synthetic lethal with loss of the RB1 tumor suppressor gene. Cancer Discovery, 9, 248–263. https://doi.org/10.1158/2159-8290.CD-18-0469
Hanahan, D., & Weinberg, R. A. (2011). Hallmarks of cancer: The next generation. Cell, 144, 646–674. https://doi.org/10.1016/J.CELL.2011.02.013
Helleday, T. (2011). The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Molecular Oncology, 5, 387–393. https://doi.org/10.1016/j.molonc.2011.07.001
Hennessy, K. M., Lee, A., Chen, E., & Botstein, D. (1991). A group of interacting yeast DNA replication genes. Genes & Development, 5, 958–969. https://doi.org/10.1101/GAD.5.6.958
Hu, K., Li, K., Lv, J., Feng, J., Chen, J., Wu, H., Cheng, F., Jiang, W., Wang, J., Pei, H., et al. (2020). Suppression of the SLC7A11/glutathione axis causes synthetic lethality in KRAS-mutant lung adenocarcinoma. The Journal of Clinical Investigation, 130, 1752–1766. https://doi.org/10.1172/JCI124049
Hu, Y., & Guo, M. (2020). Synthetic lethality strategies: Beyond BRCA1/2 mutations in pancreatic cancer. Cancer Science, 111, 3111–3121. https://doi.org/10.1111/CAS.14565
Huang, S., Pettaway, C. A., Uehara, H., Bucana, C. D., & Fidler, I. J. (2001). Blockade of NF-kappaB activity in human prostate cancer cells is associated with suppression of angiogenesis, invasion, and metastasis. Oncogene, 20, 4188–4197. https://doi.org/10.1038/SJ.ONC.1204535
Jin, R., Yamashita, H., Yu, X., Wang, J., Franco, O. E., Wang, Y., Hayward, S. W., & Matusik, R. J. (2015). Inhibition of NF-kappa B signaling restores responsiveness of castrate-resistant prostate cancer cells to anti-androgen treatment by decreasing androgen receptor-variant expression. Oncogene, 34, 3700–3710. https://doi.org/10.1038/ONC.2014.302
Kaiser, C. A., & Schekman, R. (1990). Distinct sets of SEC genes govern transport vesicle formation and fusion early in the secretory pathway. Cell, 61, 723–733. https://doi.org/10.1016/0092-8674(90)90483-U
Kajino-Sakamoto, R., Fujishita, T., Taketo, M. M., & Aoki, M. (2021). Synthetic lethality between MyD88 loss and mutations in Wnt/β-catenin pathway in intestinal tumor epithelial cells. Oncogene, 40, 408–420. https://doi.org/10.1038/s41388-020-01541-3
Karin, M., & Greten, F. R. (2005). NF-kappaB: Linking inflammation and immunity to cancer development and progression. Nature Reviews Immunology, 5, 749–759. https://doi.org/10.1038/NRI1703
Kategaya, L., Perumal, S. K., Hager, J. H., & Belmont, L. D. (2019). Werner syndrome helicase is required for the survival of cancer cells with microsatellite instability. Iscience, 13, 488–497. https://doi.org/10.1016/J.ISCI.2019.02.006
Koh, S. B., Wallez, Y., Dunlop, C. R., de Quirós, B., Fernández, S., Bapiro, T. E., Richards, F. M., & Jodrell, D. I. (2018). Mechanistic distinctions between CHK1 and WEE1 inhibition guide the scheduling of triple therapy with gemcitabine. Cancer Research, 78, 3054–3066. https://doi.org/10.1158/0008-5472.CAN-17-3932
Koppula, P., Zhang, Y., Zhuang, L., & Gan, B. (2018). Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Communications, 38, 12. https://doi.org/10.1186/S40880-018-0288-x
Kwok, M., Davies, N., Agathanggelou, A., Smith, E., Oldreive, C., Petermann, E., Stewart, G., Brown, J., Lau, A., Pratt, G., et al. (2016). ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood, 127, 582–595. https://doi.org/10.1182/BLOOD-2015-05-644872
Kwok, M., Davies, N., Agathanggelou, A., Smith, E., Petermann, E., Yates, E., Brown, J., Lau, A., & Stankovic, T. (2015). Synthetic lethality in chronic lymphocytic leukaemia with DNA damage response defects by targeting the ATR pathway. Lancet (london, England), 385(Suppl 1), S58. https://doi.org/10.1016/S0140-6736(15)60373-7
Lissanu Deribe, Y., Sun, Y., Terranova, C., Khan, F., Martinez-Ledesma, J., Gay, J., Gao, G., Mullinax, R. A., Khor, T., Feng, N., et al. (2018). Mutations in the SWI/SNF complex induce a targetable dependence on oxidative phosphorylation in lung cancer. Nature Medicine, 24, 1047–1057. https://doi.org/10.1038/S41591-018-0019-5
Lord, C. J., & Ashworth, A. (2017). PARP inhibitors: Synthetic lethality in the clinic. Science, 355(6330), 1152–1158.
Lucchesi, J. C. (1968). Synthetic lethality and semi-lethality among functionally related mutants of Drosophila melanfgaster. Genetics, 59, 37–44.
Lv, J., Wang, J., Chang, S., Liu, M., & Pang, X. (2016). The greedy nature of mutant RAS: A boon for drug discovery targeting cancer metabolism? Acta Biochimica Et Biophysica Sinica (shanghai), 48, 17–26. https://doi.org/10.1093/ABBS/GMV102
Macheret, M., & Halazonetis, T. D. (2015). DNA replication stress as a hallmark of cancer. Annual Review of Pathology: Mechanisms of Disease, 10, 425–448. https://doi.org/10.1146/ANNUREV-PATHOL-012414-040424
Maiani, E., Milletti, G., Nazio, F., Holdgaard, S. G., Bartkova, J., Rizza, S., Cianfanelli, V., Lorente, M., Simoneschi, D., Di Marco, M., et al. (2021). AMBRA1 regulates cyclin D to guard S-phase entry and genomic integrity. Nature, 592, 799–803. https://doi.org/10.1038/S41586-021-03422-5
Manning, A. L., & Dyson, N. J. (2012). RB: Mitotic implications of a tumour suppressor. Nature Reviews Cancer, 12, 220–226. https://doi.org/10.1038/NRC3216
McGowan, C. H., & Russell, P. (1993). Human Wee1 kinase inhibits cell division by phosphorylating p34cdc2 exclusively on Tyr15. EMBO Journal, 12, 75–85.
Mengwasser, K. E., Adeyemi, R. O., Leng, Y., Choi, M. Y., Clairmont, C., D’Andrea, A. D., & Elledge, S. J. (2019). Genetic screens reveal FEN1 and APEX2 as BRCA2 synthetic lethal targets. Molecular Cell, 73, 885. https://doi.org/10.1016/J.MOLCEL.2018.12.008
Meyer, S. N., Scuoppo, C., Vlasevska, S., Bal, E., Holmes, A. B., Holloman, M., Garcia-Ibanez, L., Nataraj, S., Duval, R., Vantrimpont, T., et al. (2019). Unique and shared epigenetic programs of the CREBBP and EP300 acetyltransferases in germinal center B cells reveal targetable dependencies in lymphoma. Immunity, 51, 535-547.e9. https://doi.org/10.1016/J.IMMUNI.2019.08.006
Millington, G. W. M. (2013). Mutations of the BRAF gene in human cancer, by Davies et al. (Nature 2002; 417: 949-54). Clinical and Experimental Dermatology, 38, 222–223. https://doi.org/10.1111/CED.12015
Morel, D., Almouzni, G., Soria, J. C., & Postel-Vinay, S. (2017). Targeting chromatin defects in selected solid tumors based on oncogene addiction, synthetic lethality and epigenetic antagonism. Annals of Oncology, 28, 254–269. https://doi.org/10.1093/ANNONC/MDW552
Muzny, D. M., Bainbridge, M. N., Chang, K., Dinh, H. H., Drummond, J. A., Fowler, G., Kovar, C. L., Lewis, L. R., Morgan, M. B., Newsham, I. F., et al. (2012). Comprehensive molecular characterization of human colon and rectal cancer. Nature, 487, 330–337. https://doi.org/10.1038/nature11252
Myers, S. H., Ortega, J. A., & Cavalli, A. (2020). Synthetic Lethality through the Lens of Medicinal Chemistry. Journal of Medicinal Chemistry, 63, 14151–14183. https://doi.org/10.1021/ACS.JMEDCHEM.0C00766
O’Neil, N. J., Bailey, M. L., & Hieter, P. (2017). Synthetic lethality and cancer. Nature Reviews Genetics, 18, 613–623. https://doi.org/10.1038/nrg.2017.47
Ogiwara, H., Sasaki, M., Mitachi, T., Oike, T., Higuchi, S., Tominaga, Y., & Kohno, T. (2016). Targeting p300 addiction in CBP-deficient cancers causes synthetic lethality by apoptotic cell death due to abrogation of MYC expression. Cancer Discovery, 6, 430–445. https://doi.org/10.1158/2159-8290.CD-15-0754
Ogiwara, H., Takahashi, K., Sasaki, M., Kuroda, T., Yoshida, H., Watanabe, R., Maruyama, A., Makinoshima, H., Chiwaki, F., Sasaki, H., et al. (2019). Targeting the vulnerability of glutathione metabolism in ARID1A-deficient cancers. Cancer Cell, 35, 177-190.e8. https://doi.org/10.1016/J.CCELL.2018.12.009
Pavlova, N. N., & Thompson, C. B. (2016). The emerging Hallmarks of cancer metabolism. Cell Metabolism, 23, 27–47. https://doi.org/10.1016/J.CMET.2015.12.006
Pilié, P. G., Tang, C., Mills, G. B., & Yap, T. A. (2019). State-of-the-art strategies for targeting the DNA damage response in cancer. Nature Reviews Clinical Oncology, 16, 81–104. https://doi.org/10.1038/S41571-018-0114-Z
Reaper, P. M., Griffiths, M. R., Long, J. M., Charrier, J. D., MacCormick, S., Charlton, P. A., Golec, J. M. C., & Pollard, J. R. (2011). Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nature Chemical Biology, 7, 428–430. https://doi.org/10.1038/NCHEMBIO.573
Robson, M., Im, S. A., Senkus, E., Xu, B., Domchek, S. M., Masuda, N., Delaloge, S., Li, W., Tung, N., Armstrong, A., et al. (2017). Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. New England Journal of Medicine, 377, 523–533. https://doi.org/10.1056/NEJMOA1706450
Russell, M. R., Levin, K., Rader, J., Belcastro, L., Li, Y., Martinez, D., Pawel, B., Shumway, S. D., Maris, J. M., & Cole, K. A. (2013). Combination therapy targeting the Chk1 and Wee1 kinases shows therapeutic efficacy in neuroblastoma. Cancer Research, 73, 776–784. https://doi.org/10.1158/0008-5472.CAN-12-2669
Ruzankina, Y., Schoppy, D. W., Asare, A., Clark, C. E., Vonderheide, R. H., & Brown, E. J. (2009). Tissue regenerative delays and synthetic lethality in adult mice after combined deletion of Atr and Trp53. Nature Genetics, 41, 1144–1149. https://doi.org/10.1038/NG.441
Sandhu, S. K., Schelman, W. R., Wilding, G., Moreno, V., Baird, R. D., Miranda, S., Hylands, L., Riisnaes, R., Forster, M., Omlin, A., et al. (2013). The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: A phase 1 dose-escalation trial. The Lancet Oncology, 14, 882–892.
Sangster-Guity, N., Conrad, B. H., Papadopoulos, N., & Bunz, F. (2011). ATR mediates cisplatin resistance in a p53 genotype-specific manner. Oncogene, 30, 2526–2533. https://doi.org/10.1038/ONC.2010.624
Shen, J., Peng, Y., Wei, L., Zhang, W., Yang, L., Lan, L., Kapoor, P., Ju, Z., Mo, Q., Shih, I.-M., et al. (2015). ARID1A deficiency impairs the DNA damage checkpoint and sensitizes cells to PARP inhibitors. Cancer Discovery, 5, 752–767. https://doi.org/10.1158/2159-8290.CD-14-0849
Sherr, C. J., & McCormick, F. (2002). The RB and p53 pathways in cancer. Cancer Cell, 2, 103–112. https://doi.org/10.1016/S1535-6108(02)00102-2
Shiloh, Y., & Ziv, Y. (2013). The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nature Reviews Molecular Cell Biology, 14, 197–210. https://doi.org/10.1038/nrm3546
Takeda, K., Kaisho, T., & Akira, S. (2003). Toll-like receptors. Annual Review of Immunology, 21, 335–376. https://doi.org/10.1146/ANNUREV.IMMUNOL.21.120601.141126
Toledo, L. I., Altmeyer, M., Rask, M. B., Lukas, C., Larsen, D. H., Povlsen, L. K., Bekker-Jensen, S., Mailand, N., Bartek, J., & Lukas, J. (2013). ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell, 155, 1088. https://doi.org/10.1016/J.CELL.2013.10.043
van Harten, A. M., Buijze, M., van der Mast, R., Rooimans, M. A., Martens-de Kemp, S. R., Bachas, C., Brink, A., Stigter-van Walsum, M., Wolthuis, R. M., & Brakenhoff, R. H. (2019). Targeting the cell cycle in head and neck cancer by Chk1 inhibition: A novel concept of bimodal cell death. Oncogenesis. https://doi.org/10.1038/S41389-019-0147-X
van Wietmarschen, N., Sridharan, S., Nathan, W. J., Tubbs, A., Chan, E. M., Callen, E., Wu, W., Belinky, F., Tripathi, V., Wong, N., et al. (2020). Repeat expansions confer WRN dependence in microsatellite-unstable cancers. Nature, 586, 292–298. https://doi.org/10.1038/S41586-020-2769-8
Varshney, D., Spiegel, J., Zyner, K., Tannahill, D., & Balasubramanian, S. (2020). The regulation and functions of DNA and RNA G-quadruplexes. Nature Reviews Molecular Cell Biology, 21, 459. https://doi.org/10.1038/S41580-020-0236-X
Walker, G. J., Flores, J. F., Glendening, J. M., Lin, A., Markl, I. D., & Fountain, J. W. (1998). Virtually 100% of melanoma cell lines harbor alterations at the DNA level within CDKN2A, CDKN2B, or one of their downstream targets. Genes, Chromosomes & Cancer, 22, 157–163. https://doi.org/10.1002/(sici)1098-2264(199806)22:2%3c157::aid-gcc11%3e3.0.co;2-n
Wang, K., Kan, J., Yuen, S. T., Shi, S. T., Chu, K. M., Law, S., Chan, T. L., Kan, Z., Chan, A. S., Tsui, W. Y., et al. (2011). Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nature Genetics, 43, 1219–1223. https://doi.org/10.1038/NG.982
Wang, T., Yu, H., Hughes, N. W., Liu, B., Kendirli, A., Klein, K., Chen, W. W., Lander, E. S., & Sabatini, D. M. (2017). Gene essentiality profiling reveals gene networks and synthetic lethal interactions with oncogenic Ras. Cell, 168, 890-903.e15. https://doi.org/10.1016/J.CELL.2017.01.013
Weber, A. M., Bokobza, S. M., Devery, A. M., & Ryan, A. J. (2013). Abstract B91: Combined ATM and ATR kinase inhibition selectively kills p53-mutated non-small cell lung cancer (NSCLC) cells. Molecular Cancer Therapeutics, 12, B91–B91. https://doi.org/10.1158/1535-7163.TARG-13-B91
Wiegand, K. C., Shah, S. P., Al-Agha, O. M., Zhao, Y., Tse, K., Zeng, T., Senz, J., McConechy, M. K., Anglesio, M. S., Kalloger, S. E., et al. (2010). arid1a mutations in endometriosis-associated ovarian carcinomas. New England Journal of Medicine, 363, 1532–1543. https://doi.org/10.1056/nejmoa1008433
Williamson, C. T., Miller, R., Pemberton, H. N., Jones, S. E., Campbell, J., Konde, A., Badham, N., Rafiq, R., Brough, R., Gulati, A., et al. (2016). ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nature Communication, 7, 1–13. https://doi.org/10.1038/ncomms13837
Wright, S., & Dobzhansky, T. (1946). Genetics of natural populations; experimental reproduction of some of the changes caused by natural selection in certain populations of Drosophila pseudoobscura. Genetics, 31, 125–156.
Wu, C., Lyu, J., Yang, E. J., Liu, Y., Zhang, B., & Shim, J. S. (2018). Targeting AURKA-CDC25C axis to induce synthetic lethality in ARID1A-deficient colorectal cancer cells. Nature Communications. https://doi.org/10.1038/s41467-018-05694-4
Xu, H., Di Antonio, M., McKinney, S., Mathew, V., Ho, B., O’Neil, N. J., Santos, N. D., Silvester, J., Wei, V., Garcia, J., et al. (2017). CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nature Communications. https://doi.org/10.1038/NCOMMS14432
Yamada, L., Saito, M., Thar Min, A. K., Saito, K., Ashizawa, M., Kase, K., Nakajima, S., Onozawa, H., Okayama, H., Endo, H., et al. (2021). Selective sensitivity of EZH2 inhibitors based on synthetic lethality in ARID1A-deficient gastric cancer. Gastric Cancer, 24, 60–71. https://doi.org/10.1007/S10120-020-01094-0
Yan, H., Parsons, D. W., Jin, G., McLendon, R., Rasheed, B. A., Yuan, W., Kos, I., Batinic-Haberle, I., Jones, S., Riggins, G. J., et al. (2009). IDH1 and IDH2 mutations in gliomas. New England Journal of Medicine, 360, 765–773. https://doi.org/10.1056/NEJMOA0808710
Yu, D., Liu, Y., Zhou, Y., Ruiz-Rodado, V., Larion, M., Xu, G., & Yang, C. (2020). Triptolide suppresses IDH1-mutated malignancy via Nrf2-driven glutathione metabolism. Proceedings of the National Academy of Sciences, 117, 9964–9972. https://doi.org/10.1073/PNAS.1913633117
Zeman, M. K., & Cimprich, K. A. (2014). Causes and consequences of replication stress. Nature Cell Biology, 16, 2–9. https://doi.org/10.1038/NCB2897
Zhao, D., Cai, L., Lu, X., Liang, X., Li, J., Chen, P., Ittmann, M., Shang, X., Jiang, S., Li, H., et al. (2020). Chromatin regulator CHD1 remodels the immunosuppressive tumor microenvironment in PTEN-deficient prostate cancer. Cancer Discovery, 10, 1374–1387. https://doi.org/10.1158/2159-8290.CD-19-1352
Zhao, D., Lu, X., Wang, G., Lan, Z., Liao, W., Li, J., Liang, X., Chen, J. R., Shah, S., Shang, X., et al. (2017). Synthetic essentiality of chromatin remodelling factor CHD1 in PTEN-deficient cancer. Nature, 542, 484–488. https://doi.org/10.1038/NATURE21357
Funding
This study was supported by grants from the National Natural Science Foundation of China (82172820, 81771332); the Fundamental Research for the Central University, the Natural Science Foundation of Shanghai (22ZR1466200, 22ZR1451200).
Author information
Authors and Affiliations
Department of Neurosurgery, Shanghai East Hospital, School of Medicine, Tongji University, 150 Jimo Road, Shanghai, 200120, China
Ying Pang, Meng Cheng, Mu Chen, Kaijun Zhao, Yuanyuan Yin, Min Liu, Jing Zhang & Chunlong Zhong
Institute for Advanced Study, Tongji University, 1239 Siping Road, Shanghai, 200092, China
Jing Zhang
Corresponding authors
Correspondence to Jing Zhang or Chunlong Zhong.
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article
Cite this article
Pang, Y., Cheng, M., Chen, M. et al. Synthetic lethality in personalized cancer therapy. GENOME INSTAB. DIS. 4, 121–135 (2023). https://doi.org/10.1007/s42764-022-00080-3
Received26 May 2022
Revised16 July 2022
Accepted16 August 2022
Published26 August 2022
Issue DateApril 2023
DOIhttps://doi.org/10.1007/s42764-022-00080-3
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
Synthetic lethality
Cancer therapy
Genome instability
DNA damage response
用户登录
还没有账号?
立即注册