






Clonal evolution in leukemia: preleukemia, evolutionary models, and clinical implications
Review Article
Xu-Dong He, Meng-Fang Xia, Ji-Yuan Teng, Bin-Bing S. Zhou & Qian-Fei Wang
Genome Instability & Disease 4, 227–238 (2023)
Abstract
Leukemia is a polyclonal and progressive disease with drastic intra-clone heterogeneity. During the early stages of disease development, it is mostly shaped by the deterministic effects of key initiating events, which could establish the roles of genomic instability and cell plasticity in clonal evolution. Later, preleukemic cells acquire successive genetic mutations, undergoing distinct evolutionary trajectories. In this review, we summarize the current understanding of how genetic lesions define distinct clonal architectures. We further highlight two classical evolutionary models and their relevant prognostic implications. Given that drug selection pressure remains a major driving force of relapse, we also discuss recurrent patterns of clonal evolution under chemotherapy and targeted therapy. Understanding the rationale for directing distinct clonal evolution patterns will be instrumental in the development of different therapeutic strategies to prevent or overcome drug resistance and relapse during disease progression.
Introduction
Human leukemias are a group of hematopoietic malignancies initiated by a clonally expanded hematopoietic stem cell (HSC) population. At an early stage, stimulus in the microenvironment such as inflammation and infection can drive abnormal propagation of HSCs that carrying mutations in a unique set of genes. This specific group of mutated cells outgrowths its counterpart and results in clonal hematopoiesis. Later, clonal expanded HSCs acquire multiple genetic mutations sequentially and are transformed into leukemia, which is manifested by uncontrolled growth of immature blood cells in the bone marrow (Ferrando & López-Otín, 2017).
Recent studies have identified leukemia as aggregates of diverse cell populations with intra-tumor heterogeneity (Hausser & Alon, 2020). Malignant transformation and progression of leukemia cells are characterized as a dynamic evolution process driven by evolutionary forces such as selection pressure and genetic drift, known as clonal evolution (Greaves & Maley, 2012; McGranahan & Swanton, 2017). During this process, different sub-cell populations undergo expansion and diversification through genetic mutation acquisition and environmental selection. A clone involved in evolution represents a group of cells with the same origin and competitiveness (Duchmann et al., 2021). The most common way to define a clone is in terms of genetic identity, while the accumulation of driver mutations tends to alter the fitness of clones considerably.
In clinical practice, there are multiple methods available for subtyping and risk stratification in leukemia, including immunophenotypic classification, molecular genetic classification, and chromosomal genetic classification. Currently, from the perspective of molecular genetics, disease subtype classification and risk stratification of leukemia is mostly dependent on single genetic abnormalities involved in leukemia initiation and transformation (Döhner et al., 2022). However, an individual molecular event is not sufficient to recapitulate the full genetic landscape and the stage of leukemia development (Döhner et al., 2015). The initiating mutation determines the trajectory of further development through the selection of subsequent mutations in a given order to achieve optimal development (Levine et al., 2019). It has been shown that the same mutation combination with different order can lead to completely disparate disease phenotype.
Clonal architecture represents the genetic identity and proportion of subclones in leukemia at a single time point. Progression in multi-stages constitutes the clonal architecture of leukemia, reflecting the process of clonal evolution. Several clinical cohort studies have demonstrated the prognostic values of clonal architectures (Benard et al., 2021; Cerrano et al., 2021; Metzeler et al., 2016). These results suggest that the clonal architecture must be considered to assess the clinical significance of a mutation. In addition, the mechanism of clonal evolution during drug treatment has also been widely studied. Treatment options, including chemotherapy and targeted therapy, could induce distinct evolutionary patterns of leukemia from diagnosis to relapse (Ding et al., 2012; Morita et al., 2020; Peretz et al., 2021). These clonal evolution processes reflect different evolutionary driving forces of leukemia progression under different drug treatments, which might be of great significance in treatment optimization.
In summary, clonal evolution plays an indispensable role in the initiation, transition, and progression of leukemia. Therefore, the exploration of clonal trajectories and underlying driving selection forces in leukemia is important for understanding leukemia progression and making clinical treatment decisions. This review highlights studies examining clonal evolution patterns in leukemia, in which we discuss how clonal evolution shapes the hematopoietic malignancies and affects clinical therapeutic options.
Mutation accumulations from preleukemia to leukemia transformation
Numerous studies in hematologic malignancies have demonstrated that leukemia develops from an early preleukemic state, characterized by the clonal expansion of functionally normal HSCs with leukemia-associated mutations. The pool of clonally expanded HSCs, also known as preleukemia clones, still maintain the potential for normal hematopoietic differentiation and have a propensity for malignant transformation. Over time, preleukemia clone can develop into myeloid or lymphoid leukemia after the acquisition of leukemia-transforming mutations. Somatic mutations detected in the blood or bone marrow with a variant allele frequency of 2% or higher in the absence of hematologic malignancies, are termed clonal hematopoiesis of indeterminate potential (CHIP) (Busque et al., 2018). It has been found that CHIP is associated with the development of blood disorders, cardiovascular diseases, and chronic obstructive pulmonary diseases (Genovese et al., 2014; Jaiswal et al., 2014).
Mutations found in preleukemia cells can occur in genes with diverse functions, including epigenetic modifiers, splicing regulators, tumor suppressors, and signal transducers (DNMT3A, TET2, ASXL1, SRSF2, TP53, JAK2, etc.) (Corces-Zimmerman et al., 2014; Jan et al., 2012; Koeffler & Leong, 2017; Rothenberg-Thurley et al., 2018). In addition to somatic mutations, germline mutations in genes such as CEBPA, RUNX1 and GATA2 are associated with the preleukemia state (Brown et al., 2020; Hahn et al., 2011; Simon et al., 2020; Smith et al., 2004). The acquisition of preleukemic mutations in HSCs was found to confer a self-renewal advantage during leukemogenesis (Challen et al., 2012; Damm et al., 2014; Moran-Crusio et al., 2011). Compared to healthy people, individuals bearing these mutations have a greater possibility to progress into leukemia later (Koeffler & Leong, 2017).
Depending on the specific gene involved, the initiating mutations of leukemia can have a strong impact on either the genetic or epigenetic landscape down the road. The landscaping effects of initiating mutations on leukemia transformation can be diverse (Fig. 1). For example, mutations involved in DNA repair pathways such as TP53 could induce genomic instability and accelerate subsequent leukemic progression through large genetic rearrangement including fusion genes and chromothripsis (Fig. 1b) (Baslan et al., 2022; Mantovani et al., 2019). HSCs with TP53 inactivation are more likely to develop therapy-related acute myeloid leukemia (tAML) than de novo acute myeloid leukemia (AML) (Lal et al., 2017; Wong et al., 2015; Zebisch et al., 2016). Mechanistically, TP53 loss of function mutations lead not only to defective DNA repair but also to increased chemoresistance (Gibson et al., 2017; Wong et al., 2015). In acute lymphoblastic leukemia (ALL), chemotherapy can drive mutagenesis and the acquisition of drug-resistant mutations such as TP53 mutations leading to relapse (Li et al., 2020). In particular, thiopurine and mismatch repair (MMR) deficiency can cooperate to generate hypermutation and fuel TP53 mutagenesis during ALL relapse (Yang et al., 2021).
Fig. 1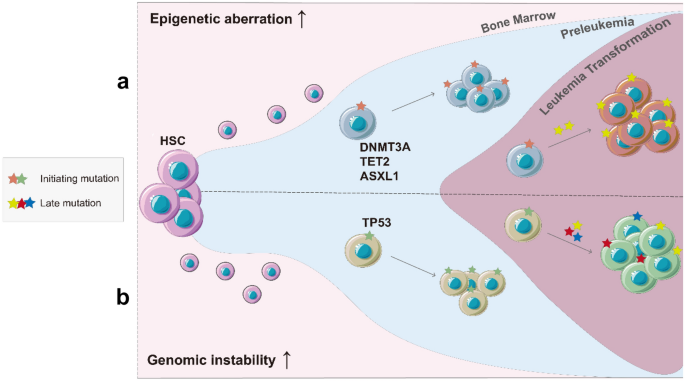
The distinct landscaping effects of initiating mutations. a (Top) Mutations of epigenetic modifiers induced leukemia initiation. b (Bottom) Genomic instability-driven leukemia initiation
From a different aspect of the landscaping effects, initiating mutations involving epigenetic modifiers and splicing complexes might increase epigenetic diversity, rather than direct mutagenesis, and facilitate leukemic transformation and progression (Fig. 1a). For example, DNMT3A and TET2 mutations can affect DNA methylation regulation and generate unique methylation patterns during transformation (Koya et al., 2016; Rasmussen et al., 2015; Russler-Germain et al., 2014; Spencer et al., 2017). Furthermore, mutations in the histone modifier ASXL1 have been shown to cause dysregulation in inflammatory responses that may promote leukemia transition (Sallman & List, 2019). In chronic lymphocytic leukemia (CLL), the occurrence of epigenetically related mutations is associated with locally disordered DNA methylation, which leads to increased intratumor heterogeneity (Landau et al., 2014). CLL patients harboring the DNMT3AQ153, TET1N789I, and IDH1S210N mutations were found to be associated with adverse clinical outcomes (Landau et al., 2014). These recent findings suggest that disordered epigenetic patterns, like genomic instability, also enhance the intratumor complexity and enable cancer cells to follow clonal trajectories during disease progression.
However, only a small proportion of CHIP individuals eventually develop leukemia, suggesting that initiating genetic events alone are not sufficient for leukemia transformation. In the classic two-hit model proposed by Gary Gilliland in 2002, full onset of leukemia requires secondary genetic aberrations, which would endow an additional proliferative and survival advantage (Gilliland & Griffin, 2002). It may take many years for preleukemia to proceed into leukemia, and has been observed from large retrospective clinical trials that there are stable recurrent mutation combinations during clonal evolution (Cancer Genome Atlas Research Network et al., 2013; Papaemmanuil et al., 2016; Tyner et al., 2018).
Intriguingly, the initiating mutations play an important role in specific evolutionary trajectories, which in other words, the order of mutation accumulations during leukemia formation impacts the eventual leukemia phenotypes (Kandoth et al., 2013; Levine et al., 2019). For example, TET2-JAK2V617F double-mutant are recurrent in 10% of myeloproliferative neoplasm (MPN) patients. Among these patients, JAK2 are detected at early stages in half of the double-mutant patients and the remaining half are TET2 first. TET2 first patients have a higher proportion of myeloid progenitors, while JAK2 first patients yield increased megakaryocyte and erythrocyte progenitors (Ortmann et al., 2015). In accordance with clinical outcomes, JAK2 mutant-first patients are more inclined to develop polycythemia vera instead of essential thrombocythemia, more possible of thrombosis and more sensitive to the JAK2 inhibitor than TET2 mutant-first patients (Ortmann et al., 2015). Thus, the choice and order of mutations can lead to divergent MPN disease phenotypes. A similar situation has been observed in a DNMT3A and JAK2 double-mutant setting; JAK2V617F arose in wild-type DNMT3A patients and had an increased possibility to induce polycythemia vera compared to those arising on DNMT3A mutant background (Nangalia et al., 2015). Thus, the landscaping mutations lay the groundwork for the selection of later mutations, which lead to divergent leukemia phenotypes depending on the order of events.
Linear vs branched: distinct evolutionary models
The development of evolutionary trajectories leads to the generation of subclones with unique genetic identities. By resolving the genetic identities of all subclones in the clonal architecture, evolutionary trajectories can be traced and defined. Therefore, precise reconstruction of the clonal architecture is instrumental in understanding the evolutionary process of leukemia.
The variant allele frequency (VAF) of mutations is the proportion of variant sequence reads relative to the total number of sequence reads. The deduction of clonal architecture from early studies was based on clustering variants with similar VAFs using whole-genome sequencing (WGS) data of bulk samples (Ding et al., 2012; Welch et al., 2012). This method has limitations in analyzing complex clonal structure, especially dealing with mutations with low VAFs. Other studies used single-cell colony culture for mutation analysis at subclone levels (Hirsch et al., 2016), but this method cannot measure the precise proportion of each clone in the original tumor simultaneously. Therefore, none of these approaches are sufficient to accurately describe the clonal architecture. Recent development of single-cell DNA profiling has provided a high-resolution approach to recapitulate leukemia evolution paths, which has further overcome difficulties encountered in bulk sequencing or colony culture of clinical samples (Pellegrino et al., 2018). Emerging studies of clonal evolution have revealed that tumors have distinct evolutionary models with different clonal selection processes and clonal architectures. In particular, two main types of evolutionary models have been observed in leukemia, namely linear and branched evolution (Miles et al., 2020; Morita et al., 2020) (Fig. 2).
Fig. 2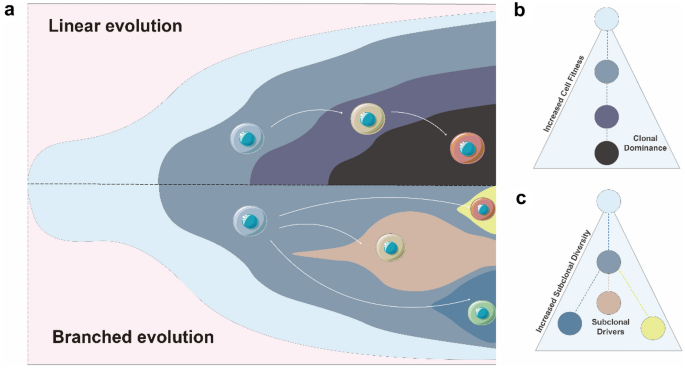
Two main types of evolutionary models in leukemia. a The illustrations of linear evolution (top) and branched evolution (bottom) concept. b and c Characteristics of two evolutionary models. See text for detailed descriptions
The linear evolution model was originally proposed by Peter Nowell in 1976 (Greaves & Maley, 2012). In the model, the appearance and expansion of new clones are driven step by step through environmental selection, with a sequential accumulation of new somatic mutations conferring greater selective advantages than old clones (Fig. 2b). The daughter clone with additional somatic mutations outcompetes the ancestral clone leading to the dominant clone, a process known as clonal sweep (McGranahan & Swanton, 2017). Afterwards, new mutations resulting in increased clone fitness are more likely to occur again in the dominant clone, so that the daughter clones expand to become the dominant clone and continue to accumulate new mutations under specific environmental pressure. Finally, leukemia cells tend to expand and evolve in a linear manner, which could eventually result in lower subclone complexity and clearer clonal dominance (Morita et al., 2020). The formation of linear architectures, accompanied by the phenomenon of gradually increasing cell fitness, may suggest the significant role of natural selection. Epigenetic-regulator mutations (DNMT3A, TET2, ASXL1, IDH, IDH2) are more likely to evolve in linear model (Miles et al., 2020). Clones harboring these mutations, especially in the early stages, are likely to be the dominant clones (Klco et al., 2015), suggesting that these mutations might confer intrinsic cell fitness capable of leukemia initiation and maintenance. Alternative viewpoint argues that linear evolution could derive from a small population of cancer stem cells under genetic drift (Lyne et al., 2021). This hypothesis could explain the presence of some cases in hematological malignancies without obvious driver mutations.
In the branched evolution model, the initial founding clone evolves in divergent manners with daughter clones acquiring distinct mutations simultaneously (Ding et al., 2012; Miles et al., 2020; Morita et al., 2020; Welch et al., 2012). The accumulation of new mutations in daughter clones does not confer selective competitiveness over the other daughter clones. Therefore, these daughter clones all survive and evolve in parallel under environmental selection. Compared to linear evolution, branched evolution results in a more complicated tumor structure and increased intratumor heterogeneity (Fig. 2c). In addition, branched evolution is thought to have a higher mutation burden and more subclone diversity, possibly indicating a result of genomic instability and increased mutagenesis rate (Morita et al., 2020). Besides, a striking feature of branched evolution is that all clones in the different branches evolve continuously, where mutation burdens gradually accumulate in cancer clones over time, contributing to subclone expansion and increased clonal complexity (Greaves & Maley, 2012; McGranahan & Swanton, 2017). The evolutionary pattern resulting from neutral selection solely represents an extreme case of branched evolution, with extremely high genetic diversity (Davis et al., 2017; Ling et al., 2015). However, there may be multiple complex factors involved in the formation of branched evolution in leukemia, including abnormal epigenetic modifications, DNA replication errors, DNA repair deficiencies, and environmental pressures (Merlo et al., 2006). The mechanism underlying branched evolution is complicated and yet to be elucidated.
In CLL, most driver mutations are subclonal at diagnosis, which might be derived from the high level of stochastic methylation heterogeneity (Landau et al., 2013). Another classic example of a branched evolution model observed in leukemia is convergent evolution, in which multiple independent trajectories are carried out by driver genes with similar functions or harbored in the same pathway, leading to independent and parallel clonal expansions (Miles et al., 2020; Morita et al., 2020). Single-cell DNA sequencing of de novo AML patients revealed that many patients acquired multiple mutations in the related signaling pathway genes (including FLT3, NRAS, KIT, KRAS, which all belong to the general RTK/RAS signaling pathways) and developed a branched evolution structure. Later mutations in signaling pathways are found to be most, if not all, mutually exclusive in subclones, which indicates these mutations might be functionally redundant (Hirsch et al., 2016; Miles et al., 2020; Morita et al., 2020). The aforementioned studies suggest that subclones may co-exist and evolve in parallel, which are not simply driven by fitness selection leading to clonal sweeps and dominance.
Of note, the accumulation of new driver mutations does not necessarily confer a fitness advantage. For instance, the increase of clonal diversity in AML, based on a recent single-cell sequencing study, was not consistent with the mutation burden in the largest clone, suggesting that increased mutational burden within a dominant clone is not necessarily the determinative force of clonal dominance (Miles et al., 2020). Furthermore, the selection pressure driving clonal evolution was not always realized through clonal competition. Subclone could also cooperate to evolve by secreting extracellular signaling molecules such as inflammatory factors (Duchmann et al., 2021) or diffusible metabolites such as R-2-hydroxyglutarate (Chaturvedi et al., 2016).
Furthermore, evolutionary models have clinical and prognostic implications. In CLL, an increased number of subclonal driver mutations was associated with an inferior failure-free survival, while an increase in the number of clonal driver mutations was not associated with poor prognosis (Landau et al., 2013). Studies of AML have also suggested that clonal dominance in linear evolution was associated with poor overall survival (Cerrano et al., 2021). In the core-binding factor (CBF) subgroup of AML, the branched evolution of subclones with different signaling pathway mutations was an independent predictor of poor clinical outcome (Itzykson et al., 2018). Furthermore, the presence of multiple different FMS-like tyrosine kinase-3 internal tandem duplication (FLT3-ITD) variants with different lengths and insertion sizes, possibly in the parallel subclone, were associated with a higher risk of chemotherapy treatment failure (Rücker et al., 2021).
Clonal evolution under drug selection
When leukemia is under treatment, evolutionary driving forces is in distinction from those in the initiation and transition phases. Cytotoxic agents and targeted inhibitors become the dominative forces in clonal evolution. Leukemia often develops resistance to treatments due to intra-tumor heterogeneity and the constant clonal evolution causing therapeutic targets moving. Hence, understanding the clonal evolution processes under the action of drugs is of great significance for clinical applications. Relapse patterns can be classified into two categories: (1) the acquisition of new somatic mutations, and (2) the expansion of pre-existing minor clone (Fig. 3 and Table. 1).
Fig. 3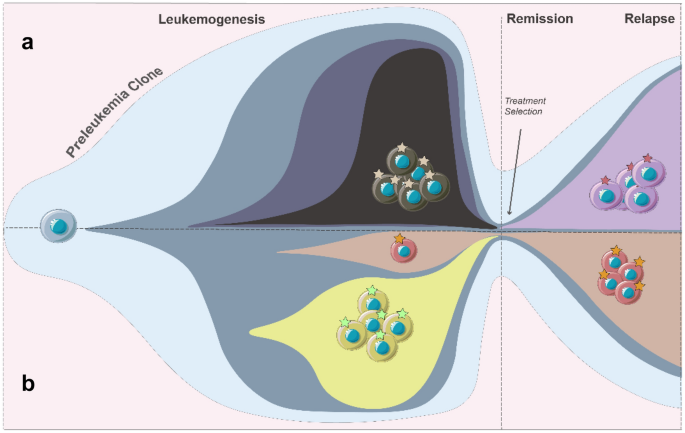
Two relapse patterns in leukemia. a The newly acquired mutation leads to drug resistance and relapse. b The minor clone independent of drug targets evolves to relapse
Treatment regimen | Target | Leukemia | Mechanism of relapse | Gene with mutations involved | References |
|---|---|---|---|---|---|
Thiopurines | DNA replication/DNA damage response pathway | ALL | Induction of drug resistance mutations | NT5C2, NR3C1, TP53 | |
Cytarabine etc | AML | Further development of preleukemic clones | DNMT3A etc | Shlush et al., (2014) | |
Imatinib | BCR-ABL1 | CML | (1) Accumulation of drug-resistant mutations; (2) Expansion of pre-existing drug-resistant clones | ABL1 | |
Retinoic acid | PML-RARA | APL | RARA | Goto et al., (2011) | |
Gilteritinib | FLT3 | AML | FLT3-TKD, NRAS, KRAS, PTPN11, KIT | McMahon et al., (2019) | |
Midostaurin | Schmalbrock et al., (2021) | ||||
Quizartinib | |||||
Crenolanib | Zhang et al., (2019) |
Table 1 Evolutionary mechanisms of drug-resistant relapse in leukemia
Chemotherapy is the major treatment strategy for leukemia with remarkable historic success. The toxicity of cytotoxic agents is most significant in S phase during the cell cycle, resulting in DNA double-strand breakages (Mills et al., 2018). Chemotherapy-induced stress is mainly genetic pressure since the toxicity of chemotherapy leads to great damage to the genome and has drastic changes in the mutation landscape, which provides a reservoir for the selection of drug-resistant clones. It was reported that multiple types of mutations are involved in a relapse of chemotherapy (Ding et al., 2012; Welch et al., 2012). Recent findings in ALL have demonstrated that chemotherapy itself could directly induce drug-resistant mutations through genomic instability during relapse clonal evolution (B. Li et al., 2020; Yang et al., 2021).
Interestingly, preleukemic clones can resist chemotherapy and lead to relapse. Early studies first revealed that several preleukemic mutations such as DNMT3A, TET2 and IDH1/2, but not late mutations such as FLT3 and NRAS, tend to be preserved at relapse, by paired sequencing of diagnostic and relapsed samples (Corces-Zimmerman et al., 2014; Jan et al., 2012). Subsequent studies have shown that preleukemic HSCs are resistant to induction chemotherapy, resulting in an increased proportion of preleukemia clones during remission achieved by chemotherapy (Shlush et al., 2014). The preleukemia population with theses mutation, which persisted at remission, were associated with a higher probability of relapse (Cocciardi et al., 2019; Greif et al., 2018; Murphy et al., 2019). These findings suggested that the preleukemic clones, with leukemia-initiating capacity, were resistant to chemotherapy. Even though the cytotoxic agents can eliminate most of the leukemia cell, the remaining preleukemic population may expand to be the dominant and restart the subsequent leukemia by accumulating new mutations.
During the past decades, the development of therapeutic agents targeting key recurrent driver mutations has improved clinical outcomes for patients with blood cancers (Cucchi et al., 2021). Successful cases include retinoic acid in treating PML-RARA positive acute promyelocytic leukemia (APL) (Degos & Wang, 2001) and imatinib in treating chronic myeloid leukemia (CML) with BCR-ABL1 fusion (Capdeville et al., 2002). More than 90% of patients who had received these therapies achieved long-term survival (Hochhaus et al., 2017; Lo-Coco et al., 2013). Despite these great successes in APL and CML, drug resistance and refractory remain major challenges for targeted therapy in the majority of leukemias due to the clonal evolution under treatment pressure (Goto et al., 2011; Hochhaus et al., 2002; Roche-Lestienne et al., 2002).
Targeted therapy-driven leukemia clonal evolution shows a unique determinative pattern. The selection force imposed by the targeted agents drives leukemic cell population through an evolutionary bottleneck. Unlike chemotherapy, mutations that mediate relapse under targeted therapeutics are highly associated with specific function pathways. As a result, targeted therapy reduces the size of clones harboring the targeted genetic variant, and promotes the proliferation of subclones that are independent of the target.
In resistant and refractory patients treated with targeted agents, the accumulation of mutations activating the downstream of the targeted pathway in a target could confer drug resistance to the targeted agents. For example, in the treatment of FLT3-ITD mutated AML with FLT3 inhibitors, FLT3-ITD mutated clones frequently developed additional point mutations in the tyrosine kinase domain of the FLT3 gene (FLT3-TKD), leading to resistance to FLT3 inhibitors, in relapsed patients (Daver et al., 2019; Peretz et al., 2021). Recent studies also found additional mutations in the RTK/RAS signaling pathways (NRAS, KRAS, PTPN11, KIT) at relapse that are highly associated with FLT3 inhibitor resistance (Peretz et al., 2021; C. C. Smith et al., 2017; McMahon et al., 2019; Zhang et al., 2019; Schmalbrock et al., 2021). These pathway-activating mutations could either arise from FLT3-ITD mutated clones or clones independent of FLT3-ITD, both of which activated the downstream of the RAS-MAPK pathway in an FLT3-mutant-independent manner.
The emergence of multiple types of signaling transducer mutations, whether at diagnose or relapse, indicates an intrinsic dependency of these AMLs on these signaling pathways. Therefore, when FLT3-ITD is inhibited by targeted agents, there is a great possibility that convergent evolution will call on other downstream candidates for replacement, maintaining the activation of the signaling pathway. This is in accordance with the expansion of subclones with RAS mutation at the relapse stage in FLT3 inhibitor-treated patients. On treatment with the FLT3 inhibitor gilteritinib, subclones harboring both FLT3-ITD and NRAS mutations were dominant at relapse (McMahon et al., 2019). However, the co-occurrence of FLT3-ITD and NRAS mutations in the same clone is rarely observed at diagnosis (Miles et al., 2020; Morita et al., 2020), supporting the expansion of FLT3-ITD and NRAS double mutant clones is driven by inhibitor-mediated selection.
Clinical implications and future perspective
Genomic studies have already revealed the roles of many recurrent driver mutations in the initiation, transformation, and progression of leukemia. However, the same mutation could play different roles in different clonal architectures or stages, suggesting the enormous heterogeneity during leukemia clonal evolution. Therefore, the exploration of genetic landscape variation alone is insufficient to thoroughly elucidate the intratumor heterogeneity and clonal dynamics observed in leukemia, and further detailed studies of distinct clonal architectures would be helpful for a better perspective on genetic landscapes and their clinical implications. Although some studies have revealed the clinical significance of certain clonal evolution mechanisms, it is tricky to directly translate the basic findings of clonal evolution studies into clinical applications.
Targeted therapeutic regimens are generally designed on the basis of detected driver mutations especially those with high VAFs, however, not all driver mutations merit the consideration of therapeutic targets. The explorations of clonal architectures have highlighted the significance of developing drugs with a broad mechanism of targeting multiple downstream signaling pathways rather than specific genetic lesions, especially in convergent evolution with subclones harboring different mutations in the same functional pathway. Despite targeted therapies for leukemia patients have made a breakthrough recently owing to the rapid development of FLT3 (Daver et al., 2019), IDH1/2 (Stein et al., 2021) and BCL-2 inhibitors (DiNardo et al., 2020), limited treatment efficacies and high recurrence rate remain as major issues urgently needed to be resolved.
Taking into account the contributions of genetic and epigenetic factors to leukemia clonal evolution, a combination of epigenetic inhibitors and chemotherapeutics might be required to eradicate the complex and heterogeneous disease. The emergence of resistant subclones evolved from initially minimal residual disease after treatment (Chen et al., 2019; Duan et al., 2014) or already resistant minority subclones can easily lead to therapeutic resistance and leukemia relapse over single therapeutics, particularly targeted monotherapy. On the other hand, complete responses to combination treatment consisting of targeted drugs and chemotherapeutic or epigenetic inhibitors (e. g. hypomethylating agents, HMAs) can be achieved in many patients (Döhner et al., 2021). The exploration of more selective inhibitors targeting epigenetic factors such as histone deacetylases (HDACs) or oncogenic factors such as the MYC family in the context of combination therapy are urgently needed in the clinical setting.
Synthetic lethality-based therapeutic strategies in leukemia exhibit robust potentials to be applied in clinical settings. It is well known that Poly ADP‐ribose polymerase (PARP) inhibitors can elicit synthetic lethality with homologous recombination (HR) deficiency in different cancer types. Several studies have explored the synthetic lethal interactions and selective vulnerabilities of current available PARP inhibitors in AML and ALL based on preclinical and clinical findings, opening a potential therapeutic window for specific disease subgroups (Padella et al., 2022). In addition, mismatch repair (MMR) defects can cause hypermutation and thiopurine resistance, leading to subsequent multi-drug resistance and relapse during ALL treatment (Yang et al., 2021), which suggests potential synthetic lethal targets in MMR-deficient ALLs.
In summary, studies on the clonal evolution of many genetic mutations enhance our understanding of the underlying mechanisms of acquired therapeutic resistance under drug treatment. There are still other questions surrounding clonal architecture levels such as the cellular hierarchy and epigenetic heterogeneity, which remains to be explored and integrated together to provide a more comprehensive perspective on the clonal evolution and potential clinical implications in leukemia (Bottomly et al., 2022; Carter & Zhao, 2021; S. Li et al., 2016; van Galen et al., 2019; Zeng et al., 2022). In the end, better-designed combination therapeutic strategies based on our understanding of leukemia clonal evolution could be much more effective to prevent relapse and eradicate complex and heterogeneous diseases.
Data availability
This article does not contain any additional data.
References
Baslan, T., Morris, J. P., Zhao, Z., Reyes, J., Ho, Y.-J., Tsanov, K. M., Bermeo, J., Tian, S., Zhang, S., Askan, G., Yavas, A., Lecomte, N., Erakky, A., Varghese, A. M., Zhang, A., Kendall, J., Ghiban, E., Chorbadjiev, L., Wu, J., & Lowe, S. W. (2022). Ordered and deterministic cancer genome evolution after p53 loss. Nature. https://doi.org/10.1038/s41586-022-05082-5. Article 7924.
Benard, B. A., Leak, L. B., Azizi, A., Thomas, D., Gentles, A. J., & Majeti, R. (2021). Clonal architecture predicts clinical outcomes and drug sensitivity in acute myeloid leukemia. Nature Communications, 12(1), 7244. https://doi.org/10.1038/s41467-021-27472-5
Bottomly, D., Long, N., Schultz, A. R., Kurtz, S. E., Tognon, C. E., Johnson, K., Abel, M., Agarwal, A., Avaylon, S., Benton, E., Blucher, A., Borate, U., Braun, T. P., Brown, J., Bryant, J., Burke, R., Carlos, A., Chang, B. H., Cho, H. J., & Tyner, J. W. (2022). Integrative analysis of drug response and clinical outcome in acute myeloid leukemia. Cancer Cell, 40(8), 850-864.e9. https://doi.org/10.1016/j.ccell.2022.07.002
Brown, A. L., Arts, P., Carmichael, C. L., Babic, M., Dobbins, J., Chong, C.-E., Schreiber, A. W., Feng, J., Phillips, K., Wang, P. P. S., Ha, T., Homan, C. C., King-Smith, S. L., Rawlings, L., Vakulin, C., Dubowsky, A., Burdett, J., Moore, S., McKavanagh, G., & Scott, H. S. (2020). RUNX1-mutated families show phenotype heterogeneity and a somatic mutation profile unique to germline predisposed AML. Blood Advances, 4(6), 1131–1144. https://doi.org/10.1182/bloodadvances.2019000901
Busque, L., Buscarlet, M., Mollica, L., & Levine, R. L. (2018). Concise review: age-related clonal hematopoiesis: stem cells tempting the devil. Stem Cells, 36(9), 1287–1294. https://doi.org/10.1002/stem.2845
Cancer Genome Atlas Research Network, Ley, T. J., Miller, C., Ding, L., Raphael, B. J., Mungall, A. J., Robertson, A. G., Hoadley, K., Triche, T. J., Laird, P. W., Baty, J. D., Fulton, L. L., Fulton, R., Heath, S. E., Kalicki-Veizer, J., Kandoth, C., Klco, J. M., Koboldt, D. C., Kanchi, K.-L., & Eley, G. (2013). Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. The New England Journal of Medicine, 368(22), 2059–2074. https://doi.org/10.1056/NEJMoa1301689
Capdeville, R., Buchdunger, E., Zimmermann, J., & Matter, A. (2002). Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nature Reviews Drug Discovery. https://doi.org/10.1038/nrd839. Article 7.
Carter, B., & Zhao, K. (2021). The epigenetic basis of cellular heterogeneity. Nature Reviews Genetics. https://doi.org/10.1038/s41576-020-00300-0. Article 4.
Cerrano, M., Duchmann, M., Kim, R., Vasseur, L., Hirsch, P., Thomas, X., Quentin, S., Pasanisi, J., Passet, M., Rabian, F., Rahmé, R., Lengliné, E., Raffoux, E., Dhédin, N., Sébert, M., Maarek, O., Raimbault, A., Celli-Lebras, K., Adès, L., & Itzykson, R. (2021). Clonal dominance is an adverse prognostic factor in acute myeloid leukemia treated with intensive chemotherapy. Leukemia. https://doi.org/10.1038/s41375-020-0932-8. Article 3.
Challen, G. A., Sun, D., Jeong, M., Luo, M., Jelinek, J., Berg, J. S., Bock, C., Vasanthakumar, A., Gu, H., Xi, Y., Liang, S., Lu, Y., Darlington, G. J., Meissner, A., Issa, J.-P.J., Godley, L. A., Li, W., & Goodell, M. A. (2012). Dnmt3a is essential for hematopoietic stem cell differentiation. Nature Genetics, 44(1), 23–31. https://doi.org/10.1038/ng.1009
Chaturvedi, A., Araujo Cruz, M. M., Jyotsana, N., Sharma, A., Goparaju, R., Schwarzer, A., Görlich, K., Schottmann, R., Struys, E. A., Jansen, E. E., Rohde, C., Müller-Tidow, C., Geffers, R., Göhring, G., Ganser, A., Thol, F., & Heuser, M. (2016). Enantiomer-specific and paracrine leukemogenicity of mutant IDH metabolite 2-hydroxyglutarate. Leukemia. https://doi.org/10.1038/leu.2016.71. Article 8.
Chen, Y.-L., Tang, C., Zhang, M.-Y., Huang, W.-L., Xu, Y., Sun, H.-Y., Yang, F., Song, L.-L., Wang, H., Mu, L.-L., Li, M.-H., Zheng, W.-W., Miao, Y., Ding, L.-X., Li, B.-S., Shen, S.-H., Liu, S.-L., Li, H., Zhu, Z.-Q., & Zhou, B.-B.S. (2019). Blocking ATM-dependent NF-κB pathway overcomes niche protection and improves chemotherapy response in acute lymphoblastic leukemia. Leukemia. https://doi.org/10.1038/s41375-019-0458-0. Article 10.
Cocciardi, S., Dolnik, A., Kapp-Schwoerer, S., Rücker, F. G., Lux, S., Blätte, T. J., Skambraks, S., Krönke, J., Heidel, F. H., Schnöder, T. M., Corbacioglu, A., Gaidzik, V. I., Paschka, P., Teleanu, V., Göhring, G., Thol, F., Heuser, M., Ganser, A., Weber, D., & Döhner, K. (2019). Clonal evolution patterns in acute myeloid leukemia with NPM1 mutation. Nature Communications, 10(1), 2031. https://doi.org/10.1038/s41467-019-09745-2
Corces-Zimmerman, M. R., Hong, W.-J., Weissman, I. L., Medeiros, B. C., & Majeti, R. (2014). Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proceedings of the National Academy of Sciences, 111(7), 2548–2553. https://doi.org/10.1073/pnas.1324297111
Cucchi, D. G. J., Polak, T. B., Ossenkoppele, G. J., Uyl–De Groot, C. A., Cloos, J., Zweegman, S., & Janssen, J. J. W. M. (2021). Two decades of targeted therapies in acute myeloid leukemia. Leukemia, 35(3), Article 3. https://doi.org/10.1038/s41375-021-01164-x
Damm, F., Mylonas, E., Cosson, A., Yoshida, K., Della Valle, V., Mouly, E., Diop, M., Scourzic, L., Shiraishi, Y., Chiba, K., Tanaka, H., Miyano, S., Kikushige, Y., Davi, F., Lambert, J., Gautheret, D., Merle-Béral, H., Sutton, L., Dessen, P., & Bernard, O. A. (2014). Acquired initiating mutations in early hematopoietic cells of CLL patients. Cancer Discovery, 4(9), 1088–1101. https://doi.org/10.1158/2159-8290.CD-14-0104
Daver, N., Schlenk, R. F., Russell, N. H., & Levis, M. J. (2019). Targeting FLT3 mutations in AML: Review of current knowledge and evidence. Leukemia, 33(2), 299–312.
Davis, A., Gao, R., & Navin, N. (2017). Tumor evolution: Linear, branching, neutral or punctuated? Biochimica Et Biophysica Acta, 1867(2), 151–161. https://doi.org/10.1016/j.bbcan.2017.01.003
Degos, L., & Wang, Z. Y. (2001). All trans retinoic acid in acute promyelocytic leukemia. Oncogene, 20(49), 7140–7145. https://doi.org/10.1038/sj.onc.1204763
DiNardo, C. D., Jonas, B. A., Pullarkat, V., Thirman, M. J., Garcia, J. S., Wei, A. H., Konopleva, M., Döhner, H., Letai, A., Fenaux, P., Koller, E., Havelange, V., Leber, B., Esteve, J., Wang, J., Pejsa, V., Hájek, R., Porkka, K., Illés, Á., & Pratz, K. W. (2020). Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. New England Journal of Medicine, 383(7), 617–629. https://doi.org/10.1056/NEJMoa2012971
Ding, L., Ley, T. J., Larson, D. E., Miller, C. A., Koboldt, D. C., Welch, J. S., Ritchey, J. K., Young, M. A., Lamprecht, T., McLellan, M. D., McMichael, J. F., Wallis, J. W., Lu, C., Shen, D., Harris, C. C., Dooling, D. J., Fulton, R. S., Fulton, L. L., Chen, K., & DiPersio, J. F. (2012). Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature, 481(7382), 506–510. https://doi.org/10.1038/nature10738
Döhner, H., Wei, A. H., Appelbaum, F. R., Craddock, C., DiNardo, C. D., Dombret, H., Ebert, B. L., Fenaux, P., Godley, L. A., Hasserjian, R. P., Larson, R. A., Levine, R. L., Miyazaki, Y., Niederwieser, D., Ossenkoppele, G., Röllig, C., Sierra, J., Stein, E. M., Tallman, M. S., & Löwenberg, B. (2022). Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood, 140(12), 1345–1377. https://doi.org/10.1182/blood.2022016867
Döhner, H., Wei, A. H., & Löwenberg, B. (2021). Towards precision medicine for AML. Nature Reviews Clinical Oncology, 18(9), 577–590. https://doi.org/10.1038/s41571-021-00509-w
Döhner, H., Weisdorf, D. J., & Bloomfield, C. D. (2015). Acute myeloid leukemia. New England Journal of Medicine, 373(12), 1136–1152. https://doi.org/10.1056/NEJMra1406184
Duan, C.-W., Shi, J., Chen, J., Wang, B., Yu, Y.-H., Qin, X., Zhou, X.-C., Cai, Y.-J., Li, Z.-Q., Zhang, F., Yin, M.-Z., Tao, Y., Mi, J.-Q., Li, L.-H., Enver, T., Chen, G.-Q., & Hong, D.-L. (2014). Leukemia propagating cells rebuild an evolving niche in response to therapy. Cancer Cell, 25(6), 778–793. https://doi.org/10.1016/j.ccr.2014.04.015
Duchmann, M., Laplane, L., & Itzykson, R. (2021). Clonal architecture and evolutionary dynamics in acute myeloid leukemias. Cancers, 13(19), 4887. https://doi.org/10.3390/cancers13194887
Ferrando, A. A., & López-Otín, C. (2017). Clonal evolution in leukemia. Nature Medicine. https://doi.org/10.1038/nm.4410. Article 10.
Genovese, G., Kähler, A. K., Handsaker, R. E., Lindberg, J., Rose, S. A., Bakhoum, S. F., Chambert, K., Mick, E., Neale, B. M., Fromer, M., Purcell, S. M., Svantesson, O., Landén, M., Höglund, M., Lehmann, S., Gabriel, S. B., Moran, J. L., Lander, E. S., Sullivan, P. F., & McCarroll, S. A. (2014). Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. New England Journal of Medicine, 371(26), 2477–2487. https://doi.org/10.1056/NEJMoa1409405
Gibson, C. J., Lindsley, R. C., Tchekmedyian, V., Mar, B. G., Shi, J., Jaiswal, S., Bosworth, A., Francisco, L., He, J., Bansal, A., Morgan, E. A., Lacasce, A. S., Freedman, A. S., Fisher, D. C., Jacobsen, E., Armand, P., Alyea, E. P., Koreth, J., Ho, V., & Ebert, B. L. (2017). Clonal hematopoiesis associated with adverse outcomes after autologous stem-cell transplantation for lymphoma. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology, 35(14), 1598–1605. https://doi.org/10.1200/JCO.2016.71.6712
Gilliland, D. G., & Griffin, J. D. (2002). The roles of FLT3 in hematopoiesis and leukemia. Blood, 100(5), 1532–1542. https://doi.org/10.1182/blood-2002-02-0492
Goto, E., Tomita, A., Hayakawa, F., Atsumi, A., Kiyoi, H., & Naoe, T. (2011). Missense mutations in PML-RARA are critical for the lack of responsiveness to arsenic trioxide treatment. Blood, 118(6), 1600–1609. https://doi.org/10.1182/blood-2011-01-329433
Greaves, M., & Maley, C. C. (2012). Clonal evolution in cancer. Nature. https://doi.org/10.1038/nature10762. Article 7381.
Greif, P. A., Hartmann, L., Vosberg, S., Stief, S. M., Mattes, R., Hellmann, I., Metzeler, K. H., Herold, T., Bamopoulos, S. A., Kerbs, P., Jurinovic, V., Schumacher, D., Pastore, F., Bräundl, K., Zellmeier, E., Ksienzyk, B., Konstandin, N. P., Schneider, S., Graf, A., & Spiekermann, K. (2018). Evolution of cytogenetically normal acute myeloid leukemia during therapy and relapse: An exome sequencing study of 50 patients. Clinical Cancer Research, 24(7), 1716–1726. https://doi.org/10.1158/1078-0432.CCR-17-2344
Hahn, C. N., Chong, C.-E., Carmichael, C. L., Wilkins, E. J., Brautigan, P. J., Li, X.-C., Babic, M., Lin, M., Carmagnac, A., Lee, Y. K., Kok, C. H., Gagliardi, L., Friend, K. L., Ekert, P. G., Butcher, C. M., Brown, A. L., Lewis, I. D., To, L. B., Timms, A. E., & Scott, H. S. (2011). Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nature Genetics. https://doi.org/10.1038/ng.913. Article 10.
Hausser, J., & Alon, U. (2020). Tumour heterogeneity and the evolutionary trade-offs of cancer. Nature Reviews Cancer. https://doi.org/10.1038/s41568-020-0241-6. Article 4.
Hirsch, P., Zhang, Y., Tang, R., Joulin, V., Boutroux, H., Pronier, E., Moatti, H., Flandrin, P., Marzac, C., Bories, D., Fava, F., Mokrani, H., Betems, A., Lorre, F., Favier, R., Féger, F., Mohty, M., Douay, L., Legrand, O., & Delhommeau, F. (2016). Genetic hierarchy and temporal variegation in the clonal history of acute myeloid leukaemia. Nature Communications. https://doi.org/10.1038/ncomms12475. Article 1.
Hochhaus, A., Kreil, S., Corbin, A. S., La Rosée, P., Müller, M. C., Lahaye, T., Hanfstein, B., Schoch, C., Cross, N. C. P., Berger, U., Gschaidmeier, H., Druker, B. J., & Hehlmann, R. (2002). Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia. https://doi.org/10.1038/sj.leu.2402741. Article 11.
Hochhaus, A., Larson, R. A., Guilhot, F., Radich, J. P., Branford, S., Hughes, T. P., Baccarani, M., Deininger, M. W., Cervantes, F., Fujihara, S., Ortmann, C.-E., Menssen, H. D., Kantarjian, H., O’Brien, S. G., & Druker, B. J. (2017). Long-term outcomes of imatinib treatment for chronic myeloid leukemia. New England Journal of Medicine, 376(10), 917–927. https://doi.org/10.1056/NEJMoa1609324
Itzykson, R., Duployez, N., Fasan, A., Decool, G., Marceau-Renaut, A., Meggendorfer, M., Jourdan, E., Petit, A., Lapillonne, H., Micol, J.-B., Cornillet-Lefebvre, P., Ifrah, N., Leverger, G., Dombret, H., Boissel, N., Haferlach, T., & Preudhomme, C. (2018). Clonal interference of signaling mutations worsens prognosis in core-binding factor acute myeloid leukemia. Blood, 132(2), 187–196. https://doi.org/10.1182/blood-2018-03-837781
Jaiswal, S., Fontanillas, P., Flannick, J., Manning, A., Grauman, P. V., Mar, B. G., Lindsley, R. C., Mermel, C. H., Burtt, N., Chavez, A., Higgins, J. M., Moltchanov, V., Kuo, F. C., Kluk, M. J., Henderson, B., Kinnunen, L., Koistinen, H. A., Ladenvall, C., Getz, G., & Ebert, B. L. (2014). Age-related clonal hematopoiesis associated with adverse outcomes. New England Journal of Medicine, 371(26), 2488–2498. https://doi.org/10.1056/NEJMoa1408617
Jan, M., Snyder, T. M., Corces-Zimmerman, M. R., Vyas, P., Weissman, I. L., Quake, S. R., & Majeti, R. (2012). Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Science Translational Medicine, 4(149), 149ra118-149ra118. https://doi.org/10.1126/scitranslmed.3004315
Kandoth, C., McLellan, M. D., Vandin, F., Ye, K., Niu, B., Lu, C., Xie, M., Zhang, Q., McMichael, J. F., Wyczalkowski, M. A., Leiserson, M. D. M., Miller, C. A., Welch, J. S., Walter, M. J., Wendl, M. C., Ley, T. J., Wilson, R. K., Raphael, B. J., & Ding, L. (2013). Mutational landscape and significance across 12 major cancer types. Nature, 502(7471), 333–339. https://doi.org/10.1038/nature12634
Klco, J. M., Miller, C. A., Griffith, M., Petti, A., Spencer, D. H., Ketkar-Kulkarni, S., Wartman, L. D., Christopher, M., Lamprecht, T. L., Helton, N. M., Duncavage, E. J., Payton, J. E., Baty, J., Heath, S. E., Griffith, O. L., Shen, D., Hundal, J., Chang, G. S., Fulton, R., & Ley, T. J. (2015). Association between mutation clearance after induction therapy and outcomes in acute myeloid leukemia. JAMA, 314(8), 811–822. https://doi.org/10.1001/jama.2015.9643
Koeffler, H. P., & Leong, G. (2017). Preleukemia: One name, many meanings. Leukemia. https://doi.org/10.1038/leu.2016.364. Article 3.
Koya, J., Kataoka, K., Sato, T., Bando, M., Kato, Y., Tsuruta-Kishino, T., Kobayashi, H., Narukawa, K., Miyoshi, H., Shirahige, K., & Kurokawa, M. (2016). DNMT3A R882 mutants interact with polycomb proteins to block haematopoietic stem and leukaemic cell differentiation. Nature Communications, 7(1), 10924. https://doi.org/10.1038/ncomms10924
Lal, R., Lind, K., Heitzer, E., Ulz, P., Aubell, K., Kashofer, K., Middeke, J. M., Thiede, C., Schulz, E., Rosenberger, A., Hofer, S., Feilhauer, B., Rinner, B., Svendova, V., Schimek, M. G., Rücker, F. G., Hoefler, G., Döhner, K., Zebisch, A., & Sill, H. (2017). Somatic TP53 mutations characterize preleukemic stem cells in acute myeloid leukemia. Blood, 129(18), 2587–2591. https://doi.org/10.1182/blood-2016-11-751008
Landau, D. A., Carter, S. L., Stojanov, P., McKenna, A., Stevenson, K., Lawrence, M. S., Sougnez, C., Stewart, C., Sivachenko, A., Wang, L., Wan, Y., Zhang, W., Shukla, S. A., Vartanov, A., Fernandes, S. M., Saksena, G., Cibulskis, K., Tesar, B., Gabriel, S., & Wu, C. J. (2013). Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell, 152(4), 714–726. https://doi.org/10.1016/j.cell.2013.01.019
Landau, D. A., Clement, K., Ziller, M. J., Boyle, P., Fan, J., Gu, H., Stevenson, K., Sougnez, C., Wang, L., Li, S., Kotliar, D., Zhang, W., Ghandi, M., Garraway, L., Fernandes, S. M., Livak, K. J., Gabriel, S., Gnirke, A., Lander, E. S., & Wu, C. J. (2014). Locally disordered methylation forms the basis of intra-tumor methylome variation in chronic lymphocytic leukemia. Cancer Cell, 26(6), 813–825. https://doi.org/10.1016/j.ccell.2014.10.012
Levine, A. J., Jenkins, N. A., & Copeland, N. G. (2019). The roles of initiating truncal mutations in human cancers: The order of mutations and tumor cell type matters. Cancer Cell, 35(1), 10–15. https://doi.org/10.1016/j.ccell.2018.11.009
Li, B., Brady, S. W., Ma, X., Shen, S., Zhang, Y., Li, Y., Szlachta, K., Dong, L., Liu, Y., Yang, F., Wang, N., Flasch, D. A., Myers, M. A., Mulder, H. L., Ding, L., Liu, Y., Tian, L., Hagiwara, K., Xu, K., & Zhang, J. (2020). Therapy-induced mutations drive the genomic landscape of relapsed acute lymphoblastic leukemia. Blood, 135(1), 41–55. https://doi.org/10.1182/blood.2019002220
Li, S., Garrett-Bakelman, F. E., Chung, S. S., Sanders, M. A., Hricik, T., Rapaport, F., Patel, J., Dillon, R., Vijay, P., Brown, A. L., Perl, A. E., Cannon, J., Bullinger, L., Luger, S., Becker, M., Lewis, I. D., To, L. B., Delwel, R., Löwenberg, B., & Mason, C. E. (2016). Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nature Medicine, 22(7), 792–799. https://doi.org/10.1038/nm.4125
Ling, S., Hu, Z., Yang, Z., Yang, F., Li, Y., Lin, P., Chen, K., Dong, L., Cao, L., Tao, Y., Hao, L., Chen, Q., Gong, Q., Wu, D., Li, W., Zhao, W., Tian, X., Hao, C., Hungate, E. A., & Wu, C.-I. (2015). Extremely high genetic diversity in a single tumor points to prevalence of non-Darwinian cell evolution. Proceedings of the National Academy of Sciences, 112(47), E6496–E6505. https://doi.org/10.1073/pnas.1519556112
Lo-Coco, F., Avvisati, G., Vignetti, M., Thiede, C., Orlando, S. M., Iacobelli, S., Ferrara, F., Fazi, P., Cicconi, L., Di Bona, E., Specchia, G., Sica, S., Divona, M., Levis, A., Fiedler, W., Cerqui, E., Breccia, M., Fioritoni, G., Salih, H. R., & Platzbecker, U. (2013). Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. New England Journal of Medicine, 369(2), 111–121. https://doi.org/10.1056/NEJMoa1300874
Lyne, A.-M., Laplane, L., & Perié, L. (2021). To portray clonal evolution in blood cancer, count your stem cells. Blood, 137(14), 1862–1870. https://doi.org/10.1182/blood.2020008407
Mantovani, F., Collavin, L., & Del Sal, G. (2019). Mutant p53 as a guardian of the cancer cell. Cell Death & Differentiation. https://doi.org/10.1038/s41418-018-0246-9. Article 2.
McGranahan, N., & Swanton, C. (2017). Clonal heterogeneity and tumor evolution: Past, present, and the future. Cell, 168(4), 613–628. https://doi.org/10.1016/j.cell.2017.01.018
McMahon, C. M., Ferng, T., Canaani, J., Wang, E. S., Morrissette, J. J. D., Eastburn, D. J., Pellegrino, M., Durruthy-Durruthy, R., Watt, C. D., Asthana, S., Lasater, E. A., DeFilippis, R., Peretz, C. A. C., McGary, L. H. F., Deihimi, S., Logan, A. C., Luger, S. M., Shah, N. P., Carroll, M., & Perl, A. E. (2019). Clonal selection with RAS pathway activation mediates secondary clinical resistance to selective FLT3 inhibition in acute myeloid leukemia. Cancer Discovery, 9(8), 1050–1063. https://doi.org/10.1158/2159-8290.cd-18-1453
Merlo, L. M. F., Pepper, J. W., Reid, B. J., & Maley, C. C. (2006). Cancer as an evolutionary and ecological process. Nature Reviews Cancer. https://doi.org/10.1038/nrc2013. Article 12.
Metzeler, K. H., Herold, T., Rothenberg-Thurley, M., Amler, S., Sauerland, M. C., Görlich, D., Schneider, S., Konstandin, N. P., Dufour, A., Bräundl, K., Ksienzyk, B., Zellmeier, E., Hartmann, L., Greif, P. A., Fiegl, M., Subklewe, M., Bohlander, S. K., Krug, U., Faldum, A., On behalf of the AMLCG Study Group. (2016). Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood, 128(5), 686–698. https://doi.org/10.1182/blood-2016-01-693879
Miles, L. A., Bowman, R. L., Merlinsky, T. R., Csete, I. S., Ooi, A. T., Durruthy-Durruthy, R., Bowman, M., Famulare, C., Patel, M. A., Mendez, P., Ainali, C., Demaree, B., Delley, C. L., Abate, A. R., Manivannan, M., Sahu, S., Goldberg, A. D., Bolton, K. L., Zehir, A., & Levine, R. L. (2020). Single-cell mutation analysis of clonal evolution in myeloid malignancies. Nature, 587(7834), 477–482. https://doi.org/10.1038/s41586-020-2864-x
Mills, C. C., Kolb, E. A., & Sampson, V. B. (2018). Development of chemotherapy with cell cycle inhibitors for adult and pediatric cancer therapy. Cancer Research, 78(2), 320–325. https://doi.org/10.1158/0008-5472.CAN-17-2782
Moran-Crusio, K., Reavie, L., Shih, A., Abdel-Wahab, O., Ndiaye-Lobry, D., Lobry, C., Figueroa, M. E., Vasanthakumar, A., Patel, J., Zhao, X., Perna, F., Pandey, S., Madzo, J., Song, C., Dai, Q., He, C., Ibrahim, S., Beran, M., Zavadil, J., & Levine, R. L. (2011). Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell, 20(1), 11–24. https://doi.org/10.1016/j.ccr.2011.06.001
Morita, K., Wang, F., Jahn, K., Hu, T., Tanaka, T., Sasaki, Y., Kuipers, J., Loghavi, S., Wang, S. A., Yan, Y., Furudate, K., Matthews, J., Little, L., Gumbs, C., Zhang, J., Song, X., Thompson, E., Patel, K. P., Bueso-Ramos, C. E., & Takahashi, K. (2020). Clonal evolution of acute myeloid leukemia revealed by high-throughput single-cell genomics. Nature Communications, 11(1), 5327. https://doi.org/10.1038/s41467-020-19119-8
Murphy, T., Zou, J., Daher-Reyes, G. S., Arruda, A., Gupta, V., McNamara, C. J., Minden, M. D., Schimmer, A. D., Sibai, H., Yee, K. W. L., Korulla, M., Stockley, T., Kamel-Reid, S., Maze, D., Tierens, A., Bratman, S. V., Schuh, A. C., & Chan, S. M. (2019). Impact of preleukemic mutations and their persistence on hematologic recovery after induction chemotherapy for AML. Blood Advances, 3(15), 2307–2311. https://doi.org/10.1182/bloodadvances.2019000306
Nangalia, J., Nice, F. L., Wedge, D. C., Godfrey, A. L., Grinfeld, J., Thakker, C., Massie, C. E., Baxter, J., Sewell, D., Silber, Y., Campbell, P. J., & Green, A. R. (2015). DNMT3A mutations occur early or late in patients with myeloproliferative neoplasms and mutation order influences phenotype. Haematologica. https://doi.org/10.3324/haematol.2015.129510. Article 11.
Ortmann, C. A., Kent, D. G., Nangalia, J., Silber, Y., Wedge, D. C., Grinfeld, J., Baxter, E. J., Massie, C. E., Papaemmanuil, E., Menon, S., Godfrey, A. L., Dimitropoulou, D., Guglielmelli, P., Bellosillo, B., Besses, C., Döhner, K., Harrison, C. N., Vassiliou, G. S., Vannucchi, A., & Green, A. R. (2015). Effect of mutation order on myeloproliferative neoplasms. New England Journal of Medicine, 372(7), 601–612. https://doi.org/10.1056/nejmoa1412098
Padella, A., Ghelli Luserna Di Rorà, A., Marconi, G., Ghetti, M., Martinelli, G., & Simonetti, G. (2022). Targeting PARP proteins in acute leukemia: DNA damage response inhibition and therapeutic strategies. Journal of Hematology & Oncology. https://doi.org/10.1186/s13045-022-01228-0
Papaemmanuil, E., Gerstung, M., Bullinger, L., Gaidzik, V. I., Paschka, P., Roberts, N. D., Potter, N. E., Heuser, M., Thol, F., Bolli, N., Gundem, G., Van Loo, P., Martincorena, I., Ganly, P., Mudie, L., McLaren, S., O’Meara, S., Raine, K., Jones, D. R., & Campbell, P. J. (2016). Genomic classification and prognosis in acute myeloid leukemia. New England Journal of Medicine, 374(23), 2209–2221. https://doi.org/10.1056/nejmoa1516192
Pellegrino, M., Sciambi, A., Treusch, S., Durruthy-Durruthy, R., Gokhale, K., Jacob, J., Chen, T. X., Geis, J. A., Oldham, W., Matthews, J., Kantarjian, H., Futreal, P. A., Patel, K., Jones, K. W., Takahashi, K., & Eastburn, D. J. (2018). High-throughput single-cell DNA sequencing of acute myeloid leukemia tumors with droplet microfluidics. Genome Research, 28(9), 1345–1352. https://doi.org/10.1101/gr.232272.117
Peretz, C. A. C., McGary, L. H. F., Kumar, T., Jackson, H., Jacob, J., Durruthy-Durruthy, R., Levis, M. J., Perl, A., Huang, B. J., & Smith, C. C. (2021). Single-cell DNA sequencing reveals complex mechanisms of resistance to quizartinib. Blood Advances, 5(5), 1437–1441. https://doi.org/10.1182/bloodadvances.2020003398
Rasmussen, K. D., Jia, G., Johansen, J. V., Pedersen, M. T., Rapin, N., Bagger, F. O., Porse, B. T., Bernard, O. A., Christensen, J., & Helin, K. (2015). Loss of TET2 in hematopoietic cells leads to DNA hypermethylation of active enhancers and induction of leukemogenesis. Genes & Development, 29(9), 910–922. https://doi.org/10.1101/gad.260174.115
Roche-Lestienne, C., Soenen-Cornu, V., Grardel-Duflos, N., Laï, J.-L., Philippe, N., Facon, T., Fenaux, P., & Preudhomme, C. (2002). Several types of mutations of the Abl gene can be found in chronic myeloid leukemia patients resistant to STI571, and they can pre-exist to the onset of treatment. Blood, 100(3), 1014–1018. https://doi.org/10.1182/blood.v100.3.1014
Rothenberg-Thurley, M., Amler, S., Goerlich, D., Köhnke, T., Konstandin, N. P., Schneider, S., Sauerland, M. C., Herold, T., Hubmann, M., Ksienzyk, B., Zellmeier, E., Bohlander, S. K., Subklewe, M., Faldum, A., Hiddemann, W., Braess, J., Spiekermann, K., & Metzeler, K. H. (2018). Persistence of pre-leukemic clones during first remission and risk of relapse in acute myeloid leukemia. Leukemia. https://doi.org/10.1038/s41375-018-0034-z. Article 7.
Rücker, F. G., Du, L., Luck, T. J., Benner, A., Krzykalla, J., Gathmann, I., Voso, M. T., Amadori, S., Prior, T. W., Brandwein, J. M., Appelbaum, F. R., Medeiros, B. C., Tallman, M. S., Savoie, L., Sierra, J., Pallaud, C., Sanz, M. A., Jansen, J. H., Niederwieser, D., & Döhner, K. (2021). Molecular landscape and prognostic impact of FLT3-ITD insertion site in acute myeloid leukemia: RATIFY study results. Leukemia. https://doi.org/10.1038/s41375-021-01323-0
Russler-Germain, D. A., Spencer, D. H., Young, M. A., Lamprecht, T. L., Miller, C. A., Fulton, R., Meyer, M. R., Erdmann-Gilmore, P., Townsend, R. R., Wilson, R. K., & Ley, T. J. (2014). The R882H DNMT3A mutation associated with AML dominantly inhibits WT DNMT3A by blocking its ability to form active tetramers. Cancer Cell, 25(4), 442–454. https://doi.org/10.1016/j.ccr.2014.02.010
Sallman, D. A., & List, A. (2019). The central role of inflammatory signaling in the pathogenesis of myelodysplastic syndromes. Blood, 133(10), 1039–1048. https://doi.org/10.1182/blood-2018-10-844654
Schmalbrock, L. K., Dolnik, A., Cocciardi, S., Sträng, E., Theis, F., Jahn, N., Panina, E., Blätte, T. J., Herzig, J., Skambraks, S., Rücker, F. G., Gaidzik, V. I., Paschka, P., Fiedler, W., Salih, H. R., Wulf, G., Schroeder, T., Lübbert, M., Schlenk, R. F., & Bullinger, L. (2021). Clonal evolution of acute myeloid leukemia with FLT3-ITD mutation under treatment with midostaurin. Blood, 137(22), 3093–3104. https://doi.org/10.1182/blood.2020007626
Shlush, L. I., Zandi, S., Mitchell, A., Chen, W. C., Brandwein, J. M., Gupta, V., Kennedy, J. A., Schimmer, A. D., Schuh, A. C., Yee, K. W., McLeod, J. L., Doedens, M., Medeiros, J. J. F., Marke, R., Kim, H. J., Lee, K., McPherson, J. D., Hudson, T. J., Pan-Leukemia Gene Panel Consortium, T. H., & Dick, J. E. (2014). Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. https://doi.org/10.1038/nature13038. Article 7488.
Simon, L., Spinella, J.-F., Yao, C.-Y., Lavallée, V.-P., Boivin, I., Boucher, G., Audemard, E., Bordeleau, M.-E., Lemieux, S., Hébert, J., & Sauvageau, G. (2020). High frequency of germline RUNX1 mutations in patients with RUNX1-mutated AML. Blood, 135(21), 1882–1886. https://doi.org/10.1182/blood.2019003357
Smith, C. C., Paguirigan, A., Jeschke, G. R., Lin, K. C., Massi, E., Tarver, T., Chin, C.-S., Asthana, S., Olshen, A., Travers, K. J., Wang, S., Levis, M. J., Perl, A. E., Radich, J. P., & Shah, N. P. (2017). Heterogeneous resistance to quizartinib in acute myeloid leukemia revealed by single-cell analysis. Blood, 130(1), 48–58. https://doi.org/10.1182/blood-2016-04-711820
Smith, M. L., Cavenagh, J. D., Lister, T. A., & Fitzgibbon, J. (2004). Mutation of CEBPA in familial acute myeloid leukemia. New England Journal of Medicine, 351(23), 2403–2407. https://doi.org/10.1056/NEJMoa041331
Spencer, D. H., Russler-Germain, D. A., Ketkar, S., Helton, N. M., Lamprecht, T. L., Fulton, R. S., Fronick, C. C., O’Laughlin, M., Heath, S. E., Shinawi, M., Westervelt, P., Payton, J. E., Wartman, L. D., Welch, J. S., Wilson, R. K., Walter, M. J., Link, D. C., DiPersio, J. F., & Ley, T. J. (2017). CpG island hypermethylation mediated by DNMT3A Is a consequence of AML progression. Cell, 168(5), 801-816.e13. https://doi.org/10.1016/j.cell.2017.01.021
Stein, E. M., DiNardo, C. D., Fathi, A. T., Mims, A. S., Pratz, K. W., Savona, M. R., Stein, A. S., Stone, R. M., Winer, E. S., Seet, C. S., Döhner, H., Pollyea, D. A., McCloskey, J. K., Odenike, O., Löwenberg, B., Ossenkoppele, G. J., Patel, P. A., Roshal, M., Frattini, M. G., & Tallman, M. S. (2021). Ivosidenib or enasidenib combined with intensive chemotherapy in patients with newly diagnosed AML: A phase 1 study. Blood, 137(13), 1792–1803. https://doi.org/10.1182/blood.2020007233
Tyner, J. W., Tognon, C. E., Bottomly, D., Wilmot, B., Kurtz, S. E., Savage, S. L., Long, N., Schultz, A. R., Traer, E., Abel, M., Agarwal, A., Blucher, A., Borate, U., Bryant, J., Burke, R., Carlos, A., Carpenter, R., Carroll, J., Chang, B. H., & Druker, B. J. (2018). Functional genomic landscape of acute myeloid leukaemia. Nature, 562(7728), 526–531. https://doi.org/10.1038/s41586-018-0623-z
van Galen, P., Hovestadt, V., Wadsworth, M. H., II., Hughes, T. K., Griffin, G. K., Battaglia, S., Verga, J. A., Stephansky, J., Pastika, T. J., Lombardi Story, J., Pinkus, G. S., Pozdnyakova, O., Galinsky, I., Stone, R. M., Graubert, T. A., Shalek, A. K., Aster, J. C., Lane, A. A., & Bernstein, B. E. (2019). Single-cell RNA-Seq reveals AML hierarchies relevant to disease progression and immunity. Cell, 176(6), 1265-1281.e24. https://doi.org/10.1016/j.cell.2019.01.031
Welch, J. S., Ley, T. J., Link, D. C., Miller, C. A., Larson, D. E., Koboldt, D. C., Wartman, L. D., Lamprecht, T. L., Liu, F., Xia, J., Kandoth, C., Fulton, R. S., McLellan, M. D., Dooling, D. J., Wallis, J. W., Chen, K., Harris, C. C., Schmidt, H. K., Kalicki-Veizer, J. M., & Wilson, R. K. (2012). The origin and evolution of mutations in acute myeloid leukemia. Cell, 150(2), 264–278. https://doi.org/10.1016/j.cell.2012.06.023
Wong, T. N., Ramsingh, G., Young, A. L., Miller, C. A., Touma, W., Welch, J. S., Lamprecht, T. L., Shen, D., Hundal, J., Fulton, R. S., Heath, S., Baty, J. D., Klco, J. M., Ding, L., Mardis, E. R., Westervelt, P., DiPersio, J. F., Walter, M. J., Graubert, T. A., & Wilson, R. K. (2015). The role of TP53 mutations in the origin and evolution of therapy-related AML. Nature, 518(7540), 552–555. https://doi.org/10.1038/nature13968
Yang, F., Brady, S. W., Tang, C., Sun, H., Du, L., Barz, M. J., Ma, X., Chen, Y., Fang, H., Li, X., Kolekar, P., Pathak, O., Cai, J., Ding, L., Wang, T., von Stackelberg, A., Shen, S., Eckert, C., Klco, J. M., & Zhou, B.-B.S. (2021). Chemotherapy and mismatch repair deficiency cooperate to fuel TP53 mutagenesis and ALL relapse. Nature Cancer, 2(8), 819–834. https://doi.org/10.1038/s43018-021-00230-8
Zebisch, A., Lal, R., Müller, M., Lind, K., Kashofer, K., Girschikofsky, M., Fuchs, D., Wölfler, A., Geigl, J. B., & Sill, H. (2016). Acute myeloid leukemia with TP53 germ line mutations. Blood, 128(18), 2270–2272. https://doi.org/10.1182/blood-2016-08-732610
Zeng, A. G. X., Bansal, S., Jin, L., Mitchell, A., Chen, W. C., Abbas, H. A., Chan-Seng-Yue, M., Voisin, V., van Galen, P., Tierens, A., Cheok, M., Preudhomme, C., Dombret, H., Daver, N., Futreal, P. A., Minden, M. D., Kennedy, J. A., Wang, J. C. Y., & Dick, J. E. (2022). A cellular hierarchy framework for understanding heterogeneity and predicting drug response in acute myeloid leukemia. Nature Medicine. https://doi.org/10.1038/s41591-022-01819-x. Article 6.
Zhang, H., Savage, S., Schultz, A. R., Bottomly, D., White, L., Segerdell, E., Wilmot, B., McWeeney, S. K., Eide, C. A., Nechiporuk, T., Carlos, A., Henson, R., Lin, C., Searles, R., Ho, H., Lam, Y. L., Sweat, R., Follit, C., Jain, V., & Tyner, J. W. (2019). Clinical resistance to crenolanib in acute myeloid leukemia due to diverse molecular mechanisms. Nature Communications, 10(1), 244. https://doi.org/10.1038/s41467-018-08263-x
Acknowledgements
This work was supported by grants from the National Key R&D Program of China (No. 2021YFC2500300; No. 2021YFA1100800 to B-BSZ), and the National Natural Science Foundation of China (No. 81890992 to Q-FW; No. 81970141 to B-BSZ).
Author information
Xu-Dong He, Meng-Fang Xia and Ji-Yuan Teng contributed equally to this work.
Authors and Affiliations
CAS Key Laboratory of Genomic and Precision Medicine, Beijing Institute of Genomics, Chinese Academy of Sciences and China National Center for Bioinformation, Beijing, 100101, China
Xu-Dong He, Meng-Fang Xia & Qian-Fei Wang
University of Chinese Academy of Sciences, Beijing, 100049, China
Xu-Dong He, Meng-Fang Xia & Qian-Fei Wang
Key Laboratory of Pediatric Hematology and Oncology Ministry of Health, Pediatric Translational Medicine Institute, Department of Hematology and Oncology, Shanghai Children’s Medical Center, School of Medicine, Shanghai Jiao Tong University, Shanghai, 200127, China
Ji-Yuan Teng & Bin-Bing S. Zhou
Department of Pharmacology and Chemical Biology, School of Basic Medicine and Collaborative Innovation Center for Translational Medicine, School of Medicine, Shanghai Jiao Tong University, Shanghai, 200025, China
Ji-Yuan Teng & Bin-Bing S. Zhou
Contributions
X-DH, M-FX and J-YT were responsible for literature research and paper writing. M-FX contributed to figure preparation. Q-FW and B-BSZ provided helpful suggestions for conceptual explanation and paper writing.
Corresponding authors
Correspondence to Bin-Bing S. Zhou or Qian-Fei Wang.
Ethics declarations
Conflict of interest
The authors declare that there is no competing interest.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article
Cite this article
He, XD., Xia, MF., Teng, JY. et al. Clonal evolution in leukemia: preleukemia, evolutionary models, and clinical implications. GENOME INSTAB. DIS. 4, 227–238 (2023). https://doi.org/10.1007/s42764-023-00104-6
Received09 April 2023
Revised16 June 2023
Accepted29 June 2023
Published20 July 2023
Issue DateAugust 2023
DOIhttps://doi.org/10.1007/s42764-023-00104-6
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable linkKeywords
Leukemia
Clonal evolution
Evolutionary model
Drug therapy
用户登录
还没有账号?
立即注册