






In Silico design and characterization of RAD51 protein inhibitors targeting homologous recombination repair for cancer therapy
Original Research Paper
Harsimrat Kaur, Harsimran Kaur, Preeti Rajesh & Changanamkandath Rajesh
Genome Instability & Disease 4, 289–302 (2023)
Abstract
Cancer occurs when a portion of the body's cells begin to divide uncontrollably and spread into surrounding tissues. Most common reason for growth is genetic mutations. DNA Double Strand Breaks (DSB) brought about by ionizing radiation or other cancer-causing chemicals are one of the unavoidable changes bringing about carcinogenesis. Efficient DNA repair is generally gainful for living beings but in cancer therapy efficient DNA repair challenges the activity of radio and chemotherapies based on DNA damaging agents with cells becoming resistant to drugs. DSB repair pathways therefore serve as critical components for tumor suppression. Cells accomplish error-free repair of DNA DSBs by homologous recombination (HR) repair pathway. Mammalian proteins involved in HR include BRCA1, BRCA2, RAD51 and the RAD51 paralogs. Targeting the function of these proteins can, therefore, offer solution to resistance to anti-cancer treatment, thereby improving the efficiency of chemotherapy and preventing reoccurrence of tumors. RAD51 and its paralogs are central players of the pathway and are targeted for functional disruption. Sequence analysis of RAD51 proteins demonstrates regions within the ATPase domain that are conserved across species and amongst various paralogs. Multiple CHK1/CHK2 phosphorylation sites are present in all the paralogs with at least one of the site in close proximity to the ATPase domain. Inhibitors are identified that bind to the ATP-binding pocket of RAD51 paralogs thereby affecting its hydrolysis. The RAD51 protein interaction with BRCA2 protein is another important target through peptides designed against evolutionarily conserved regions of BRCA2, BRC4. Combinatorial targeting of the function of proteins involved in HR in combination with DNA damaging agents can, therefore, offer solution to radio and chemoresistance, thereby improving the efficiency of chemotherapy and preventing re-occurrence of tumors.
Introduction
Cancer is a global health threat. As per the World Health Organization (WHO), cancer is a worldwide pandemic with 14 million cases diagnosed in 2012 and 23 million new cases expected by 2030 (National Cancer Institute, 2018). These calamitous numbers warrant the development of innovative strategies to combat this deadly disease. Currently, first-line treatments for cancer include surgery, radiation, chemotherapy and in specific cases hormonal therapy. However, the mainstay chemotherapy and radiotherapy treatments have debilitating side effects and toxicity (Begg et al., 2011; Vanneman & Dranoff, 2012; Zamarin & Postow, 2015). In addition, resistance developed by the cancer cells to therapy presents a major obstacle in cancer treatment. Cancer can develop therapeutic resistance to chemotherapy, radiotherapy and targeted therapies, through different mechanisms at the outset of treatment, or after an initial clinical response. Specific genetic and epigenetic alterations in the cancer cell and/or the microenvironment in which the cancer cell resides contribute to the emergence of resistance (Dean et al., 2005; Donnenberg & Donnenberg, 2005; Indran et al., 2011; Park et al., 2018). It is imperative to discover more effective treatments that can defeat cancer more efficiently and have fewer side effects.
Cancer cells harbor numerous genetic alterations, which are responsible for cancer development and promote cancer progression but is also the primary reason for the death of these cells by chemotherapy and radiotherapy. It is crucial to understand the molecular mechanisms of DNA damage and repair to generate innovative intervention strategies. One such potential approach in therapy is to block the ability of a cancer cell to recognize and repair the damaged DNA in front-line cancer treatments; chemotherapy and radiation (Kelley & Fishel, 2008). DNA damage is an inevitable process. It is not only caused by cancer treatments but arises as a result of normal cellular processes. Reactive oxygen species generated by cellular metabolism can damage DNA bases, stalled replication forks causing double-stranded break in the DNA. Cells need to constantly repair these damages in order to maintain stable genomes thus preventing cancer occurrence and aging (Hoeijmakers, 2001; Jackson & Bartek, 2009; Kanaar et al., 1998). To accomplish this, most cells utilize two main DSB repair pathways: non-homologous end-joining (NHEJ) and homologous recombination (HR) (Aparicio et al., 2014). In NHEJ, DSB ends are trimmed and ligated, and NHEJ is, therefore, error prone, while HR uses a homologous sequence (typically the sister chromatid) as a repair template and is generally accurate (Nickoloff et al., 2017).
Mammalian proteins involved in HR repair include BRCA1, BRCA2, RAD51 (a RecA homolog) and the RAD51 paralogs (RAD51B, RAD51C, RAD51D, XRCC2 and XRCC3). The RAD51 protein along with its paralogs plays a crucial role in the HR pathway (Davies et al., 2001; Friedberg & Friedberg, 2006; Smiraldo et al., 2005). RAD51-mediated HR is the dominant pathway for restarting stalled or broken replication forks, which if processed improperly by NHEJ cause genome instability and neoplastic transformation (Nickoloff et al., 2017). Detection of DSBs by the DNA damage sensors leads to the activation of damage response signaling and recruitment of Checkpoint kinase 1 (CHK1) which regulates HR and cell-cycle checkpoints (Sørensen et al., 2005). While efficient DNA repair is indispensable in normal cells, it counteracts the DNA-damaging action of radio- and chemotherapies with cancer cells becoming resistant to drugs (Christodoulopoulos et al., 1999). Over-expression of RAD51 has been correlated to resistance to anticancer treatment and to the level of cancer advancement (Hannay et al., 2007; Klein, 2008). Thus, these HR proteins and their interacting partners make excellent potential molecular targets. Specific inhibition of these targets and their protein–protein interactions (PPIs) can potentially enhance the current standard of cancer therapy.
In the present study, chemical inhibitors and peptides have been used as ligands that bind to HR protein sites and thus inhibit the repair of DNA damage generated by chemotherapeutic agents. In silico analysis of RAD51 and its paralog proteins identified the ATPase domains, DNA binding domains and protein interacting domains of these proteins as potential drug targets. In this study, we have designed peptides and discussed other chemical inhibitors to block the functioning of RAD51 and its associated proteins for potential combinatorial chemotherapeutic application.
Materials and methodology
Protein sequences and structures
The protein sequences for human RAD51 and its paralogs i.e. RAD51B, RAD51C, RAD51D, XRCC2 and XRCC3 were retrieved from the UniProt database (The UniProt Consortium). The structure for human RAD51 was retrieved from the Protein Data Bank (PDB ID: 1N0W) (Pellegrini et al., 2002). Structures of all the RAD51 and its paralog structures are not available, so the sequences retrieved from the UniProt Database were used for the structure prediction. The raw amino acid sequences of RAD51 and its paralogs proteins were uploaded in FASTA format to I-TASSER and RaptorX web servers, both of which use the threading approach to predict protein structures (Yang et al., 2015). I-TASSER (Iterative Threading ASSEmbly Refinement) server (as ‘Zhang-Server’) and RaptorX are web-based services for the prediction of protein structures and functions and automatically generated high-quality models of the 3D structure of proteins from their amino acid sequences. Tertiary structures were predicted in PDB format. The results generated five top models for each entry and the one with the highest confidence score (c-score) represented the best model and was selected for this study. ProQ- Protein Quality Predictor and ProSA program (Protein Structure Analysis) were used for refinement and validation of the predicted structures (Wallner & Elofsson, 2003; Wiederstein & Sippl, 2007). The protein sequences of these proteins were also analyzed for the CHK1 and CHK2 consensus phosphorylation site motif (RXXS/T) using the SCANSITE program (Obenauer et al., 2003).
Sequence alignment
Retrieved sequences were aligned using the T-COFFEE program to find the conserved region. It efficiently combined the output of various Multiple Sequence Alignment (MSA) packages into one final MSA (Stothard, 2000). The alignments were further analyzed for the similarity between RAD51 and its paralog proteins using the online tool ‘Ident and Sim’ from SMS: Sequence Manipulation Suite (Bioinformatics.org) (Stothard, 2000).
Structural analysis of protein interaction
The protein–protein interaction sites were analyzed for use as targets for inhibition using the available crystalline structure of HR protein RAD51. The PDB ID 1N0W that has RAD51 and BRCA2 crystallized together was used for the study. To look for the region where they interact in the crystallized structure, the software LIGPLOT + was used (Laskowski & Swindells, 2011). The potential binding sites data was also retrieved from I-TASSER along with the structure prediction data. To countercheck the predicted ligand binding sites, the 3DLigandSite tool was used (Wass et al., 2010).
Chemical and peptide inhibitors
The available chemical inhibitors were studied from the literature for their ability to inhibit ATPase activity. The PDB structures of the ATP site inhibitors were retrieved using the online tool CORINA by converting SDF format to PDB format. The 3D format was retrieved from the PubChem database. Using the Peptide Generator Tool from GenScript, peptides from the BRCA2 interacting sequence were retrieved. BRC repeat 4 (BRC4) was used as a ligand molecule for studying the interaction of BRCA2 with RAD51. The peptide structure was generated using the PEP-FOLD3 online web tool (http://bioserv.rpbs.univ-paris-diderot.fr/services/PEP-FOLD3/) (Lamiable et al., 2016). PEP-FOLD is a de novo approach aimed at predicting peptide structures from amino acid sequences.
Docking studies of inhibitors
A Molecular Graphics Laboratory (MGL) tool (The Scripps Research Institute, La Jolla, CA, USA) was used for the visualization and analysis of molecular structures. AutoDockTools (ADT) was used to prepare the molecule for docking. AutoDock Vina (The Scripps Research Institute) docking tool was used to predict the interaction between small molecules, such as drug compounds or substrates, with the three-dimensional structure of the protein. Docking between HR repair protein (RAD51: Receptor) and chemical inhibitors and peptides ligands was performed using AutoDock Vina (Trott & Olson, 2010). Docked structures were visualized using the open source, molecular visualization system PyMol 1.7.4 (Schrödinger, LLC, Cambridge, MA, USA). Input comprises receptor, ligand and docking box, while output is a list of poses ranked by DG, the predicted binding energy in kcal/mol.
The receptor and ligand molecules were prepared for docking using AutoDockTools. RAD51 molecule was opened using ADT, after which water molecules and other bound ligand molecules were removed. The binding site was defined after adding hydrogen molecules to the RAD51 receptor molecule to describe the receptor cavity. Receptor and ligand molecules were saved as PDBQT.
Results
Sequence analysis identify putative sites to target within the RAD51 proteins
RAD51 recombinase and its human paralogs (HsRAD51B, HsRAD51C, HsRAD51D, HsXRCC2 and HsXRCC3) form several multimeric complexes to facilitate the process of homologous recombination. Multiple sequence alignment of protein sequences of human HsRAD51 and its paralogs with those of mouse (Mus musculus), zebrafish (Danio rerio), Arabidopsis and bovine (Bos taurus) species revealed > 90% similarity of human RAD51 to mouse, zebrafish, and bovine species. The nucleotide sequence similarity in Arabidoposis was slightly low at around 80% (Kaur & Rajesh, 2016)). Protein complexes of these paralogs bind to single-stranded or double-stranded DNA and exhibit ATP hydrolysis activity. Therefore, as expected, the RAD51 proteins in all the species share a highly conserved ATPase domain, central to which is the presence of two separate sequence motifs known as the Walker A and Walker B adenosine nucleotide binding motifs. HsRAD51 and HsRAD51D also share a small Linker region, L1 that also exhibited a high amount of similarity (Fig. 1).
Fig. 1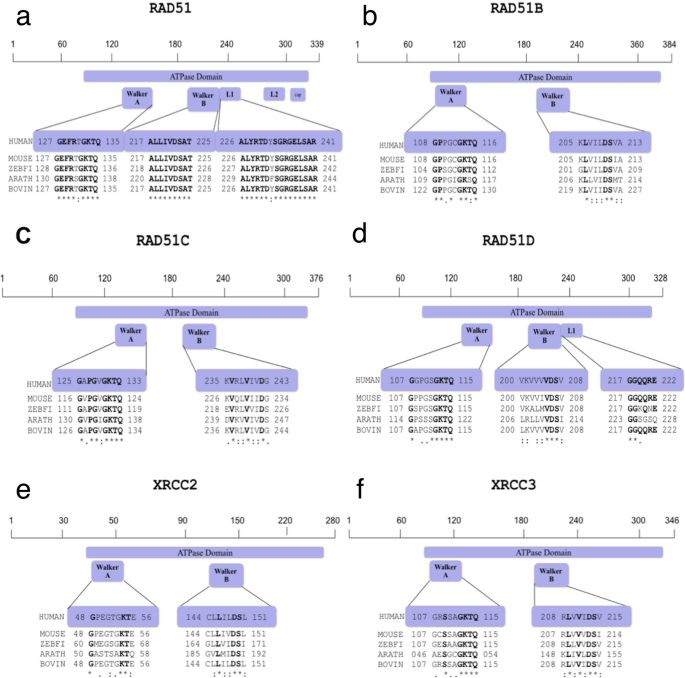
Inter-species Sequence Alignment of RAD51 Protein and Its Human Paralogs. Protein sequences retrieved from Uniprotein Database were used for interspecies comparison and identification of conserved regions as targets. (a) Homo sapiens (Hs)RAD51 (Q06609) was aligned to Mus musculus (Mm)RAD51 (Q08297), Danio rerio (Dr)RAD51 (Q5TYR0), Arabidopsis thaliana, (At)RAD51 (P94102), Bos taurus, (Bt)RAD51 (Q2KJ94); (b) HsRAD51B (O15315) was aligned to MmRAD51B (O35719), DrRAD51B (F1QJ27), AtRAD51B (Q9SK02), BtRAD51B (E1BE93); (c) HsRAD51C (O43502) was aligned to MmRAD51C (Q924H5), DrRAD51C (F1QQC9), AtRAD51C (Q8GXF0), BtRAD51C (F1MW99), (d) HsRAD51D (O75771) was aligned to MmRAD51D (O55230), DrRAD51D (F1R474), AtRAD51D (Q9LQQ2), BtRAD51D (Q2HJ51); (e) HsXRCC2 (O43543) was aligned to MmXRCC2 (Q9CX47), DrXRCC2 (F1Q9N3), AtXRCC2 (Q682D3), BtXRCC2 (A7MB17); (f) HsXRCC3 (O43542) was aligned to MmXRCC3 (Q9CXE6), DrXRCC3 (Q5BL30), AtXRCC3 (Q9FKM5) and BtXRCC3 (Q08DH8). The ATPase domains with motifs walker A and walker B and Linker regions (L1 and L2) are highlighted in purple with the conserved residues marked in bold font. The asterisk (*) signifies the conserved residues amongst sequences of all species analysed
Multiple sequence alignment of human HsRAD51 with its paralogs (HsRAD51B, HsRAD51C, HsRAD51D, HsXRCC2 and HsXRCC3) identified a set of conserved regions within all the paralogs based on the number of identical and similar amino acids (Fig. 2a). The protein sequences of the five paralog proteins when compared to RAD51 showed 30–40% overall sequence similarity. However, the 8 bp Walker A motif of the paralogs was approximately 50% similar to that of RAD51 while the 5 bp walker B motif is 100% similar among all the paralogs. The Walker A signature sequence was (G/A)XXXXGK(T/S), where “X” can be any amino acid and the Walker B signature sequence was ΦΦΦΦD, has four hydrophobic residues (indicated by “Φ”) followed by an aspartate carboxyl group (delToro et al., 2016) Fig. 2a). No deviation from the classical Walker sequences was observed in any of the 6 proteins. Glycine was observed at position 1 and GKT were the amino acids at position 6, 7 and 8. Similarity index of 50% is attributed to the difference in the variable quartet. In all the 6 proteins, this region differed in sequence composition, which could account for the differences in the nucleotide recognition and/or catalytic mechanisms in these proteins. Walker B motif in all 6 proteins is composed of 3 hydrophobic amino acids leucine, isoleucine and valine in unique arrangements followed by the aspartic acid (Fig. 2a).
Fig. 2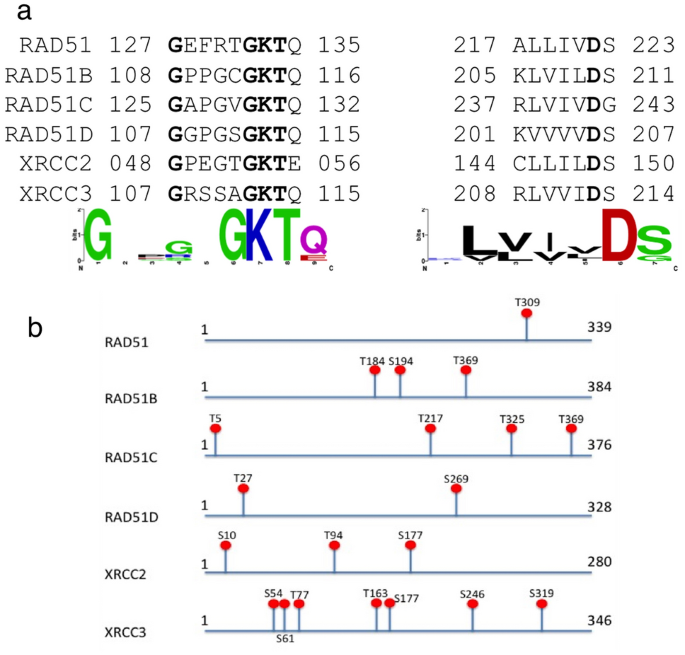
Putative Functional Targets of HsRAD51 Protein Functional targets for inhibition of HR activity were identified on HsRAD51 proteins based on sequence analysis. (a) Graphical representation of sequence alignment and consensus sequence of HsRAD51 (Q06609) with respect to its paralogs, HsRAD51B (O15315), HsRAD51C (O43502), HsRAD51D (O75771) HsXRCC2 (O43543) and HsXRCC3 (O43542). The motifs walker A and walker B are shown among the paralogs with the conserved residues in bold. Graphical representation of the consensus sequence of walker A and walker B (Sequence Logo) is also depicted. (b) Predicted Serine/Threonine phosphorylation sites (S/T) in Human RAD51 protein and its paralogs as predicted by SCANSITE2.0
To understand their functional regulation, we screened the proteins sequences of these proteins for the CHK1 and CHK2 consensus phosphorylation site motif (RXXS/T) using the SCANSITE program (Obenauer et al., 2003). CHK1/CHK2 mediated phosphorylation is important for the transition of the hBRCA2-RAD51 complex from an inactive form in undamaged cells to an active form in damaged cells (Bahassi et al., 2008). Our results demonstrated that all 6 paralogs harbor the CHK1/2 consensus phosphorylation motifs at multiple sites. T309 (RAD51); T184, S194, S240 (RAD51B); T5, T217, T325, T369 (RAD51C), T27, S269 (RAD51D), S10, T94, S177 (XRCC2) and S54, S61, T77, T163, S177, S246, S319 (XRCC3) are the potential phosphorylation sites (Fig. 2b). Each paralog had at least one potential site within or adjacent to the conserved ATPase domain that could be affected on targeting the site for inhibition.
Structural modeling of RAD51 and paralogs
The complete three-dimensional (3-D) structures of RAD51 and its paralogs are not currently available in the PDB structure databases and have been a hindrance to the studies related to this protein. We predicted the 3-D models of RAD51 and its paralog proteins using I-TASSER and RaptorX web servers, both of which use the threading approach to predict protein structures (Källberg et al., 2012). I-TASSER uses the best-aligned template obtained by searching against the Protein Data Bank database and is one of the most successful servers in the community-wide CASP (Critical Assessment of Protein Structure Prediction) experiments. 3D structure prediction by I-TASSER generates five top models with C-scores ranging from − 5 to 2. The one with the highest C-score represents the best model.
The highest value for predicted models of RAD51, RAD51B, RAD51C, RAD51D, XRCC2 and XRCC3 were −0.28, −0.84, −1.10, 0.30, −0.30 and 0.09, respectively. The best-selected 3-D models for proteins based on the C-score were consistent with secondary structure elements generated by RaptorX, which performs threading template identification using a nonlinear alignment score. The results retrieved from both servers were analyzed using ProQ and ProSA web servers. The ProQ analysis of the quality of the protein model gave LG score of more than 4 for structures predicted by I-TASSER whereas less than 4 for those predicted by RaptorX. An LG score for proteins of more than 4 is considered as extremely good models and less than 4 corresponds to very good models. The ProSA web server for checking errors in 3D structures gave a better Z score for I-TASSER-predicted models than RaptorX-predicted models. Based on these analyses, the models generated by I-TASSER were selected and used for further analysis and target docking. They also served as templates to determine the inhibitor effect of various targets.
Binding site prediction of RAD51 proteins reveal ATPase domain as a potential binding site
The modeled structures of RAD51 and paralogs were submitted to the 3DLigandSite server to predict the cluster of ligands and the potential binding sites associated with these clusters. The server found the ligands bound to the structures similar to the query and superimposed them onto the model to predict the binding site. ATP, ADP and Mg2+ ligands were bound to the structures of RAD51 and its paralogs. The predicted binding sites for each protein were found to be predominantly part of the ATP binding site domain and the WALKER A motif with very few amino acid sites outside this region for each protein (Fig. 3). The specific Walker A motif predicted as potential ligand sites were Phe129-Gln135 for HsRAD51, Pro110-Thr115 for HsRAD51B, Gly128-Gln133 for HsRAD51C, Gly110-Gln115 for HsRAD51D, Pro49-Glu56 for HsXRCC2 and Arg108-Gln115 for HsXRCC3, respectively (Fig. 1a–f and Fig. 3a–f). Most of these amino acids represented conserved residues among different species as well as among the paralogs (Figs. 1 and 2a). HsXRCC3 showed another small three amino acid region Ile332-Ala334 with potential binding capacity to these ligands. Interestingly, there were also potential binding sites near the previously predicted phosphorylation sites outside the ATPase domain namely Arg310 in HsRAD51 (Close to Thr309), Ile37 in HsRAD51B (Close to Thr369), Trp317 in HsRAD51C (Close to Thr325), Asp310 in HsRAD51D (Close to S269), Arg91 in HsXRCC2 (Close to T94) and Arg313 in HsXRCC3 (Close to Ser319) (Fig. 2b). The ligand binding motifs within the ATPase Walker A region suggests it to present the best potential target for inhibition, not only due to its conserved sequence but also based on the binding potential.
Fig. 3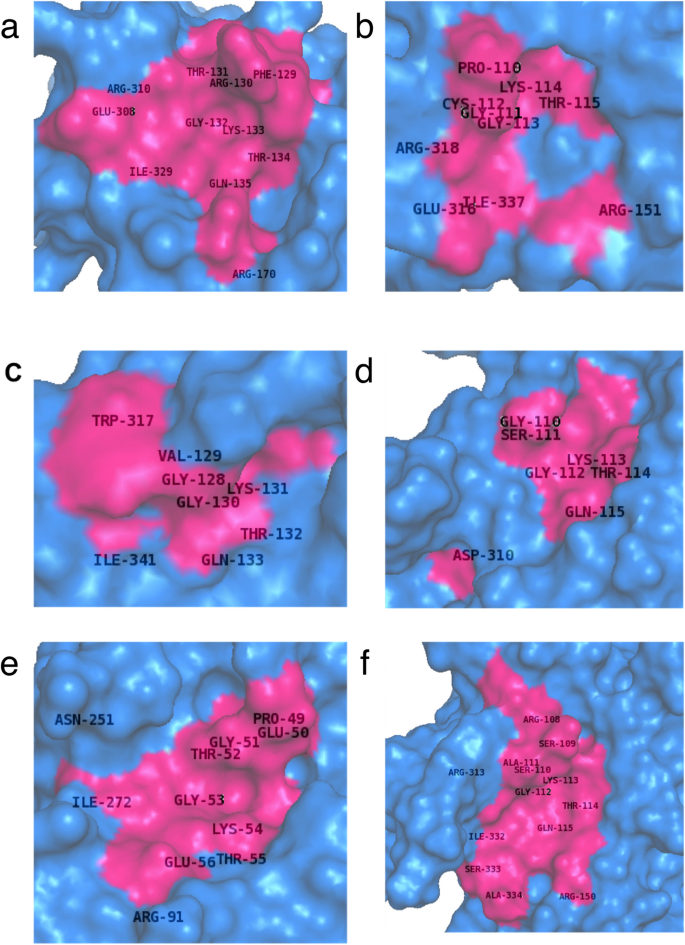
Predicting the Ligand Binding Sites of RAD51 and its Paralogs Pictorial representation of the potential binding sites of various RAD51 proteins as predicted by the software 3DLigandSite are shown. The structures generated using I-TASSER of various human RAD51 proteins HsRAD51 (a), HsRAD51B (b), HsRAD51C (c), HsRAD51D (d), HsXRCC2 (e), HsXRCC3 (f) were used for the prediction of ligand binding sites. The surface model of the RAD51 and its paralogs are depicted in blue while the pink region corresponds to the software predicted sites. The amino acid residues depicted for each protein along with their amino acid location are the ones predicted by 3D-LigandSite as potential binding sites for the ligands
Docking studies identify CB-5083 as a potential inhibitor of RAD51 protein function
Over ten small-molecule inhibitors targeting the ATP binding pocket were identified based on the literature search that included Apoptolidin, Bafilomycin A1, Brefeldin, Bufalin, CB-5083, ICRF-193, Monastrol, Oligomycin, Sarcophine and Sodium orthovandate. The molecular docking of RAD51 proteins with these chosen ligands was performed using AutoDock Vina, which generated nine different poses for each ligand with each paralog. For each inhibitor, the docking results were compared with the 3DLigandSite results (Results not shown). Of all these ligands analyzed, CB-5083, demonstrated the best results. CB5083 docked with the HsRAD51 residues Phe129, Arg130, Gln135, Arg170, Lys304, Glu308, Arg310 and Ile329 (binding energy -5.8 kcal/mol) (Fig. 4a). It was an exact match to the 3DLigandSite predicted model as well. The residues Phe129, Arg130 and Gln135 are conserved residues of the Walker A motif (Fig. 2a). Similar results were obtained for other paralogs as well with CB5083 docking with conserved Walker A residues Lys114, Thr115 and Gln116 of HsRAD51B in addition to Glu144, Arg151, Gln258, Glu316 and Ile337 outside the motif (Fig. 4b). With HsRAD51C, CB5083 docked with Pro127, Gly128, Lys131, Thr132, Gln133 within Walker A conserved region and Glu161, Ser163 and Gln285 outside (Fig. 4c). HsRAD51D CB5083 docking with conserved residues of Walker A Gly110, Gly112, Lys113, Thr114 and Gln115 as well as Asp206 and Asp207 of Walker B motif with only Ile311 and Thr248 outside the ATPase domain (Fig. 4d). The docking within HsXRCC2 is also observed with variable Glu50 and Gly51 and conserved Lys54, Thr55 and Glu56 within the Walker A and His86 and Ile198 outside the domain (Fig. 4e). It was only with HsXRCC3 that the CB5083 docked outside the Walker although still within the ATPase domain with residues Arg231, Ala232, Ala283, Leu284, Gly285 and Ile286 (Fig. 4f). These data suggest that inhibitory RAD51 binding compounds, as CB5083 could interfere with ATP binding and hydrolysis in multiple RAD51 proteins and could therefore interfere with their function on DNA repair.
Fig. 4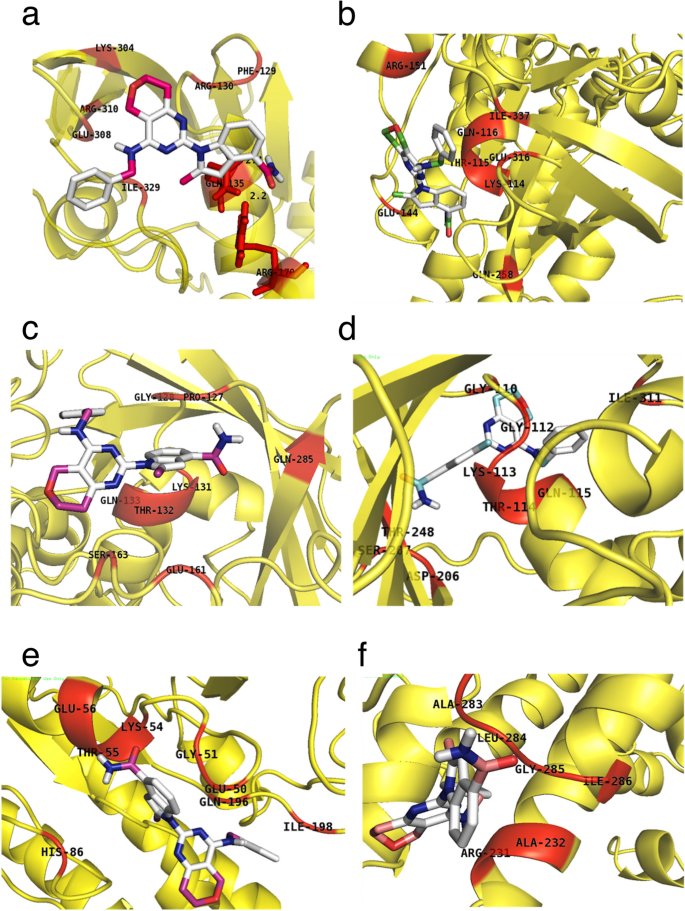
The Docked Model of RAD51 Protein and Ligand CB-5083 Pictorial representation of docked structure of CB-5083 (ball and stick model) with different RAD51 proteins (Cartoon model) (a) HsRAD51 (b) HsRAD51B (c) HsRAD51C (d), HsRAD51D (e) HsXRCC2 and (f) HsXRCC3, respectively. The amino acid residues for each protein along with their amino acid location are the ones predicted as potential binding sites of ligand, CB5083 on docking
Peptide inhibitors to interfere functional interaction of RAD51-BRCA2
Interaction network of RAD51 and its paralogs proteins were mapped by searching the STRING [Search Tool for the Retrieval of Interacting Genes/Proteins] database version 10.5 with a confidence cutoff of 0.9 (Fig. 5a). These proteins directly or indirectly interact with a number of proteins including the DNA damage sensors and effectors like Mre11 and CHK1, with proteins involved in other DNA repair pathways MSH2, MSH3, MSH6 and EXO1 as well as Cell cycle and DNA metabolism proteins as MCM2, BLM, CDK2, RPA2, etc. These protein–protein interactions (PPIs) can be very important for various cellular functions. Inhibition of these PPIs could offer potential targets for small-molecule intervention in cancer with deregulated HR. One very important PPI in the process of HR is the interaction between the proteins RAD51 and BRCA2. The BRCA2–RAD51 interaction promotes the localization of the protein complex to its site of function in the cell nucleus and targets RAD51 to sites of DNA damage where it assembles in microscopic foci (Davies et al., 2001).
Fig. 5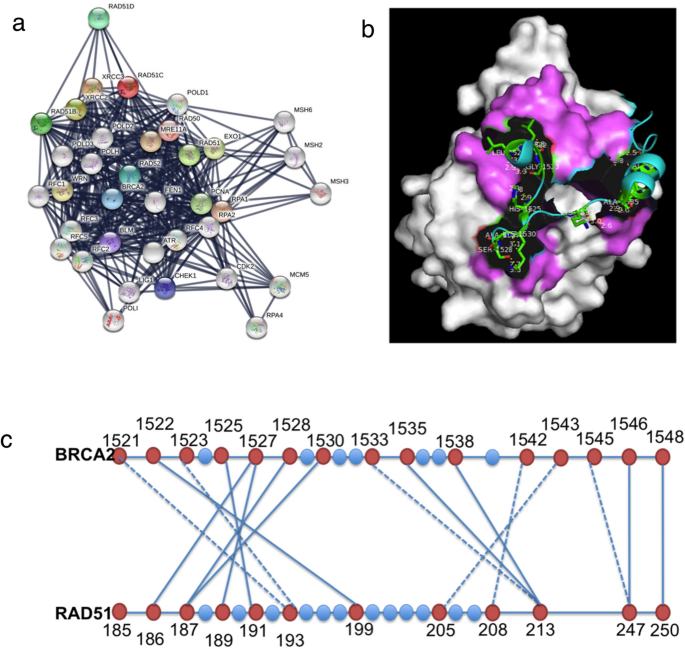
RAD51 Protein Interactome and BRCA2-RAD51 Binding (a) Interactome of RAD51 protein and its paralogs were mapped by searching the STRING database version 10.5 with a confidence cutoff of 0.900. In the resulting protein association network, proteins are presented as nodes that are connected by lines whose thickness represents the confidence level(0.4–0.9) (b) The BRCA2 BRC repeat (cartoon model) and the pink region (surface model) indicates the interacting region of RAD51 (1N0W) as shown in PyMol view. (c) The schematic representation of residues 1521–1548 of BRCA2 interacting with RAD51 residues via water (dashed line) or directly through hydrogen bond (solid line)
The 1.7 A˚ crystal structure of the RAD51-BRCA2 complex between an evolutionarily conserved sequence in BRCA2 (the BRC 4 repeat) and the RecA-homology domain of RAD51 [PDB: 1N0W] has been reported previously (Pellegrini et al., 2002). It was demonstrated that BRC4 remains in continuous contact with RAD51 over a stretch of 28 amino acids (Leu 1521 to Glu 1548) as shown in the PyMol view (Fig. 5b). DIMPLOT analysis of this interaction revealed a region in the ATPase domain of RAD51 (Ala186 to Arg 250), which makes polar contact with the BRC4 domain either via direct hydrogen bond or an intermediate water molecule. A total of 16 points of contact have been identified with 10 hydrogen bond interactions and 6 via intermediate water molecules (Fig. 5c).
To target the RAD51-BRCA2 interaction, we used the BRC4 repeat sequence as a reference to design peptides. The first peptide we chose was a 27 amino acid region [LGFHTASGKKVKIAKESLDKVKNLFDE; Peptide A] of the BRC4 sequence. A second shorter 12 amino acid peptide was designed [FHTAGGGSLFDE; Peptide B] combining the 2 motifs FHTA and LFDE at the N terminus and C terminus of the BRC4 sequence, respectively, with a universal spacer sequence (Rajendra & Venkitaraman, 2010). The physicochemical characteristics (chemical formula, molecular mass, hydrophobicity, pI and charge at pH 8) of the 2 peptides were estimated using the GenScript online tool. Structural evaluation for the 2 peptides was performed using RAMPAGE and viewed as the Ramachandran plot. The analysis for the modeled peptides revealed that 100% of the residues for peptide A and 90% for peptide B localized within the allowed regions of the plot suggesting good quality of the models for both peptides (Fig. 6a–c).
Fig. 6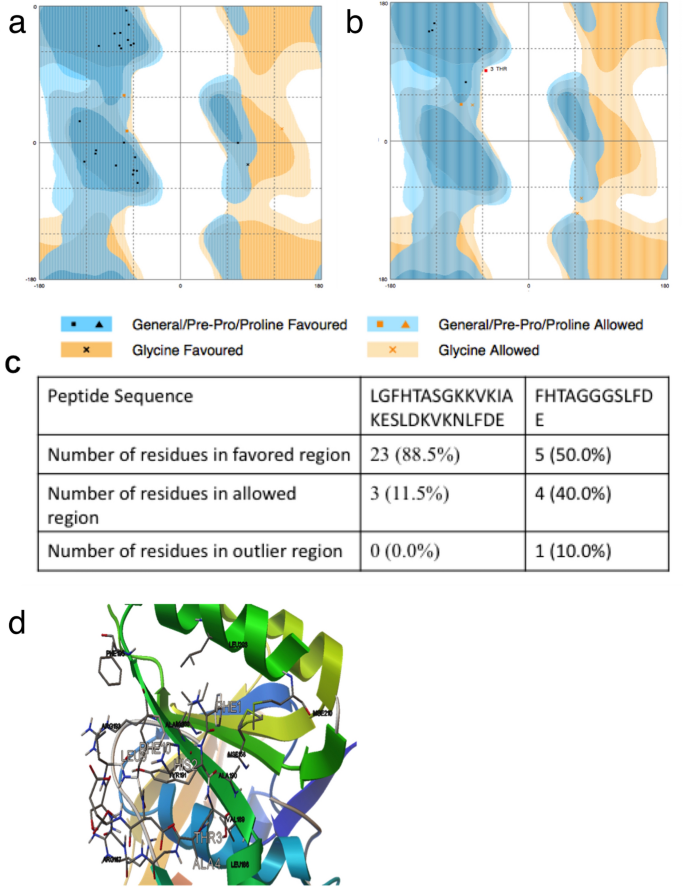
The Docked Model of RAD51 Protein and Peptide inhibitors (a) Pictorial representation of Ramachandran plot of the 27 Amino Acid peptide A (LGFHTASGKKVKIAKESLDKVKNLFDE) using online tool RAMPAGE. (b) Pictorial graphs of Ramachandran plot of the 12 Amino Acid peptide B (FHTAGGGSLFDE) using online tool RAMPAGE. (c) Table summarizing the docking results of the Ramachandran Plot demonstrating the number of residues in the favored and allowed region and the residues in the outlier region. (d) The docked model of RAD51 (cartoon) and peptide (Ball and Stick) visualized using PyMol. The amino acid residues and location of RAD51 protein along with the amino acid and location of the potential binding sites of the peptide on docking are also indicated
Since the results from the two peptides were comparable, we chose the smaller peptide B for further development and targeting studies. The molecular docking of RAD51 with peptide B (Pep12) was performed by AutoDock Vina, which generated nine different poses. The pose with the lowest energy value (−5.3 kcal/mol) was considered as the best-fit model. The docked model was visualized using PyMol (Fig. 6d). RAD51 binding to pep12 involved hydrogen bonds between substrate and RAD51 residues. Of these 12 residues, four were involved with BRCA2 binding, confirming the localization of pep12 binding within the BRCA2 binding site. Pep12 docking with RAD51 displayed nine different areas of interaction; with the lowest binding energy measured at −5.3 kcal/mol. Residues involved in interactions between pep12 and RAD51 were Met-158, Ile-160, Arg-167, Leu-186, Val-189, Ala-190, Tyr-191, Ala-192, Arg-193, Phe-195, Leu-203, Mse-210. Of these four residues, i.e. Leu-186, Val-189, Tyr-191, Arg-193 were located in the BRCA2 binding site. This confirms the interaction of the pep12 molecule with the BRCA2 binding site. The peptide binds with the residues Leu186, Val189, Tyr191 and Arg193 similar to the residues shown by the DIMPLOT.
Discussion
The human RAD51 protein (HsRAD51) is crucial for DNA repair processes based on homologous recombination between damaged loci and their undamaged copies in the sister chromatid. The protein and its paralogs, RAD51B, RAD51C, RAD51D, XRCC2 and XRCC3 are involved in the repair of a double-stranded break, the most severe form of DNA damage (Friedberg & Friedberg, 2006; Smiraldo et al., 2005). Efficient DNA repair is usually beneficial for living organisms. However, in the case of cancer cells, their efficient DNA repair opposes the action of radio- and chemotherapies based on DNA-damaging agents (Christodoulopoulos et al., 1999). RAD51 over-expression is reported in cancer cells, thereby resulting in resistance to anticancer treatment and a degree of recurrence of cancer (Hannay et al., 2007; Klein, 2008).
Identification of regions of the protein that are conserved among HR proteins across species can be an important tool to narrow down significant targets for functional intervention. The conserved domains in the DNA binding region and the Walker A and Walker B domains of the ATPase site have been reported previously (Gruver et al., 2005; Kaur & Rajesh, 2016). Specifically, the sequence GXXXXGKTQ/X was predicted as the most conserved region among human HR proteins as well as by interspecies comparisons. In RAD51, phenylalanine at position 129 [GEFRTGTK] in the variable quartet is shown to have an important role in the mechanism of RAD51. Mutation of HsRAD51 F129 affects ATP turnover and it has been suggested that cation-induced conformational transitions are defective in HsRAD51(F129V) which affects the ability of the mutant proteins to interact appropriately with DNA and to properly catalyze ATP hydrolysis (Amunugama & Fishel, 2011). CHK1 and CHK2 are also prime effector kinases in the DNA damage response. Phosphorylation of HR proteins via these kinases could prove to be an important mechanism in regulating their sub-cellular re-localization and assembly into various complexes. Each paralog has at least one potential site within or adjacent to the conserved ATPase domain. It has been demonstrated that phosphorylation of RAD51 on Thr 309 in a CHK1-dependent manner recruits it to DNA repair foci to promote homologous recombination repair and cell survival (Bahassi et al., 2008). Investigation of the additional phosphorylation sites of these paralogs would clarify how the functions of these proteins are regulated and can be targeted for therapeutic purposes.
Several small molecules have are reported that target various DNA repair pathways (Hengel et al., 2017). Of these, the ones targeting the ATPase domains were selected for the analysis. The chemical inhibitor i.e. CB-5083, was identified as the most potent ligand of the ATP binding site consisting of the highly conserved amino acids Phe129, Arg130 and Gln135 of the Walker A motif. The compound has been developed as an anti-tumor models across a variety of tumor models. The use of the CB5083 as an inhibitor could also have an effect on these phosophorylation sites due to the proximity to the binding residues of these inhibitors. It has, however, recently failed Clinical Trials at Phase 1 due to off-target effects necessitating a need to modify the inhibitor or use specifically targeted delivery methods (Tang et al., 2019).
Proteins seldom act alone and hold structural domains that allow their interaction with and bind to specific sequences on other protein. Protein–protein interactions (PPIs) organize their protein components to build molecular machinery, which carries out a variety of diverse molecular processes within a cell. The RAD51 paralogs interact to form at least two stable complexes (a dimer consisting of RAD51C–XRCC3 and a larger complex containing RAD51B, RAD51C, RAD51D and XRCC2) that assist the RAD51 strand transfer during HR (Masson et al., 2001). They are also reported to form complexes with several other proteins such as ssDNA/RNA processing, Helicase, Nucleotide synthesis, Transcription, Cell division, proliferation and developmental proteins, Cellular trafficking, Cell signaling, Protein synthesis, folding, and modification proteins. (Rajesh et al., 2009). It has also been demonstrated that the association of the RAD51 protein complex with SFPQ is essential for homologous recombination repair of DNA damage and maintaining genome integrity (Rajesh et al., 2011a, 2011b). Recent crystal structure of SFPQ has also been generated and can be used as a target for disruption of interaction with RAD51 proteins that can affect HR (Lee et al., 2015). RAD51D is also known to mediate the processing of 6-thioguanine and O6 methylguanine lesions downstream of mismatch repair suggesting a possible role in the Mismatch Repair pathway as well (Rajesh et al., 2010, 2011a, 2011b; Wyatt et al., 2018). The breast cancer susceptibility protein BRCA2 controls the functioning of RAD51, a recombinase enzyme, in pathways for DNA repair by homologous recombination. The interaction between BRCA2 and RAD51 is critical for the biological functions of both molecules (Scully & Livingston, 2000; Venkitaraman, 2002). Discrete nuclear foci containing RAD51 usually accumulate within the nucleus of mammalian cells exposed to DNA damage. RAD51 foci fail to form in BRCA2-deficient cells suggesting that BRCA2 may transport RAD51 to sites where DNA damage is processed by recombination (Chen et al., 1999a, 1999b; Yu et al., 2000). The RAD51 proteins are also shown to interact with several other DNA binding proteins, so disruption of these interactions will also lead to the inactivity of other proteins in various DNA repair pathways as well as other DNA modification functions. Disruption of these protein–protein interactions using peptides can result in a wide range of effector functions. Peptide libraries provide a powerful tool for targeting protein–protein interactions and other biochemical as well as pharmaceutical applications. The peptide on binding to homologous repair proteins will lead to the hindrance of the HR repair pathway by functional interruption of the HR activity. Therefore, this could lead to the effective killing of the tumor cells. BRC4 is a peptide of 28 amino acid residues (Leu 1521 to Glu 1548) that remains in continuous contact with RAD51. The amino acid sequence of the peptide is NH2–FHTAGGGSLFDE–COOH (Pep12) and used in the present study to generate the peptide model. ß-strand region surrounding the BRC4 residues Phe and Ala in the motif Phe-X-X-Ala (where X is any amino acid) structurally mimics RAD51 residues essential for the protomer–protomer interface, based on structural homology to the ADP-bound RecA filament (Story & Steitz, 1992). The binding site for a conserved module, Phe-X-X-Ala, in BRC repeats is positioned at the protomer–protomer interface and possesses filament-inhibitory potential (Short et al., 2016). Therefore, the targeting of the ‘FXXA’ motif leads to two functional inhibitions. The FXXA motif is important for RAD51 nucleo-filament formation and its interaction with the tumor suppressor protein BRCA2 (Scott et al., 2016). The two modules comprise the sequence FXXA, known to inhibit RAD51 assembly by blocking the oligomerization interface, and a previously unrecognized tetramer with the consensus sequence LFDE, which binds to a RAD51 pocket distinct from this interface. The LFDE motif is essential in BRC repeats for modes of RAD51 binding both permissive and inhibitory to RAD51 oligomerization. Targeted insertion of point mutations in RAD51 that disrupt the LFDE-binding pocket impair its assembly at DNA damage sites in living cells (Rajendra & Venkitaraman, 2010). The RAD51 binding to Pep12 provides similarly targeted insertion involving hydrogen bonds between substrate and RAD51 residues. Of these 12 residues, four are involved with BRCA2 binding, confirming the localization of pep12 binding within the BRCA2 binding site. Therefore, we can postulate that the binding of pep12 in the BRCA2 binding site might affect the binding of its natural substrate, BRCA2, and, therefore, interfere in HR repair mechanism.
Conclusions and future scope
Overcoming resistance to chemotherapeutic agents is one of the major hurdles in eradicating cancer. Combinatorial approaches targeting multiple pathways would be the way forward in this field. The work proposes one such approach where the HR DNA repair pathway is targeted mostly by functional inhibition of RAD51 proteins either directly via its ATPase activity or via its interaction BRCA2 which is important to initiate the repair cascade. This would effectively prevent the repair of damage generated by currently used radio and chemotherapy resulting in efficient cell killing (Fig. 7). Such drugs would be extremely helpful in increasing the efficacy of current treatment options for tumors with elevated homologous recombination. The additional drugs used and off target effect of these drugs as the CB5083 that is investigated in this study will need to be circumvented by designing a specific mechanism to target these drugs directly to the tumor site or cancer cells. Several nanotechnology-based drug targeting mechanisms are currently in place and being used to passively and actively target the drugs (Kaur et al., 2019).
Fig. 7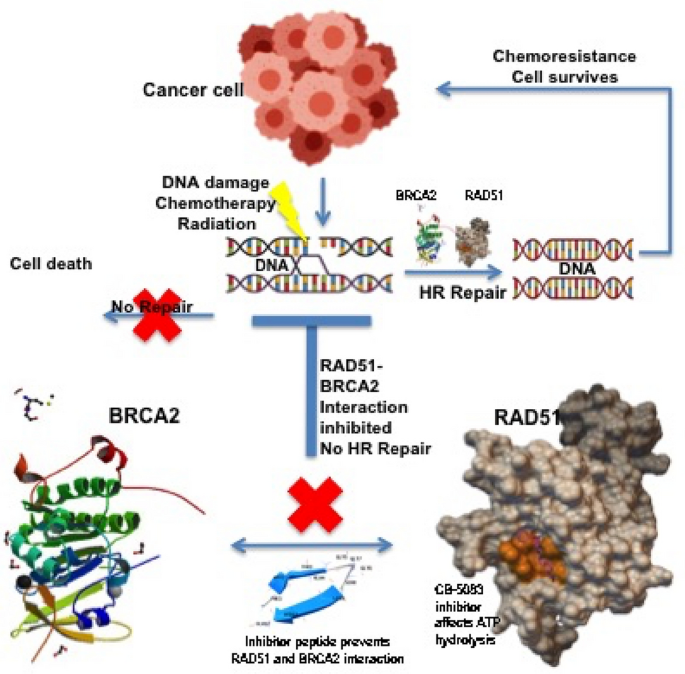
Schematic Representation of the proposed mechanism that will enhance the chemotherapeutic regimen by preventing chemo-resistance The model displays how chemotherapy or radiotherapy induces DNA damage in cancer cells that can result in cell death and cancer cure (Top left panel). However, in case of effective repair (HR Repair) by BRCA2 recruitment of RAD51 the DNA damage is repaired and the cancer cell survives and develops chemoresistance (Top right panel). The inhibitor peptide will prevent the interaction of RAD51 and BRCA2, thus preventing recruitment, while any RAD51 recruited is functionally inhibited by CB-5083 inhibitor by preventing ATP hydrolysis. This, therefore, can enhance the chemotherapeutic effectiveness
The use of such targeted nanogels with a combination of these drugs will be useful in designing the most efficient mechanism for the effective treatment of cancer. The mechanism can safely supplement the already used therapeutic regimens or can be used by adding drugs within the mix to induce DNA damage.
Data availability
All data generated or analysed during this study are included in this published article. However, any data or analysis from the current study can be made available from the corresponding author upon reasonable request.
References
Amunugama, R., & Fishel, R. (2011). Subunit interface residues F129 and H294 of human RAD51 are essential for recombinase function. PLoS ONE, 6(8), e23071. https://doi.org/10.1371/journal.pone.0023071.
Aparicio, T., Baer, R., & Gautier, J. (2014). DNA double-strand break repair pathway choice and cancer. DNA Repair, 19, 169–175. https://doi.org/10.1016/j.dnarep.2014.03.014.
Bahassi, E. M., Ovesen, J. L., Riesenberg, A. L., Bernstein, W. Z., Hasty, P. E., & Stambrook, P. J. (2008). The checkpoint kinases Chk1 and Chk2 regulate the functional associations between hBRCA2 and Rad51 in response to DNA damage. Oncogene, 27(28), 3977–3985. https://doi.org/10.1038/onc.2008.17.
Begg, A. C., Stewart, F. A., & Vens, C. (2011). Strategies to improve radiotherapy with targeted drugs. Nature Reviews Cancer, 11(4), 239–253. https://doi.org/10.1038/nrc3007.
Chen, C. F., Chen, P. L., Zhong, Q., Sharp, Z. D., & Lee, W. H. (1999a). Expression of BRC repeats in breast cancer cells disrupts the BRCA2-Rad51 complex and leads to radiation hypersensitivity and loss of G(2)/M checkpoint control. The Journal of Biological Chemistry, 274(46), 32931–32935. https://doi.org/10.1074/jbc.274.46.32931.
Chen, G., Yuan, S. S., Liu, W., Xu, Y., Trujillo, K., Song, B., Cong, F., Goff, S. P., Wu, Y., Arlinghaus, R., Baltimore, D., Gasser, P. J., Park, M. S., Sung, P., & Lee, E. Y. (1999b). Radiation-induced assembly of Rad51 and Rad52 recombination complex requires ATM and c-Abl. The Journal of Biological Chemistry, 274(18), 12748–12752. https://doi.org/10.1074/jbc.274.18.12748.
Christodoulopoulos, G., Malapetsa, A., Schipper, H., Golub, E., Radding, C., & Panasci, L. C. (1999). Chlorambucil induction of HsRad51 in B-cell chronic lymphocytic leukemia. Clinical Cancer Research, 5(8), 2178–2184.
Davies, A. A., Masson, J. Y., McIlwraith, M. J., Stasiak, A. Z., Stasiak, A., Venkitaraman, A. R., & West, S. C. (2001). Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Molecular Cell, 7(2), 273–282. https://doi.org/10.1016/s1097-2765(01)00175-7.
Dean, M., Fojo, T., & Bates, S. (2005). Tumour stem cells and drug resistance. Nature Reviews Cancer, 5(4), 275–284. https://doi.org/10.1038/nrc1590.
delToro, D., Ortiz, D., Ordyan, M., Sippy, J., Oh, C.-S., Keller, N., Feiss, M., Catalano, C. E., & Smith, D. E. (2016). Walker-A motif acts to coordinate ATP hydrolysis with motor output in viral DNA packaging. Journal of Molecular Biology, 428(13), 2709–2729. https://doi.org/10.1016/j.jmb.2016.04.029.
Donnenberg, V. S., & Donnenberg, A. D. (2005). Multiple drug resistance in cancer revisited: The cancer stem cell hypothesis. Journal of Clinical Pharmacology, 45(8), 872–877. https://doi.org/10.1177/0091270005276905.
Friedberg, E. C., & Friedberg, E. C. (Eds.). (2006). DNA repair and mutagenesis (2nd ed.). ASM Press.
Gruver, A. M., Miller, K. A., Rajesh, C., Smiraldo, P. G., Kaliyaperumal, S., Balder, R., Stiles, K. M., Albala, J. S., & Pittman, D. L. (2005). The ATPase motif in RAD51D is required for resistance to DNA interstrand crosslinking agents and interaction with RAD51C. Mutagenesis, 20(6), 433–440. https://doi.org/10.1093/mutage/gei059.
Hannay, J. A. F., Liu, J., Zhu, Q.-S., Bolshakov, S. V., Li, L., Pisters, P. W. T., Lazar, A. J. F., Yu, D., Pollock, R. E., & Lev, D. (2007). Rad51 overexpression contributes to chemoresistance in human soft tissue sarcoma cells: A role for p53/activator protein 2 transcriptional regulation. Molecular Cancer Therapeutics, 6(5), 1650–1660. https://doi.org/10.1158/1535-7163.MCT-06-0636.
Hengel, S. R., Spies, M. A., & Spies, M. (2017). Small-molecule inhibitors targeting DNA repair and DNA repair deficiency in research and cancer therapy. Cell Chemical Biology, 24(9), 1101–1119. https://doi.org/10.1016/j.chembiol.2017.08.027.
Hoeijmakers, J. H. (2001). Genome maintenance mechanisms for preventing cancer. Nature, 411(6835), 366–374. https://doi.org/10.1038/35077232.
Indran, I. R., Tufo, G., Pervaiz, S., & Brenner, C. (2011). Recent advances in apoptosis, mitochondria and drug resistance in cancer cells. Biochimica Et Biophysica Acta, 1807(6), 735–745. https://doi.org/10.1016/j.bbabio.2011.03.010.
Jackson, S. P., & Bartek, J. (2009). The DNA-damage response in human biology and disease. Nature, 461(7267), 1071–1078. https://doi.org/10.1038/nature08467.
Källberg, M., Wang, H., Wang, S., Peng, J., Wang, Z., Lu, H., & Xu, J. (2012). Template-based protein structure modeling using the RaptorX web server. Nature Protocols, 7(8), 1511–1522. https://doi.org/10.1038/nprot.2012.085.
Kanaar, R., Hoeijmakers, J. H., & van Gent, D. C. (1998). Molecular mechanisms of DNA double strand break repair. Trends in Cell Biology, 8(12), 483–489. https://doi.org/10.1016/s0962-8924(98)01383-x.
Kaur, H., & Rajesh, C. (2016). Identification of conserved domains of proteins involved in homologous recombination repair. Recent Advances in Emerging Technologies, 39–51.
Kaur, H., Kaur, H., Rajesh, P., & Rajesh, C. (2019). Nanotechnology based drug delivery system for cancer therapy. International Journal of Nanobiotechnology, 5(2), 8–36.
Kelley, M. R., & Fishel, M. L. (2008). DNA repair proteins as molecular targets for cancer therapeutics. Anti-Cancer Agents in Medicinal Chemistry, 8(4), 417–425. https://doi.org/10.2174/187152008784220294.
Klein, H. L. (2008). The consequences of Rad51 overexpression for normal and tumor cells. DNA Repair, 7(5), 686–693. https://doi.org/10.1016/j.dnarep.2007.12.008.
Lamiable, A., Thévenet, P., Rey, J., Vavrusa, M., Derreumaux, P., & Tufféry, P. (2016). PEP-FOLD3: Faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Research, 44(W1), W449-454. https://doi.org/10.1093/nar/gkw329.
Laskowski, R. A., & Swindells, M. B. (2011). LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. Journal of Chemical Information and Modeling, 51(10), 2778–2786. https://doi.org/10.1021/ci200227u.
Lee, M., Sadowska, A., Bekere, I., Ho, D., Gully, B. S., Lu, Y., Iyer, K. S., Trewhella, J., Fox, A. H., & Bond, C. S. (2015). The structure of human SFPQ reveals a coiled-coil mediated polymer essential for functional aggregation in gene regulation. Nucleic Acids Research, 43(7), 3826–3840. https://doi.org/10.1093/nar/gkv156.
Masson, J. Y., Tarsounas, M. C., Stasiak, A. Z., Stasiak, A., Shah, R., McIlwraith, M. J., Benson, F. E., & West, S. C. (2001). Identification and purification of two distinct complexes containing the five RAD51 paralogs. Genes & Development, 15(24), 3296–3307. https://doi.org/10.1101/gad.947001.
National Cancer Institute (NCI) (2018). https://www.Cancer.Gov/about-Cancer/Understanding/Statistics.
Nickoloff, J. A., Jones, D., Lee, S.-H., Williamson, E. A., & Hromas, R. (2017). Drugging the cancers addicted to DNA repair. Journal of the National Cancer Institute. https://doi.org/10.1093/jnci/djx059.
Obenauer, J. C., Cantley, L. C., & Yaffe, M. B. (2003). Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Research, 31(13), 3635–3641. https://doi.org/10.1093/nar/gkg584.
Park, N. H., Cheng, W., Lai, F., Yang, C., Florez de Sessions, P., Periaswamy, B., Wenhan Chu, C., Bianco, S., Liu, S., Venkataraman, S., Chen, Q., Yang, Y. Y., & Hedrick, J. L. (2018). Addressing drug resistance in cancer with macromolecular chemotherapeutic agents. Journal of the American Chemical Society, 140(12), 4244–4252. https://doi.org/10.1021/jacs.7b11468.
Pellegrini, L., Yu, D. S., Lo, T., Anand, S., Lee, M., Blundell, T. L., & Venkitaraman, A. R. (2002). Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature, 420(6913), 287–293. https://doi.org/10.1038/nature01230.
Rajendra, E., & Venkitaraman, A. R. (2010). Two modules in the BRC repeats of BRCA2 mediate structural and functional interactions with the RAD51 recombinase. Nucleic Acids Research, 38(1), 82–96. https://doi.org/10.1093/nar/gkp873.
Rajesh, C., Baker, D. K., Pierce, A. J., & Pittman, D. L. (2011a). The splicing-factor related protein SFPQ/PSF interacts with RAD51D and is necessary for homology-directed repair and sister chromatid cohesion. Nucleic Acids Research, 39(1), 132–145. https://doi.org/10.1093/nar/gkq738.
Rajesh, C., Gruver, A. M., Basrur, V., & Pittman, D. L. (2009). The interaction profile of homologous recombination repair proteins RAD51C, RAD51D and XRCC2 as determined by proteomic analysis. Proteomics, 9(16), 4071–4086. https://doi.org/10.1002/pmic.200800977.
Rajesh, P., Litvinchuk, A. V., Pittman, D. L., & Wyatt, M. D. (2011b). The homologous recombination protein RAD51D mediates the processing of 6-thioguanine lesions downstream of mismatch repair. Molecular Cancer Research MCR, 9(2), 206–214. https://doi.org/10.1158/1541-7786.MCR-10-0451.
Rajesh, P., Rajesh, C., Wyatt, M. D., & Pittman, D. L. (2010). RAD51D protects against MLH1-dependent cytotoxic responses to O(6)-methylguanine. DNA Repair, 9(4), 458–467. https://doi.org/10.1016/j.dnarep.2010.01.009.
Scott, D. E., Marsh, M., Blundell, T. L., Abell, C., & Hyvönen, M. (2016). Structure-activity relationship of the peptide binding-motif mediating the BRCA2:RAD51 protein-protein interaction. FEBS Letters, 590(8), 1094–1102. https://doi.org/10.1002/1873-3468.12139.
Scully, R., & Livingston, D. M. (2000). In search of the tumour-suppressor functions of BRCA1 and BRCA2. Nature, 408(6811), 429–432. https://doi.org/10.1038/35044000.
Short, J. M., Liu, Y., Chen, S., Soni, N., Madhusudhan, M. S., Shivji, M. K. K., & Venkitaraman, A. R. (2016). High-resolution structure of the presynaptic RAD51 filament on single-stranded DNA by electron cryo-microscopy. Nucleic Acids Research, 44(19), 9017–9030. https://doi.org/10.1093/nar/gkw783.
Smiraldo, P. G., Gruver, A. M., Osborn, J. C., & Pittman, D. L. (2005). Extensive chromosomal instability in Rad51d-deficient mouse cells. Cancer Research, 65(6), 2089–2096. https://doi.org/10.1158/0008-5472.CAN-04-2079.
Sørensen, C. S., Hansen, L. T., Dziegielewski, J., Syljuåsen, R. G., Lundin, C., Bartek, J., & Helleday, T. (2005). The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nature Cell Biology, 7(2), 195–201. https://doi.org/10.1038/ncb1212.
Story, R. M., & Steitz, T. A. (1992). Structure of the recA protein-ADP complex. Nature, 355(6358), 374–376. https://doi.org/10.1038/355374a0.
Stothard, P. (2000). The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. BioTechniques, 28(6), 1102–1104. https://doi.org/10.2144/00286ir01.
Tang, W. K., Odzorig, T., Jin, W., & Xia, D. (2019). Structural basis of p97 inhibition by the site-selective anticancer compound CB-5083. Molecular Pharmacology, 95(3), 286–293. https://doi.org/10.1124/mol.118.114256.
Trott, O., & Olson, A. J. (2010). AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 31(2), 455–461. https://doi.org/10.1002/jcc.21334.
Vanneman, M., & Dranoff, G. (2012). Combining immunotherapy and targeted therapies in cancer treatment. Nature Reviews Cancer, 12(4), 237–251. https://doi.org/10.1038/nrc3237.
Venkitaraman, A. R. (2002). Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell, 108(2), 171–182. https://doi.org/10.1016/s0092-8674(02)00615-3.
Wallner, B., & Elofsson, A. (2003). Can correct protein models be identified? Protein Science A Publication of the Protein Society, 12(5), 1073–1086. https://doi.org/10.1110/ps.0236803.
Wass, M. N., Kelley, L. A., & Sternberg, M. J. E. (2010). 3DLigandSite: Predicting ligand-binding sites using similar structures. Nucleic Acids Research. https://doi.org/10.1093/nar/gkq406.
Wiederstein, M., & Sippl, M. J. (2007). ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Research. https://doi.org/10.1093/nar/gkm290.
Wyatt, M. D., Reilly, N. M., Patel, S., Rajesh, P., Schools, G. P., Smiraldo, P. G., & Pittman, D. L. (2018). Thiopurine-induced mitotic catastrophe in Rad51d-deficient mammalian cells. Environmental and Molecular Mutagenesis, 59(1), 38–48. https://doi.org/10.1002/em.22138.
Yang, J., Yan, R., Roy, A., Xu, D., Poisson, J., & Zhang, Y. (2015). The I-TASSER suite: Protein structure and function prediction. Nature Methods, 12(1), 7–8. https://doi.org/10.1038/nmeth.3213.
Yu, V. P., Koehler, M., Steinlein, C., Schmid, M., Hanakahi, L. A., van Gool, A. J., West, S. C., & Venkitaraman, A. R. (2000). Gross chromosomal rearrangements and genetic exchange between nonhomologous chromosomes following BRCA2 inactivation. Genes & Development, 14(11), 1400–1406.
Zamarin, D., & Postow, M. A. (2015). Immune checkpoint modulation: Rational design of combination strategies. Pharmacology & Therapeutics, 150, 23–32. https://doi.org/10.1016/j.pharmthera.2015.01.003.
Funding
This study was supported by the Department of Science Technology-Science and Engineering Research Board (DST-SERB), Govt. of India, YSS/2015/001281, Preeti Rajesh, SB/YS/LS-168/2013, Harsimran Kaur.
Author information
Authors and Affiliations
Department of Biotechnology, Sri Guru Granth Sahib World University, Fatehgarh Sahib, Punjab, 140406, India
Harsimrat Kaur
Department of Biotechnology, Chandigarh University, NH-95 Chandigarh-Ludhiana Highway, Mohali, 140413, India
Harsimran Kaur & Preeti Rajesh
School of Bioengineering Sciences and Research, MIT-Art, Design and Technology University, Loni Kalbhor, Pune, Maharashtra, 412201, India
Changanamkandath Rajesh
Corresponding authors
Correspondence to Preeti Rajesh or Changanamkandath Rajesh.
Ethics declarations
Conflict of interest
The authors declare no conflict of interest with respect to research, authorship and /or publication of this manuscript.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article
Cite this article
Kaur, H., Kaur, H., Rajesh, P. et al. In Silico design and characterization of RAD51 protein inhibitors targeting homologous recombination repair for cancer therapy. GENOME INSTAB. DIS. 4, 289–302 (2023). https://doi.org/10.1007/s42764-023-00106-4
Received04 July 2023
Revised23 August 2023
Accepted03 September 2023
Published20 September 2023
Issue DateOctober 2023
DOIhttps://doi.org/10.1007/s42764-023-00106-4
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable link
用户登录
还没有账号?
立即注册