






Chemotherapy-induced neuronal DNA damage: an intriguing toolbox to elucidate DNA repair mechanisms in the brain
Review Article
Genome Instability & Disease (2023)
Abstract
The accumulation of DNA damage and subsequent decline in cellular functions are significant factors contributing to aging and-age associated neurodegeneration. Mutations in the genes involved in DNA repair pathways have been linked to premature aging and age-related neurodegeneration. Although DNA repair mechanisms are remarkably preserved across species, our current knowledge of neuronal-specific DNA repair pathways largely stems from investigations carried out on cellular and animal models, mainly focusing on cancer-related research. While DNA repair mechanisms are generally efficient in correcting damage caused by internal and external sources, the regulation of these mechanisms in post-mitotic neuronal cells, which are non-dividing, is not well understood. Studies utilizing autopsy brain samples have identified specific types of DNA damage and repair proteins in human neurons. However, these findings are inadequate to fully understand the regulatory aspects of neuronal-specific DNA repair pathways. This understanding is crucial for developing mechanism-based drugs that can prevent neuronal cell death, a characteristic feature of neurodegenerative diseases. As a result, further research is required to understand the intricate regulation of the DNA repair mechanisms involved in maintaining genome integrity in neurons. Several chemotherapeutic drugs cause DNA damage or impede cell division and cell death. Drugs that are primarily known to induce DNA damage in dividing cells can also damage neuronal DNA. In this context, we propose that it may be worthwhile to consider the DNA damage response induced by chemotherapy in cancer survivors as a tool to understand definite neuronal DNA repair mechanisms.
Introduction
Neurodegenerative diseases (NDDs) such as Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD), and amyotrophic lateral sclerosis (ALS) have complex pathogenic mechanisms involving multiple factors. These conditions are often a result of a combination of genetic mutations, impaired DNA repair, mitochondrial dysfunction, metabolic stress, environmental factors, and aging. The progressive loss of neuronal cells is a prominent hallmark shared by the majority of these disorders. Extensive research has established a strong connection between impaired DNA damage response (DDR) and repair processes and the development of NDDs (Hegde et al., 2017; Hoeijmakers, 2009; McKinnon, 2017; Welch & Tsai, 2022). Mature neurons are particularly vulnerable to DNA damage due to their high oxidative metabolism, enhanced transcriptional activity, reduced repair capacity, and prolonged lifespan. Free radicals produced by oxidative stress and cellular metabolism can degrade DNA and create protein-DNA cross-links, among other types of DNA damage (McKinnon, 2009). Additionally, endogenous DNA damage can occur as a byproduct of glutamate receptor activation (Didier et al., 1996; Yang et al., 2010). These damages can affect active transcription and induce genomic instability by activating apoptosis or senescence (McKinnon, 2009; Schumacher et al., 2021). A progressive decline in DNA repair capacity with aging has also been proposed to contribute to the accumulation of unrepaired DNA lesions in neurons, ultimately playing a role in the aging process (Lu et al., 2004). Therefore, it is vital to understand the complex mechanisms underlying neuronal cell death, and studying molecular pathways involved in DNA repair which offers promising prospects for the development of future therapeutic or preventive strategies targeting NDDs. In the context of NDDs, this review aims to provide a comprehensive overview of the current knowledge regarding DNA damage and repair mechanisms from studies using both human brain samples and samples from cancer survivors. Finally, we will emphasize the importance of conducting further research on human brain samples to gain a better understanding of the specific DDR and repair pathways. Such investigations have immense potential for facilitating the development of therapeutic interventions aimed at preventing neuronal cell death and neurodegeneration.
The frequency of DNA damage in neurons depends on various factors including age, genetic makeup, and exposure to damaging agents. Additionally, neurons in different regions of the brain may experience varying levels of DNA damage due to differences in metabolic activity, resulting in reactive oxygen species (ROS) and vulnerability to specific environmental factors. The most common repair pathways in neuronal cells include double-strand break (DSB) repair, single-strand break (SSB) repair, base excision repair (BER), and nucleotide excision repair (NER).
DSBs are among the most severe forms of DNA damage, as they can lead to genomic instability, cell death, or mutations if not properly repaired. Cells have evolved several mechanisms to repair DSBs, which are broadly divided into two pathways, HR and NHEJ. Dividing cells predominantly rely on error-free and efficient DNA HR pathways to repair DSBs that occur during replication, or as a product of other repair pathways. HR employs an undamaged sister chromatid as a template for accurate repair, whereas NHEJ rejoins broken DNA ends without extensive homology, which can result in the loss or alteration of a few nucleotides at the repair site (Branzei & Foiani, 2008; Huang & Zhou, 2021). HR is a complex process that involves several steps and proteins. HR usually operates during the S and G2 phases of the cell cycle, when sister chromatids are available as templates. Damaged DNA strands are processed by factors such as the MRN complex (Mre11-Rad50-Nbs1), CtlP, exonuclease 1 (Exo1), and Dna2 to generate 3′-single-stranded overhangs. This ssDNA tail invades regions on a sister chromatid to form a structure known as a displacement loop (D-loop). Rad51 and related proteins play important roles in this strand invasion process. DNA polymerase δ or Ɛ are recruited to the D-loop structure and synthesize the new strand using undamaged strand as template. This results in the formation of Holliday junction. These structures are resolved by enzymes, such as GEN1 or Mus81-Eme1. Finally, DNA ligases join the gap, ensuring the continuity of the DNA strand (Branzei & Foiani, 2008). In NHEJ, the broken DNA ends are recognized and bound by the Ku70/Ku80 heterodimer. Ku proteins help stabilize the broken end and protect them from degradation by exonucleases. The broken ends are processed to remove any damaged bases. The DNA-dependent protein kinase catalytic subunit (DNA-PKCs), a serine/threonine kinase, forms a complex with Ku proteins. DNA-PKCs are activated upon binding to their DNA ends. This results in the recruitment of various DNA repair factors to the site of damage. DNA ligase IV, along with XRCC4 and XLF, catalyzes ligation of the processed DNA ends. This step rejoins the broken DNA strands. The final step involves resolution of any DNA flap or structure that may have formed during the repair process. NHEJ is error-prone mechanisms that can operate throughout the cell cycle. NHEJ may result in small insertions or deletions at the repair junction owing to inaccurate processing of DNA ends, potentially leading to mutations (Branzei & Foiani, 2008).
SSBs may arise from exposure to radiation, ROS, or certain chemicals. These breaks are generally less severe than double-strand breaks (DSBs) but can be converted to DSBs if left unrepaired. SSB repair involves detection and correction of damage to one of the two complementary strands of the DNA double helix. The first step involves the recognition of SSB by poly(ADP-ribose) polymerase 1 (PARP-1). Once activated, PARP-1 generates poly(ADP-ribose) (PAR) chains that recruit repair proteins such as XRCC1 to the break site. After removing the damaged single-stranded DNA, the resulting gap is filled with DNA polymerase β. Finally, the DNA ligase III seals the nick. Upon completion of the repair process, enzymes such as poly(ADP-ribose) glycosylase (PARG) or ADP-ribosylhydrolase remove PAR chains by terminating PARP-1 activity (Branzei & Foiani, 2008; Huang & Zhou, 2021).
BER repairs small and non-helix-distorting lesions in the DNA. These lesions usually arise from spontaneous deamination of bases, oxidation or alkylation of bases, or incorrect incorporation of bases during DNA replication. Oxidative damage, such as the formation of 8-oxoguanine, is commonly repaired via the BER pathway. BER is essential for maintaining genome integrity and preventing the accumulation of mutations that can lead to diseases such as cancer and NDD. BER is initiated by the recognition of damaged bases by specialized enzymes such as DNA glycosylases. These enzymes remove the damaged base by cleaving the bond between the damaged base and sugar-phosphate backbone. This results in the formation of an apurinic/apyrimidinic (AP) site, known as the abasic site. The enzyme AP endonuclease cleaves DNA at the AP site, creating a gap in the DNA strand with the 5-deoxyribose phosphate (dRP) group. The dRP group is then removed either by a dRP lyase or endonuclease. DNA polymerase then fills the gap using an undamaged complementary strand as a template. Finally, DNA ligase seals the nick and restores the integrity of the DNA strand (Branzei & Foiani, 2008; Huang & Zhou, 2021).
NER repairs a wide range of DNA damage, including UV-induced photoadducts, chemical adducts, and other bulky lesions (Krasikova et al., 2021). In global genome NER (GG-NER), DNA damage is recognized by the XPC-Rad23B-CETN2 complex, which scans the DNA for distortions or structural changes in the DNA strand. In transcription-coupled NER (TC-NER), RNA Polymerase II stalling at a DNA lesion signals damage. The TFIIH complex, which contains the helicase enzymes XPB and XPD, proteins then unwinds DNA within the vicinity of damage. Two incisions are made near the damage site to create a small DNA fragment containing the damage. In GG-NER, XPF and XPG endonucleases makes the incision on the 5′ and 3′ sides on the lesion, respectively. In TC-NER, CSB and CSA proteins perform the incision step. The excised DNA fragment was then removed, leaving an ssDNA gap. DNA polymerase such as DNA polymerase δ or Ɛ along with PCNA then fills the gap. Finally, DNA ligase 1 joins the newly synthesized strand (Branzei & Foiani, 2008; Huang & Zhou, 2021). Neuron-specific DNA repair is discussed in the next section, with particular emphasis on the defective repair mechanisms observed in NDD.
Neuronal DNA repair
Most evidence supporting DNA repair mechanisms in neurons comes from studies conducted on dividing cells and model organisms. In general, certain DNA repair mechanisms exhibit varying degrees of functionality in dividing and non-dividing cells. However, non-dividing cells generally display a lower DNA repair efficiency than dividing cells. This reduced repair capacity may be attributed to decreased expression or activity of certain repair enzymes. Several studies have indicated a decline in DNA repair efficiency with age, in which specific repair pathways have been identified as being affected (Gupta et al., 2021; Shackelford, 2006). Dividing cells follow a well-defined cell cycle consisting of distinct phases (G1, S, G2, and M), which enables accurate DNA replication and repair (Ciccia & Elledge, 2010; Petsalaki & Zachos, 2020). On the other hand, mature neurons are primarily non-dividing post-mitotic cells. As a result, they lack cell cycle checkpoints and the DNA repair machinery associated with DNA replication. Upon detecting DNA damage, cells activate a robust DDR pathway that involves a series of sequential and coordinated chromatin remodeling events facilitated by post-translational modifications. These modifications facilitate the recruitment of DNA repair factors to damaged sites (Dabin et al., 2023; Urulangodi & Mohanty, 2020). DDR pathways possess cell cycle checkpoints that monitor DNA integrity before progressing through each phase. When DNA damage is detected, these checkpoints can delay or arrest the cell cycle, allowing DNA repair or triggering cell death if the damage is irreversible (Branzei & Foiani, 2008; Huang & Zhou, 2021). Neurons, on the other hand, have minimal DDR, allowing them to tolerate certain levels of DNA damage without causing cell death, but potentially leading to the accumulation of DNA lesions over time (Shadfar et al., 2022). Our understanding of complex mechanisms responsible for maintaining genome stability, enabling DNA repair processes, and supporting the longevity of neurons continues to progress steadily. More research is needed to understand the unique vulnerabilities of neurons to DNA damage, as well as how impaired DNA repair may contribute to NDDs.
Repair of DSBs Homologous recombination (HR) and non-homologous end-joining (NHEJ) are the primary repair pathways for DSBs. In neuronal cells, NHEJ is often prevalent (Yamamoto et al., 2007), whereas HR is typically more active in dividing cells, possibly because dividing cells have sister chromatids, which non-dividing neurons lack. Early studies on hereditary diseases, such as Ataxia Telangiectasia (AT), Werner’s syndrome, and Bloom syndrome, provided evidence for the significance of double-strand break (DSB) repair in neurons (Cunniff et al., 2017; Goto, 1997; Shibata & Jeggo, 2021). These studies revealed that mutations in factors required for DSB repair, specifically ATM, WRN, and BLM, result in DNA breaks, neurodegeneration, and premature aging (Progeria syndrome) (Coppedé, 2010). Excessive transcription in neurons leads to the accumulation of unresolved R-loops, resulting in the formation of DNA breaks (Brambati et al., 2020; Huertas & Aguilera, 2003). The precise mechanism by which R-loops contribute to neurodegeneration is unknown. Recent studies have observed the accumulation of R-loops and DNA damage in ALS patients with loss-of-function mutations in TDP-43, along with defective NHEJ repair (Giannini et al., 2020; Mitra et al., 2019). SSB repair: another common form of DNA damage is single-strand break (SSB), which can result directly from ROS or indirectly as an intermediate during BER (Leandro et al., 2015). This repair is essential to prevent the SSBs from progressing to more dangerous and mutagenic DSBs. Repair of oxidative DNA damage by BER: this type of repair involves the sequential removal of the damaged base by a specific DNA glycosylase enzyme, followed by excision of the resulting abasic site and subsequent DNA synthesis and ligation steps. Some familial forms of ALS are linked to mutations in genes involved in BER. For example, mutations in the C9orf72 gene in familial ALS, can lead to the accumulation of DNA damage, due to impaired BER (Farg et al., 2017). Spinocerebellar ataxias (SCAs) are associated with mutations in genes in the BER pathway. For example, mutations in ATXN1 gene result in oxidative DNA damage and impaired BER, leading to the CAG repeat expansion seen in SCA1 (Sullivan et al., 2019). NER: in non-dividing cells, NER is another important mechanism for repairing bulky DNA lesions induced by UV radiation or certain chemicals (Krasikova et al., 2021). TC-NER is reported to be the most prominent pathway in mature neurons. NER recognizes and removes damaged DNA segments, followed by resynthesis and ligation. Initial evidence emphasizing the significance of NER in neurons came from studies on genetic diseases, specifically Xeroderma pigmentosum (XP) and Cockayne syndrome (CS), where mutations affecting these pathways were observed. In the case of XP, mutations have been identified in in NER pathway genes including XPA, XPC, XPD, and XPG. CS is associated with the presence of mutations in NER genes CSA and CSB. Similarly, Trichothiodystrophy (TTD) is characterized by mutations in NER genes XPD and XPB, while cerebrooculofacioskeletal syndrome (COFS) have mutations in CSA and CSB. Defects in TC-NER have also been reported in the development of HD and ALS.
Neurodegenerative diseases such as AD and PD have been linked to mitochondrial DNA damage (Bender et al., 2006; Santos et al., 2013). Although mitochondria produce ATP for cellular functions, they are also sites of ROS production. Consequently, mitochondrial function and repair capacity decline with age, contributing to the development of NDD (de Souza-Pinto et al., 2008; Stefanatos & Sanz, 2018). Mitochondrial oxidative DNA damage is mostly repaired by BER (Karahalil et al., 2002). In non-dividing cells, a specialized repair mechanism called translesion synthesis (TLS) can be employed to bypass the unrepaired lesions caused by DNA-damaging agents (Li et al., 2022). TLS utilizes specialized DNA polymerases that can replicate across damaged DNA templates; however, this process is prone to error. Neurons also have specialized transcription-coupled repair mechanisms to efficiently repair transcription-blocking lesions and to maintain the integrity of the neuronal genome (Pani & Nudler, 2017). Overall, it appears that neurons possess distinct and unique DNA repair pathways along with specialized regulatory mechanisms, in addition to common repair pathways. However, further detailed investigations are necessary to determine the existence and precise mechanisms in neurons from the human brain and their direct link to neurodegeneration.
DNA repair hotspots in neurons
A study conducted on terminally differentiated neuronal cells demonstrated that DNA repair efficiency is higher in transcribed regions than in inactive regions within neurons (Nouspikel & Hanawalt, 2000). This indicates that neurons protect their genome and longevity through specialized mechanisms that prioritize DNA repair in highly transcribed genes, where there is a higher likelihood of mutations. Supporting this notion, investigations using postmortem human brain samples revealed reduced expression of numerous genes after the age of 40, possibly due to increased mutations in the promoter regions of these genes and diminished BER capacity (Lu et al., 2004). More recent studies using human embryonic stem cell-derived neurons have discovered the existence of DNA repair hotspots that safeguard regions of active transcription (Reid et al., 2021). Employing a similar experimental approach, researchers observed SSBs localized at enhancers, CpG islands, and DNA methylation sites (Wu et al., 2021). Moreover, it has been demonstrated that DNA demethylation mediated by TET enzymes are responsible for increased SSBs at enhancer regions in neurons derived from induced pluripotent stem cells (iPSCs) (Wang et al., 2022). Single-cell DNA sequence data have also confirmed the presence of DNA damage in the enhancer and promoter regions (Luquette et al., 2022). These findings suggest that in cases of defective repair, mutations may accumulate specifically in the transcribed regions, consequently affecting gene expression patterns and potentially aiding in the development of NDDs.
Neuronal DNA damage and repair: evidence from studies using human brain samples
In this section, we will discuss different types of DNA damage, defective DNA repair pathways, and the detection of mitochondrial and nuclear mutations in postmortem brain tissue samples from various NDD and aging populations (Fig. 1).
Fig. 1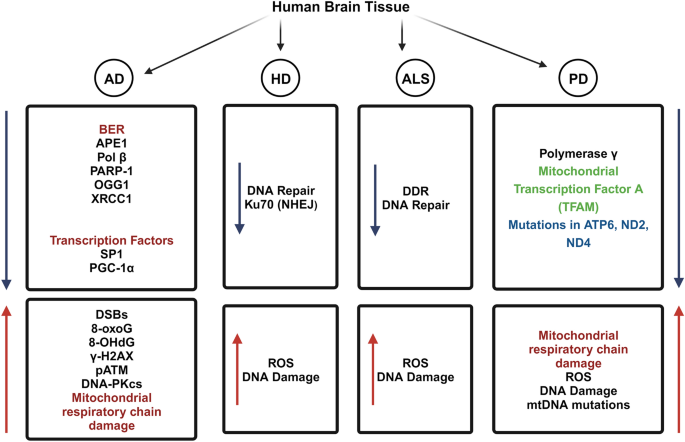
Decreased DNA repair pathways and increased mitochondrial and nuclear mutations in postmortem brain tissue samples from Alzheimer’s disease, Parkinson’s disease, Huntington’s disease and Amyotrophic Lateral sclerosis. The blue arrow denotes a decrease in the expression of various DDR and DNA repair factors, as well as factors that regulate the expression of genes involved in DNA repair. The red arrow indicates an increase in the accumulation of DNA damage, mitochondrial damage, and mutations. The figure was generated with BioRender (https://www.biorender.com/)
Defective DNA repair in Alzheimer’s disease
A study was conducted to determine the potential role of early DNA DSBs in the pathogenesis of AD neurodegeneration (Shanbhag et al., 2019). Immunofluorescence was used to analyze the presence of DSBs by measuring the levels of phosphorylated histone H2AX (γH2AX), a well-established marker of DNA damage, in postmortem brain tissue from patients with AD and healthy controls. AD patients showed an increase in γH2AX levels within the neurons, suggesting the accumulation of DSBs. Notably, neuronal DNA damage occurred in the early stages of AD, even before the appearance of amyloid plaques and neurofibrillary tangles, the hallmark pathological features of the disease. Additionally, a decrease in the expression of proteins involved in DNA repair pathways, including ATM and DNA-PKcs, was observed in the brains of patients with AD (Shanbhag et al., 2019). These findings imply that neuron’s impaired DNA repair capacity may contribute to the accumulation of DNA damage seen in AD. Interestingly, they found a positive correlation between γH2AX levels and markers of synaptic loss and neurodegeneration in AD brains, indicating a potential link between DNA damage and neuronal dysfunction. Another study found a higher level of DNA DSBs in hippocampal regions associated with AD pathology in postmortem human brain samples from patients with AD and age-matched controls. They employed techniques such as ELISA and qRT-PCR (Thadathil et al., 2021). The study highlighted the role of oxidative stress in promoting DNA damage and suggested that DSBs may contribute to cognitive impairment in AD. Recently, the landscape of DSBs in postmortem brain tissue from individuals with AD and non-demented individuals were published (Zhang et al., 2023a, 2023b). Using immunofluorescence and confocal microscopy, the distribution of γH2AX foci was found to be distinct in AD, exhibiting a clustered pattern primarily localized to the neuronal nuclei. These clustered DSBs have been found in a variety of brain regions affected by AD pathology, including the hippocampus and cerebral cortex. In addition, it was increased in AD brains, suggesting an activated DDR pathway in response to DSBs (Zhang et al., 2023a, 2023b). The presence of clustered DSBs coincided with the accumulation of hyperphosphorylated tau protein in the neuronal nuclei, indicating a possible interplay between DNA damage and tau pathology in AD.
In a separate study, DNA repair enzymes involved in BER were analyzed using postmortem brain tissue samples from three distinct groups: patients with AD, individuals with amnestic mild cognitive impairment (aMCI), and healthy controls. This study revealed a significant decline in the activity of various BER enzymes, such as DNA glycosylases and apurinic/apyrimidinic endonuclease 1 (APE1), in both AD and aMCI brain samples. Further analysis demonstrated that reduced activity of BER enzymes correlated with increased DNA damage in the form of oxidative DNA lesions, specifically 8-oxoguanosine (8-oxoG) and 8-hydroxydeoxyguanosine (8-OHdG). In addition, the levels of BER-associated proteins, including DNA polymerase β (Pol β) and poly (ADP-ribose) polymerase 1 (PARP-1) were significantly lower in the brains of patients with AD and aMCI (Weissman et al., 2007). A significant alteration in the BER pathway was observed in both brain tissue and blood samples obtained from AD patients when compared to healthy controls using quantitative real-time polymerase chain reaction (qRT-PCR) and immunohistochemistry techniques (Lillenes et al., 2016). Specifically, the expression levels of several BER enzymes, including OGG1, APE1, and XRCC1, were reduced in AD brain tissue, indicating impaired DNA repair capacity. Similarly, blood samples from patients with AD exhibited lower expression levels of BER-associated genes. Furthermore, an increase in DNA damage markers, such as 8-oxoG, was observed in both AD brain tissue and blood samples. The accumulation of DNA damage suggests an inadequate repair response in AD, potentially contributing to disease progression and cognitive impairment. Interestingly, this study found a positive correlation between the DNA repair profile in blood samples and brain tissue, indicating that blood-based analyses may provide valuable insights into DNA repair deficiencies in AD. These results suggest that blood samples could be used as a substitute for brain tissue, allowing for non-invasive assessments of DNA repair capacity in people with AD (Lillenes et al., 2016).
In a comparative study, qPCR was used to check the expression of the NER gene in postmortem brain tissue samples from the temporal cortex and blood samples from people with AD. The study revealed that NER gene expression was significantly higher in brain tissue samples from AD patients compared to controls (Jensen et al., 2018). In contrast, blood samples from individuals with AD showed lower NER gene expression. The increased NER expression in the brains of AD patients suggests an adaptive response to DNA damage and may reflect an attempt to counteract the accumulation of genetic lesions associated with the disease. The contrasting results in blood samples, with decreased NER expression in AD patients, raise questions about the systemic impact of AD on DNA repair processes. It is possible that peripheral blood cells exhibit different gene expression patterns than brain tissue due to various factors such as cell type-specific responses or systemic effects of AD pathology. Further research is required to understand the underlying reasons for this disparity. However, the observed differences in NER expression between brain tissue and blood suggest that the results should be carefully analyzed when generalizing findings from blood-based studies to brain-related pathologies, such as AD. These results highlight the importance of investigating tissue-specific mechanisms and considering the limitations of peripheral samples in understanding central nervous system (CNS) disorders. The results presented in this study are in stark contrast to numerous other studies that have reported a decrease in DNA repair activities.
Genes involved in various cellular processes, such as DNA repair, oxidative stress response, and synaptic function, were found to be altered in the aged individuals. This was identified using microarray analysis in postmortem brain tissue samples from people ranging in age from 26 to 106 years (Lu et al., 2004). Moreover, it was found that the extent of DNA damage positively correlated with the dysregulation of genes involved in DNA repair and the oxidative stress response. In addition, the transcription factor Sp1 levels decrease with age, potentially impairing the transcriptional activation of target genes involved in DNA repair and antioxidant defense. Similarly, decreased PGC-1α levels may compromise the mitochondrial function and increase oxidative stress.
Nuclear and mitochondrial somatic mutations in Alzheimer’s disease
Whole-exome sequencing using genomic DNA extracted from the brain tissues of AD patients and controls revealed the presence of somatic mutations in the brains of AD patients (Park et al., 2019). These mutations were predominantly found in genes associated with various cellular processes, including neuronal function, synaptic plasticity, and immune response. Importantly, several of the identified mutations have been linked to the dysregulation of tau phosphorylation, a critical process implicated in AD pathology (Park et al., 2019). Furthermore, the study demonstrated a positive correlation between the burden of brain somatic mutations and age, suggesting that these mutations may accumulate over time and contribute to the aging process and the increased susceptibility to AD. A comprehensive analysis of somatic mitochondrial DNA (mtDNA) point mutations in the brain tissue of elderly individuals with and without AD revealed a significantly higher burden of mutations (Lin et al., 2012). Importantly, the burden of mtDNA mutations was correlated with age, indicating the accumulation of these mutations over time. Moreover, AD patients exhibited a higher burden of mtDNA mutations than non-demented individuals of similar age, suggesting a potential role of mtDNA mutations in AD pathogenesis. A negative correlation was found between the mtDNA mutation burden and the activity of mitochondrial respiratory chain complexes, indicating that the accumulation of mtDNA mutations is associated with impaired mitochondrial function (Lin et al., 2012). It was also found that brain tissue with a higher burden of mtDNA mutations showed decreased levels of mitochondrial translation products, suggesting that these mutations may disrupt protein synthesis within the mitochondria. In addition, a higher density of mtDNA mutations, such as in the hippocampus and entorhinal cortex, was observed in regions vulnerable to AD pathology, providing evidence for the association between mtDNA mutations and AD-related neurodegeneration. Polymerase chain reaction (PCR) and Southern blot analysis revealed a significant increase in the levels of mtDNA deletions in postmortem brain samples from AD patients. The mtDNA deletion levels in the AD brains were approximately five times higher than those observed in the control brains (Corral-Debrinski et al., 1994). The temporal cortex, a brain region severely affected by AD, had the highest levels of mtDNA deletions when compared to the other regions studied, including the cerebellum and frontal cortex.
Mitochondrial DNA damage in Parkinson’s disease
The presence of mtDNA deletions and rearrangements were analyzed in brain tissue samples from patients with PD and related neurodegenerative disorders, such as dementia with Lewy bodies (DLB), AD, and progressive supranuclear palsy (PSP). Using PCR and Southern blot analysis, they detected mtDNA deletions and rearrangements in a significant proportion of PD and DLB cases, with a higher prevalence than that of AD and PSP (Gu et al., 2002). The observed mtDNA abnormalities were mostly found in the substantia nigra, a brain region critically affected in PD. Interestingly, it was found that patients with an earlier age of onset showed a higher frequency and greater extent of mtDNA abnormalities, suggesting a possible link between mitochondrial dysfunction and disease severity. To investigate the functional consequences of mtDNA deletions/rearrangements, immunohistochemical staining of the mitochondrial respiratory chain complex subunits in brain tissue samples was performed. They observed reduced staining intensity for specific subunits in regions harboring mtDNA abnormalities, indicating impaired mitochondrial function in affected brain regions. Moreover, the study examined the presence of mitochondrial proteins involved in mtDNA maintenance and repair, such as polymerase γ and mitochondrial transcription factor A (TFAM). Decreased levels of both polymerase γ and TFAM in brain regions with mtDNA deletions/rearrangements suggest an impairment in mtDNA replication and maintenance mechanisms. In another study, mtDNA deletions were detected by performing PCR and quantitative analysis in the substantia nigra neurons of postmortem brain samples of healthy individuals, aged controls, and PD patients (Bender et al., 2006). The study revealed significantly higher levels of mtDNA deletions in both healthy aging individuals and PD patients than in age-matched controls. Importantly, the levels of mtDNA deletions were positively correlated with age, indicating an accumulation of these deletions over time. The levels of mtDNA deletions were inversely correlated with respiratory chain complex activities, suggesting that the accumulation of mtDNA deletions leads to mitochondrial dysfunction in the substantia nigra neurons. Furthermore, substantia nigra neurons were found to have significantly higher levels of mtDNA deletions than other regions, including the frontal cortex and cerebellum, highlighting the selective vulnerability of substantia nigra neurons to mtDNA deletions. The occurrence of somatic mtDNA mutations in postmortem brain tissue samples from individuals with early-stage PD and incidental Lewy body disease (iLBD), a condition characterized by the presence of Lewy bodies without clinical PD symptoms, has been reported (Lin et al., 2002). This study revealed a significantly higher burden of somatic mtDNA mutations in the brain tissue of both early-stage PD patients and iLBD patients than in healthy controls. Moreover, the mutation burden was positively correlated with age, suggesting an accumulation of these mutations over time. The mutation burden was also found to be associated with reduced mitochondrial respiratory chain complex activity and increased levels of oxidative stress markers in the brain tissue (Lin et al., 2002). The researchers observed a higher density of mtDNA mutations in regions vulnerable to Lewy body pathology, such as the substantia nigra and locus coeruleus. They discovered that a higher mtDNA mutation burden was associated with more severe motor symptoms and cognitive decline in PD patients. A large-scale genetic analysis using genome-wide association study (GWAS) identified mutations in several genes related to mitochondrial function that were associated with an increased risk of PD (Billingsley et al., 2019). These genes included mitochondrial ATP6, ND2, and ND4, which encode components of the mitochondrial electron transport chain. In addition, they found that some variants were associated with altered expression levels of mitochondrial genes in the postmortem brain tissue (Billingsley et al., 2019).
DNA damage in Huntington's disease
Quantitative polymerase chain reaction (PCR) was used to measure mtDNA damage in postmortem brain tissue samples from HD patients and control subjects (Polidori et al., 1999). When compared to the control group, HD patients had significantly higher levels of 8-OHdG in the mtDNA of the parietal cortex. HD patients exhibit increased oxidative damage in affected brain regions, particularly in the basal ganglia. Oxidative damage and impaired mitochondrial function lead to an energy deficit in the brains of HD patients, contributing to metabolic dysfunction (Browne et al., 1997). Using HD patient-derived cells, it was shown that the mutant HTT protein disrupted the function of Ku70, a crucial protein involved in DNA repair, leading to increased genomic instability in HD. It was demonstrated that mutant HTT interacts with Ku70 and forms nuclear aggregates in HD patient-derived cells, implying that mutant HTT may play a role in disrupting DNA repair (Enokido et al., 2010). Mutant HTT impairs the recruitment of Ku70 to DSBs, a critical step in the DNA repair process. This impairment results in defective repair of DSBs, leading to accumulation of DNA damage. Furthermore, they observed that mutant HTT enhanced the phosphorylation of Ku70 at serine 51, which negatively regulated its DNA-binding ability. This aberrant phosphorylation is mediated by the activation of the stress-activated protein kinase c-Jun N-terminal kinase (JNK), which is dysregulated in HD.
Defective DNA damage response in Amyotrophic lateral sclerosis
The progressive loss of specific neuronal populations defines ALS. Some cases of ALS have been linked to mutations in the fused-in-sarcoma (FUS) gene. FUS protein is predominantly localized in the nucleus, and mutations in the NLS region disrupt its nuclear import, resulting in cytoplasmic aggregation. In a study, the effects of FUS-NLS mutations on DDR signaling and its implications for neurodegeneration were investigated using patient-derived induced pluripotent stem cells (iPSCs) and CRISPR/Cas9 genome editing (Naumann et al., 2018). It was found that cells expressing mutant FUS exhibited impaired DDR signaling and increased sensitivity to DNA damage compared to cells expressing wild-type FUS. Further analysis revealed that impaired DDR signaling in FUS-NLS-mutant cells was associated with decreased recruitment of DNA repair proteins to the site of DNA damage (Naumann et al., 2018). This compromised repair process resulted in increased accumulation of DNA damage and genomic instability. Moreover, the mutant FUS protein interacts with key DDR components, suggesting a direct role in impairing DDR signaling.
The data presented above suggest that DNA damage and impaired DNA repair may play a role in the pathogenesis of NDDs. The observed correlation between mutation burden, in particular mtDNA, mitochondrial dysfunction, and NDDS suggests a potential role of mtDNA mutations in these diseases. In the following sections, we will begin by discussing the current knowledge regarding the molecular link between cancer and neurodegeneration. Subsequently, the impact of cancer treatment, particularly chemotherapy, on brain tissues, supported by evidence from human studies will be discussed.
Intersection of cancer and neurodegeneration
Neurodegeneration primarily involves the degeneration and loss of specific neurons in the brain, whereas cancer is characterized by increased cell division and deficiency in programmed cell death (Seo & Park, 2020). Initially, these two conditions may appear completely contrasting or unrelated. However, subsequent epidemiological studies have indicated that they possess certain factors that suggest overlapping molecular mechanisms. The following shared factors and processes may contribute to both these conditions.
1.
Mutations in specific genes resulting from unrepaired DNA lesions contribute to the development of both conditions, and the specific mechanisms involved can vary between these conditions. For instance, mutations in DDR gene ATM have been detected in many cancers and was responsible for neuronal cell damage in ataxia-telangiectasia (Ball & Xiao, 2005). PD patients have been reported to have an increased risk of developing melanoma but no other types of cancer. Familial melanoma cases showed mutations in Cyclin-Dependent Kinase Inhibitor 2A (CDKN2A) and Cyclin-Dependent Kinase 4 (CDK4). Additionally, individuals with a family history of melanoma had a two-fold higher risk of developing PD than those without such a history (Gao et al., 2009). Genome-wide association studies (GWAS) have further revealed mutations in the melanocortin 1 receptor (MC1R), tyrosinase (TYR), and Phospholipase A2 (PLA2G6) genes that were associated with both melanoma risk and early onset PD (Bishop et al., 2009; Magrinelli et al., 2022; Paisan-Ruiz et al., 2009). These findings require further investigation to establish meaningful associations between these disease categories. Certain mutations associated with PD, such as PARK2 (parkin), PINK1, and LRRK2, have also been identified in cancer cases (Greenman et al., 2007; Klein et al., 2007; Veeriah et al., 2010). Somatic mutations and deletions in PARK2 have been detected in glioblastoma, colon, and lung cancers. However, there was a notable distinction between PD and cancer in terms of the mutation patterns. PD cases typically exhibit homozygous or compound heterozygous mutations in PARK2, whereas cancer cases predominantly exhibit heterozygous mutations. Consequently, to understand the connection between these mutations and the development of cancer and NDDs, it is crucial to compare the frequency and types of mutations, including heterozygous, compound heterozygous, and homozygous mutations, as well as their germline or somatic nature. Generally, germline mutations are associated with neurodegeneration, whereas somatic mutations are linked to cancer (e.g., ATM and PARK2).
2.
Inflammatory pathways promote tumor growth and progression, as well as the progression of NDDS such as AD and PD. Inflammatory processes in the tumor microenvironment can promote cell proliferation, angiogenesis, tissue remodeling, and immune evasion. Furthermore, chronic inflammation can cause DNA damage, genomic instability, and the activation of signaling pathways that aid in tumor survival and progression. Similarly, inflammation is a prominent feature in NDDs, including AD, PD, and ALS (Liu & Wang, 2017; Morgan et al., 2022; Zhang et al., 2023a, 2023b). When inflammatory pathways in the brain are activated, pro-inflammatory molecules such as cytokines and chemokines are released, which contribute to neuronal damage and cell death. Additionally, certain chemotherapeutic drugs or their byproducts have the potential to initiate inflammation, resulting in damage to the blood–brain barrier and the induction of neuronal damage (Fig. 2).
3.
Oxidative stress serves as a link between cancer and neurodegenerative disease. In cancer, increased ROS levels can promote tumor development and progression. ROS contribute to mitochondrial and nuclear DNA damage, genomic instability, and mutations, which are key factors in the initiation and growth of cancer cells. Similarly, excessive ROS production and impaired antioxidant defense mechanisms play crucial roles in NDDs. NDDs such as AD, PD, and HT are characterized by oxidative damage. (Hegde et al., 2012).
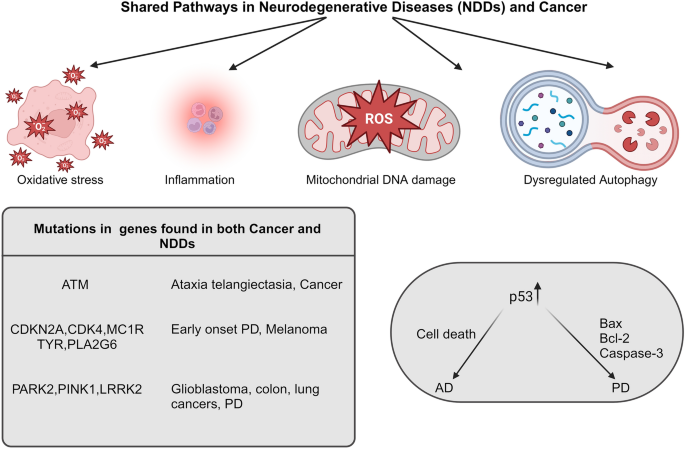
Convergent Molecular Pathways in Neurodegenerative Diseases (NDDs) and Cancer. This diagram explains the intersecting molecular pathways and mechanisms found across a spectrum of neurodegenerative diseases (including Alzheimer's disease, Parkinson's disease, etc.,) and cancer. While p53 is frequently mutated in numerous cancer types, it exhibits elevated expression levels in the brains of individuals with PD and AD. This expression may explain the increased neuronal cell death and the progression of PD and AD. The image was created with BioRender (https://www.biorender.com/)
Some evidence suggests an inverse relationship between cancer occurrence and age-related neurodegeneration. (Ibanez et al., 2014). For example, individuals with cancers, such as breast, lung, and prostate cancer, may have a lower risk of developing AD (Roe, et al., 2010). In contrast, some studies have found that people with neurodegenerative disorders have a lower risk of developing certain cancers. A similar inverse relationship has been observed between cancer and HD (Sørensen et al., 1999). However, mutations in DNA repair genes, such as BRCA1 and BRCA2, not only increase the risk of breast and ovarian cancer, but have also been linked to an increased risk of AD in animal models (Suberbielle et al., 2015). Although the precise mechanisms underlying these inverse correlations remain unknown, several hypotheses have been proposed. One possibility is that there are shared biological pathways involved in cancer and neurodegeneration, and alterations in these pathways may result in a protective effect against one disease, while increasing susceptibility to others. Mutations in the tumor suppressor gene p53 are strongly linked to various types of cancers, and extensive research has elucidated their molecular mechanisms in promoting cancer. Unlike in cancer, no mutations in the p53 gene have been identified in NDD. However, in the context of AD, studies have demonstrated increased expression and activity of p53 in the brain, along with elevated levels of cell death (Anderson et al., 2000; Cenini et al., 2008; Kitamura et al., 1997). Increased p53 expression has also been found in the brains of PD patients, along with p53-dependent apoptotic pathways involving proteins such as Bcl-2 associated X (Bax) and caspase-3 (Burguillos et al., 2011; Mogi et al., 2007).
Dysfunctional cyclin dependent kinase (CDK) complexes have been associated with the initiation and progression of cancer. Interestingly, increased CDK4 expression and activity has also been detected in the brains of individuals with AD (McShea et al., 1997). In vitro studies have shown that increased CDK4 activity can result in tau protein phosphorylation and caspase-3 activation, ultimately leading to neuronal cell death. Furthermore, investigations using postmortem brain samples from PD patients and experimental models have revealed abnormal expression of mitosis-associated proteins, such as the E2F-1 transcription factor, and dysregulation of cell cycle regulators in neurons (Höglinger et al., 2007). These alterations have been linked to neuronal cell death in PD. Parkin, a protein encoded by PARK2, acts as an E3 ubiquitin ligase and targets Cyclin E for proteasomal degradation (Kitada et al., 1998). Mutations in PARK2, as observed in PD, result in impaired degradation of cyclin E mediated by parkin. Although these findings were observed in cell culture experiments, this impairment has been shown to result in apoptosis. Moreover, Cyclin E expression in animal models of AD has been linked to cell cycle activation and neurodegeneration (Lee et al., 2017). Another cyclin, Cyclin F, forms a ubiquitin ligase complex known as SCF (SCF-cyclin F) that plays a role in cell cycle regulation. Altered Cyclin F expression has been reported in various cancers. In NDDs, mutations in Cyclin F have been found in ALS and frontotemporal dementia (FTD) (Williams et al., 2016). These mutations cause an abnormal accumulation of ubiquitinated TDP-43, a pathological feature of ALS and FTD.
There is a positive correlation between peptidyl-prolyl cis/trans isomerase (PIN1), a regulator of cyclin D1, and the expression and progression of various cancers. However, an inverse correlation between PIN1 and AD has been found in the AD brain (Liou et al., 2003). Unlike in AD, PIN1 was found to be elevated in the PD brain and is involved in Lewy body formation by enhancing aggregation of α-synuclein (Ghosh et al., 2013). The opposing expression levels of PIN1 in AD and PD should be substantiated by further experiments to understand the specific roles of these two diseases. Reduced activity of the serine/threonine protein phosphatase PP2A has been associated with both tumors and AD. In AD, reduced PP2A levels cause hyperphosphorylation of tau (Sontag et al., 2004). PIN1, a regulator of cyclin D1, is positively correlated with the progression of various cancers. However, an inverse correlation has been observed in the context of AD, as PIN1 expression is reduced in the AD brain (Liou et al., 2003). In contrast, elevated levels of PIN1 have been found in PD brains and have been linked to the formation of Lewy bodies by promoting the aggregation of α-synuclein. (Ghosh et al., 2013). Furthermore, reduced activity of the serine/threonine protein phosphatase PP2A has been associated with both tumors and AD (Chen et al., 2005). Similarly, a decrease in PP2A activity has been reported in PD (Park et al., 2016). These findings suggest the potential involvement of many shared pathways in the pathogenesis of NDDs and cancer. However, it is important to note that each disease has its own distinct molecular mechanisms and pathological processes. More research is needed to fully understand the underlying mechanisms and to investigate potential therapeutic interventions targeting these shared pathways.
The effect of chemotherapy on the central nervous system
The side effects of chemotherapy in cancer survivors have been extensively documented in literature. These effects include accelerated aging, neurological issues, increased risk of secondary malignancies, and toxicity to important organs, such as the kidney, liver, brain, and heart (Bhatia et al., 2022; Cupit-Link, et al., 2017; Guida et al., 2019; Shafqat et al., 2022). In pediatric survivors, this is particularly significant as it can impact their quality of life in later years, depending on the type of genotoxic agents used, dosage administered, type of damage inflicted, and affected tissue. In general, chemotherapeutic drugs either induce DNA damage or inhibit cell division and promote cell death. Notably, many chemotherapeutic drugs targeting DNA also damage healthy cells (Nonnekens & Hoeijmakers, 2017). High doses of these drugs administered in clinical settings cause DNA damage that exceeds the capacity of cellular DNA repair mechanisms, ultimately leading to cell death. The literature has extensively documented acute and chronic neurological complications caused by chemotherapeutic drugs (Macdonald, 1991; Sioka & Kyritsis, 2009; Zukas & Schiff, 2018). The majority of neurotoxicity was observed in the peripheral nervous system (PNS) rather than in the CNS, primarily due to the mode of administration and the protective blood–brain barrier. For instance, methotrexate, used in the treatment of childhood leukemia, can cross the blood–brain barrier and lead to complications, ranging from learning disabilities to dementia (van der Plas et al., 2015). Short-term symptoms, such as seizures, are induced by many chemotherapeutic drugs and typically subside shortly after discontinuation of drug administration. The long-term effects of these drugs on the CNS depend on factors such as the dosage, duration of administration, and route of administration (van den Boogaard et al., 2022). The specific damage and impact of chemotherapy on brain cells can vary depending on the type of the drug, the dosage, and individual patient’s body response. Preventing chemotherapy-induced brain damage is a complex challenge, and while there is no way to completely eliminate the risk, there are strategies that may help to reduce such damage. Neuroprotective agents like antioxidant supplements such as vitamin C, E or other natural products may be considered to protect the brain from oxidative damage. However, excessive antioxidant supplementation could potentially interfere with the effectiveness of chemotherapy (Wieland et al., 2021). In addition, the dosage and schedule of chemotherapy may be adjusted to reduce the impact on CNS. For example, by lowering the dose, extending the treatment schedule, or using alternative drugs with less neurological side effects.
Various alkylating agents can lead to neurotoxicity (Magge & DeAngelis, 2015). Platinum compounds like cisplatin, carboplatin, and oxaliplatin can bind to DNA covalently and form intrastrand or interstrand crosslinks. If these crosslinks are not properly repaired, they can lead to the development of DSBs, which in turn can result in cell death, aging, and development of secondary cancers. Moreover, platinum compounds also elevate the production of ROS, causing further damage to the mitochondria and inducing cellular stress (Kelland, 2007). However, the neurotoxic effects of platinum compounds typically affect the PNS, particularly the dorsal root ganglia (DRG) of the primary neurons. Chronic neurotoxicity can only occur at high doses (Magge & DeAngelis, 2015). Additionally, studies have shown that oxaliplatin can cause blood–brain barrier damage in cellular experimental models, providing a possible explanation for CNS toxicity observed in cancer patients as a result of drug exposure (Branca et al., 2018). Chronic leukoencephalopathy has been reported in individuals treated with the topoisomerase inhibitor, etoposide. Varying degrees of neurotoxicity can be caused by vinca alkaloids, such as vincristine, and the proteasome inhibitor bortezomib. Table 1 summarizes the effect of various commonly used chemotherapy drugs on CNS. Exposing rat primary neurons to doxorubicin treatment led to an increase in DSBs and a reduction in the repair factor BRCA1 (Manchon et al., 2016). A substantial amount of research has been conducted on chemotherapy-induced cognitive impairment (CICI), commonly known as “chemobrain,” using animal cancer models, cellular models, and genetically modified animal models (Bagnall-Moreau et al. 2019; John et al., 2021). Although the exact mechanism is unknown, it has been proposed that chemotherapeutic drugs may directly or indirectly damage neurons via mechanisms such as the generation of ROS, increased blood–brain barrier permeability and neuroinflammation, gene polymorphisms in susceptible genes, epigenetic alterations, telomere shortening, mitochondrial damage, and disruption of neurotransmitter balance (Fig. 3) (Flanary & Streit, 2004; Ren et al., 2019; Taillibert et al., 2016). Although there is clinical evidence supporting the presence of chemobrain in cancer patients, studying the exact mechanisms of this condition through direct examination of human brain samples is challenging because of the limited availability of brain tissue samples from living patients.

Table 1 List of commonly used therapeutic drugs, their corresponding target genes, and associated acute and chronic CNS conditions are shown
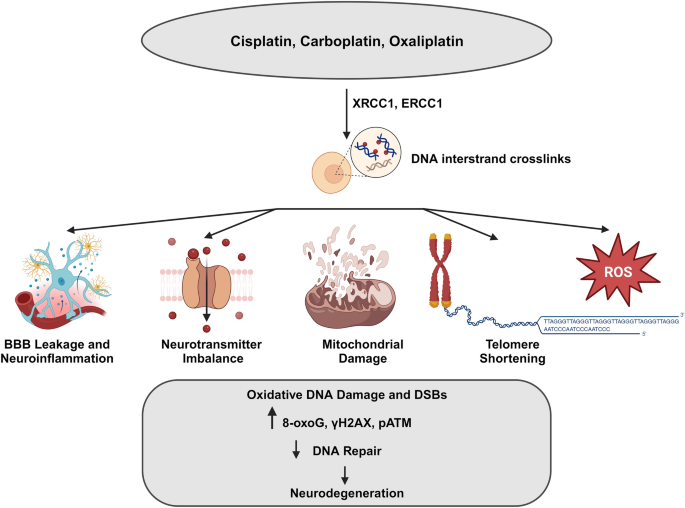
Schematic representation of direct and indirect actions of chemotherapeutic drugs on brain. Platinum-based compounds such as cisplatin, oxaliplatin, and carboplatin exert their effects by inducing DNA damage through crosslinking, which subsequently affects both mitochondrial and nuclear DNA. Additionally, these compounds can indirectly impact neurons by disrupting the blood–brain barrier (BBB), leading to increased permeability, resulting in neuroinflammation, increased production of reactive oxygen species (ROS), and oxidative damage. The image was created with BioRender (https://www.biorender.com/)
Future research should prioritize the development of innovative strategies aimed at investigating specific types of neuronal damage caused by different classes of drugs and their corresponding clinical symptoms. This knowledge contributes to our understanding of the nature of DNA damage and specific repair mechanisms in neurons, as well as the differences in DNA repair machines between neurons and other tissues. To tackle this challenge, one potential approach would involve evaluating the molecular signatures present in postmortem brain samples or archived brain bank samples obtained from individuals who have previously undergone treatment with chemotherapeutic drugs. In the next section, studies conducted with this objective will be discussed.
Chemotherapy induced DNA damages in the neurons: insights from cancer survivors brain samples
A recent study looked into the effects of chemotherapy on oxidative stress and DNA damage in frontal cortex cortical neurons. This study utilized brain tissue samples obtained from autopsies and included three distinct study groups. The first group consisted of individuals who had undergone chemotherapy as treatment for cancer. The second group comprised cancer patients who had not received chemotherapy, whereas the third group consisted of individuals with no history of cancer or chemotherapy treatment. Within the chemotherapy group, the researchers further categorized the samples based on the time interval between the last administration of chemotherapy and the individual's death as well as the total number of chemotherapy cycles. The results of the study revealed significant differences between the groups. Patients undergoing chemotherapy exhibited increased levels of oxidative stress markers, such as 8-oxoG, a biomarker of DNA damage caused by ROS (Fig. 3). Additionally, they observed higher levels of DNA damage in the cortical neurons of the chemotherapy patients than in the control groups. This is indicated by an increase in γH2AX and phosphorylated ATM (pATM), which are markers of DNA DSBs (Torre et al., 2021).
The long-term effects of chemotherapy on brain structure and function in breast cancer survivors, a population that often experiences cognitive impairment, have been reported. The relationship between these changes and oxidative DNA damage was examined. The study involved breast cancer survivors who had undergone chemotherapy, and a control group of healthy women (Conroy et al., 2013). Magnetic resonance imaging (MRI) was used to evaluate the brain structure, focusing on the gray matter volume. Functional MRI (fMRI) scans were also performed to assess brain activity during a working memory task. Breast cancer survivors exhibited lower gray matter volume in regions of brain involved in memory and attention. Additionally, the fMRI results indicated altered brain activation patterns during the working memory task in the survivor group. Survivors with a longer interval since chemotherapy completion showed less severe brain structural changes compared to those with a shorter interval. This suggests a potential recovery or normalization of brain structure over time following chemotherapy. A positive correlation between 8-OHdG, a marker of oxidative DNA damage levels, and the severity of brain structural changes was seen in breast cancer survivors.
A study looked at the relationship between oxidative stress and executive function in children receiving chemotherapy for acute lymphoblastic leukemia (ALL). The study includes paediatric ALL patients who were about to undergo chemotherapy and measured markers of oxidative stress, including lipid peroxidation and total antioxidant capacity, in the patient’s blood samples (Caron et al., 2009). When compared to healthy controls, ALL had significantly higher levels of oxidative stress. And the higher oxidative stress was associated with poorer performance on tests assessing executive functions such as attention, working memory, and cognitive flexibility. Based on these results, it was proposed that chemotherapy-related oxidative stress may impair the functioning of brain regions involved in executive functions, leading to cognitive deficits (Caron et al., 2009). These findings highlight the significance of considering the long-term effects of chemotherapy on mitochondrial and nuclear DNA damage, alterations in DNA repair mechanisms, and cognitive and other brain functions.
Conclusions
Neurodegeneration can arise from various factors, such as genetic mutations, mitochondrial dysfunction, metabolic stress, exposure to environmental toxins, accumulation of DNA damage associated with natural aging, and impaired DNA repair. However, the existing treatments for these diseases are limited, as there are no available therapies capable of effectively slowing down or halting the loss of neurons. Therefore, using chemotherapy-induced neuronal DNA damage as a research tool to study neuronal DNA repair mechanisms, we can expand the understanding of how neurons safeguard genomic integrity and respond to genotoxic stress. This knowledge, which provides insights into the unique DNA repair mechanisms operating in neurons, holds significant implications for the treatment of NDDs. By quantifying the extent and kinetics of DNA repair, the repair capacity of neurons can be determined in comparison to dividing cells. This analysis will enable the identification of differences in repair efficiency between these cell types, thereby providing valuable information on their respective abilities to repair DNA damage. Furthermore, this knowledge also will assist in the design of strategies aimed at developing approaches to mitigate DNA damage in neurons and reduce chemotherapy-induced neurotoxicity. Neurodegenerative diseases like AD and PD are characterized by compromised DNA repair in neurons. Investigating the DNA damage caused by chemotherapy can provide valuable insights into how these diseases affect DNA repair processes, ultimately leading to neuronal dysfunction and cell death.
Data availability
All the raw data generated in this study are available from corresponding authors on request.
Abbreviations
NDD:
Neurodegenerative diseases
AD:
Alzheimer's disease
PD:
Parkinson's disease
HD:
Huntington's disease
ALS:
Amyotrophic lateral sclerosis
FTD:
Frontotemporal dementia
DDR:
DNA damage response
HR:
Homologous recombination
NHEJ:
Non-homologous end joining
BER:
Base excision repair
NER:
Nucleotide excision repair
SSB:
Single-strand breaks
DSB:
Double-strand breaks
APE1:
Apurinic/apyrimidinic endonucleases 1
8-oxoG:
8-Oxoguanosine
8-OHdG:
8-Hydroxydeoxyguanosine
PARP-1:
Poly-(ADP-ribose) polymerase 1
AT:
Ataxia telangiectasia
ATM:
Ataxia telangiectasia mutated
XP:
Xeroderma pigmentosum
CS:
Cockayne’s syndrome
TLS:
Translesion synthesis
PLA2G6:
Phospholipase A2 gene
CICI:
Chemotherapy-induced cognitive impairment
References
Anderson, A. J., Stoltzner, S., Lai, F., Su, J., & Nixon, R. A. (2000). Morphological and biochemical assessment of DNA damage and apoptosis in Down syndrome and Alzheimer disease, and effect of postmortem tissue archival on TUNEL. Neurobiology of Aging, 21(4), 511–524. https://doi.org/10.1016/s0197-4580(00)00126-3
Bagnall-Moreau, C., Chaudhry, S., Salas-Ramirez, K., Ahles, T., & Hubbard, K. (2019). Chemotherapy-induced cognitive impairment is associated with increased inflammation and oxidative damage in the hippocampus. Molecular Neurobiology, 56(10), 7159–7172. https://doi.org/10.1007/s12035-019-1589-z
Ball, L. G., & Xiao, W. (2005). Molecular basis of ataxia telangiectasia and related diseases. Acta Pharmacologica Sinica, 26(8), 897–907. https://doi.org/10.1111/j.1745-7254.2005.00165.x
Bender, A., Krishnan, K. J., Morris, C. M., Taylor, G. A., Reeve, A. K., Perry, R. H., Jaros, E., Hersheson, J. S., Betts, J., Klopstock, T., Taylor, R. W., & Turnbull, D. M. (2006). High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nature Genetics, 38(5), 515–517. https://doi.org/10.1038/ng1769
Bhatia, R., Holtan, S., Jurdi, N. E., Prizment, A., & Blaes, A. (2022). Do cancer and cancer treatments accelerate aging? Current Oncology Reports, 24(11), 1401–1412. https://doi.org/10.1007/s11912-022-01311-2
Billingsley, K. J., Barbosa, I. A., Bandrés-Ciga, S., Quinn, J. P., Bubb, V. J., Deshpande, C., Botia, J. A., Reynolds, R. H., Zhang, D., Simpson, M. A., Blauwendraat, C., Gan-Or, Z., Gibbs, J. R., Nalls, M. A., & Singleton, A. (2019). Mitochondria function associated genes contribute to Parkinson’s Disease risk and later age at onset. NPJ Parkinson’s Disease, 5, 8. https://doi.org/10.1038/s41531-019-0080-x
Bishop, D. T., Demenais, F., Iles, M. M., Harland, M., Taylor, J. C., Corda, E., Randerson-Moor, J., Aitken, J. F., Avril, M. F., Azizi, E., Bakker, B., Bianchi-Scarrà, G., Bressac-de Paillerets, B., Calista, D., Cannon-Albright, L. A., Chin-A-Woeng, T., Debniak, T., Galore-Haskel, G., Ghiorzo, P., et al. (2009). Genome-wide association study identifies three loci associated with melanoma risk. Nature Genetics, 41(8), 920–925. https://doi.org/10.1038/ng.411
Brambati, A., Zardoni, L., Nardini, E., Pellicioli, A., & Liberi, G. (2020). The dark side of RNA:DNA hybrids. Mutation Research, Reviews in Mutation Research, 784, 108300. https://doi.org/10.1016/j.mrrev.2020.108300
Branca, J. J. V., Maresca, M., Morucci, G., Becatti, M., Paternostro, F., Gulisano, M., Ghelardini, C., Salvemini, D., Di Ceseare Mannelli, L., & Pacini, A. (2018). Oxaliplatin-induced blood brain barrier loosening: a new point of view on chemotherapy-induced neurotoxicity. Oncotarget, 9(34), 23426–23438. https://doi.org/10.18632/oncotarget.25193
Branzei, D., & Foiani, M. (2008). Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Bio, 9(4), 297–308. https://doi.org/10.1038/nrm2351
Browne, S. E., Bowling, A. C., MacGarvey, U., Baik, M. J., Berger, S. C., Muqit, M. M., Bird, E. D., & Beal, M. F. (1997). Oxidative damage and metabolic dysfunction in Huntington’s disease: selective vulnerability of the basal ganglia. Annals of Neurology, 41(5), 646–653. https://doi.org/10.1002/ana.410410514
Burguillos, M. A., Deierborg, T., Kavanagh, E., Persson, A., Hajji, N., Garcia-Quintanilla, A., Cano, J., Brundin, P., Englund, E., Venero, J. L., & Joseph, B. (2011). Caspase signalling controls microglia activation and neurotoxicity. Nature, 472(7343), 319–324. https://doi.org/10.1038/nature09788
Caron, J. E., Krull, K. R., Hockenberry, M., Jain, N., Kaemingk, K., & Moore, I. M. (2009). Oxidative stress and executive function in children receiving chemotherapy for acute lymphoblastic leukemia. Pediatric Blood & Cancer, 53(4), 551–556. https://doi.org/10.1002/pbc.22128
Cenini, G., Sultana, R., Memo, M., & Butterfield, D. A. (2008). Elevated levels of pro-apoptotic p53 and its oxidative modification by the lipid peroxidation product, HNE, in brain from subjects with amnestic mild cognitive impairment and Alzheimer’s disease. Journal of Cellular and Molecular Medicine, 12(3), 987–994. https://doi.org/10.1111/j.1582-4934.2008.00163.x
Chen, W., Arroyo, J. D., Timmons, J. C., Possemato, R., & Hahn, W. C. (2005). Cancer-associated PP2A alpha subunits induce functional haploin sufficiency and tumorigenicity. Cancer Research, 65(18), 8183–8192. https://doi.org/10.1158/0008-5472.CAN-05-1103
Ciccia, A., & Elledge, S. J. (2010). The DNA damage response: making it safe to play with knives. Molecular Cell, 40(2), 179–204. https://doi.org/10.1016/j.molcel.2010.09.019
Conroy, S. K., McDonald, B. C., Smith, D. J., Moser, L. R., West, J. D., Kamendulis, L. M., Klaunig, J. E., Champion, V. L., Unverzagt, F. W., & Saykin, A. J. (2013). Alterations in brain structure and function in breast cancer survivors: effect of post-chemotherapy interval and relation to oxidative DNA damage. Breast Cancer Research and Treatment, 137(2), 493–502. https://doi.org/10.1007/s10549-012-2385-x
Coppedè, F. (2010). Mutations involved in premature-ageing syndromes. The Application of Clinical Genetics, 14, 279–295. https://doi.org/10.2147/TACG.S273525
Corral-Debrinski, M., Horton, T., Lott, M. T., Shoffner, J. M., McKee, A. C., Beal, M. F., Graham, B. H., & Wallace, D. C. (1994). Marked changes in mitochondrial DNA deletion levels in Alzheimer brains. Genomics, 23(2), 471–476. https://doi.org/10.1006/geno.1994.1525
Cunniff, C., Bassetti, J. A., & Ellis, N. A. (2017). Bloom’s Syndrome: clinical spectrum, molecular pathogenesis, and cancer predisposition. Molecular Syndromology, 8(1), 4–23. https://doi.org/10.1159/000452082
Cupit-Link, M. C., Kirkland, J. L., Ness, K. K., Armstrong, G. T., Tchkonia, T., LeBrasseur, N. K., Armenian, S. H., Ruddy, K. J., & Hashmi, S. K. (2017). Biology of premature ageing in survivors of cancer. ESMO Open, 2, e000250. https://doi.org/10.1136/esmoopen-2017-000250
Dabin, J., Mori, M., & Polo, S. E. (2023). The DNA damage response in the chromatin context: a coordinated process. Current Opinion in Cell Biology, 82, 102176. https://doi.org/10.1016/j.ceb.2023.102176
de Souza-Pinto, N. C., Wilson, D. M., 3rd., Stevnsner, T. V., & Bohr, V. A. (2008). Mitochondrial DNA, base excision repair and neurodegeneration. DNA Repair (amst), 7(7), 1098–1109. https://doi.org/10.1016/j.dnarep.2008.03.011
Didier, M., Bursztajn, S., Adamec, E., Passani, L., Nixon, R. A., Coyle, J. T., Wei, J. Y., & Berman, S. A. (1996). DNA strand breaks induced by sustained glutamate excitotoxicity in primary neuronal cultures. The Journal of Neuroscience, 16(7), 2238–2250. https://doi.org/10.1523/JNEUROSCI.16-07-02238.1996
Enokido, Y., Tamura, T., Ito, H., Arumughan, A., Komuro, A., Shiwaku, H., Sone, M., Foulle, R., Sawada, H., Ishiguro, H., Ono, T., Murata, M., Kanazawa, I., Tomilin, N., Tagawa, K., Wanker, E. E., & Okazawa, H. (2010). Mutant huntingtin impairs Ku70-mediated DNA repair. The Journal of Cell Biology, 189, 425–443. https://doi.org/10.1083/jcb.200905138
Farg, M. A., Konopka, A., Soo, K. Y., Ito, D., & Atkin, J. D. (2017). The DNA damage response (DDR) is induced by the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Human Molecular Genetics, 26(15), 2882–2896. https://doi.org/10.1093/hmg/ddx170
Flanary, B. E., & Streit, W. J. (2004). Progressive telomere shortening occurs in cultured rat microglia, but not astrocytes. Glia, 45(1), 75–88. https://doi.org/10.1002/glia.10301
Gao, X., Simon, K. C., Han, J., Schwarzschild, M. A., & Ascherio, A. (2009). Family history of melanoma and Parkinson disease risk. Neurology, 73(16), 1286–1291. https://doi.org/10.1212/WNL.0b013e3181bd13a1
Ghosh, A., Saminathan, H., Kanthasamy, A., Anantharam, V., Jin, H., Sondarva, G., Harischandra, D. S., Qian, Z., Rana, A., & Kanthasamy, A. G. (2013). The peptidyl-prolyl isomerase Pin1 up-regulation and proapoptotic function in dopaminergic neurons: relevance to the pathogenesis of Parkinson disease. The Journal of Biological Chemistry, 288(30), 21955–21971. https://doi.org/10.1074/jbc.M112.444224
Giannini, M., Bayona-Feliu, A., Sproviero, D., Barroso, S. I., Cereda, C., & Aguilera, A. (2020). TDP-43 mutations link amyotrophic lateral sclerosis with R-loop homeostasis and R loop-mediated DNA damage. PLoS Genetics, 16(12), e1009260. https://doi.org/10.1371/journal.pgen.1009260
Goto, M. (1997). Hierarchical deterioration of body systems in Werner’s syndrome: implications for normal ageing. Mechanisms of Ageing and Development, 98(3), 239–254. https://doi.org/10.1016/s0047-6374(97)00111-5
Greenman, C., Stephens, P., Smith, R., Dalgliesh, G. L., Hunter, C., Bignell, G., Davies, H., Teague, J., Butler, A., Stevens, C., Edkins, S., O’Meara, S., Vastrik, I., Schmidt, E. E., Avis, T., Barthorpe, S., Bhamra, G., Buck, G., Choudhury, B., et al. (2007). Patterns of somatic mutation in human cancer genomes. Nature, 446(7132), 153–158. https://doi.org/10.1038/nature05610
Gu, G., Reyes, P. E., Golden, G. T., Woltjer, R. L., Hulette, C., Montine, T. J., & Zhang, J. (2002). Mitochondrial DNA deletions/rearrangements in Parkinson disease and related neurodegenerative disorders. Journal of Neuropathology and Experimental Neurology, 61(7), 634–639. https://doi.org/10.1093/jnen/61.7.634
Guida, J. L., Ahles, T. A., Belsky, D., Campisi, J., Cohen, H. J., DeGregori, J., Fuldner, R., Ferrucci, L., Gallicchio, L., Gavrilov, L., Gavrilova, N., Green, P. A., Jhappan, C., Kohanski, R., Krull, K., Mandelblatt, J., Ness, K. K., O’Mara, A., Price, N., et al. (2019). Measuring aging and identifying aging phenotypes in cancer survivors. Journal of the National Cancer Institute, 111, 1245–1254. https://doi.org/10.1093/jnci/djz136
Gupta, S., You, P., SenGupta, T., Nilsen, H., & Sharma, K. (2021). Crosstalk between different DNA repair pathways contributes to neurodegenerative diseases. Biology (basel), 10(2), 163. https://doi.org/10.3390/biology10020163
Hegde, M. L., Bohr, V. A., & Mitra, S. (2017). DNA damage responses in central nervous system and age-associated neurodegeneration. Mechanisms of Ageing and Development, 161(Pt A), 1–3. https://doi.org/10.1016/j.mad.2017.01.010
Hegde, M. L., Mantha, A. K., Hazra, T. K., Bhakat, K. K., Mitra, S., & Szczesny, B. (2012). Oxidative genome damage and its repair: Implications in aging and neurodegenerative diseases. Mechanisms of Ageing and Development, 133(4), 157–168. https://doi.org/10.1016/j.mad.2012.01.005
Hoeijmakers, J. H. J. (2009). DNA damage, aging, and cancer. The New England Journal of Medicine, 361, 1475–1485. https://doi.org/10.1056/NEJMra0804615
Höglinger, G. U., Breunig, J. J., Depboylu, C., Rouaux, C., Michel, P. P., Alvarez-Fischer, D., Boutillier, A. L., Degregori, J., Oertel, W. H., Rakic, P., Hirsch, E. C., & Hunot, S. (2007). The pRb/E2F cell-cycle pathway mediates cell death in Parkinson’s disease. Proceedings of the National Academy of Sciences of the United States of America, 104(9), 3585–3590. https://doi.org/10.1073/pnas.0611671104
Huang, R., & Zhou, P. K. (2021). DNA damage repair: Historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal Transduction and Targeted Therapy, 6(1), 254. https://doi.org/10.1038/s41392-021-00648-7
Huertas, P., & Aguilera, A. (2003). Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Molecular Cell, 12(3), 711–721. https://doi.org/10.1016/j.molcel.2003.08.010
Ibanez, K., Boullosa, C., Tabares-Seisdedos, R., Baudot, A., & Valencia, A. (2014). Molecular evidence for the inverse comorbidity between central nervous system disorders and cancers detected by transcriptomic meta-analyses. PLoS Genetics, 10(2), e1004173. https://doi.org/10.1371/journal.pgen.1004173
Iyama, T., & Wilson, D. M., 3rd. (2013). DNA repair mechanisms in dividing and non-dividing cells. DNA Repair (amst), 12(8), 620–636. https://doi.org/10.1016/j.dnarep.2013.04.015
Jensen, H. L. B., Lillenes, M. S., Rabano, A., Günther, C. C., Riaz, T., Kalayou, S. T., Ulstein, I. D., & Bøhmer, T. (2018). Expression of nucleotide excision repair in Alzheimer’s disease is higher in brain tissue than in blood. Neuroscience Letters, 672, 53–58. https://doi.org/10.1016/j.neulet.2018.02.043
John, J., Kinra, M., Mudgal, J., Viswanatha, G. L., & Nandakumar, K. (2021). Animal models of chemotherapy-induced cognitive decline in preclinical drug development. Psychopharmacology (berl), 238(11), 3025–3053. https://doi.org/10.1007/s00213-021-05977-7
Karahalil, B., Hogue, B. A., de Souza-Pinto, N. C., & Bohr, V. A. (2002). Base excision repair capacity in mitochondria and nuclei: Tissue-specific variations. FASEB Journal, 16(14), 1895–1902. https://doi.org/10.1096/fj.02-0463com
Kelland, L. (2007). The resurgence of platinum-based cancer chemotherapy. Nature Reviews Cancer, 7(8), 573–584. https://doi.org/10.1038/nrc2167
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., Yokochi, M., Mizuno, Y., & Shimizu, N. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature, 392(6676), 605–608. https://doi.org/10.1038/33416
Kitamura, Y., Shimohama, S., Kamoshima, W., Matsuoka, Y., Nomura, Y., & Taniguchi, T. (1997). Changes of p53 in the brains of patients with Alzheimer’s disease. Biochemical and Biophysical Research Communications, 232(2), 418–421. https://doi.org/10.1006/bbrc.1997.6301
Klein, C., Lohmann-Hedrich, K., Rogaeva, E., Schlossmacher, M. G., & Lang, A. E. (2007). Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurology, 6(7), 652–662. https://doi.org/10.1016/S1474-4422(07)70174-6
Krasikova, Y., Rechkunova, N., & Lavrik, O. (2021). Nucleotide excision repair: from molecular defects to neurological abnormalities. International Journal of Molecular Sciences, 22(12), 6220. https://doi.org/10.3390/ijms22126220
Leandro, G. S., Sykora, P., & Bohr, V. A. (2015). The impact of base excision DNA repair in age-related neurodegenerative diseases. Mutation Research., 776, 31–39. https://doi.org/10.1016/j.mrfmmm.2014.12.011
Lee, K. H., Lee, S. J., Lee, H. J., Choi, G. E., Jung, Y. H., Kim, D. I., Gabr, A. A., Ryu, J. M., & Han, H. J. (2017). Amyloid beta1-42 (Abeta1-42) induces the CDK2-mediated phosphorylation of tau through the activation of the mTORC1 signaling pathway while promoting neuronal cell death. Frontiers in Molecular Neuroscience, 10, 229. https://doi.org/10.3389/fnmol.2017.00229
Li, X., Cao, G., Liu, X., Tang, T. S., Guo, C., & Liu, H. (2022). Polymerases and DNA repair in neurons: implications in neuronal survival and neurodegenerative diseases. Frontiers in Cellular Neuroscience, 16, 852002. https://doi.org/10.3389/fncel.2022.852002
Lillenes, M. S., Rabano, A., Støen, M., Riaz, T., Misaghian, D., Møllersen, L., Esbensen, Y., Günther, C. C., Selnes, P., Stenset, V. T., Fladby, T., & Tønjum, T. (2016). Altered DNA base excision repair profile in brain tissue and blood in Alzheimer’s disease. Molecular Brain, 9(1), 61. https://doi.org/10.1186/s13041-016-0237-z
Lin, M. T., Cantuti-Castelvetri, I., Zheng, K., Jackson, K. E., Tan, Y. B., Arzberger, T., Lees, A. J., Betensky, R. A., Beal, M. F., & Simon, D. K. (2012). Somatic mitochondrial DNA mutations in early Parkinson and incidental Lewy body disease. Annals of Neurology, 71(6), 850–854. https://doi.org/10.1002/ana.23568
Lin, M. T., Simon, D. K., Ahn, C. H., Kim, L. M., & Beal, M. F. (2002). High aggregate burden of somatic mtDNA point mutations in aging and Alzheimer’s disease brain. Human Molecular Genetics, 11(2), 133–145. https://doi.org/10.1093/hmg/11.2.133
Liou, Y. C., Sun, A., Ryo, A., Zhou, X. Z., Yu, Z. X., Huang, H. K., Uchida, T., Bronson, R., Bing, G., Li, X., Hunter, T., & Lu, K. P. (2003). Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature, 424(6948), 556–561. https://doi.org/10.1038/nature01832
Liu, J., & Wang, F. (2017). Role of neuroinflammation in amyotrophic lateral sclerosis: cellular mechanisms and therapeutic implications. Frontiers in Immunology, 8, 1005. https://doi.org/10.3389/fimmu.2017.01005
Lu, T., Pan, Y., Kao, S. Y., Li, C., Kohane, I., Chan, J., & Yankner, B. A. (2004). Gene regulation and DNA damage in the ageing human brain. Nature, 429(6994), 883–891. https://doi.org/10.1038/nature02661
Luquette, L. J., Miller, M. B., Zhou, Z., Bohrson, C. L., Zhao, Y., Jin, H., Gulhan, D., Ganz, J., Bizzotto, S., Kirkham, S., Hochepied, T., Libert, C., Galor, A., Kim, J., Lodato, M. A., Garaycoechea, J. I., Gawad, C., West, J., Walsh, C. A., & Park, P. J. (2022). Single-cell genome sequencing of human neurons identifies somatic point mutation and indel enrichment in regulatory elements. Nature Genetics, 54(10), 1564–1571. https://doi.org/10.1038/s41588-022-01180-2
Macdonald, D. R. (1991). Neurologic complications of chemotherapy. Neurologic Clinics, 9(4), 955–967.
Magge, R. S., & DeAngelis, L. M. (2015). The double-edged sword: neurotoxicity of chemotherapy. Blood Reviews, 29(2), 93–100. https://doi.org/10.1016/j.blre.2014.09.012
Magrinelli, F., Mehta, S., Di Lazzaro, G., Latorre, A., Edwards, M. J., Balint, B., Basu, P., Kobylecki, C., Groppa, S., Hegde, A., Mulroy, E., Estevez-Fraga, C., Arora, A., Kumar, H., Schneider, S. A., Lewis, P. A., Jaunmuktane, Z., Revesz, T., Gandhi, S., et al. (2022). Dissecting the phenotype and genotype of PLA2G6-related parkinsonism. Movement Disorders, 37(1), 148–161. https://doi.org/10.1002/mds.28807
Manchon, J. F., Dabaghian, Y., Uzor, N. E., Kesler, S. R., Wefel, J. S., & Tsvetkov, A. S. (2016). Levetiracetam mitigates doxorubicin-induced DNA and synaptic damage in neurons. Scientific Reports, 6, 25705. https://doi.org/10.1038/srep25705
McKinnon, P. J. (2009). DNA repair deficiency and neurological disease. Nature Reviews Neuroscience, 10(2), 100–112. https://doi.org/10.1038/nrn2559
McKinnon, P. J. (2017). Genome integrity and disease prevention in the nervous system. Genes & Development, 31(12), 1180–1194. https://doi.org/10.1101/gad.301325.117
McShea, A., Harris, P. L., Webster, K. R., Wahl, A. F., & Smith, M. A. (1997). Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer’s disease. The American Journal of Pathology, 150(6), 1933–1939.
Mitra, J., Guerrero, E. N., Hegde, P. M., Liachko, N. F., Wang, H., Vasquez, V., Gao, J., Pandey, A., Taylor, J. P., Kraemer, B. C., Wu, P., Boldogh, I., Garruto, R. M., Mitra, S., & RaoHegde, K. S. M. L. (2019). Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proceedings of the National Academy of Sciences of the United States of America, 116(10), 4696–4705. https://doi.org/10.1073/pnas.1818415116
Mogi, M., Kondo, T., Mizuno, Y., & Nagatsu, T. (2007). p53 protein, interferon-gamma, and NF-kappa B levels are elevated in the parkinsonian brain. Neuroscience Letters, 414(1), 94–97. https://doi.org/10.1016/j.neulet.2006.12.003
Morgan, S. L., Naderi, P., Koler, K., Pita-Juarez, Y., Prokopenko, D., Vlachos, I. S., Tanzi, R. E., Bertram, L., & Hide, W. A. (2022). Most pathways can be related to the pathogenesis of Alzheimer’s disease. Frontiers in Aging Neuroscience, 14, 846902. https://doi.org/10.3389/fnagi.2022.846902
Naumann, M., Pal, A., Goswami, A., Lojewski, X., Japtok, J., Vehlow, A., Naujock, M., Günther, R., Jin, M., Stanslowsky, N., Reinhardt, P., Sterneckert, J., Frickenhaus, M., Pan-Montojo, F., Storkebaum, E., Poser, I., Freischmidt, A., Weishaupt, J. H., Holzmann, K., et al. (2018). Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nature Communications, 9(1), 335. https://doi.org/10.1038/s41467-017-02299-1
Nonnekens, J., & Hoeijmakers, J. H. (2017). After surviving cancer, what about late life effects of the cure? EMBO Molecular Medicine, 9, 4–6. https://doi.org/10.15252/emmm.201607062
Nouspikel, T., & Hanawalt, P. C. (2000). Terminally differentiated human neurons repair transcribed genes but display attenuated global DNA repair and modulation of repair gene expression. Molecular and Cellular Biology, 20(5), 1562–1570. https://doi.org/10.1128/MCB.20.5.1562-1570.2000
Paisan-Ruiz, C., Bhatia, K. P., Li, A., Hernandez, D., Davis, M., Wood, N. W., Hardy, J., Houlden, H., Singleton, A., & Schneider, S. A. (2009). Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Annals of Neurology, 65(1), 19–23. https://doi.org/10.1002/ana.21415
Pani, B., & Nudler, E. (2017). Mechanistic insights into transcription coupled DNA repair. DNA Repair (amst), 56, 42–50. https://doi.org/10.1016/j.dnarep.2017.06.006
Park, H. J., Lee, K. W., Park, E. S., Oh, S., Yan, R., Zhang, J., Beach, T. G., Adler, C. H., Voronkov, M., Braithwaite, S. P., Stock, J. B., & Mouradian, M. M. (2016). Dysregulation of protein phosphatase 2A in Parkinson disease and dementia with Lewy bodies. Annals of Clinical and Translational Neurology, 3(10), 769–780. https://doi.org/10.1002/acn3.337
Park, J. S., Lee, J., Jung, E. S., Kim, M. H., Kim, I. B., Son, H., Kim, S., Kim, S., Park, Y. M., Mook-Jung, I., Yu, S. J., & Lee, J. H. (2019). Brain somatic mutations observed in Alzheimer’s disease associated with aging and dysregulation of tau phosphorylation. Nature Communications, 10(1), 3090. https://doi.org/10.1038/s41467-019-11000-7
Petsalaki, E., & Zachos, G. (2020). DNA damage response proteins regulating mitotic cell division: double agents preserving genome stability. FEBS Journal, 287(9), 1700–1721. https://doi.org/10.1111/febs.15240
Polidori, M. C., Mecocci, P., Browne, S. E., Senin, U., & Beal, M. F. (1999). Oxidative damage to mitochondrial DNA in Huntington’s disease parietal cortex. Neuroscience Letters, 272, 53–56. https://doi.org/10.1016/s0304-3940(99)00578-9
Reid, D. A., Reed, P. J., Schlachetzki, J. C. M., Nitulescu, I. I., Chou, G., Tsui, E. C., Jones, J. R., Chandran, S., Lu, A. T., McClain, C. A., Ooi, J. H., Wang, T. W., Lana, A. J., Linker, S. B., Ricciardulli, A. S., Lau, S., Schafer, S. T., Horvath, S., Dixon, J. R., et al. (2021). Incorporation of a nucleoside analog maps genome repair sites in postmitotic human neurons. Science, 372(6537), 91–94. https://doi.org/10.1126/science.abb9032
Ren, X., Boriero, D., Chaiswing, L., Bondada, S., St Clair, D. K., & Butterfield, D. A. (2019). Plausible biochemical mechanisms of chemotherapy-induced cognitive impairment (“chemobrain”), a condition that significantly impairs the quality of life of many cancer survivors. Biochimica Et Biophysica Acta, Molecular Basis of Disease, 1865(6), 1088–1097. https://doi.org/10.1016/j.bbadis.2019.02.007
Roe, C. M., Fitzpatrick, A. L., Xiong, C., Sieh, W., Kuller, L., Miller, J. P., Williams, M. M., Kopan, R., Behrens, M. I., & Morris, J. C. (2010). Cancer linked to Alzheimer disease but not vascular dementia. Neurology, 74(2), 106–112. https://doi.org/10.1212/WNL.0b013e3181c91873
Santos, R. X., Correia, S. C., Zhu, X., Smith, M. A., Moreira, P. I., Castellani, R. J., Nunomura, A., & Perry, G. (2013). Mitochondrial DNA oxidative damage and repair in aging and Alzheimer’s disease. Antioxidants & Redox Signaling, 18(18), 2444–2457. https://doi.org/10.1089/ars.2012.5039
Schumacher, B., Pothof, J., Vijg, J., & Hoeijmakers, J. H. J. (2021). The central role of DNA damage in the ageing process. Nature, 592(7856), 695–703. https://doi.org/10.1038/s41586-021-03307-7
Seo, J., & Park, M. (2020). Molecular crosstalk between cancer and neurodegenerative diseases. Cellular and Molecular Life Sciences, 77(14), 2659–2680. https://doi.org/10.1007/s00018-019-03428-3
Shackelford, D. A. (2006). DNA end joining activity is reduced in Alzheimer’s disease. Neurobiology of Aging, 27, 596–605. https://doi.org/10.1016/j.neurobiolaging.2005.03.009
Shadfar, S., Brocardo, M., & Atkin, J. D. (2022). he Complex mechanisms by which neurons die following DNA damage in neurodegenerative diseases. International Journal of Molecular Sciences, 23(5), 2484. https://doi.org/10.3390/ijms23052484
Shafqat, S., Arana Chicas, E., Shafqat, A., & Hashmi, S. K. (2022). The Achilles’ heel of cancer survivors: fundamentals of accelerated cellular senescence. The Journal of Clinical Investigation, 132(13), e158452. https://doi.org/10.1172/JCI158452
Shanbhag, N. M., Evans, M. D., Mao, W., Nana, A. L., Seeley, W. W., Adame, A., Rissman, R. A., Masliah, E., & Mucke, L. (2019). Early neuronal accumulation of DNA double strand breaks in Alzheimer’s disease. Acta Neuropathologica Communications, 7(1), 77. https://doi.org/10.1186/s40478-019-0723-5
Shibata, A., & Jeggo, P. A. (2021). ATM’s role in the repair of DNA double-strand breaks. Genes (basel), 12(9), 1370. https://doi.org/10.3390/genes12091370
Sioka, C., & Kyritsis, A. P. (2009). Central and peripheral nervous system toxicity of common chemotherapeutic agents. Cancer Chemotherapy and Pharmacology, 63(5), 761–767. https://doi.org/10.1007/s00280-008-0876-6
Sontag, E., Luangpirom, A., Hladik, C., Mudrak, I., Ogris, E., Speciale, S., & White, C. L., 3rd. (2004). Altered expression levels of the protein phosphatase 2A ABalphaC enzyme are associated with Alzheimer disease pathology. Journal of Neuropathology and Experimental Neurology, 63(4), 287–301. https://doi.org/10.1093/jnen/63.4.287
Sørensen, S. A., Fenger, K., & Olsen, J. H. (1999). Significantly lower incidence of cancer among patients with Huntington disease: an apoptotic effect of an expanded polyglutamine tract? Cancer, 86(7), 1342–1346.
Stefanatos, R., & Sanz, A. (2018). The role of mitochondrial ROS in the aging brain. FEBS Letters, 592(5), 743–758. https://doi.org/10.1002/1873-3468.12902
Suberbielle, E., Djukic, B., Evans, M., Kim, D. H., Taneja, P., Wang, X., Finucane, M., Knox, J., Ho, K., Devidze, N., Masliah, E., & Mucke, L. (2015). DNA repair factor BRCA1 depletion occurs in Alzheimer brains and impairs cognitive function in mice. Nature Communications, 6, 8897. https://doi.org/10.1038/ncomms9897
Sullivan, R., Yau, W. Y., O’Connor, E., & Houlden, H. (2019). Spinocerebellar ataxia: An update. Journal of Neurology, 266(2), 533–544. https://doi.org/10.1007/s00415-018-9076-4
Taillibert, S., Le Rhun, E., & Chamberlain, M. C. (2016). Chemotherapy-related neurotoxicity. Current Neurology and Neuroscience Reports., 16(9), 81. https://doi.org/10.1007/s11910-016-0686-x
Thadathil, N., Delotterie, D. F., Xiao, J., Hori, R., McDonald, M. P., & Khan, M. M. (2021). DNA double-strand break accumulation in Alzheimer’s disease: evidence from experimental models and postmortem human brains. Molecular Neurobiology, 58(1), 118–131. https://doi.org/10.1007/s12035-020-02109-8
Torre, M., Dey, A., Woods, J. K., & Feany, M. B. (2021). Elevated oxidative stress and DNA damage in cortical neurons of chemotherapy patients. Journal of Neuropathology and Experimental Neurology, 80(7), 705–712. https://doi.org/10.1093/jnen/nlab074
Urulangodi, M., & Mohanty, A. (2020). DNA damage response and repair pathway modulation by non-histone protein methylation: Implications in neurodegeneration. J Cell Communication and Signaling, 14(1), 31–45. https://doi.org/10.1007/s12079-019-00538-2
van den Boogaard, W. M. C., Komninos, D. S. J., & Vermeij, W. P. (2022). Chemotherapy side-effects: not all DNA damage is equal. Cancers (basel), 14(3), 627. https://doi.org/10.3390/cancers14030627
van der Plas, E., Nieman, B. J., Butcher, D. T., Hitzler, J. K., Weksberg, R., Ito, S., & Schachar, R. (2015). Neurocognitive late effects of chemotherapy in survivors of acute lymphoblastic leukemia: Focus on methotrexate. Journal of Canadian Academy of Child and Adolescent Psychiatry, 24(1), 25–32.
Veeriah, S., Taylor, B. S., Meng, S., Fang, F., Yilmaz, E., Vivanco, I., Janakiraman, M., Schultz, N., Hanrahan, A. J., Pao, W., Ladanyi, M., Sander, C., Heguy, A., Holland, E. C., Paty, P. B., Mischel, P. S., Liau, L., Cloughesy, T. F., Mellinghoff, I. K., et al. (2010). Somatic mutations of the Parkinson’s disease-associated gene PARK2 in glioblastoma and other human malignancies. Nature Genetics, 42(1), 77–82. https://doi.org/10.1038/ng.491
Wang, D., Wu, W., Callen, E., Pavani, R., Zolnerowich, N., Kodali, S., Zong, D., Wong, N., Noriega, S., Nathan, W. J., Matos-Rodrigues, G., Chari, R., Kruhlak, M. J., Livak, F., Ward, M., Caldecott, K., Di Stefano, B., & Nussenzweig, A. (2022). Active DNA demethylation promotes cell fate specification and the DNA damage response. Science, 378(6623), 983–989. https://doi.org/10.1126/science.add9838
Weissman, L., Jo, D. G., Sørensen, M. M., de Souza-Pinto, N. C., Markesbery, W. R., Mattson, M. P., & Bohr, V. A. (2007). Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucleic Acids Research, 35(16), 5545–5555. https://doi.org/10.1093/nar/gkm605
Welch, G., & Tsai, L. H. (2022). Mechanisms of DNA damage-mediated neurotoxicity in neurodegenerative disease. EMBO Reports, 23, e54217. https://doi.org/10.15252/embr.202154217
Wieland, L. S., Moffet, I., Shade, S., Emadi, A., Knott, C., Gorman, E. F., & D’Adamo, C. (2021). Risks and benefits of antioxidant dietary supplement use during cancer treatment: Protocol for a scoping review. British Medical Journal Open, 11(4), e047200. https://doi.org/10.1136/bmjopen-2020-047200
Williams, K. L., Topp, S., Yang, S., Smith, B., Fifita, J. A., Warraich, S. T., Zhang, K. Y., Farrawell, N., Vance, C., Hu, X., Chesi, A., Leblond, C. S., Lee, A., Rayner, S. L., Sundaramoorthy, V., Dobson-Stone, C., Molloy, M. P., van Blitterswijk, M., Dickson, D. W., et al. (2016). CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nature Communications, 7, 11253. https://doi.org/10.1038/ncomms11253
Wu, W., Hill, S. E., Nathan, W. J., Paiano, J., Callen, E., Wang, D., Shinoda, K., van Wietmarschen, N., Colón-Mercado, J. M., Zong, D., De Pace, R., Shih, H. Y., Coon, S., Parsadanian, M., Pavani, R., Hanzlikova, H., Park, S., Jung, S. K., McHugh, P. J., et al. (2021). Neuronal enhancers are hotspots for DNA single-strand break repair. Nature, 593(7859), 440–444. https://doi.org/10.1038/s41586-021-03468-5
Yamamoto, A., Nakamura, Y., Kobayashi, N., Iwamoto, T., Yoshioka, A., Kuniyasu, H., Kishimoto, T., & Mori, T. (2007). Neurons and astrocytes exhibit lower activities of global genome nucleotide excision repair than do fibroblasts. DNA Repair (amst)., 6(5), 649–657. https://doi.org/10.1016/j.dnarep.2006.12.006
Yang, J. L., Tadokoro, T., Keijzers, G., Mattson, M. P., & Bohr, V. A. (2010). Neurons efficiently repair glutamate-induced oxidative DNA damage by a process involving CREB-mediated up-regulation of apurinic endonuclease 1. The Journal of Biological Chemistry, 285(36), 28191–28199. https://doi.org/10.1074/jbc.M109.082883
Zhang, W., Xiao, D., Mao, Q., & Xia, H. (2023a). Role of neuroinflammation in neurodegeneration development. Signal Transduction and Targeted Therapy, 8(1), 267. https://doi.org/10.1038/s41392-023-01486-5
Zhang, X., Liu, Y., Huang, M., Gunewardena, S., Haeri, M., Swerdlow, R. H., & Wang, N. (2023b). Landscape of double-stranded DNA breaks in postmortem brains from Alzheimer’s disease and non-demented individuals. Journal of Alzheimer’s Disease. https://doi.org/10.3233/JAD-230316
Zukas, A. M., & Schiff, D. (2018). Neurological complications of new chemotherapy agents. Neuro-Oncology, 20(1), 24–36. https://doi.org/10.1093/neuonc/nox115
Acknowledgements
The authors thank Dr. Srinivas Gopala, Dr. Cibin T Raghavan, and all other members of the Department of Biochemistry, SCTIMST, for constant discussion and inputs during the preparation of this review article.
Funding
No extramural funding was received for this study.
Author information
Authors and Affiliations
Department of Biochemistry, Sree Chitra Tirunal Institute for Medical Sciences and Technology (SCTIMST), Medical College POST, Thiruvananthapuram, Kerala, 695011, India
Aishwarya Babu & Madhusoodanan Urulangodi
Corresponding author
Correspondence to Madhusoodanan Urulangodi.
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article
Cite this article
Babu, A., Urulangodi, M. Chemotherapy-induced neuronal DNA damage: an intriguing toolbox to elucidate DNA repair mechanisms in the brain. GENOME INSTAB. DIS. (2023). https://doi.org/10.1007/s42764-023-00110-8
Received27 July 2023
Revised27 September 2023
Accepted08 October 2023
Published06 November 2023
DOIhttps://doi.org/10.1007/s42764-023-00110-8
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable link
用户登录
还没有账号?
立即注册