






The role of H3K27me3 methylation in cancer development
Review Article
Published: 04 January 2024
Longjiang Di & Wei-Guo Zhu,
Genome Instability & Disease Volume 5, pages 17–34, (2024)
Abstract
Although the importance of histone methylation in epigenetics was first suggested more than 50 years ago, research into histone modifications conducted in the past decade has led to an exponential increase in our understanding of histone H3 modifications. In particular, the involvement of H3 histone 27 lysine trimethylation in the development of various cancer phenotypes has been demonstrated. Unlike mutations in the DNA sequence, such epigenetic changes are reversible, suggesting that inhibitors of H3 histone 27 amino acid methylation enzymes could be used as anti-cancer agents. Here, we outline the regulatory functions of H3 histone 27 lysine trimethylation carried out by different enzymes, in carcinogenesis. We describe the role of H3 histone 27 lysine trimethylation as an important epigenetic regulatory mechanism in the development of various cancers via effects on inflammation, DNA damage repair, cell proliferation, cell metastasis, regulatory cell death, ferroptosis, and angiogenesis. Finally, we focus specifically on H3 histone 27 lysine trimethylation regulators and their future development as anti-cancer drugs.
Introduction
Cancer is a complex genomic disease with a high mortality rate (Nowell, 1976). Mutations in certain genes, including proto-oncogenes and tumor suppressor genes, play a key role in driving the development and progression of cancer (Vogelstein et al., 2013). Moreover, the increase in genetic research in recent years has highlighted the important contribution of epigenetic alterations to the mechanisms of cancer development (Chabanon et al., 2020). Changes in chromatin structure can lead to alterations in gene expression via proto-oncogene activation or tumor-suppressor silencing (Dawson & Kouzarides, 2012), for example, increased levels of H3K14me3 in cervical cancer cells lead to the activation of ATR, resulting in their resistance to replicative stress and affecting the prognosis of tumor patients (Zhu et al., 2021). SETD2 deficiency downregulates H3K36me3 enrichment and cholesterol efflux gene expression, leading to hepatocarcinogenesis (Li et al., 2021). Emerging evidence has highlighted a potential role for epigenetics in future therapeutic strategies for tumors. It has been found that more than 80% of patients with diffuse intrinsic pontine glioma (DIPG), a fatal malignant childhood tumor, have a heterozygous point mutation (H3K27M) at the histone H3K27 locus, which leads to a reduction in H3K27me3 levels. As surgical resection and conventional radiotherapy approaches are ineffective in treating DIPG, elevating intracellular H3K27me3 levels to inhibit tumor growth could represent a novel strategy for treating DIPG. Recent studies have identified a previously unknown mechanism via which α-ketoglutarate maintains the epigenetic status of low H3K27me3 levels. Based on this epigenetic mechanism, targeting α-ketoglutarate generation provide a new strategy for the treatment of DIPG (Chung et al., 2020).
Eukaryotic chromatin is made up of DNA and its major protein components, histones. This group of highly conserved proteins includes the core histones H2A, H2B, H3, and H4, as well as the splice histone H1. One pair of each core histone assembles with 146 base pairs of DNA to form nucleosomes, which comprise the basic units of chromatin (Kornberg, 1974; Margueron & Reinberg, 2010). When cells are stimulated by factors such as radiation, they initiate a rapid DNA damage response triggering various processes, including chromatin remodeling, cell cycle arrest, autophagy, apoptosis, and senescence (Harper & Elledge, 2007; Jackson & Bartek, 2009; Polo & Jackson, 2011; Soria et al., 2012). Dysregulation of these processes, resulting in cellular dysfunction and altered gene expression, is central to cancer development (Cacabelos et al., 2016; Sharma et al., 2010). By affecting chromatin structure, histone modification plays a key regulatory role in these crucial cellular processes. Important histone modifications include acetylation, ubiquitination, threonylation, glycosylation, phosphorylation, and methylation (Kouzarides, 2007). Methylation and acetylation, the most common types of modifications, were first observed to be involved in the regulation of RNA synthesis more than 50 years ago (Allfrey et al., 1964).
Over the years, our understanding of histone modifications has deepened. Methylation of histones mainly occurs at the positively charged amino acids lysine (K) and arginine (R), which are abundant within histone proteins. The most widely studied histone methylation sites include the histone H3 lysines 4, 9, 27, and 36 (H3K4, H3K9, H3K27, and H3K36). Methylation modifications of H3K4 and H3K36 mediate transcriptional activation, while methylation modifications of H3K9 and H3K27 mediate transcriptional repression. Different histone lysine residues have different—sometimes opposing—functions when methylated, and different degrees of methylation (mono-, di-, or tri-methylation) on the same residue may cause dramatic functional differences (Barski et al., 2007; Heintzman et al., 2009). In addition, histones can be methylated on arginine residues, a modification that can play a positive or negative role in transcription (Bedford & Clarke, 2009). To date, more than 20 methylation marks on lysine and arginine residues have been identified (Mosammaparast & Shi, 2010). Methylation of lysine residues in histone tails, in particular, has been a focus of epigenetics research in recent years (Fig. 1).
Fig. 1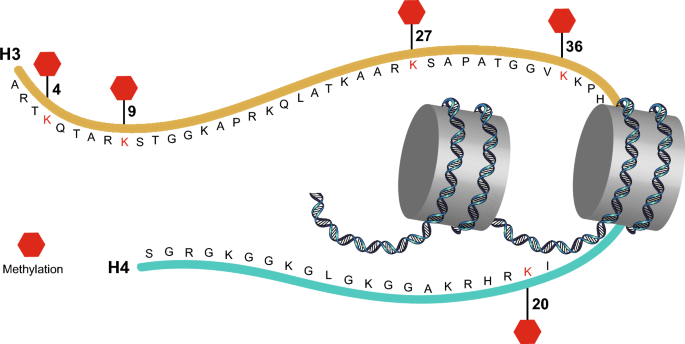
Methylation sites of the major histone lysine residues
In general, methylation is considered to be more complex than other forms of histone modification. Methyl groups are turned over more slowly than other modifications—indeed, histone methylation was initially believed to be irreversible (Byvoet et al., 1972); the later discovery of demethylases such as lysine-specific demethylase 1A (KDM1A) revealed that histone methylation is, in fact, reversible (Shi et al., 2004). A large number of specific histone methylation-modifying enzymes are now known to mediate the addition or removal of methyl groups from various lysine residues on histones, catalyzing post-translational events. This specificity reflects the fact that enzymes that catalyze histone methylation tend to produce well-defined modifying functions (Xiao et al., 2003). The discovery of enzymes capable of producing a diversity of stable and altered methylation states has helped to establish the increasingly important regulatory role of H3 histone methylation in oncogenesis. In particular, aberrant epigenetic regulation of histone H3K27 modification seems to be involved in the activation of multiple oncogenic and tumor suppressor signaling pathways and the progression of many types of cancer (Ezponda & Licht, 2014; Li et al., 2017). Below, we delineate the mechanisms that can lead to the altered methylation of histone H3K27.
Generation and elimination of the H3K27 methyl marker
During the past decade, the enzymes associated with histone H3K27 methylation have been studied to uncover the mechanisms contributing to genetic epistasis regulation. The enzymes responsible for catalyzing the methylation/demethylation of histone H3K27 (histone methyltransferases and demethylases) are considered to be potential therapeutic targets for the clinical treatment of cancer (Shi & Whetstine, 2007; Yoshimi & Kurokawa, 2011). The main H3K27 methyltransferases include the PRC2 complex (which consists of the SET structural domain-containing histone methyltransferases EZH2 or EZH1 and core accessory proteins), as well as the proteins ATXR5, ATXR6, TXR1, EZL2, EHMT2, and EHMT1. The major H3K27 demethylases are UTX/KMD6A, JMJD3/KDM6B, and KIAA1718/KDM7A.
The enzymes mediating H3K27 methylation differ between species. In Arabidopsis, the methyltransferases ATXR5, ATXR6, MEDEA, CLF, and SWINGER play important roles in H3K27 methylation (Makarevich et al., 2006). Inactivation of ATXR5 and ATXR6 leads to a significant reduction in H3K27 methylation (Jacob et al., 2009). MEDEA, CLF, and SWINGER reduce gene expression by increasing H3K27 methylation (Gu et al., 2014; Schubert et al., 2006). In the unicellular eukaryote Tetrahymena thermophila, H3K27 methylation was significantly reduced by deletion of TXR1 (Zhang et al., 2013); however, H3K27 methylation status was not completely lost when TXR1 was inhibited, demonstrating the existence of other methyltransferases or pathways regulating H3K27 methylation. Gao et al. and others also identified EZL1, EZL2, and EZL3 (homologs of the Drosophila EZ proteins) as methyltransferases in Tetrahymena (Gao et al., 2013; Liu et al., 2007; Zhang et al., 2014). EZL2 was expressed at significant levels, indicating its potential importance as a regulator of H3K27 methylation (Gao et al., 2013).
In mammalian cells, EZH1 was the first enzyme reported to methylate H3K27, and EZH1 and EZH2 were subsequently shown to form part of distinct PRC2 complexes. The PRC2 complex consists of four core protein subunits, including EZH2 (or EZH1), zeste 12 inhibitor (SUZ12), embryonic ectodermal development (EED), and retinoblastoma protein-associated protein 46/48 (RbAp46/48). Shen et al. observed significant reductions in H3K27me2 and H3K27me3, as well as partial reductions in H3K27me1, in Ezh2−/− embryonic stem cells (ESCs) (Shen et al., 2008), while knockdown of Ezh1 in Ezh2−/− ESCs completely suppressed H3K27me1. Genome-wide analysis showed that cancer-associated overexpression of EZH2 resulted in increased H3K27me3 levels and repression of tumor suppressor genes (Deb et al., 2014; Kim & Yu, 2012). In addition, Pasini et al. demonstrated the specific loss of H3K27me2 and H3K27me3 in Suz12-deficient embryos (Pasini et al., 2004). Similarly, Montgomery et al. reported that EED knockdown reduced H3K27me2 and H3K27me3 levels (Montgomery et al., 2005). G9a/EHMT2 and GLP/EHMT1 have also been identified as H3K27 methyltransferases in vitro and in vivo (Mozzetta et al., 2014, 2015; Tachibana et al., 2005; Wu et al., 2011). Removal of the H3K27 methyl moiety is performed by the histone demethylases UTX/KMD6A and JMJD3/KDM6B (Lan et al., 2007; Lee et al., 2007; Tsukada et al., 2006), as demonstrated in ESC differentiation experiments conducted by Agger et al. (2007). De Santa et al. also identified KDM6B as an inducible enzyme that catalyzed H3K27me3 removal and altered macrophage transdifferentiation (De Santa et al., 2007). Notably, although KDM6A and KDM6B contributed significantly to H3K27me2 and H3K27me3 demethylation, these enzymes did not show significant H3K27me1 demethylation activity. Another enzyme, KDM7A, was later identified as a histone demethylase capable of acting on both H3K27me1 and H3K27me2 (Huang et al., 2010; Lin et al., 2010; Yang et al., 2010). The overall production and elimination of H3K27 methyl marks reflect the balance between methyltransferases and demethylases. Moreover, the responses of these enzymes to various signaling cascades affect the activation/repression of oncogenes at different stages of a tumor’s development.
Different H3K27 methylation-related enzymes differ in the degree of methylation they can achieve. Monomethylation of H3K27 (addition of me1) is mainly carried out by ATXR5, ATXR6, TXR1, EZH1, EZH2, EHMT1, and EHMT2, while the methyltransferases capable of converting H3K27me1 to H3K27me2 are MEA, CLF, SWN, EZL2, EZH2, Suz12, and EED (Fig. 2). Demethylation of H3K27me1 and H3K27me2 is mainly performed by KDM6A, KDM6B, and KDM7A. In addition, MEA, CLF, SWN, EZL2, EZH2, Suz12, and EED are the major methyltransferases that convert H3K27me2 to H3K27me3, while demethylation of H3K27me3 is mostly carried out by KDM6A and KDM6B (Fig. 2).
Fig. 2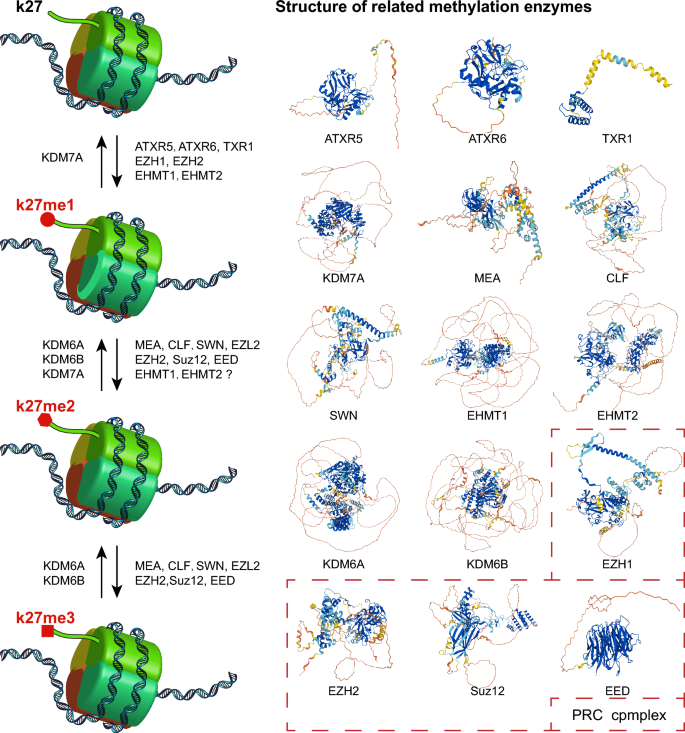
Enzymes involved in the methylation modification of histone H3K27
Although there have been many studies investigating H3K27me3 and the repression of gene expression, the functions of H3K27me1 and H3K27me2 in gene regulation remain poorly understood. Ferrari et al. demonstrated that H3K27me1 accumulates within transcribed genes, promotes transcription, and is regulated by Set2-dependent deposition of H3K36me3 (Ferrari et al., 2014). H3K27me2 is present on approximately 70% of total histone H3 and is distributed within large chromatin domains that exert a protective function by preventing the initiation of non-cell-type-specific enhancers. Considering the key role played by H3K27me3 in the regulation of gene expression and signaling pathways, the rest of this review will focus on new findings regarding H3K27me3 in cancer-related signatures, including cellular inflammation, DNA damage and repair, cell metastasis, proliferation, invasion, and apoptosis/ferroptosis (Table 1).
Signature | Description | No. of genes |
|---|---|---|
Angiogenesis | Angiogenesis ensures that cancer cells receive continuous supplies of oxygen and other nutrients | 73 |
Apoptosis | Cancer cell resistance to apoptosis leads to the persistence of grossly abnormal cells in tissues | 66 |
DNA damage | Unrepaired DNA damage accumulates in replicating cells, potentially contributing to cancer progression | 110 |
DNA repair | Deficits in DNA repair disrupt genomic integrity and may lead to carcinogenesis | 119 |
Inflammation | Chronic inflammation can cause between 15 and 25% of human cancers | 112 |
Invasion | Invasion is a critical carcinogenic event whereby cancer cells escape from their primary sites and spread into the blood or lymphatic vessels | 97 |
Metastasis | Metastasis promotes the malignant transformation of cancer and causes most cancer deaths | 166 |
Proliferation | Cell proliferation is responsible for tumor progression | 88 |
Table 1 Number of genes involved in key cancer signatures
H3K27me3 and inflammation
Inflammation is a self-induced response to tissue damage and often occurs in conjunction with the clinical manifestations of disease. Inflammation can be caused by a variety of types of trauma, including physical injury, ischemic injury, infection, and exposure to toxins. Acute inflammatory responses cause a series of physiological changes, giving rise to cellular changes at the damage site and immune stress in the body. If inflammation persists for physical reasons, acute inflammation becomes chronic, a process that leads to gradual cellular mutation; continued mutation is often a prerequisite for oncogenesis. Inflammation is caused by the activation of macrophages and the resulting production of inflammatory cytokines, including interleukin-1β (IL-1β), tumor necrosis factor-alpha (TNFα), and IL-10 (Arango Duque & Descoteaux, 2014). In human primary macrophages, lipopolysaccharide (LPS) was shown to stimulate KDM6B expression, promoting macrophage polarization toward a pro-inflammatory M1 phenotype via the NF-κB signaling pathway. By contrast, GSK-J4, an inhibitor of KDM6B, was shown to inhibit LPS-induced TNFα release in macrophages and attenuate the inflammatory response by promoting H3K27me3 accumulation (Kruidenier et al., 2012). In addition, during bacterial infection, deficiency in the m6A reader YTH-domain family 2 stabilizes KDM6B protein, promoting H3K27me3 demethylation and subsequently enhancing the transcription of the pro-inflammatory cytokines IL-6, IL-12, and ICAM (Wu et al., 2020).
The regulation of H3K27me3 status by different enzymes is closely associated with the activation of key signaling pathways that contribute to inflammation in various diseases. For example, a process of modulating KDM6B to regulate H3K27me3 methylation status and thus promote the persistence of inflammation has been demonstrated in necrotizing small bowel colitis (Ma et al., 2022), as well as during the normal physiological process of muscle repair in humans (Nakka et al., 2022). In addition, EZH-catalyzed H3K27me3 is involved in inflammatory processes associated with many diseases. For example, in bronchopulmonary dysplasia in preterm infants, EZH2-catalyzed H3K27me3 resulted in RUNX3 downregulation and caused intrauterine inflammation and infection (Zhu et al., 2015). A similar process has been identified in periodontal membrane cells in periodontal disease (Schuldt et al., 2022).
An increasing number of studies have shown that inflammation-associated DNA methylation is common in tumors, with 60% of aberrant tumor-associated methylation also present in inflamed tissues (Hahn et al., 2008). This suggests that methylation may have important regulatory effects on cancer development. To investigate this association more closely, we obtained a total of 112 genes associated with cancer inflammation from the CancerSEA bioinformatics database (http://biocc.hrbmu.edu.cn/CancerSEA) (Supplementary Table 1) and performed protein–protein interaction (PPI) network analysis for the human H3K27me3-related enzymes KDM6A, KDM6B, EZH2, SUZ12, and EED (Fig. 3). Our results showed that these proteins are closely associated with inflammogenesis genes, suggesting that regulation of H3K27me3 by these enzymes may be involved in the development of cancer-related inflammation.
Fig. 3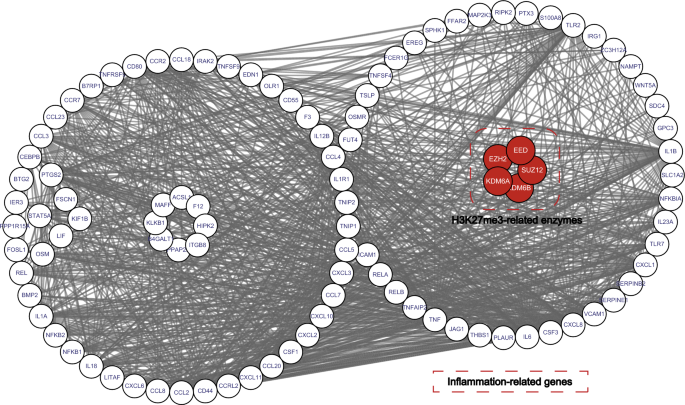
Protein–protein interaction network of related enzymes regulating H3K27me3 and cancer inflammation-related genes
H3K27me3 and DNA damage repair
DNA damage, which involves alteration of the chemical structure of DNA, frequently accumulates in cells during cancer development. DNA repair contributes to the maintenance of genomic integrity, while defective DNA repair often leads to cancer progression. In clinical practice, conventional chemotherapeutic agents usually act by targeting tumor cells to induce DNA damage and cell death (Cheung-Ong et al., 2013; Espinosa et al., 2003; Nitiss, 2009); however, H3K27me3-labeled regions may not be accessible to such agents because of their compact chromatin structure (Pang et al., 2015). Decompaction of chromatin may therefore enhance the therapeutic effects of cytotoxic drugs. Consistent with this notion, inhibition of EZH2 in elderly patients with acute myeloid leukemia (AML) resulted in the decompaction of H3K27me3-labeled chromatin, thereby enhancing chromatin accessibility and promoting chemotherapy-induced DNA damage/leukemia suppression (Porazzi et al., 2022). This finding indicates that lowering the dose of chemotherapeutic agents in AML may be possible, which could reduce treatment-related side effects (Porazzi et al., 2022). Other studies have shown that the H3K27M mutation in a subset of pediatric high-grade gliomas reduced overall histone H3K27 trimethylation by affecting the enzymatic activity of EZH2, leading to enhanced DNA damage repair in tumor cells, thereby promoting tumorigenicity of cancer cells in vivo and resistance to tumor radiotherapy (Bender et al., 2013; Lewis et al., 2013). In glioblastoma, mutations in the telomerase reverse transcriptase promoter were shown to stimulate EZH2 expression and pharmacologically inhibit lipid accumulation and carcinogenesis by regulating H3K27 trimethylation (Ahmad et al., 2017). In addition, in various cancers such as hepatocellular carcinoma (HCC), triple-negative breast cancer, small cell lung cancer, and bone marrow tumors, H3K27me3 is influenced by protein modifications/gene mutations, with concomitant effects on DNA damage/repair-related pathways and tumorigenesis (Gao et al., 2017; Gardner et al., 2017; Imgruet et al., 2021; Yamamoto et al., 2019). For example, in small cell lung cancers that rapidly develop chemoresistance to cisplatin and etoposide, significant inhibition of SLFN11 (a factor associated with defective DNA damage repair) expression was shown, while silencing of SLFN11 expression in vivo was associated with significant deposition of H3K27me3; thus, combining an H3K27me3 inhibitor with standard cytotoxic therapies could improve small cell lung cancer chemotherapeutic efficacy (Gardner et al., 2017).
PPI network validation for genes related to cancer DNA damage and repair (obtained from the CancerSEA bioinformatics database, as before; Supplementary Table 1) revealed close associations with the human H3K27me3-related enzymes KDM6A, KDM6B, EZH2, SUZ12, and EED (Fig. 4). These findings suggest that regulation of H3K27me3 by these enzymes is involved in DNA damage and repair in cancer cells. These results are highly relevant to the study of the pharmacological modulation of H3K27 trimethylation in clinical treatment for cancer. Indeed, many anti-cancer drugs are known to induce DNA breakage by modulating H3K27me3 levels. For example, Pang et al. discovered that the anthracycline aclarubicin expelled histones from H3K27me3-labeled heterochromatin, exerting an important regulatory effect on diffuse large B-cell lymphoma cells with elevated H3K27me3 levels (Pang et al., 2015). Tang et al. showed that clinical treatment with metformin significantly reduced histone H3K27me3 levels and thus played an anti-tumor role in ovarian cancer (Tang et al., 2018). Similarly-acting drugs include tanshinone I, which is effective against AML (Huang et al., 2021), and sorafenib, which is effective against HCC (Kusakabe et al., 2021). Thus, interestingly, although anti-cancer drugs can produce differing genomic effects in different classes of tumors, regulating the DNA damage and repair effects caused by H3K27me3 seems to represent a common pathway for many agents.
Fig. 4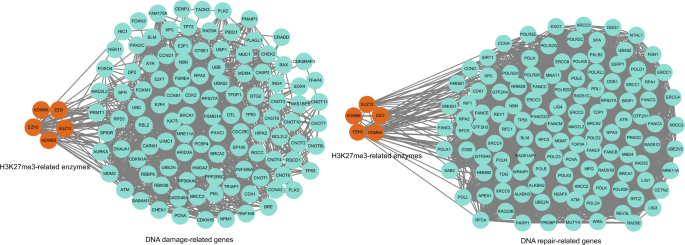
Protein–protein interaction network of related enzymes regulating H3K27me3 and cancer DNA damage or repair-related genes
H3K27me3 and cell proliferation, metastasis, and invasion
Aberrant proliferation is a hallmark of carcinogenesis. As an early step in tumor development, the strict regulatory mechanisms that control survival and proliferation in normal cells are lost in tumor cells, allowing cancer cells to survive beyond their normal lifespan and proliferate aberrantly. These changes involve the altered expression of cell cycle-related proteins and changes in many signal transduction pathways resulting from the dysregulation of epigenetic mechanisms (Feitelson et al., 2015). Following tumor development, cancer cells not only remain in a state of abnormal proliferation but also undergo metastasis and invasion. In most cancers, Metastasis involves the malignant transformation of cancer cells and their spread to other sites, increasing the risk of mortality. Invasive cancer cells spread from their primary site into the blood or lymphatic vessels and participate in tumorigenesis steps—including epithelial-to-mesenchymal transition, angiogenesis, and the triggering of autophagy—by producing excessive levels of hormones, inflammatory factors, and other cytokines (Jedinak et al., 2010; Qian & Pollard, 2010). Dysregulation of epigenetic mechanisms may act as an important driver of certain tumorigenesis mechanisms (Timp & Feinberg, 2013). For example, epigenetic changes driven by alterations in H3K27me3 can result in altered signaling pathways that allow cells to resist normal apoptotic mechanisms and promote their proliferation (Hanahan & Weinberg, 2011). Thus, H3K27me3 represents a meaningful target for the development of new therapeutic agents that act prior to or at the earliest stages of malignant transformation.
In recent years, research into the involvement of H3K27me3 in the proliferation, metastasis, and invasion of tumor cells has progressed rapidly. For example, in 2017, Wu et al. demonstrated that the long non-coding RNA (lncRNA) CASC9 negatively regulated PDCD4 expression in esophageal squamous cell carcinoma by recruiting EZH2 and subsequently altering the levels of H3K27me3, thereby promoting cancer cell proliferation (Wu et al., 2017). In 2019, Ma et al. found that in human glioma specimens, β-catenin/USP1/EZH2 promoted cell proliferation in vitro and tumor formation in vivo; moreover, USP1-mediated stabilization of EZH2 and promoted its recruitment to the CDKN1B, RUNX3, and HOXA5 promoters, leading to H3K27me3 enrichment and repression of target gene expression (Ma et al., 2019). In the same year, Yang et al. found that HOXD-AS1 inhibited the proliferation and migration of colorectal cancer (CRC) cells in vitro, as well as CRC tumorigenesis and metastasis in vivo, by suppressing HOXD3-mediated activation of the MAPK/AKT signaling pathway; specifically, HOXD-AS1 inhibited HOXD3 transcription by inducing the accumulation of H3K27me3 at the HOXD3 promoter (Yang et al., 2019). In 2020, Ricci et al. revealed that the proto-oncogene Bmi1 mediated tumor proliferation, metastasis, and invasion in a mouse model of glioblastoma-initiating cells via the EfnA5 gene, regulated by H3K27me3 (Ricci et al., 2020). In 2021, Wang et al. discovered that the lncRNA DNM3OS was significantly upregulated in HCC compared with its expression in adjacent normal liver tissue; tumor-associated mesenchymal stem cells accelerated HCC growth and metastasis via the DNM3OS/KDM6B/TIAM1 axis, whereby the binding of DNM3OS to KDM6B induced its expression by reducing H3K27me3 at the TIAM1 promoter (Wang et al., 2021). Yang et al. demonstrated that a combined intervention using inhibitors of PRMT5 (GSK591) and EZH2 (GSK126) synergistically inhibited the growth of CRC cells/xenografts in vitro and in vivo by reducing EZH2 binding and H3K27me3 deposition (Yang et al., 2021). In addition, in 2022, Ying et al. found that the silencing of zinc finger protein 280C in CRC cells inhibited cell proliferation, clone formation, cell migration, xenograft growth, and liver metastasis, for which regulation by H3K27me3 was essential (Ying et al., 2022). Finally, Peralta-Arrieta et al. showed that MEOX2/GLI1 increased tumor cell proliferation and resistance to anti-cancer chemotherapeutic agents in human lung cancer; this effect was mediated via aberrant epigenetic regulation of the EGFR/AKT/ERK signaling pathway, including reduced binding of histone H3K27me3 to EGFR regulatory regions (Peralta-Arrieta et al., 2022). These studies, combined with bioinformatics analyses (Fig. 5), have identified H3K27me3 regulation as a key factor in tumor cell proliferation, metastasis, and invasion, which may have implications for cancer treatment. Despite the improved efficacy of anti-cancer treatment in recent years, the prognosis for many patients with cancer remains poor. Cancer therapy often relies on cytotoxic drugs that kill highly proliferative tumor cells but also affect rapidly proliferating normal cells in the gastrointestinal tract, hair, and epithelium. Moreover, most targets of oncology drugs are multidirectional and can regulate different signaling pathways, thus these drugs can have potentially leading to irreversible side effects. By contrast, regulation of H3K27me3 may help to delay cancer onset and/or reverse cancer cell proliferation and reduce the toxicity associated with current anti-cancer drugs.
Fig. 5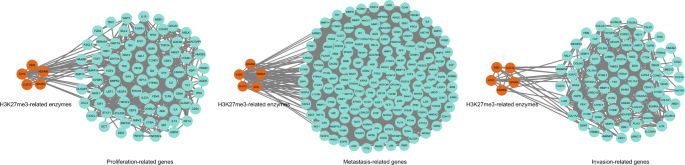
Protein–protein interaction network of related enzymes regulating H3K27me3 and cancer cell proliferation-, metastasis-, and invasion-related genes
H3K27me3 and apoptosis/ferroptosis
Dysregulation of apoptosis and ferroptosis persists in cancerous tissues. Apoptosis is a natural, programmed form of cell death induced by a variety of cellular signals. The process of apoptosis includes cell breakage and shrinkage, chromatin condensation, and apoptosome production (Saraste & Pulkki, 2000), as well as nuclear fragmentation. The apoptotic process depends on extrinsic and intrinsic pathways. The intrinsic pathway is triggered when cells are exposed to certain harmful stimuli that cause DNA damage, such as radiation, toxins, or hypoxia, resulting in the formation of pores in the mitochondrial membrane via Bcl-2-associated X protein (BAX) and Bcl-2 antagonist killer 1 (BAK). The resulting massive release of cytochrome c activates effector caspases that break up the nucleus and degrade cytoskeletal elements, leading to the eventual formation of apoptotic bodies (Kalkavan et al., 2018; Kharbanda et al., 1997; Zhang et al., 2017). By contrast, the exogenous pathway depends on ligand-receptor interactions, such as TNFα binding to the TNFα receptor and Fas ligand (FasL) binding to the Fas receptor, which induce apoptosis by activating caspases (Lavrik et al., 2005; Strasser et al., 2009). Pro- and anti-apoptotic proteins, including BAX, BAK, Bcl-2, Bcl-xL, p53, and TNF-related apoptosis-inducing ligands (TRAIL), have been shown to regulate the apoptotic process by influencing mitochondrial membrane impermeability, cytochrome c release, and intracellular Fas signaling (el-Deiry, 1998; Kale et al., 2018; Nakano & Vousden, 2001; Rathore et al., 2017; Yuan et al., 2018).
The H3K27me3 marker has been found to play a key role in apoptosis. A systematic examination of the relationship between 1923 apoptosis-related genes and the associated histone modifications during neuronal development, by Jiang et al., showed that downregulation of apoptotic genes could be attributed to reduced H3K4me3 signals at promoters, while upregulation of apoptotic genes resulted from the loss of H3K27me3 marks (Jiang et al., 2021). As expected, many other studies have shown the regulation of apoptosis by H3K27me3. For example, miR-143 activates the BAX-dependent caspase 3 signaling pathway to promote apoptosis, and H3K27me3 can inhibit miR-143 transcription by affecting the recruitment of transcription factors and binding proteins, thereby affecting the process of apoptosis (Zhong et al., 2020); subsequently, increased H3K27me3 labeling combined with diminished apoptosis caused unstable cell growth, leading to rapid proliferation and promoting the initiation of the malignant process. This finding demonstrates the key role played by H3K27me3 in the regulatory processes of tumor cells. Another study reported that nuclear receptor-binding SET structural domain protein 1 (NSD1) increased the viability of breast cancer cells and inhibited apoptosis via the NSD1/H3K27me3/Wnt10b/β-catenin signaling pathway, thereby causing resistance to drugs such as paclitaxel (Chen et al., 2022). Evasion of apoptosis is one of the characteristics of cancer cell formation. Indeed, many treatment-resistant cancers have shown an association between H3K27me3 and the ability of tumor cells to evade apoptosis, including glioblastoma, oral cancer, AML, cervical cancer, B-cell lymphoma, CRC, breast cancer, and ovarian cancer (Akpa et al., 2019; Benard et al., 2014; Chu et al., 2020; Dev et al., 2021; Huang & Ling, 2017; Li et al., 2012; Lin et al., 2019; Zhao et al., 2021). Bioinformatics analysis conducted using the CancerSEA database (http://biocc.hrbmu.edu.cn/CancerSEA), as before (Supplementary Table 1), also showed associations between KDM6A, KDM6B, EZH2, SUZ12, and EED and multiple cancer-related apoptosis genes (Fig. 6). These findings confirm that H3K27me3, which is regulated by these enzymes, modulates the resistance of cancer cells to apoptosis.
Fig. 6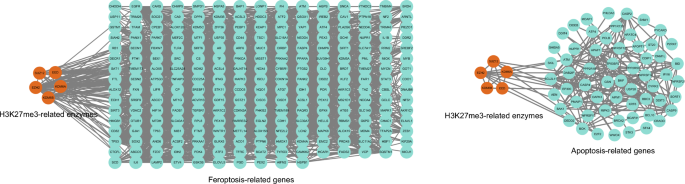
Protein–protein interaction network of related enzymes regulating H3K27me3 and cancer apoptosis-, ferroptosis-related genes
Ferroptosis is a novel, iron-regulated form of programmed cell death—distinct from apoptosis, cell necrosis, and autophagy—that was initially defined as iron-dependent cell death induced by erastin and RAS-selective lethal factor 3 (Dixon et al., 2012). In the presence of divalent iron or ester oxygenase, the levels of unsaturated fatty acids in cell membranes increase and lipid peroxides and reactive oxygen species (ROS) gradually accumulate, resulting in cell death. Morphological features of ferroptosis include mitochondrial shrinkage, mitochondrial membrane condensation, and reduction or elimination of mitochondrial cristae (Agmon et al., 2018); these features are caused by excessive ROS production and lipid peroxide accumulation. Ferroptosis is also induced by the inhibition of GPX4, a core antioxidant enzyme, because of the resulting accumulation of cytotoxic lipid peroxides.
Some studies have shown that H3K27me3 may play a role in diseases related to ferroptosis in various organs, including the liver, kidney, and heart. For example, hepatitis B virus (HBV) X protein promotes ferroptosis in acute liver failure via EZH2/H3K27me3-mediated inhibition of SLC7A11 (Liu et al., 2021), while palmitic acid stimulation of cardiomyocytes decreases H3K27me3 levels and promotes ferroptosis. Moreover, GSK-J4, a potent dual inhibitor of the H3K27me3/me2 demethylases JMJD3/KDM6B and UTX/KDM6A, was shown to reduce ferroptosis in cardiomyocytes by preserving H3K27me3 levels (Xu et al., 2022). H3K27me3 is also involved in the pathophysiological processes of tumors via the regulation of ferroptosis. For example, in melanoma, UNC 1999, an inhibitor of EZH1 and EZH2, downregulates the levels of H3K27me3 and inhibits cancer cell growth via the ferroptosis pathway (Hou et al., 2022). Ferroptosis plays different roles in different diseases, and H3K27me3 can influence disease progression by inhibiting or promoting ferroptosis, providing a promising target for the treatment of many diseases. As before, using ferroptosis-related regulators and markers from a ferroptosis database (http://www.zhounan.org/ferrdb) (Supplementary Table 2) to conduct PPI network analysis, we confirmed that the H3K27me3-associated enzymes KDM6A, KDM6B, EZH2, SUZ12, and EED are closely associated with ferroptosis-related genes in humans (Fig. 6). These findings indicate that H3K27me3 may play an important role in the regulation of ferroptosis in cancer cells.
H3K27me3 and angiogenesis
The survival and proliferation of cancer cells require the establishment of a blood supply to meet their demand for oxygen and nutrients and perform other metabolic functions (Potente et al., 2011). The formation of this vascular network occurs via angiogenesis. Vascular homeostasis is regulated by a large number of pro-angiogenic and anti-angiogenic factors. Tumors will remain dormant if these factors are in balance. Conversely, when homeostasis is imbalanced and pro-angiogenic signaling dominates, tumors are released from dormancy and new blood vessels are generated, leading to the rapid growth of the associated cancer cells. In 2003, anti-angiogenic therapies were successfully used to treat cancer in clinical trials and were subsequently approved by the US Food and Drug Administration (Hurwitz et al., 2004). This clinical process has accelerated research into the mechanisms of tumor angiogenesis and the associated signaling pathways. Studies have shown that in the immediate early stages of cancer, the basal lamina isolates the tumor from normal tissue containing blood vessels (Bluff et al., 2009; Bossi et al., 1995). As the tumor progresses from a benign to a malignant stage, the cancer cells become more aggressive, inducing a powerful angiogenic response that triggers the development of an actively growing and infiltrating vascular network (Bergers & Benjamin, 2003).
The extent of tumor vascularization can vary considerably as a result of regulation by tumor-inducing drivers, tumor-associated stromal cells, bioactive products, and other signaling molecules (Blouw et al., 2003; Bluff et al., 2009; Fukumura et al., 1997; Jubb et al., 2011; Monsky et al., 2002; Morrissey et al., 2008). Notably, the key drivers of angiogenesis, i.e., pro-angiogenic factors, can be triggered by epigenetic alterations in tumor cells. Among these, H3K27me3 has an important regulatory role. Recently, the combination of EZH and H3K27me3 was found to regulate TPM1 to promote cancer cell proliferation and angiogenesis in CRC (Liang et al., 2021). EZH2 and H3K27me3 were also found to be involved in the proliferation, angiogenesis, and metastasis of HCC cells via the downregulation of miR-138-5p (Bai et al., 2022). Several studies have shown that the involvement of H3K27me3 in angiogenesis also synergistically affects the proliferation, migration, and invasion of various tumor cell types, including prostate, breast, and ovarian cancer cells (Jain et al., 2020; Li & Zhang, 2013; Lyu et al., 2013; Ning & Ma, 2019; Xu et al., 2017). Analysis of cancer angiogenesis-related genes in the CancerSEA database (http://biocc.hrbmu.edu.cn/CancerSEA) (Supplementary Table 1) indicated their association with H3K27me3-related enzymes (Fig. 7), demonstrating the involvement of H3K27me3 in tumor angiogenesis.
Fig. 7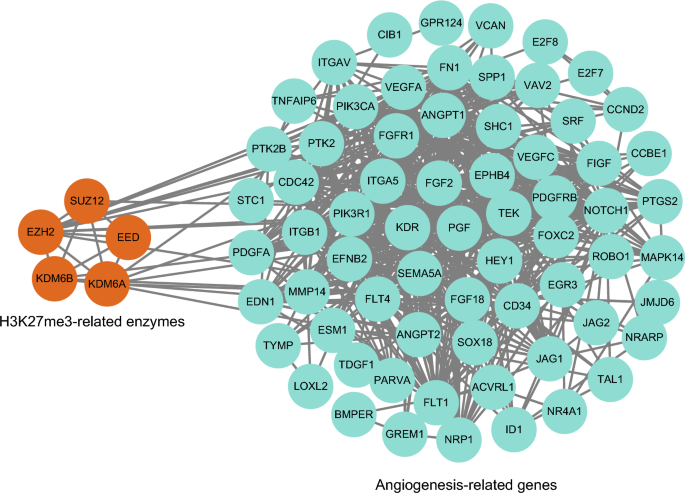
Protein–protein interaction network of related enzymes regulating H3K27me3 and cancer angiogenesis-related genes
H3K27me3 and autophagy
Autophagy is a physiological process involving the degradation of cytoplasmic macromolecules, aggregated proteins, damaged organelles, and pathogens by lysosomes (Cuervo, 2004; Klionsky, 2007; Levine, 2007; Levine & Klionsky, 2004; Levine & Kroemer, 2008; Mizushima et al., 2011; Rogov et al., 2014). There are three autophagic pathways: macroautophagy (also referred to simply as autophagy), microautophagy, and molecular chaperone-mediated autophagy (Glick et al., 2010; Xie & Klionsky, 2007). Macroautophagy occurs as a physiological stress response and consists of several steps, including induction, assembly, and formation of autophagosomes; docking and fusion of autophagosomes with lysosomal membranes; and degradation of autophagosomal contents (Hansen & Johansen, 2011; Mizushima, 2007) (Fig. 8). Damaged membrane-bound organelles such as mitochondria and other entities such as invading pathogens are processed in this way (Galluzzi et al., 2017; Levine & Kroemer, 2019). In microautophagy, cellular material is sequestered via invagination of late endosomal or lysosomal membranes (Mejlvang et al., 2018; Sahu et al., 2011; Uytterhoeven et al., 2015). By contrast, chaperone-mediated autophagy involves the recognition of KFERQ-containing proteins by the heat shock cognate 71 kDa protein HSC70/HSPA8 and binding to lysosomal-associated membrane protein 2A, followed by entry into the lysosome and degradation (Kaushik & Cuervo, 2018).
Fig. 8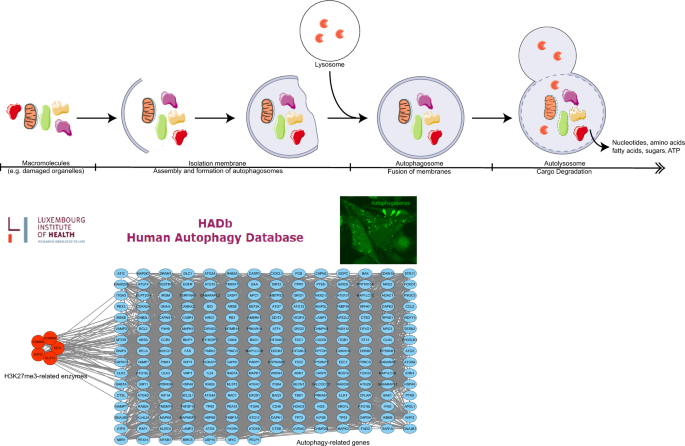
The process of autophagy and the protein–protein interaction network of related enzymes regulating H3K27me3 and cancer autophagy-related genes
Autophagy is essential for cellular homeostasis in vivo, acting to maintain the metabolism and development of cells under endogenous or exogenous stress. In addition, the elimination of damaged proteins and organelles by autophagy maintains protein quality and quantity (Mizushima & Komatsu, 2011; Rabinowitz & White, 2010). In recent years, the various regulatory roles of autophagy have been studied in depth. In healthy physiological states, autophagy has been demonstrated to be directly involved in the regulation of multiple processes, including developmental mechanisms (Allen & Baehrecke, 2020; Mizushima & Levine, 2010), maintenance of stem cell self-renewal activity (Chen et al., 2018; Dong et al., 2021), and cell differentiation and survival (Boya et al., 2018; Clarke & Simon, 2019). Autophagic mechanisms are also involved in intercellular communication, mediating the secretion of non-classical proteins (Ponpuak et al., 2015; Zahoor & Farhan, 2018), regulating immune cell function (Deretic, 2021), and maintaining the integrity of tissue barriers (Galluzzi & Green, 2019; Levine & Kroemer, 2019).
Autophagy regulation plays a complex role during the development of cancer. For example, dysfunctional regulation of autophagic cell death can contribute to the development of some human cancers (Amaravadi et al., 2011). Interestingly, the potential ability of autophagy to regulate cell death makes it a therapeutic target for cancer (Mathew et al., 2007; White & DiPaola, 2009). In liver cancer, elevated levels of hyperhomocysteine (Hcy) can cause persistent liver injury, which increases the incidence of hepatocellular carcinoma. Studies have reported that Hcy inhibits cystic fibrosis transmembrane conductance regulator (CFTR) expression through the interaction between H3K27me3 and DNA methylation, thereby activating autophagy and leading to persistent liver injury (Yang et al., 2018). Used in myeloma therapy, chidamide is a novel inhibitor of the histone deacetylase benzamide with potent antimyeloma activity. The autophagy-inhibitory effect of chidamide in myeloma cells is mediated primarily by significant downregulation of the histone deacetylase SIRT1 and upregulation of the histone methyltransferase EZH2, which lead to the acetylation of histone H4 lysine 16 (H4K16ac) and elevated overall levels of histone H3 lysine 27 trimethylation (H3K27me3) in the promoter region of the autophagy-associated gene LC3B, which enhances the therapeutic efficacy of antimyeloma therapy (Xu et al., 2020). In the past decade, many research groups have identified a multilevel role for H3K27me3 in regulating autophagy during tumor processes in various cancers, including head and neck cancers, myeloma, and glioblastoma (Lee et al., 2021; Liu et al., 2018). In addition, we downloaded 231 genes associated with autophagy (Supplementary Table 1) from the autophagy database (http://www.autophagy.lu/clustering/index.html). Our PPI network analysis showed that autophagy genes were closely related to H3K27me3 methylation-related enzymes (Fig. 8), confirming the role of H3K27me3 in autophagic processes.
Future prospects for H3K27me3
Histone methylation is a relatively stable epigenetic mark that is often involved in tumorigenesis, with alterations in H3K27 trimethylation being particularly critical for cancer development. The discovery of H3K27 histone methyltransferases and demethylases provides a theoretical basis for the use of H3K27 trimethylation as a therapeutic target for cancers, and the future may see the discovery of additional methylation-modifying enzymes that can be used as new therapeutic targets for anti-tumor drugs. Currently, drugs targeting H3K27me3 modification are mainly focused on anti-tumor therapy for DIPG. The small molecule ONC201, a dopamine receptor D2 (DRD2) antagonist, has been shown to be effective in treating DIPG by altering H3K27me3 activity, in both phase 2 clinical and phase 3 placebo-controlled trials for the treatment of H3K27M-DIPG (Venneti et al., 2023). Drugs targeting H3K27me3 can help overcome the limitations of single-target therapy for patients with cancer and increase patients’ chances of survival. However, while overcoming the problem of a single target, multi-target histone epitope inhibition may increase drug toxicity, so it will be important to find ways to increase the specificity of such drugs if they are to be successfully used in cancer therapy.
In this review, we have detailed the current understanding of the role H3K27 trimethylation plays in carcinogenesis. However, several questions regarding the dynamics of H3K27 methylation remain to be explored. For example, how do the complex regulatory mechanisms of H3K27me3 play a role in cancer progression and what impact do non-coding RNAs have on the relationship between H3K27me3 and cancer development? The major signaling pathways involving H3K27me3 during tumorigenesis also need to be elucidated. A clearer understanding of these unresolved areas will help us to better understand the epigenetic regulatory role of histone modifications in cancer.
Data availability
All the raw data generated in this study are available from corresponding authors on request.
Change history
12 March 2024
A Correction to this paper has been published: https://doi.org/10.1007/s42764-024-00127-7
Agger, K., Cloos, P. A., Christensen, J., Pasini, D., Rose, S., Rappsilber, J., Issaeva, I., Canaani, E., Salcini, A. E., & Helin, K. (2007). UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature, 449(7163), 731–734. https://doi.org/10.1038/nature06145
Agmon, E., Solon, J., Bassereau, P., & Stockwell, B. R. (2018). Modeling the effects of lipid peroxidation during ferroptosis on membrane properties. Scientific Reports, 8(1), 5155. https://doi.org/10.1038/s41598-018-23408-0
Ahmad, F., Patrick, S., Sheikh, T., Sharma, V., Pathak, P., Malgulwar, P. B., Kumar, A., Joshi, S. D., Sarkar, C., & Sen, E. (2017). Telomerase reverse transcriptase (TERT)—Enhancer of zeste homolog 2 (EZH2) network regulates lipid metabolism and DNA damage responses in glioblastoma. Journal of Neurochemistry, 143(6), 671–683. https://doi.org/10.1111/jnc.14152
Akpa, C. A., Kleo, K., Lenze, D., Oker, E., Dimitrova, L., & Hummel, M. (2019). DZNep-mediated apoptosis in B-cell lymphoma is independent of the lymphoma type, EZH2 mutation status and MYC, BCL2 or BCL6 translocations. PLoS ONE, 14(8), e0220681. https://doi.org/10.1371/journal.pone.0220681
Allen, E. A., & Baehrecke, E. H. (2020). Autophagy in animal development. Cell Death and Differentiation, 27(3), 903–918. https://doi.org/10.1038/s41418-020-0497-0
Allfrey, V. G., Faulkner, R., & Mirsky, A. E. (1964). Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proceedings of the National Academy of Sciences of the United States of America, 51(5), 786–794. https://doi.org/10.1073/pnas.51.5.786
Amaravadi, R. K., Lippincott-Schwartz, J., Yin, X. M., Weiss, W. A., Takebe, N., Timmer, W., DiPaola, R. S., Lotze, M. T., & White, E. (2011). Principles and current strategies for targeting autophagy for cancer treatment. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research, 17(4), 654–666. https://doi.org/10.1158/1078-0432.CCR-10-2634
Arango Duque, G., & Descoteaux, A. (2014). Macrophage cytokines: Involvement in immunity and infectious diseases. Frontiers in Immunology, 5, 491. https://doi.org/10.3389/fimmu.2014.00491
Bai, B., Liu, Y., Fu, X. M., Qin, H. Y., Li, G. K., Wang, H. C., & Sun, S. L. (2022). Dysregulation of EZH2/miR-138-5p axis contributes to radiosensitivity in hepatocellular carcinoma cell by downregulating hypoxia-inducible factor 1 alpha (HIF-1α). Oxidative Medicine and Cellular Longevity, 2022, 7608712. https://doi.org/10.1155/2022/7608712
Barski, A., Cuddapah, S., Cui, K., Roh, T. Y., Schones, D. E., Wang, Z., Wei, G., Chepelev, I., & Zhao, K. (2007). High-resolution profiling of histone methylations in the human genome. Cell, 129(4), 823–837. https://doi.org/10.1016/j.cell.2007.05.009
Bedford, M. T., & Clarke, S. G. (2009). Protein arginine methylation in mammals: Who, what, and why. Molecular Cell, 33(1), 1–13. https://doi.org/10.1016/j.molcel.2008.12.013
Benard, A., Janssen, C. M., van den Elsen, P. J., van Eggermond, M. C., Hoon, D. S., van de Velde, C. J., & Kuppen, P. J. (2014). Chromatin status of apoptosis genes correlates with sensitivity to chemo-, immune- and radiation therapy in colorectal cancer cell lines. Apoptosis : An International Journal on Programmed Cell Death, 19(12), 1769–1778. https://doi.org/10.1007/s10495-014-1042-8
Bender, S., Tang, Y., Lindroth, A. M., Hovestadt, V., Jones, D. T., Kool, M., Zapatka, M., Northcott, P. A., Sturm, D., Wang, W., Radlwimmer, B., Højfeldt, J. W., Truffaux, N., Castel, D., Schubert, S., Ryzhova, M., Seker-Cin, H., Gronych, J., Johann, P. D., Stark, S., et al. (2013). Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell, 24(5), 660–672. https://doi.org/10.1016/j.ccr.2013.10.006
Bergers, G., & Benjamin, L. E. (2003). Tumorigenesis and the angiogenic switch. Nature Reviews Cancer, 3(6), 401–410. https://doi.org/10.1038/nrc1093
Blouw, B., Song, H., Tihan, T., Bosze, J., Ferrara, N., Gerber, H. P., Johnson, R. S., & Bergers, G. (2003). The hypoxic response of tumors is dependent on their microenvironment. Cancer Cell, 4(2), 133–146. https://doi.org/10.1016/s1535-6108(03)00194-6
Bluff, J. E., Menakuru, S. R., Cross, S. S., Higham, S. E., Balasubramanian, S. P., Brown, N. J., Reed, M. W., & Staton, C. A. (2009). Angiogenesis is associated with the onset of hyperplasia in human ductal breast disease. British Journal of Cancer, 101(4), 666–672. https://doi.org/10.1038/sj.bjc.6605196
Bossi, P., Viale, G., Lee, A. K., Alfano, R., Coggi, G., & Bosari, S. (1995). Angiogenesis in colorectal tumors: Microvessel quantitation in adenomas and carcinomas with clinicopathological correlations. Cancer Research, 55(21), 5049–5053.
Boya, P., Codogno, P., & Rodriguez-Muela, N. (2018). Autophagy in stem cells: Repair, remodelling and metabolic reprogramming. Development (Cambridge, England), 145(4), dev146506. https://doi.org/10.1242/dev.146506
Byvoet, P., Shepherd, G. R., Hardin, J. M., & Noland, B. J. (1972). The distribution and turnover of labeled methyl groups in histone fractions of cultured mammalian cells. Archives of Biochemistry and Biophysics, 148(2), 558–567. https://doi.org/10.1016/0003-9861(72)90174-9
Cacabelos, R., Torrellas, C., Carrera, I., Cacabelos, P., Corzo, L., Fernández-Novoa, L., Tellado, I., Carril, J. C., & Aliev, G. (2016). Novel therapeutic strategies for dementia. CNS & Neurological Disorders Drug Targets, 15(2), 141–241. https://doi.org/10.2174/1871527315666160202121548
Chabanon, R. M., Morel, D., & Postel-Vinay, S. (2020). Exploiting epigenetic vulnerabilities in solid tumors: Novel therapeutic opportunities in the treatment of SWI/SNF-defective cancers. Seminars in Cancer Biology, 61, 180–198. https://doi.org/10.1016/j.semcancer.2019.09.018
Chen, X., He, Y., & Lu, F. (2018). Autophagy in stem cell biology: A perspective on stem cell self-renewal and differentiation. Stem Cells International, 2018, 9131397. https://doi.org/10.1155/2018/9131397
Chen, Y., Li, X., Xu, J., Xiao, H., Tang, C., Liang, W., Zhu, X., Fang, Y., Wang, H., & Shi, J. (2022). Knockdown of nuclear receptor binding SET domain-containing protein 1 (NSD1) inhibits proliferation and facilitates apoptosis in paclitaxel-resistant breast cancer cells via inactivating the Wnt/β-catenin signaling pathway. Bioengineered, 13(2), 3526–3536. https://doi.org/10.1080/21655979.2021.2018973
Cheung-Ong, K., Giaever, G., & Nislow, C. (2013). DNA-damaging agents in cancer chemotherapy: Serendipity and chemical biology. Chemistry & Biology, 20(5), 648–659. https://doi.org/10.1016/j.chembiol.2013.04.007
Chu, X., Zhong, L., Yu, L., Xiong, L., Li, J., Dan, W., Ye, J., Liu, C., Luo, X., & Liu, B. (2020). GSK-J4 induces cell cycle arrest and apoptosis via ER stress and the synergism between GSK-J4 and decitabine in acute myeloid leukemia KG-1a cells. Cancer Cell International, 20, 209. https://doi.org/10.1186/s12935-020-01297-6
Chung, C., Sweha, S. R., Pratt, D., Tamrazi, B., Panwalkar, P., Banda, A., Bayliss, J., Hawes, D., Yang, F., Lee, H. J., Shan, M., Cieslik, M., Qin, T., Werner, C. K., Wahl, D. R., Lyssiotis, C. A., Bian, Z., Shotwell, J. B., Yadav, V. N., Koschmann, C., et al. (2020). Integrated metabolic and epigenomic reprograming by H3K27M mutations in diffuse intrinsic pontine gliomas. Cancer Cell, 38(3), 334–3499. https://doi.org/10.1016/j.ccell.2020.07.008
Clarke, A. J., & Simon, A. K. (2019). Autophagy in the renewal, differentiation and homeostasis of immune cells. Nature Reviews Immunology, 19(3), 170–183. https://doi.org/10.1038/s41577-018-0095-2
Cuervo, A. M. (2004). Autophagy: In sickness and in health. Trends in Cell Biology, 14(2), 70–77. https://doi.org/10.1016/j.tcb.2003.12.002
Dawson, M. A., & Kouzarides, T. (2012). Cancer epigenetics: From mechanism to therapy. Cell, 150(1), 12–27. https://doi.org/10.1016/j.cell.2012.06.013
De Santa, F., Totaro, M. G., Prosperini, E., Notarbartolo, S., Testa, G., & Natoli, G. (2007). The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell, 130(6), 1083–1094. https://doi.org/10.1016/j.cell.2007.08.019
Deb, G., Singh, A. K., & Gupta, S. (2014). EZH2: Not EZHY (easy) to deal. Molecular Cancer Research MCR, 12(5), 639–653. https://doi.org/10.1158/1541-7786.MCR-13-0546
Deretic, V. (2021). Autophagy in inflammation, infection, and immunometabolism. Immunity, 54(3), 437–453. https://doi.org/10.1016/j.immuni.2021.01.018
Dev, A., Sardoiwala, M. N., Kushwaha, A. C., Karmakar, S., & Choudhury, S. R. (2021). Genistein nanoformulation promotes selective apoptosis in oral squamous cell carcinoma through repression of 3PK-EZH2 signalling pathway. Phytomedicine : International Journal of Phytotherapy and Phytopharmacology, 80, 153386. https://doi.org/10.1016/j.phymed.2020.153386
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., Patel, D. N., Bauer, A. J., Cantley, A. M., Yang, W. S., Morrison, B., 3rd., & Stockwell, B. R. (2012). Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell, 149(5), 1060–1072. https://doi.org/10.1016/j.cell.2012.03.042
Dong, S., Wang, Q., Kao, Y. R., Diaz, A., Tasset, I., Kaushik, S., Thiruthuvanathan, V., Zintiridou, A., Nieves, E., Dzieciatkowska, M., Reisz, J. A., Gavathiotis, E., D’Alessandro, A., Will, B., & Cuervo, A. M. (2021). Chaperone-mediated autophagy sustains haematopoietic stem-cell function. Nature, 591(7848), 117–123. https://doi.org/10.1038/s41586-020-03129-z
El-Deiry, W. S. (1998). Regulation of p53 downstream genes. Seminars in Cancer Biology, 8(5), 345–357. https://doi.org/10.1006/scbi.1998.0097
Espinosa, E., Zamora, P., Feliu, J., & González Barón, M. (2003). Classification of anticancer drugs—A new system based on therapeutic targets. Cancer Treatment Reviews, 29(6), 515–523. https://doi.org/10.1016/s0305-7372(03)00116-6
Ezponda, T., & Licht, J. D. (2014). Molecular pathways: Deregulation of histone h3 lysine 27 methylation in cancer-different paths, same destination. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 20(19), 5001–5008. https://doi.org/10.1158/1078-0432.CCR-13-2499
Feitelson, M. A., Arzumanyan, A., Kulathinal, R. J., Blain, S. W., Holcombe, R. F., Mahajna, J., Marino, M., Martinez-Chantar, M. L., Nawroth, R., Sanchez-Garcia, I., Sharma, D., Saxena, N. K., Singh, N., Vlachostergios, P. J., Guo, S., Honoki, K., Fujii, H., Georgakilas, A. G., Bilsland, A., Amedei, A., et al. (2015). Sustained proliferation in cancer: Mechanisms and novel therapeutic targets. Seminars in Cancer Biology, 35(Suppl), S25–S54. https://doi.org/10.1016/j.semcancer.2015.02.006
Ferrari, K. J., Scelfo, A., Jammula, S., Cuomo, A., Barozzi, I., Stützer, A., Fischle, W., Bonaldi, T., & Pasini, D. (2014). Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Molecular Cell, 53(1), 49–62. https://doi.org/10.1016/j.molcel.2013.10.030
Fukumura, D., Yuan, F., Monsky, W. L., Chen, Y., & Jain, R. K. (1997). Effect of host microenvironment on the microcirculation of human colon adenocarcinoma. The American Journal of Pathology, 151(3), 679–688.
Galluzzi, L., Baehrecke, E. H., Ballabio, A., Boya, P., Bravo-San Pedro, J. M., Cecconi, F., Choi, A. M., Chu, C. T., Codogno, P., Colombo, M. I., Cuervo, A. M., Debnath, J., Deretic, V., Dikic, I., Eskelinen, E. L., Fimia, G. M., Fulda, S., Gewirtz, D. A., Green, D. R., Hansen, M., et al. (2017). Molecular definitions of autophagy and related processes. The EMBO Journal, 36(13), 1811–1836. https://doi.org/10.15252/embj.201796697
Galluzzi, L., & Green, D. R. (2019). Autophagy-independent functions of the autophagy machinery. Cell, 177(7), 1682–1699. https://doi.org/10.1016/j.cell.2019.05.026
Gao, S. B., Li, K. L., Qiu, H., Zhu, L. Y., Pan, C. B., Zhao, Y., Wei, S. H., Shi, S., Jin, G. H., & Xue, L. X. (2017). Enhancing chemotherapy sensitivity by targeting PcG via the ATM/p53 pathway. American Journal of Cancer Research, 7(9), 1874–1883.
Gao, S., Xiong, J., Zhang, C., Berquist, B. R., Yang, R., Zhao, M., Molascon, A. J., Kwiatkowski, S. Y., Yuan, D., Qin, Z., Wen, J., Kapler, G. M., Andrews, P. C., Miao, W., & Liu, Y. (2013). Impaired replication elongation in Tetrahymena mutants deficient in histone H3 Lys 27 monomethylation. Genes & Development, 27(15), 1662–1679. https://doi.org/10.1101/gad.218966.113
Gardner, E. E., Lok, B. H., Schneeberger, V. E., Desmeules, P., Miles, L. A., Arnold, P. K., Ni, A., Khodos, I., de Stanchina, E., Nguyen, T., Sage, J., Campbell, J. E., Ribich, S., Rekhtman, N., Dowlati, A., Massion, P. P., Rudin, C. M., & Poirier, J. T. (2017). Chemosensitive relapse in small cell lung cancer proceeds through an EZH2-SLFN11 axis. Cancer Cell, 31(2), 286–299. https://doi.org/10.1016/j.ccell.2017.01.006
Glick, D., Barth, S., & Macleod, K. F. (2010). Autophagy: Cellular and molecular mechanisms. The Journal of Pathology, 221(1), 3–12. https://doi.org/10.1002/path.2697
Gu, X., Xu, T., & He, Y. (2014). A histone H3 lysine-27 methyltransferase complex represses lateral root formation in Arabidopsis thaliana. Molecular Plant, 7(6), 977–988. https://doi.org/10.1093/mp/ssu035
Hahn, M. A., Hahn, T., Lee, D. H., Esworthy, R. S., Kim, B. W., Riggs, A. D., Chu, F. F., & Pfeifer, G. P. (2008). Methylation of polycomb target genes in intestinal cancer is mediated by inflammation. Cancer Research, 68(24), 10280–10289. https://doi.org/10.1158/0008-5472.CAN-08-1957
Hanahan, D., & Weinberg, R. A. (2011). Hallmarks of cancer: The next generation. Cell, 144(5), 646–674. https://doi.org/10.1016/j.cell.2011.02.013
Hansen, T. E., & Johansen, T. (2011). Following autophagy step by step. BMC Biology, 9, 39. https://doi.org/10.1186/1741-7007-9-39
Harper, J. W., & Elledge, S. J. (2007). The DNA damage response: Ten years after. Molecular Cell, 28(5), 739–745. https://doi.org/10.1016/j.molcel.2007.11.015
Heintzman, N. D., Hon, G. C., Hawkins, R. D., Kheradpour, P., Stark, A., Harp, L. F., Ye, Z., Lee, L. K., Stuart, R. K., Ching, C. W., Ching, K. A., Antosiewicz-Bourget, J. E., Liu, H., Zhang, X., Green, R. D., Lobanenkov, V. V., Stewart, R., Thomson, J. A., Crawford, G. E., Kellis, M., et al. (2009). Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature, 459(7243), 108–112. https://doi.org/10.1038/nature07829
Hou, C., Xiao, L., Ren, X., Cheng, L., Guo, B., Zhang, M., & Yan, N. (2022). EZH2-mediated H3K27me3 is a predictive biomarker and therapeutic target in uveal melanoma. Frontiers in Genetics, 13, 1013475. https://doi.org/10.3389/fgene.2022.1013475
Huang, C., Xiang, Y., Wang, Y., Li, X., Xu, L., Zhu, Z., Zhang, T., Zhu, Q., Zhang, K., Jing, N., & Chen, C. D. (2010). Dual-specificity histone demethylase KIAA1718 (KDM7A) regulates neural differentiation through FGF4. Cell Research, 20(2), 154–165. https://doi.org/10.1038/cr.2010.5
Huang, J. P., & Ling, K. (2017). EZH2 and histone deacetylase inhibitors induce apoptosis in triple negative breast cancer cells by differentially increasing H3 Lys27 acetylation in the BIM gene promoter and enhancers. Oncology Letters, 14(5), 5735–5742. https://doi.org/10.3892/ol.2017.6912
Huang, Y., Yu, S. H., Zhen, W. X., Cheng, T., Wang, D., Lin, J. B., Wu, Y. H., Wang, Y. F., Chen, Y., Shu, L. P., Wang, Y., Sun, X. J., Zhou, Y., Yang, F., Hsu, C. H., & Xu, P. F. (2021). Tanshinone I, a new EZH2 inhibitor restricts normal and malignant hematopoiesis through upregulation of MMP9 and ABCG2. Theranostics, 11(14), 6891–6904. https://doi.org/10.7150/thno.53170
Hurwitz, H., Fehrenbacher, L., Novotny, W., Cartwright, T., Hainsworth, J., Heim, W., Berlin, J., Baron, A., Griffing, S., Holmgren, E., Ferrara, N., Fyfe, G., Rogers, B., Ross, R., & Kabbinavar, F. (2004). Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. The New England Journal of Medicine, 350(23), 2335–2342. https://doi.org/10.1056/NEJMoa032691
Imgruet, M. K., Lutze, J., An, N., Hu, B., Khan, S., Kurkewich, J., Martinez, T. C., Wolfgeher, D., Gurbuxani, S. K., Kron, S. J., & McNerney, M. E. (2021). Loss of a 7q gene, CUX1, disrupts epigenetically driven DNA repair and drives therapy-related myeloid neoplasms. Blood, 138(9), 790–805. https://doi.org/10.1182/blood.2020009195
Jackson, S. P., & Bartek, J. (2009). The DNA-damage response in human biology and disease. Nature, 461(7267), 1071–1078. https://doi.org/10.1038/nature08467
Jacob, Y., Feng, S., LeBlanc, C. A., Bernatavichute, Y. V., Stroud, H., Cokus, S., Johnson, L. M., Pellegrini, M., Jacobsen, S. E., & Michaels, S. D. (2009). ATXR5 and ATXR6 are H3K27 monomethyltransferases required for chromatin structure and gene silencing. Nature Structural & Molecular Biology, 16(7), 763–768. https://doi.org/10.1038/nsmb.1611
Jain, P., Ballare, C., Blanco, E., Vizan, P., & Di Croce, L. (2020). PHF19 mediated regulation of proliferation and invasiveness in prostate cancer cells. eLife, 9, e51373. https://doi.org/10.7554/eLife.51373
Jedinak, A., Dudhgaonkar, S., & Sliva, D. (2010). Activated macrophages induce metastatic behavior of colon cancer cells. Immunobiology, 215(3), 242–249. https://doi.org/10.1016/j.imbio.2009.03.004
Jiang, W., Chen, L., & Zheng, S. (2021). Global reprogramming of apoptosis-related genes during brain development. Cells, 10(11), 2901. https://doi.org/10.3390/cells10112901
Jubb, A. M., Cesario, A., Ferguson, M., Congedo, M. T., Gatter, K. C., Lococo, F., Mulè, A., & Pezzella, F. (2011). Vascular phenotypes in primary non-small cell lung carcinomas and matched brain metastases. British Journal of Cancer, 104(12), 1877–1881. https://doi.org/10.1038/bjc.2011.147
Kale, J., Osterlund, E. J., & Andrews, D. W. (2018). BCL-2 family proteins: Changing partners in the dance towards death. Cell Death and Differentiation, 25(1), 65–80. https://doi.org/10.1038/cdd.2017.186
Kalkavan, H., & Green, D. R. (2018). MOMP, cell suicide as a BCL-2 family business. Cell Death and Differentiation, 25(1), 46–55. https://doi.org/10.1038/cdd.2017.179
Kaushik, S., & Cuervo, A. M. (2018). The coming of age of chaperone-mediated autophagy. Nature Reviews Molecular Cell Biology, 19(6), 365–381. https://doi.org/10.1038/s41580-018-0001-6
Kharbanda, S., Pandey, P., Schofield, L., Israels, S., Roncinske, R., Yoshida, K., Bharti, A., Yuan, Z. M., Saxena, S., Weichselbaum, R., Nalin, C., & Kufe, D. (1997). Role for Bcl-xL as an inhibitor of cytosolic cytochrome C accumulation in DNA damage-induced apoptosis. Proceedings of the National Academy of Sciences of the United States of America, 94(13), 6939–6942. https://doi.org/10.1073/pnas.94.13.693
Kim, J., & Yu, J. (2012). Interrogating genomic and epigenomic data to understand prostate cancer. Biochimica Et Biophysica Acta, 1825(2), 186–196. https://doi.org/10.1016/j.bbcan.2011.12.003
Klionsky, D. J. (2007). Autophagy: From phenomenology to molecular understanding in less than a decade. Nature Reviews Molecular Cell Biology, 8(11), 931–937. https://doi.org/10.1038/nrm2245
Kornberg, R. D. (1974). Chromatin structure: A repeating unit of histones and DNA. Science (New York, NY), 184(4139), 868–871. https://doi.org/10.1126/science.184.4139.868
Kouzarides, T. (2007). Chromatin modifications and their function. Cell, 128(4), 693–705. https://doi.org/10.1016/j.cell.2007.02.005
Kruidenier, L., Chung, C. W., Cheng, Z., Liddle, J., Che, K., Joberty, G., Bantscheff, M., Bountra, C., Bridges, A., Diallo, H., Eberhard, D., Hutchinson, S., Jones, E., Katso, R., Leveridge, M., Mander, P. K., Mosley, J., Ramirez-Molina, C., Rowland, P., Schofield, C. J., et al. (2012). A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature, 488(7411), 404–408. https://doi.org/10.1038/nature11262
Kusakabe, Y., Chiba, T., Oshima, M., Koide, S., Rizq, O., Aoyama, K., Ao, J., Kaneko, T., Kanzaki, H., Kanayama, K., Maeda, T., Saito, T., Nakagawa, R., Kobayashi, K., Kiyono, S., Nakamura, M., Ogasawara, S., Suzuki, E., Nakamoto, S., Yasui, S., et al. (2021). EZH1/2 inhibition augments the anti-tumor effects of sorafenib in hepatocellular carcinoma. Scientific Reports, 11(1), 21396. https://doi.org/10.1038/s41598-021-00889-0
Lan, F., Bayliss, P. E., Rinn, J. L., Whetstine, J. R., Wang, J. K., Chen, S., Iwase, S., Alpatov, R., Issaeva, I., Canaani, E., Roberts, T. M., Chang, H. Y., & Shi, Y. (2007). A histone H3 lysine 27 demethylase regulates animal posterior development. Nature, 449(7163), 689–694. https://doi.org/10.1038/nature06192
Lavrik, I., Golks, A., & Krammer, P. H. (2005). Death receptor signaling. Journal of Cell Science, 118(Pt 2), 265–267. https://doi.org/10.1242/jcs.01610
Lee, M., Nam, H. Y., Kang, H. B., Lee, W. H., Lee, G. H., Sung, G. J., Han, M. W., Cho, K. J., Chang, E. J., Choi, K. C., Kim, S. W., & Kim, S. Y. (2021). Epigenetic regulation of p62/SQSTM1 overcomes the radioresistance of head and neck cancer cells via autophagy-dependent senescence induction. Cell Death & Disease, 12(3), 250. https://doi.org/10.1038/s41419-021-03539-5
Lee, M. G., Villa, R., Trojer, P., Norman, J., Yan, K. P., Reinberg, D., Di Croce, L., & Shiekhattar, R. (2007). Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science (New York, NY), 318(5849), 447–450. https://doi.org/10.1126/science.1149042
Levine, B. (2007). Cell biology: Autophagy and cancer. Nature, 446(7137), 745–747. https://doi.org/10.1038/446745a
Levine, B., & Klionsky, D. J. (2004). Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Developmental Cell, 6(4), 463–477. https://doi.org/10.1016/s1534-5807(04)00099-1
Levine, B., & Kroemer, G. (2008). Autophagy in the pathogenesis of disease. Cell, 132(1), 27–42. https://doi.org/10.1016/j.cell.2007.12.018
Levine, B., & Kroemer, G. (2019). Biological functions of autophagy genes: A disease perspective. Cell, 176(1–2), 11–42. https://doi.org/10.1016/j.cell.2018.09.0
Lewis, P. W., Müller, M. M., Koletsky, M. S., Cordero, F., Lin, S., Banaszynski, L. A., Garcia, B. A., Muir, T. W., Becher, O. J., & Allis, C. D. (2013). Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science (New York, NY), 340(6134), 857–861. https://doi.org/10.1126/science.1232245
Li, H., Cai, Q., Wu, H., Vathipadiekal, V., Dobbin, Z. C., Li, T., Hua, X., Landen, C. N., Birrer, M. J., Sánchez-Beato, M., & Zhang, R. (2012). SUZ12 promotes human epithelial ovarian cancer by suppressing apoptosis via silencing HRK. Molecular Cancer Research: MCR, 10(11), 1462–1472. https://doi.org/10.1158/1541-7786.MCR-12-0335
Li, H., & Zhang, R. (2013). Role of EZH2 in epithelial ovarian cancer: From biological insights to therapeutic target. Frontiers in Oncology, 3, 47. https://doi.org/10.3389/fonc.2013.00047
Li, J., Xi, Y., Li, W., McCarthy, R. L., Stratton, S. A., Zou, W., Li, W., Dent, S. Y., Jain, A. K., & Barton, M. C. (2017). TRIM28 interacts with EZH2 and SWI/SNF to activate genes that promote mammosphere formation. Oncogene, 36(21), 2991–3001. https://doi.org/10.1038/onc.2016.453
Li, X. J., Li, Q. L., Ju, L. G., Zhao, C., Zhao, L. S., Du, J. W., Wang, Y., Zheng, L., Song, B. L., Li, L. Y., Li, L., & Wu, M. (2021). Deficiency of histone methyltransferase SET domain-containing 2 in liver leads to abnormal lipid metabolism and HCC. Hepatology (Baltimore, MD), 73(5), 1797–1815. https://doi.org/10.1002/hep.31594
Liang, W., Wu, J., & Qiu, X. (2021). LINC01116 facilitates colorectal cancer cell proliferation and angiogenesis through targeting EZH2-regulated TPM1. Journal of Translational Medicine, 19(1), 45. https://doi.org/10.1186/s12967-021-02707-7
Lin, H., Guo, Q., Lu, S., Chen, J., Li, X., Gong, M., Tang, L., & Wen, J. (2019). LncRNA SUMO1P3 promotes proliferation and inhibits apoptosis in colorectal cancer by epigenetically silencing CPEB3. Biochemical and Biophysical Research Communications, 511(2), 239–245. https://doi.org/10.1016/j.bbrc.2019.02.006
Lin, H., Wang, Y., Wang, Y., Tian, F., Pu, P., Yu, Y., Mao, H., Yang, Y., Wang, P., Hu, L., Lin, Y., Liu, Y., Xu, Y., & Chen, C. D. (2010). Coordinated regulation of active and repressive histone methylations by a dual-specificity histone demethylase ceKDM7A from Caenorhabditis elegans. Cell Research, 20(8), 899–907. https://doi.org/10.1038/cr.2010.84
Liu, C., Fu, H., Liu, X., Lei, Q., Zhang, Y., She, X., Liu, Q., Liu, Q., Sun, Y., Li, G., & Wu, M. (2018). LINC00470 coordinates the epigenetic regulation of ELFN2 to distract GBM cell autophagy. Molecular Therapy: THe Journal of the American Society of Gene Therapy, 26(9), 2267–2281. https://doi.org/10.1016/j.ymthe.2018.06.019
Liu, G. Z., Xu, X. W., Tao, S. H., Gao, M. J., & Hou, Z. H. (2021). HBx facilitates ferroptosis in acute liver failure via EZH2 mediated SLC7A11 suppression. Journal of Biomedical Science, 28(1), 67. https://doi.org/10.1186/s12929-021-00762-2
Liu, Y., Taverna, S. D., Muratore, T. L., Shabanowitz, J., Hunt, D. F., & Allis, C. D. (2007). RNAi-dependent H3K27 methylation is required for heterochromatin formation and DNA elimination in Tetrahymena. Genes & Development, 21(12), 1530–1545. https://doi.org/10.1101/gad.1544207
Lyu, T., Jia, N., Wang, J., Yan, X., Yu, Y., Lu, Z., Bast, R. C., Jr., Hua, K., & Feng, W. (2013). Expression and epigenetic regulation of angiogenesis-related factors during dormancy and recurrent growth of ovarian carcinoma. Epigenetics, 8(12), 1330–1346. https://doi.org/10.4161/epi.26675
Ma, L., Lin, K., Chang, G., Chen, Y., Yue, C., Guo, Q., Zhang, S., Jia, Z., Huang, T. T., Zhou, A., & Huang, S. (2019). Aberrant activation of β-catenin signaling drives glioma tumorigenesis via USP1-mediated stabilization of EZH2. Cancer Research, 79(1), 72–85. https://doi.org/10.1158/0008-5472.CAN-18-1304
Ma, S., Xu, L., Chen, L., Sun, X., Hu, F., Gong, Y., Yang, R., Li, J., Wang, Q., Huang, S., Zhou, H., & Wang, J. (2022). Novel pharmacological inhibition of JMJD3 improves necrotizing enterocolitis by attenuating the inflammatory response and ameliorating intestinal injury. Biochemical Pharmacology, 203, 115165. https://doi.org/10.1016/j.bcp.2022.115165
Makarevich, G., Leroy, O., Akinci, U., Schubert, D., Clarenz, O., Goodrich, J., Grossniklaus, U., & Köhler, C. (2006). Different Polycomb group complexes regulate common target genes in Arabidopsis. EMBO Reports, 7(9), 947–952. https://doi.org/10.1038/sj.embor.7400760
Margueron, R., & Reinberg, D. (2010). Chromatin structure and the inheritance of epigenetic information. Nature Reviews Genetics, 11(4), 285–296. https://doi.org/10.1038/nrg2752
Mathew, R., Karantza-Wadsworth, V., & White, E. (2007). Role of autophagy in cancer. Nature Reviews Cancer, 7(12), 961–967. https://doi.org/10.1038/nrc2254
Mejlvang, J., Olsvik, H., Svenning, S., Bruun, J. A., Abudu, Y. P., Larsen, K. B., Brech, A., Hansen, T. E., Brenne, H., Hansen, T., Stenmark, H., & Johansen, T. (2018). Starvation induces rapid degradation of selective autophagy receptors by endosomal microautophagy. The Journal of Cell Biology, 217(10), 3640–3655. https://doi.org/10.1083/jcb.201711002
Mizushima, N. (2007). Autophagy: Process and function. Genes & Development, 21(22), 2861–2873. https://doi.org/10.1101/gad.1599207
Mizushima, N., & Komatsu, M. (2011). Autophagy: Renovation of cells and tissues. Cell, 147(4), 728–741. https://doi.org/10.1016/j.cell.2011.10.026
Mizushima, N., & Levine, B. (2010). Autophagy in mammalian development and differentiation. Nature Cell Biology, 12(9), 823–830. https://doi.org/10.1038/ncb0910-823
Mizushima, N., Yoshimori, T., & Ohsumi, Y. (2011). The role of Atg proteins in autophagosome formation. Annual Review of Cell and Developmental Biology, 27, 107–132. https://doi.org/10.1146/annurev-cellbio-092910-154005
Monsky, W. L., Mouta Carreira, C., Tsuzuki, Y., Gohongi, T., Fukumura, D., & Jain, R. K. (2002). Role of host microenvironment in angiogenesis and microvascular functions in human breast cancer xenografts: Mammary fat pad versus cranial tumors. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 8(4), 1008–1013.
Montgomery, N. D., Yee, D., Chen, A., Kalantry, S., Chamberlain, S. J., Otte, A. P., & Magnuson, T. (2005). The murine polycomb group protein Eed is required for global histone H3 lysine-27 methylation. Current Biology: CB, 15(10), 942–947. https://doi.org/10.1016/j.cub.2005.04.051
Morrissey, C., True, L. D., Roudier, M. P., Coleman, I. M., Hawley, S., Nelson, P. S., Coleman, R., Wang, Y. C., Corey, E., Lange, P. H., Higano, C. S., & Vessella, R. L. (2008). Differential expression of angiogenesis associated genes in prostate cancer bone, liver and lymph node metastases. Clinical & Experimental Metastasis, 25(4), 377–388. https://doi.org/10.1007/s10585-007-9116-4
Mosammaparast, N., & Shi, Y. (2010). Reversal of histone methylation: Biochemical and molecular mechanisms of histone demethylases. Annual Review of Biochemistry, 79, 155–179. https://doi.org/10.1146/annurev.biochem.78.070907.103946
Mozzetta, C., Pontis, J., & Ait-Si-Ali, S. (2015). Functional crosstalk between lysine methyltransferases on histone substrates: The case of G9A/GLP and polycomb repressive complex 2. Antioxidants & Redox Signaling, 22(16), 1365–1381. https://doi.org/10.1089/ars.2014.6116
Mozzetta, C., Pontis, J., Fritsch, L., Robin, P., Portoso, M., Proux, C., Margueron, R., & Ait-Si-Ali, S. (2014). The histone H3 lysine 9 methyltransferases G9a and GLP regulate polycomb repressive complex 2-mediated gene silencing. Molecular Cell, 53(2), 277–289. https://doi.org/10.1016/j.molcel.2013.12.005
Nakano, K., & Vousden, K. H. (2001). PUMA, a novel proapoptotic gene, is induced by p53. Molecular Cell, 7(3), 683–694. https://doi.org/10.1016/s1097-2765(01)00214-3
Nakka, K., Hachmer, S., Mokhtari, Z., Kovac, R., Bandukwala, H., Bernard, C., Li, Y., Xie, G., Liu, C., Fallahi, M., Megeney, L. A., Gondin, J., Chazaud, B., Brand, M., Zha, X., Ge, K., & Dilworth, F. J. (2022). JMJD3 activated hyaluronan synthesis drives muscle regeneration in an inflammatory environment. Science (New York, NY), 377(6606), 666–669. https://doi.org/10.1126/science.abm9735
Ning, S., & Ma, X. (2019). Dephosphorylation-induced EZH2 activation mediated RECK downregulation by ERK1/2 signaling. Journal of Cellular Physiology, 234(10), 19010–19018. https://doi.org/10.1002/jcp.28540
Nitiss, J. L. (2009). DNA topoisomerase II and its growing repertoire of biological functions. Nature Reviews. Cancer, 9(5), 327–337. https://doi.org/10.1038/nrc2608
Nowell, P. C. (1976). The clonal evolution of tumor cell populations. Science (New York, NY), 194(4260), 23–28. https://doi.org/10.1126/science.959840
Pang, B., de Jong, J., Qiao, X., Wessels, L. F., & Neefjes, J. (2015). Chemical profiling of the genome with anti-cancer drugs defines target specificities. Nature Chemical Biology, 11(7), 472–480. https://doi.org/10.1038/nchembio.1811
Pasini, D., Bracken, A. P., Jensen, M. R., Lazzerini Denchi, E., & Helin, K. (2004). Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. The EMBO Journal, 23(20), 4061–4071. https://doi.org/10.1038/sj.emboj.7600402
Peralta-Arrieta, I., Trejo-Villegas, O. A., Armas-López, L., Ceja-Rangel, H. A., Ordóñez-Luna, M. D. C., Pineda-Villegas, P., González-López, M. A., Ortiz-Quintero, B., Mendoza-Milla, C., Zatarain-Barrón, Z. L., Arrieta, O., Zúñiga, J., & Ávila-Moreno, F. (2022). Failure to EGFR-TKI-based therapy and tumoural progression are promoted by MEOX2/GLI1-mediated epigenetic regulation of EGFR in the human lung cancer. European Journal of Cancer (Oxford, England: 1990), 160, 189–205. https://doi.org/10.1016/j.ejca.2021.10.032
Polo, S. E., & Jackson, S. P. (2011). Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes & Development, 25(5), 409–433. https://doi.org/10.1101/gad.2021311
Ponpuak, M., Mandell, M. A., Kimura, T., Chauhan, S., Cleyrat, C., & Deretic, V. (2015). Secretory autophagy. Current Opinion in Cell Biology, 35, 106–116. https://doi.org/10.1016/j.ceb.2015.04.016
Porazzi, P., Petruk, S., Pagliaroli, L., De Dominici, M., Deming, D., 2nd., Puccetti, M. V., Kushinsky, S., Kumar, G., Minieri, V., Barbieri, E., Deliard, S., Grande, A., Trizzino, M., Gardini, A., Canaani, E., Palmisiano, N., Porcu, P., Ertel, A., Fortina, P., Eischen, C. M., et al. (2022). Targeting chemotherapy to decondensed H3K27me3-marked chromatin of AML cells enhances leukemia suppression. Cancer Research, 82(3), 458–471. https://doi.org/10.1158/0008-5472.CAN-21-1297
Potente, M., Gerhardt, H., & Carmeliet, P. (2011). Basic and therapeutic aspects of angiogenesis. Cell, 146(6), 873–887. https://doi.org/10.1016/j.cell.2011.08.039
Qian, B. Z., & Pollard, J. W. (2010). Macrophage diversity enhances tumor progression and metastasis. Cell, 141(1), 39–51. https://doi.org/10.1016/j.cell.2010.03.014
Rabinowitz, J. D., & White, E. (2010). Autophagy and metabolism. Science (New York, NY), 330(6009), 1344–1348. https://doi.org/10.1126/science.119349
Rathore, R., McCallum, J. E., Varghese, E., Florea, A. M., & Büsselberg, D. (2017). Overcoming chemotherapy drug resistance by targeting inhibitors of apoptosis proteins (IAPs). Apoptosis: An International Journal on Programmed Cell Death, 22(7), 898–919. https://doi.org/10.1007/s10495-017-1375-1
Ricci, B., Millner, T. O., Pomella, N., Zhang, X., Guglielmi, L., Badodi, S., Ceric, D., Gemma, C., Cognolato, E., Zhang, Y., Brandner, S., Barnes, M. R., & Marino, S. (2020). Polycomb-mediated repression of EphrinA5 promotes growth and invasion of glioblastoma. Oncogene, 39(12), 2523–2538. https://doi.org/10.1038/s41388-020-1161-3
Rogov, V., Dötsch, V., Johansen, T., & Kirkin, V. (2014). Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Molecular Cell, 53(2), 167–178. https://doi.org/10.1016/j.molcel.2013.12.014
Sahu, R., Kaushik, S., Clement, C. C., Cannizzo, E. S., Scharf, B., Follenzi, A., Potolicchio, I., Nieves, E., Cuervo, A. M., & Santambrogio, L. (2011). Microautophagy of cytosolic proteins by late endosomes. Developmental Cell, 20(1), 131–139. https://doi.org/10.1016/j.devcel.2010.12.003
Saraste, A., & Pulkki, K. (2000). Morphologic and biochemical hallmarks of apoptosis. Cardiovascular Research, 45(3), 528–537. https://doi.org/10.1016/s0008-6363(99)00384-3
Schubert, D., Primavesi, L., Bishopp, A., Roberts, G., Doonan, J., Jenuwein, T., & Goodrich, J. (2006). Silencing by plant polycomb-group genes requires dispersed trimethylation of histone H3 at lysine 27. The EMBO Journal, 25(19), 4638–4649. https://doi.org/10.1038/sj.emboj.7601311
Schuldt, L., Reimann, M., von Brandenstein, K., Steinmetz, J., Döding, A., Schulze-Späte, U., Jacobs, C., & Symmank, J. (2022). Palmitate-triggered COX2/PGE2-related hyperinflammation in dual-stressed PdL fibroblasts is mediated by repressive H3K27 trimethylation. Cells, 11(6), 955. https://doi.org/10.3390/cells1106095
Sharma, S., Kelly, T. K., & Jones, P. A. (2010). Epigenetics in cancer. Carcinogenesis, 31(1), 27–36. https://doi.org/10.1093/carcin/bgp220
Shen, X., Liu, Y., Hsu, Y. J., Fujiwara, Y., Kim, J., Mao, X., Yuan, G. C., & Orkin, S. H. (2008). EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Molecular Cell, 32(4), 491–502. https://doi.org/10.1016/j.molcel.2008.10.016
Shi, Y., Lan, F., Matson, C., Mulligan, P., Whetstine, J. R., Cole, P. A., Casero, R. A., & Shi, Y. (2004). Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell, 119(7), 941–953. https://doi.org/10.1016/j.cell.2004.12.012
Shi, Y., & Whetstine, J. R. (2007). Dynamic regulation of histone lysine methylation by demethylases. Molecular Cell, 25(1), 1–14. https://doi.org/10.1016/j.molcel.2006.12.010
Soria, G., Polo, S. E., & Almouzni, G. (2012). Prime, repair, restore: The active role of chromatin in the DNA damage response. Molecular Cell, 46(6), 722–734. https://doi.org/10.1016/j.molcel.2012.06.002
Strasser, A., Jost, P. J., & Nagata, S. (2009). The many roles of FAS receptor signaling in the immune system. Immunity, 30(2), 180–192. https://doi.org/10.1016/j.immuni.2009.01.001
Tachibana, M., Ueda, J., Fukuda, M., Takeda, N., Ohta, T., Iwanari, H., Sakihama, T., Kodama, T., Hamakubo, T., & Shinkai, Y. (2005). Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3–K9. Genes & Development, 19(7), 815–826. https://doi.org/10.1101/gad.1284005
Tang, G., Guo, J., Zhu, Y., Huang, Z., Liu, T., Cai, J., Yu, L., & Wang, Z. (2018). Metformin inhibits ovarian cancer via decreasing H3K27 trimethylation. International Journal of Oncology, 52(6), 1899–1911. https://doi.org/10.3892/ijo.2018.4343
Timp, W., & Feinberg, A. P. (2013). Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nature Reviews Cancer, 13(7), 497–510. https://doi.org/10.1038/nrc3486
Tsukada, Y., Fang, J., Erdjument-Bromage, H., Warren, M. E., Borchers, C. H., Tempst, P., & Zhang, Y. (2006). Histone demethylation by a family of JmjC domain-containing proteins. Nature, 439(7078), 811–816. https://doi.org/10.1038/nature04433
Uytterhoeven, V., Lauwers, E., Maes, I., Miskiewicz, K., Melo, M. N., Swerts, J., Kuenen, S., Wittocx, R., Corthout, N., Marrink, S. J., Munck, S., & Verstreken, P. (2015). Hsc70-4 deforms membranes to promote synaptic protein turnover by endosomal microautophagy. Neuron, 88(4), 735–748. https://doi.org/10.1016/j.neuron.2015.10.012
Venneti, S., Kawakibi, A. R., Ji, S., Waszak, S. M., Sweha, S. R., Mota, M., Pun, M., Deogharkar, A., Chung, C., Tarapore, R. S., Ramage, S., Chi, A., Wen, P. Y., Arrillaga-Romany, I., Batchelor, T. T., Butowski, N. A., Sumrall, A., Shonka, N., Harrison, R. A., de Groot, J., et al. (2023). Clinical efficacy of ONC201 in H3K27M-mutant diffuse midline gliomas is driven by disruption of integrated metabolic and epigenetic pathways. Cancer Discovery, 13(11), 2370–2393. https://doi.org/10.1158/2159-8290.CD-23-0131
Vogelstein, B., Papadopoulos, N., Velculescu, V. E., Zhou, S., Diaz, L. A., Jr., & Kinzler, K. W. (2013). Cancer genome landscapes. Science (New York, NY), 339(6127), 1546–1558. https://doi.org/10.1126/science.1235122
Wang, W., Wang, Q., Huang, D. B., Sun, Q. K., Wu, S. S., Zhao, Y. J., Jia, W., Hu, D. S., & He, Y. F. (2021). Tumor-associated mesenchymal stem cells promote hepatocellular carcinoma metastasis via a DNM3OS/KDM6B/TIAM1 axis. Cancer Letters, 503, 19–31. https://doi.org/10.1016/j.canlet.2021.01.011
White, E., & DiPaola, R. S. (2009). The double-edged sword of autophagy modulation in cancer. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 15(17), 5308–5316. https://doi.org/10.1158/1078-0432.CCR-07-5023
Wu, C., Chen, W., He, J., Jin, S., Liu, Y., Yi, Y., Gao, Z., Yang, J., Yang, J., Cui, J., & Zhao, W. (2020). Interplay of m6A and H3K27 trimethylation restrains inflammation during bacterial infection. Science Advances, 6(34), eaba0647. https://doi.org/10.1126/sciadv.aba0647
Wu, H., Chen, X., Xiong, J., Li, Y., Li, H., Ding, X., Liu, S., Chen, S., Gao, S., & Zhu, B. (2011). Histone methyltransferase G9a contributes to H3K27 methylation in vivo. Cell Research, 21(2), 365–367. https://doi.org/10.1038/cr.2010.157
Wu, Y., Hu, L., Liang, Y., Li, J., Wang, K., Chen, X., Meng, H., Guan, X., Yang, K., & Bai, Y. (2017). Up-regulation of lncRNA CASC9 promotes esophageal squamous cell carcinoma growth by negatively regulating PDCD4 expression through EZH2. Molecular Cancer, 16(1), 150. https://doi.org/10.1186/s12943-017-0715-7
Xiao, B., Jing, C., Wilson, J. R., Walker, P. A., Vasisht, N., Kelly, G., Howell, S., Taylor, I. A., Blackburn, G. M., & Gamblin, S. J. (2003). Structure and catalytic mechanism of the human histone methyltransferase SET7/9. Nature, 421(6923), 652–656. https://doi.org/10.1038/nature01378
Xie, Z., & Klionsky, D. J. (2007). Autophagosome formation: Core machinery and adaptations. Nature Cell Biology, 9(10), 1102–1109. https://doi.org/10.1038/ncb1007-1102
Xu, K., Liu, X., Wen, B., Liu, Y., Zhang, W., Hu, X., Chen, L., Hang, W., & Chen, J. (2022). GSK-J4, a specific histone lysine demethylase 6A inhibitor, ameliorates lipotoxicity to cardiomyocytes via preserving H3K27 methylation and reducing ferroptosis. Frontiers in Cardiovascular Medicine, 9, 907747. https://doi.org/10.3389/fcvm.2022.907747
Xu, L., Feng, J., Tang, H., Dong, Y., Shu, M., & Chen, X. (2020). Chidamide epigenetically represses autophagy and exerts cooperative antimyeloma activity with bortezomib. Cell Death & Disease, 11(4), 297. https://doi.org/10.1038/s41419-020-2414-3
Xu, Y., Dong, X., Qi, P., Ye, Y., Shen, W., Leng, L., Wang, L., Li, X., Luo, X., Chen, Y., Sun, P., Xiang, R., & Li, N. (2017). Sox2 Communicates with Tregs through CCL1 to promote the stemness property of breast cancer cells. Stem Cells (Dayton, Ohio), 35(12), 2351–2365. https://doi.org/10.1002/stem.2720
Yamamoto, M., Jin, C., Hata, T., Yasumizu, Y., Zhang, Y., Hong, D., Maeda, T., Miyo, M., Hiraki, M., Suzuki, Y., Hinohara, K., Rajabi, H., & Kufe, D. (2019). MUC1-C integrates chromatin remodeling and PARP1 activity in the DNA damage response of triple-negative breast cancer cells. Cancer Research, 79(8), 2031–2041. https://doi.org/10.1158/0008-5472.CAN-18-3259
Yang, A., Jiao, Y., Yang, S., Deng, M., Yang, X., Mao, C., Sun, Y., Ding, N., Li, N., Zhang, M., Jin, S., Zhang, H., & Jiang, Y. (2018). Homocysteine activates autophagy by inhibition of CFTR expression via interaction between DNA methylation and H3K27me3 in mouse liver. Cell Death & Disease, 9(2), 169. https://doi.org/10.1038/s41419-017-0216-z
Yang, L., Ma, D. W., Cao, Y. P., Li, D. Z., Zhou, X., Feng, J. F., & Bao, J. (2021). PRMT5 functionally associates with EZH2 to promote colorectal cancer progression through epigenetically repressing CDKN2B expression. Theranostics, 11(8), 3742–3759. https://doi.org/10.7150/thno.53023
Yang, M. H., Zhao, L., Wang, L., Ou-Yang, W., Hu, S. S., Li, W. L., Ai, M. L., Wang, Y. Q., Han, Y., Li, T. T., Ding, Y. Q., & Wang, S. (2019). Nuclear lncRNA HOXD-AS1 suppresses colorectal carcinoma growth and metastasis via inhibiting HOXD3-induced integrin β3 transcriptional activating and MAPK/AKT signalling. Molecular Cancer, 18(1), 31. https://doi.org/10.1186/s12943-019-0955-9
Yang, Y., Hu, L., Wang, P., Hou, H., Lin, Y., Liu, Y., Li, Z., Gong, R., Feng, X., Zhou, L., Zhang, W., Dong, Y., Yang, H., Lin, H., Wang, Y., Chen, C. D., & Xu, Y. (2010). Structural insights into a dual-specificity histone demethylase ceKDM7A from Caenorhabditis elegans. Cell Research, 20(8), 886–898. https://doi.org/10.1038/cr.2010.86
Ying, Y., Wang, M., Chen, Y., Li, M., Ma, C., Zhang, J., Huang, X., Jia, M., Zeng, J., Wang, Y., Li, L., Wang, X., Tao, Q., & Shu, X. S. (2022). Zinc finger protein 280C contributes to colorectal tumorigenesis by maintaining epigenetic repression at H3K27me3-marked loci. Proceedings of the National Academy of Sciences of the United States of America, 119(22), e2120633119. https://doi.org/10.1073/pnas.2120633119
Yoshimi, A., & Kurokawa, M. (2011). Key roles of histone methyltransferase and demethylase in leukemogenesis. Journal of Cellular Biochemistry, 112(2), 415–424. https://doi.org/10.1002/jcb.22972
Yuan, X., Gajan, A., Chu, Q., Xiong, H., Wu, K., & Wu, G. S. (2018). Developing TRAIL/TRAIL death receptor-based cancer therapies. Cancer Metastasis Reviews, 37(4), 733–748. https://doi.org/10.1007/s10555-018-9728-y
Zahoor, M., & Farhan, H. (2018). Crosstalk of autophagy and the secretory pathway and its role in diseases. International Review of Cell and Molecular Biology, 337, 153–184. https://doi.org/10.1016/bs.ircmb.2017.12.004
Zhang, C., Gao, S., Molascon, A. J., Liu, Y., & Andrews, P. C. (2014). Quantitative proteomics reveals histone modifications in crosstalk with H3 lysine 27 methylation. Molecular & Cellular Proteomics: MCP, 13(3), 749–759. https://doi.org/10.1074/mcp.M113.029025
Zhang, C., Molascon, A. J., Gao, S., Liu, Y., & Andrews, P. C. (2013). Quantitative proteomics reveals that the specific methyltransferases Txr1p and Ezl2p differentially affect the mono-, di- and trimethylation states of histone H3 lysine 27 (H3K27). Molecular & Cellular Proteomics: MCP, 12(6), 1678–1688. https://doi.org/10.1074/mcp.M112.021733
Zhang, M., Zheng, J., Nussinov, R., & Ma, B. (2017). Release of cytochrome c from Bax pores at the mitochondrial membrane. Scientific Reports, 7(1), 2635. https://doi.org/10.1038/s41598-017-02825-7
Zhao, J., Jin, W., Yi, K., Wang, Q., Zhou, J., Tan, Y., Xu, C., Xiao, M., Hong, B., Xu, F., Zhang, K., & Kang, C. (2021). Combination LSD1 and HOTAIR-EZH2 inhibition disrupts cell cycle processes and induces apoptosis in glioblastoma cells. Pharmacological Research, 171, 105764. https://doi.org/10.1016/j.phrs.2021.105764
Zhong, Y., Li, L., Chen, Z., Diao, S., He, Y., Zhang, Z., Zhang, H., Yuan, X., & Li, J. (2020). MIR143 inhibits steroidogenesis and induces apoptosis repressed by H3K27me3 in granulosa cells. Frontiers in Cell and Developmental Biology, 8, 565261. https://doi.org/10.3389/fcell.2020.565261
Zhu, Q., Yang, Q., Lu, X., Wang, H., Tong, L., Li, Z., Liu, G., Bao, Y., Xu, X., Gu, L., Yuan, J., Liu, X., & Zhu, W. G. (2021). SETD2-mediated H3K14 trimethylation promotes ATR activation and stalled replication fork restart in response to DNA replication stress. Proceedings of the National Academy of Sciences of the United States of America, 118(23), e2011278118. https://doi.org/10.1073/pnas.2011278118
Zhu, Y., Fu, J., Yang, H., Pan, Y., Yao, L., & Xue, X. (2015). Hyperoxia-induced methylation decreases RUNX3 in a newborn rat model of bronchopulmonary dysplasia. Respiratory Research, 16(1), 75. https://doi.org/10.1186/s12931-015-0239-x
School of Basic Medical Sciences, Southern Medical University, Guangzhou, 510515, China
Longjiang Di & Wei-Guo Zhu
Guangdong Key Laboratory of Genome Instability and Human Disease Prevention, and International Cancer Center, Shenzhen University School of Medicine, Shenzhen, 518055, China
Wei-Guo Zhu
Department of Biochemistry and Molecular Biology, Shenzhen University School of Medicine, Shenzhen, 518055, China
Wei-Guo Zhu
Shenzhen Bay Laboratory, Shenzhen University School of Medicine, Shenzhen, 518055, China
Wei-Guo Zhu
Marshall Laboratory of Biomedical Engineering, Shenzhen University School of Medicine, Shenzhen, 518055, China
Wei-Guo Zhu
Received15 May 2023
Revised15 November 2023
Accepted27 November 2023
Published04 January 2024
Issue DateFebruary 2024
DOIhttps://doi.org/10.1007/s42764-023-00118-0
References
Funding
National Natural Science Foundation of China [32090030, 81530074, 82002986]; Science and Technology Program of Guangdong Province in China [2017B030301016]; Guangdong Basic and Applied Basic Research Foundation [2021A1515011126].
Author information
Authors and Affiliations
Corresponding author
Correspondence to Wei-Guo Zhu.
Ethics declarations
Conflict of interest
Prof. Wei-Guo Zhu is the editor-in-chief of the journal “Genome Instability and Disease”, and he was not involved in the peer review or the decision making of this article.
Additional information
The original online version of this article was revised: In this article incorrect conflict of interest statement was given. It should have been as follows. “Prof. Wei-Guo Zhu is the editor-in-chief of the journal “Genome Instability and Disease”, and he was not involved in the peer review or the decision making of this article”.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file1 (DOC 58 KB)
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article
Cite this article
Di, L., Zhu, WG. The role of H3K27me3 methylation in cancer development. GENOME INSTAB. DIS. 5, 17–34 (2024). https://doi.org/10.1007/s42764-023-00118-0
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable link
用户登录
还没有账号?
立即注册