






Genomic instability evaluation in different cell line by random amplified polymorphic DNA-PCR analysis
Original Research Paper
Published: 04 March 2024
Arzu Erol
Genome Instability & Disease Volume 5, (2024)
Abstract
The cause of cancer is disorders and unstable genome. Some factors affect proper functioning of the DNA. These situations lead to cell death, cancer, inborn disorders, and overall functional decline contributing to aging. The integrity of the genome of all living organisms is constantly threatened by exogenous and endogenous DNA-damaging agents. An agent might interact directly with DNA and regulate the expression or directly by influencing DNA repair responses and so improve genomic stability. The development of widely applicable methods to monitor genomic instability gains importance nowadays. RAPD-PCR assay is a molecular method able to detect comparative DNA changes. This work is to show the applicability of the method to evaluate the ultimate changes caused in various cell-culture-derived DNA that might be a model for carcinogenesis, genomic instability and routine cell-culture work.
Introduction
Preserving genomic integrity is obviously important, as manifested by our genome’s investment of some 250 genes for purposes of DNA damage repair (Akgün et al., 2022), more than 230 genes for high-fidelity DNA replication, and perhaps more than 500 for chromosome segregation, cell cycle checkpoints, telomeres, centromeres, and damage sensing (Burhansand et al., 2007).
Endogenous and exogenous DNA damaging agents are constantly challenging the integrity of the genome. If DNA damage is repaired incorrectly, it can lead to genome instability, which is associated with tumor genesis in human (Katerji & Duerksen-Hughes, 2021). Eukaryotic organisms have evolved several repair and surveillance mechanisms that remove DNA damage and coordinate cell cycle progression (Hoboken et al., 2001).
The cell cycle continues by a characterized grouping of occasions where late occasions rely on fruition of early occasions (Liu et al., 2024). The point of the reliance of occasions is to disperse finish and exact copies of the genome to daughter cells (Hoboken et al., 2001). To screen this reliance, cells are furnished with the checkpoints that are set at different phases of the cell cycle. At the point when cells have DNA harms that must be repaired, cells enact DNA harm checkpoint that captures cell cycle (Hong et al., 2005).
In spite of the fact that it may appear that immediate inversion of harm would be the least complex approach to amend the harm, by and large the invert response is impractical due to the thermodynamic or active reasons. In a couple cases, the response is reversible, and in some of these cases, systems have been produced to exploit that reversibility. Albeit all, cells have a substantial number of various sorts of repair framework, each moderately particular for a specific kind of DNA harm, those repair frameworks can be gathered into four noteworthy classes: Mismatch Repair, Base Excision Repair, Nucleotide Excision Repair, and Double-strand Break Repair (Sancar et al., 2004).
There are two important types of error in DNA. DNA damages are physical abnormalities in the DNA, such as single- and double-strand breaks, 8-hydroxydeoxyguanosine residues and polycyclic aromatic hydrocarbon adducts (Senavirathne et al., 2023).
Proteins can perceive DNA harms, and in this manner, they can be effectively repaired if repetitive data, for example, the undamaged grouping in the integral DNA strand or in a homologous chromosome, are accessible for replicating (Araten et al., 2005). In the event that a cell holds DNA harm, interpretation of a quality can be forestalled, and in this way, interpretation into a protein will likewise be blocked. Replication may likewise be blocked or potentially the cell may kick the bucket. Rather than DNA harm, a transformation is an adjustment in the base grouping of the DNA (Altonen et al., 1994).
RAPD remains for Random Amplification of Polymorphic DNA. It is a kind of PCR response, yet the portions of DNA that are enhanced are arbitrary. The researcher performing RAPD (Castano & Becerril, 2004) makes a few self-assertive, short groundworks (8–12 nucleotides), then continues with the PCR utilizing a vast format of genomic DNA, trusting that parts will increase. By settling the subsequent examples, a semi-interesting profile can be gathered from a RAPD response (Atienzar & Jha, 2006).
No learning of the DNA succession for the focused on quality is required, as the groundworks will tie some place in the arrangement, however, it is not sure precisely where. This makes the strategy well known for looking at the DNA of natural frameworks that have not had the consideration of established researchers, or in a framework in which moderately few DNA arrangements are analyzed. Because of the way that it depends on an expansive, in place DNA layout arrangement, it has a few restrictions in the utilization of debased DNA tests. Its settling force is much lower than focused, species particular DNA correlation techniques, for example, short pair rehashes. As of late, RAPD is utilized to portray, and follow, the phylogeny of assorted plant and creature species (Atienzar et al., 2000).
The study we put forward here is to our knowledge a novel approach in assessment of various DNA effects: either DNA damages or mutations caused by endogenous or exogenous exposures in routine cell-culture work (Keshava et al., 1999; Keshava et al., 2001; Lery et al., 2003; Lee et al., 2007) The various cell types either cell-lines or primary cells are for the sake of modeling various aspects of a cell’s life-time exposures and events and in this context to discuss the events such as genomic instability, DNA damage, mutation, DNA repair, mutagenesis, carcinogenesis, aging and related disorders. Below is a brief information before discussion of the results obtained that is to clarify the events underlying the work starting with the importance of genomic stability, how to maintain it, what happens when it is lost, what are the causes of genomic instability, what it may lead to in a cell’s life-time exposures.
Materials and methods
Cell types
Frozen cell lines (HEK293, HUVEC and HeLa) were purchased from American Type Culture Collection (ATCC). Collected cell-lines were stored in liquid nitrogen. All cell line culture samples were collected by American Type Culture Collection (ATCC) laboratory. All the information about cell-lines are provided in Table 1 from ATTC’s website.
ATCC number | CRL-2873 | CRL-1573 | CCL-2 |
|---|---|---|---|
Designations | 293 T/17 | HUVEC-CS | HeLa |
Organism | Homo sapiens | Homo sapiens | Homo sapiens |
Source | Kidney | Umbilical vein | Cervix |
Morphology | Epithelial | Epithelial | Epithelial |
Subcultivation ratio | 1:10 to 1:20 weekly | about 36 h | 1:2 to 1:6 weekly |
Medium Renewal | Every 2 to 3 days | Every 2 to 3 days | 2 to 3 times per week |
Cell culture chemicals and consumables
Dulbecco’s Modified Eagle’s Medium (DMEM), RPMI 1640 Medium, essential amino acids, penicillin/streptomycin, Trypsin/EDTA, and fetal bovine serum (FBS and for mesenchymal cells: hMSC qualified FBS) were purchased from Biochrom, Berlin, Germany. Cell culture flasks: 15 and 50 ml polycarbonate centrifuge tubes with lids and cryotubes were from Grainer Bio-One Corp., Germany. Cell lines were cultured at 37 °C under 5% CO2 in culture medium unless otherwise specified (Caputo, 1996; Mather & Barnes, 1998).
Thawing cell lines
Previously frozen cells or cell line in liquid nitrogen was taken out from the tank, immediately soaked into pre-warmed water bath and incubated in water bath at 37 °C until totally thawed. Thawed cell gently mixed by pipetting and transferred to 15 ml tube that was containing 10 ml growth medium. Then, cells were centrifuged at 1500 rpm for 10 min and supernatant carefully aspirated. Lastly, cell pellet was resuspended in 1 ml medium and cell number was counted. After this, cells were seeded in 25 cm culture flask with 8 ml 10% FBS growth medium and were transferred to 25 cm sterile culture flask. Culture flask size was determined according to total cell number. Culture flask was incubated at CO2 incubator under humidified microaerofilic condition with 5% CO2. Following day, cells were observed under invert microscope and growth medium was refreshed.
Sub-culture of cell lines
Cultured cells were observed under microscope and if the confluency was about 90% and if there was no contamination or other abnormal formation (Chen, 1977), cells were determined to be sub-cultured. Culture medium was aspirated and cells were washed for once with pre-warmed PBS at 37 °C. After washing cells, they were detached by treatment with Trypsin/EDTA solution for 1–3 min until all cells were detached. 3 ml FBS for 25 cm sterile culture flask was added to detached cells to stop the Trypsin /EDTA activity. Then, cells were splitted or diluted depending on the purpose, transferred to new culture flasks, and incubated in 5% CO2 incubator at 37 °C.
Cryopreservation of cell lines
Medium of the cells that are 80–90% confluent was aspirated, the cells were washed with 5 ml pre-warmed PBS in 25 cm culture flask for once, treated with 1 ml Trypsin/EDTA (4 ml/25 cm), and incubated at 370 °C for 1–3 min to be detached. To inactivate Trypsin/EDTA, 3 volumes of 1 ml FBS were added. FBS plus Trypsin/EDTA mixture was transferred to 15 ml falcon tube and then was centrifuged at 1500 RPM for 5 min. After centrifugation, the cell pellet was resuspended with FBS and counted with hemocytometer. About 1.5–2 × 10 cells were added to each tube with 10% DMSO on ice and was left at − 20 °C for a while to cool down. Cryovials were transferred to − 20 °C immediately and left for 2 h, then transferred to − 80 °C and left overnight. At last, cells were transferred into liquid nitrogen.
DNA isolation
Macherey Nagel’s MN Nucleospin Tissue Kit was used and the components are as follows. Lysis Buffer, Buffer B1, Buffer B2, Wash Buffer B5, Wash Buffer BW, Elution Buffer BE, Proteinase K (lyophilized), Proteinase Buffer PB. DNA extraction was performed according to MN-Nucleospin tissue kit protocol step by step as describe below: 1.0 × 106 cells were resuspended in a final volume of 200 μl Buffer T1. 25 μl Proteinase K solution and 200 μl Buffer B3 were added. Samples were incubated at 70 °C for 10–15 min. To adjust binding condition, 210 μl ethanol (96–100%) to the sample was added and vortexed vigorously. For each sample, one NucleoSpin® Tissue Column was placed into a collection tube. The sample was loaded onto the column, was centrifuged for 1 min at 11,000g. Flow-through was discarded and the column was placed back into the collection tube. 500 μl Buffer BW was added onto the column, centrifuged for 1 min at 11,000g. Flow-through was discarded and the column was placed back into the collection tube. 600 μl Buffer B5 was added onto the column, centrifuged for 1 min at 11,000g. Flow-through was discarded and the column was placed back into the collection tube. To dry and remove the residual ethanol from membrane empty column was centrifuged for 1 min at 11,000g. NucleoSpin® Tissue Column was placed into a 1.5 ml microcentrifuge tube and 100 μl prewarmed Elution Buffer BE (70 °C) was added onto the column, incubated at room temperature for 1 min, centrifuged 1 min at 11,000g. Highly pure DNA is extracted at the end of procedure and eluted DNA concentration and purity is calculated using Qubit Fluorometer.
DNA quantification
DNA quantification was done using Qubit fluorometer and its quantification kit components. Quant-it dsDNA BR Assay Kits Components are as follows: Working solution, Standard #1 Standard #2 and assay range is 2–1000 ng with sample starting concentration range: 100 pg/μl–1 μg/μl.
To determine concentration of the original samples, the instrument read values were multiplied by the dilution factor. Alternatively, calculate sample concentration can be chosen to have the Qubit fluorometer perform this multiplication.
Polymerase chain reaction PCR
RAPD is different from conventional PCR, as it needs one primer for amplification (Jones & Kortenkamp, 2000). The size of primer is normally short (10 nucleotides), and therefore, less specific, show in Table 2. The primers can be designed without the experimenter having any genetic information for the organism being tested. More than 2000 different RAPD primers can be available commercially.
Primer | Sequence 5´ to 3´ | Primer | Sequence 5´ to 3´ |
|---|---|---|---|
OPA 01 | CAGGCCCTTC | OPB 01 | GTTTCGCTCC |
OPA 02 | TGCCGAGCTG | OPB 02 | TGA TCCCTGG |
OPA 03 | AGTCAGCCAC | OPB 03 | CA TCCCCCTG |
OPA 04 | AATCGGGCTG | OPB 04 | GGACTGGAGT |
OPA 05 | AGGGGTCTTG | OPB 05 | TGCGCCCTTC |
OPA 06 | GGTCCCTGAC | OPB 06 | TGCTCTGCCC |
OPA 07 | GAAACGGGTG | OPB 07 | GGTGACGCAG |
OPA 08 | GTGACGT AGG | OPB 08 | GTCCACACGG |
OPA 09 | GGGTAACGCC | OPB 09 | TGGGGGACTC |
OPA 10 | GTGA TCGCAG | OPB 10 | CTGCTGGGAC |
Table 2 Base sequences of 10-mer primers
The conditions of RAPD-PCR were optimized with some modifications stated in Fig. 2 as RAMD-PCR (Random Amplified Mosaic DNA-PCR). RAPD-PCR reactions were performed in reaction mixture of 25 μl containing the components in Table 3.
Reagent | Initial concentration | Final concentration | Final volume |
|---|---|---|---|
Taq buffer | 10X | 1X | 2.5 μl |
dNTP | 2 mM | 0.2 μM | 1.5 μl |
MgCl2 | 25 mM | 2 mM | 3 μl |
Primers (× 6) | 25 pmol/μl | 25 pmol | 4 μl |
dH2O | – | – | 11.9 μl |
Taq polymerase | 5 U/μl | 1U | 0.2 μl |
Template DNA | 5 μl | ||
Total volume | 25 μl |
Table 3 RAPD-PCR solutions and their initial and final concentrations with final volumes calculated accordingly
The RAPD and RAMD protocols consisted of an initial denaturing step of 5 min at 94 °C, followed by 45 cycles at 94 °C for 30 s (denaturation), 37 °C for 60 s (annealing) and 72 °C for 60 s (extension), with an additional extension period 10 min at 72 °C.
Agarose gel electrophoresis
PCR products were resolved on 2% agarose gels. Gel is prepared adding 1.6 g of powdered agarose gel into 80 ml of 0.5 × TBE buffer solution and it is boiled until the agarose is completely dissolved in the buffer solution in Table 4. 4 μl of Ethidium Bromide was added when the boiled solutions began to cool down and reach approximately 55 °C. Solution is mixed homogenously by making hand-shaking. It is directly poured into horizontal agarose gel platform and the combs having either 8 or 22 wells are placed one side of the gel. Gels are let to solidify for at least 10 min, to confirm full polymerization 45 min. Loading and visualization of the gels were done accordingly: 10 μl of PCR product was mixed with 2 μl bromophenol blue as loading dye/buffer. 12 μl PCR mixes were then loaded in each slot with appropriate micropipettes. 1 μl of a 100 bp DNA Ladder (MBI Fermentas, Hanover, MD, USA) was mixed with 1 μl deionized water and 1 μl bromo phenol blue. Then, 5 μl of this mix was put into usually the first slot as a molecular size marker. The gel was run at 95 V in 0.5X TBE buffer for 50 min. The gel was placed in Gel Doc 2000 (Bio-Rad, Milan, Italy) apparatus and the bands were detected under UV transilluminator.
10XTBE | For 1 liter: 108 g Tris base, 55 g Boric acid, 40 mls 0.5 M EDTA (pH 8.0), autoclave for 20 min |
|---|---|
6XLoading dye | 10 mM Tris–HCl (pH 7.6), 0.03% bromophenol blue, 0.03% xylene cyanol FF, 60% glycerol, 60 mM EDTA |
GeneRulerTM 100 bp DNA Ladder | 100 μl (0.5 μg/μl) 100 bp sized DNA fragments in 10 mM Tris–HCl (pH 7.6), 1 mM EDTA |
Etbr staining chemicals | 0.5 μg/ml EtBr |
Table 4 Agarose gel electrophoresis chemicals, buffers and their components and composition concentrations
The marker range used in agarose gel electrophoresis starts from 1000 bp and decreases by 100 bp. The same marker was used in the entire study.
Results
HEK293 cells were created by change of societies of typical human embryonic kidney epithelial cells. As a tentatively changed cell line, HEK293 cells are not an especially decent model for typical cells, malignancy cells, or whatever other sort of cell that is a major question of research. Nonetheless, they are to a great degree simple to work with, being direct to culture.
HUVEC are human umbilical vein endothelial cells that are widely used in research to study vascular endothelium in a cell culture model. Primary human umbilical endothelial cells in culture have a finite proliferative lifespan. They undergo permanent growth arrest, known as replicative senescence.
When replicative senescence is bypassed by transformation with viral oncogene with telomerase reverse transcriptase, HUVEC are immortalized. The HUVE cells we used in our thesis are cell-lines like the HEK293 and HeLa cells (Masters, 2002).
Growth curves were obtained for each cell type and calculated doubling times were calculated to decide for the optimum subculture timing and the cell types nature of proliferation capacity and the nature of the cell whether it is a dividing or non-dividing cell. When working with concepts such as DNA damage, mutation, genomic instability, cell division, mutagenesis, carcinogenesis, and aging, the cell’s nature of division capacity gains utmost importance. The end of log phase is nearly 72 h for HEK cells.
74 for HUVEC cells and 96 h for HeLa cells, so for the time of doubling for each cell type, which is an indication for their proliferation potency, were calculated. We expect to see more change in HEK cells’ DNA, because it replicates faster than the others. Saturation density is the highest for HEK, the second one is HUVEC and the third one is HeLa and the results were all in compromise with the expectations.
Figure 1 is for all cell types’ nearly all passages to see the total DNA variation profile. For HEK cells especially after 9th passage, the changes are more prominent. The arrows show the types of changes observed; the cumulative changes when the passages increase indicate that these are mutations. Loss of bands, appearance of bands, decrease and increase in band intensities all detectable DNA effects have been detected for all cell types, but more prominent in HEK cells, they are epithelial cells that are having the least doubling time, and reaching the highest number two times the other cell types at 72 h, highest saturation density. Some are DNA damages because reverted back in increasing number of passages, some are mutations because were stabilized in increasing number of passages, in long term.
Fig. 1
RAPD profiles with OPB7 primer for all cell types’ all subculture DNA extracted. M: DNA size marker. Arrows indicate the most prominent changes of RAPD profiles: loss of bands, appearance of bands, decreases in band intensity and increase in band intensity
In different cell types, the first passages and the last passages are compared in Fig. 2. The stabilization can be easy seen for the last passages. The homogeneity of band profiles is prominent. In all cell types, the remaining bands were quite the same, monomorphic, although the starting profile bands were much more different, polymorphic. The remaining bands at the last passages were having band sizes in between 270 and 470 bp.
Fig. 2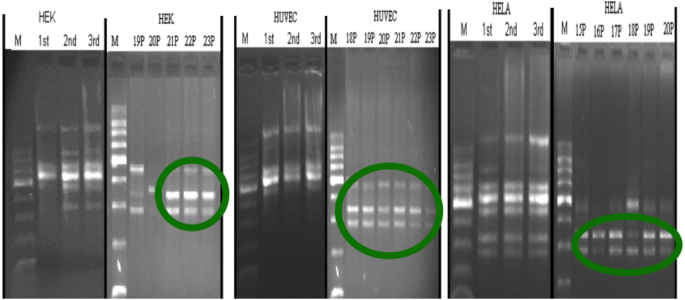
Comparable picture of the first and last passages RAPD profiles, circles indicating a pattern homogenization in the last passages no matter what the cell type was
Figure 3 shows the DNA damaging effect of glutaraldehyde which is a DNA–protein cross linker on our different cell types. The first pictures are the Glutaraldehyde exposed cells’ profiles. Profiles are for eight times RAPD-PCR of 21st passages for exposed and 22nd for the glutaraldehyde removed cell samples. For HUVEC and HeLa cells, the DNA effects are reverted back indicating that the changes are DNA damages, but for the more dividing cell, HEK cells, the effects are not reverted back, indication of mutation. The repair mechanisms cannot work properly for HEK cells and this might be an indication that the fast-dividing cells can have difficulties in repair processes which might be causing further pathology.
Fig. 3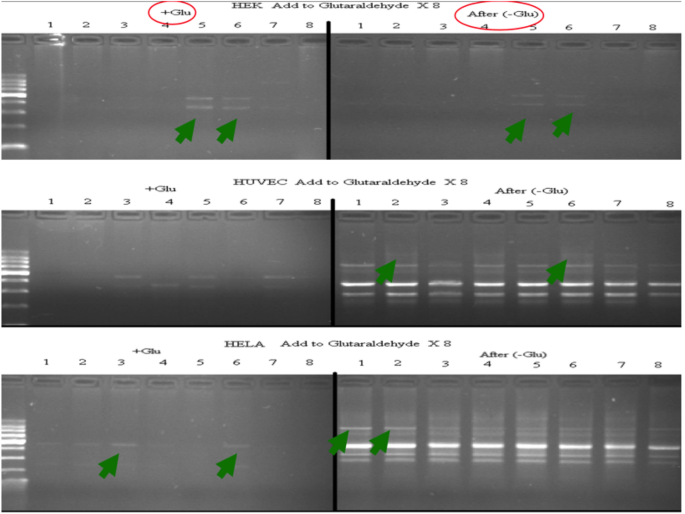
Glutaraldehyde exposed DNA × 8 RAMD profiles and after removal of glutaraldehyde, the next passages’ DNA × 8 RAMD profiles. The arrows indicate the initial and final profile changes in all cell types. + Glu is for glutaraldehyde added ones and after –glu is after removal of glutaraldehyde from the medium
The effects on DNA should be endogenous, because in cell culture, we aim to have a controlled environment for the cells to continue with the least change. There is a change, a continuous change, but it is detectable in long term rather than changes observed in each passage. The RAPD profiles obtained are in agreement with this. The RAPD-PCR profiles that can detect the intra-specific DNA variation, mosaicism of the individual passage DNA is very homogenous, monomorphic indicating there is a change and if there is a change that is DNA damage.
Discussion
In conclusion, while RAPD and related techniques have been used extensively for diverse studies, application of these techniques has also attracted criticisms with respect to its reproducibility that we have overcame in this thesis by RAPD-PCR approaches. However, it emerges that most of the criticism relates to lack of proper optimization and validation of the techniques in different cell types and species, prior to their applications under in vivo or in vitro conditions. Nevertheless, the RAPD assay and related assays offer great promise especially for the determination of genetic damage under in vivo conditions in wild species and for the evaluation of genomic instability in the process of carcinogenesis. While a large number of new technologies and assays are developing to profile the gene expression pattern either following exposure to environmental contaminants or during the process of malignant development, the RAPD-based techniques offer great promise for future and would continue to complement other new and well-established techniques in population genetics, genotoxicity and carcinogenesis studies and cell-culture work in view of this work.
Data availability
The author has no material discussed in this article.
References
Aardema, M., Gibson, D., Hu, T., Mun, G., Curren, R., Hayden, P. (2005). Addressing animal testing concerns: A novel micronucleus assay using the human 3-D skin model, EpiDerm. Presented at the 5th World Congress on Alternatives & Animal Use in the Life Sciences.
Akgün, M., Pfeifer, N., & Kohlbacher, O. (2022). Efficient privacy-preserving whole-genome variant queries. Bioinformatics (oXford, England), 38(8), 2202–2210. https://doi.org/10.1093/bioinformatics/btac070
Altonen, L. A., et al. (1994). Replication errors in benign and malignant tumors from hereditary nonpolyposis colorectal cancer patients. Cancer Research, 54, 1645–1648.
Anastasi, J., Le Beau, M. M., Vardiman, J. W., & Westbrook, C. A. (1990). Detection of numerical chromosomal abnormalities in neoplastic hematopoietic cells by in situ hybridization with a chromosome-specific probe. American Journal of Pathology, 136, 131–139.
Araten, D. J., Golde, D. W., et al. (2005). A quantitative measurement of the human somatic mutation rate. Cancer Research, 65(18), 8111–8117.
Atienzar, F., Evenden, A., et al. (2000). Optimized RAPD analysis generates high-quality genomic DNA profiles at high annealing temperature. BioTechniques, 28(1), 52–54.
Atienzar, F. A., Evenden, A. J., et al. (2002). Use of the random amplified polymorphic DNA (RAPD) assay for the detection of DNA damage and mutations: Possible implications of confounding factors. Biomarkers, 7(1), 94–101.
Atienzar, F. A., & Jha, A. N. (2006). The random amplified polymorphic DNA (RAPD) assay and related techniques applied to genotoxicity and carcinogenesis studies: A critical review. Mutation Research, 613(2–3), 76–102.
Bender, M. A., Awa, A. A., Brooks, A. L., Evans, H. J., Groer, P. G., Littlefield, L. G., Pereira, C., Preston, R. J., & Wachholz, B. W. (1988). Current status of cytogenetic procedures to detect and quantify previous exposures to radiation. Mutation Research, 196, 103–159.
Bird, A. (2007). Perceptions of epigenetics. Nature, 447, 396.
Boccia, S., et al. (2007). Polymorphisms in metabolic genes, their combination and interaction with tobacco smoke and alcohol consumption and risk of gastric cancer: a case-control study in an Italian population. BMC Cancer, 7, 206.
Bodnar, A. G., Ouellette, M., Frolkis, M., Holt, S. E., Chiu, C.-P., Morin, G. B., Harley, C. B., Shay, J. W., Lichsteiner, S., & Wright, W. E. (1998). Extension of life-span by introduction of telomerase into normal human cells. Science, 279, 349–352.
Burhansand, W., & Weinberger, C. M. (2007). Survey and summary DNA replication stress, genome ınstability and aging. Nucleic Acids Research, 35(22), 7545–7556.
Caputo, J. L. (1996). Safety procedures. In Freshney, R. I., Freshney, M. G., eds., Culture of Immortalized Cells. New York, Wiley-Liss, pp. 25–51
Castano, A., & Becerril, C. (2004). In vitro assessment of DNA damage after short- and long-term exposure to benzo(a)pyrene using RAPD and the RTG-2 fish cell line. Mutation Research, 552(1–2), 141–151.
Cervantes, R. B., Stringer, J. R., et al. (2002). Embryonic stem cells and somatic cells differ in mutation frequency and type. Proceedings of the National Academy of Sciences of the United States of America, 99(6), 3586–3590.
Chen, T. R. (1977). In situ detection of mycoplasma contamination in cell cultures by fluorescent Hoechst 33258 stain. Experimental Cell Research, 104, 255.
Dodd, I. B., Micheelsen, M. A., Sneppen, K., & Thon, G. (2007). Theoretical analysis of epigenetic cell memory by nucleosome modification. Cell, 129, 813–822.
Fenech, M. (2000). The in vitro micronucleus technique. Mutation Research, 455, 81–95.
Freshney R.I. (1994). Culture of animal cells, a manual of basic technique 3rd edition.
Hausmann, M., Dudin, G., Aten, J. A., Heilig, R., Diaz, E., & Cremer, C. (1991). Slit scan flow cytometry of isolated chromosomes following fluorescence hybridization: an approach of online screening for specific chromosomes and chromosome translocations. Zeitschrift Für Naturforschung, 46, 433–441.
Henke, J., & Henke, L. (1999). Mutation rate in human microsatellites. American Journal of Human Genetics, 64(5), 1473–1474.
Hoboken, N. J., & Garrett, M. D. (2001). Cell cycle control and cancer current science, Vol. 81, No. 5, 10, Cell Cycle Events. Science, 246, 629–634.
Hong, Y., Cervantes, R. B., et al. (2006). DNA damage response and mutagenesis in mouse embryonic stem cells. Methods in Molecular Biology, 329, 313–326.
Hong, Y., Cervantes, R. B., et al. (2007). Protecting genomic integrity in somatic cells and embryonicstem cells. Mutation Research, 614(1–2), 48–55.
Hong, Y., & Stambrook, P. J. (2004). Restoration of an absent G1 arrest and protection from apoptosis in embryonic stem cells after ionizing radiation. Proceedings of the National Academy of Sciences of the United States of America, 101(40), 14443–14448.
Honma, M., Mizusawa, H., et al. (1994). Demonstration by DNA fingerprint analysis of genomic instability in mouse BALB 3T3 cells during cell transformation. Mutation Research, 304(2), 167–179.
Horio, T., Miyauchi-Hashimoto, H., et al. (2007). Photobiological information obtained from XPA gene-deficient mice. Photochemistry and Photobiology, 83(1), 218–224.
Hou, S.-M., Yang, Ke., Nyberg, F., Hemminki, K., Pershagen, G., & Lambert, Bo. (1999). Hprt mutant frequency and aromatic DNA adduct level in non- smoking and smoking lung cancer patients and population controls. Carcinogenesis, 20(3), 437–444.
Jones, C., & Kortenkamp, A. (2000). RAPD library fingerprinting of bacterial and human DNA: Applications in mutation detection. Teratogen. Carcinogen. Mutagen., 20, 49–63.
Katerji, M., & Duerksen-Hughes, P. J. (2021). DNA damage in cancer development: Special implications in viral oncogenesis. American Journal of Cancer Research, 11(8), 3956–3979.
Keshava, C., Keshava, N., et al. (1999). Genomic instability in silica- and cadmium chloride-transformed BALB/c-3T3 and tumor cell lines by random amplified polymorphic DNA analysis. Mutation Research, 425(1), 117–123.
Keshava, N., Zhou, G., et al. (2001). Carcinogenic potential and genomic instability of beryllium sulphate in BALB/c-3T3 cells. Molecular and Cellular Biochemistry, 222(1–2), 69–76.
Kirsch-Volders, M., Elhajouji, A., Cundari, E., & Van Hummelen, P. (1997). The in vitro micronucleustest: A multi-endpoint assay to detect simultaneously mitotic delay, apoptosis, chromosome breakage, chromosome loss andnon-disjunction. Mutation Research, 392, 19–30.
Klaude, M., Eriksson, S., Nygren, J., & Ahnstrom, G. (1996). The comet assay: Mechanisms and technical considerations. Mutation Research, 363(2), 89–96.
Kondrashov, A. S., & Crow, J. F. (1993). A molecular approach to estimating the human deleterious mutation rate. Human Mutation, 2(3), 229–234.
Lee, Y. C., Yang, V. C., et al. (2007). Use of RAPD to detect sodium arsenite-induced DNA damage in human lymphoblastoid cells. Toxicology, 239(1–2), 108–115.
Lengauer, C., Kinzler, K. W., & Vogelstein, B. (1998). Genetic instabilities in human cancers. Nature, 396, 643–649.
Lery, X., LaRue, B., et al. (2003). Characterization and authentication of insect cell lines using RAPD markers. Insect Biochemistry and Molecular Biology, 33(10), 1035–1041.
Liu, X., Yan, J., & Kirschner, M. W. (2024). Cell size homeostasis is tightly controlled throughout the cell cycle. PLoS Biology, 22(1), e3002453. https://doi.org/10.1371/journal.pbio.3002453
Lloyd, D. C. (1984). An overview of radiation dosimetry by conventional cytogenetic methods. In W. G. Eisert & M. L. Mendelsohn (Eds.), Biological dosimetry, cytometric approaches to mammalian systems (pp. 3–14). Springer-Verlag.
Markowitz, S., et al. (1995). Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science, 268, 1336–1338.
Masters, J. R. (2002). HeLa cells 50 years on: The good, the bad and the ugly. Nature Reviews Cancer, 2, 315–319.
Mather, J., & Barnes, D. (1998). Animal cell culture methods, volume 57, methods in cell biology. Academic Press.
Mendelsohn, M. L. (1989). Potential DNA methods for measuring the human heritable mutation rate. Genome, 31(2), 860–863.
Morley, A. A. (1996). The estimation of in vivo mutation rate and frequency from samples of human lymphocytes. Mutation Research, 357(1–2), 167–176.
Nachtsheim, H., Vogel, F., et al. (1960). Methods and errors in research on the mutation rate of the human gene. Zeitschrift Für Menschliche Vererbungs- Und Konstitutionslehre, 35, 320–331.
Ollins, A. R., Dobson, V. L., Dusinska, M., Kennedy, G., & Stetina, R. (1997). The comet assay: What can it really tell us? Mutation Research, 375(2), 183–193.
Pearson, H. (2008). Cell biology: the cellular hullabaloo. Nature, 453(7192), 150–153.
Pinkel, D., Landegent, J., Collins, C., Fuscoe, J., Segraves, R., Lucas, J., & Gray, J. W. (1988). Fluorescence in situ hybridization with human chromosome-specific libraries: detection of trisomy 21 and translocations of chromosome 4. Proceedings of the National Academy of Sciences of the United States of America, 85, 9138–9142.
Pretlow, T. G., & Pretlow, T. P. (1989). Cell separation by gradient centrifugation methods. Methods in Enzymology, 171, 462–482.
Rizzino, A. (2002). Embryonic stem cells provide a powerful and versatile model system. Vitamins & Hormones, 64, 1–42.
Rojas, E., Lopez, M. C., & Valverde, M. (1999). Single cell gel electrophoresis: methodology and applications. Journal of Chromatography B, 722(1–2), 225–254.
Rosa, T., & Juan, P. (2007). Hypoxanthine-guanine phosophoribosyltransferase (HPRT) deficiency: Lesch-Nyhan syndrome Orphanet. Journal of Rare Diseases, 2(1), 48.
Rossi, D. J., Bryder, D., et al. (2007). Hematopoietic stem cell aging: Mechanism and consequence. Experimental Gerontology, 42(5), 385–390.
Rossi, D. J., Seita, J., et al. (2007). Hematopoietic stem cell quiescence attenuates DNA damage response and permits DNA damage accumulation during aging. Cell Cycle, 6(19), 2371–2376.
Rothblat, G. H., Cristofalo, R., & Growth, V. J. (1972). Nutrition and metabolism of cells in culture ds eds (Vol. 1–3). Academic Press.
Sancar, A., Lindsey-Boltz, L. A., Unsal-Kacmaz, K., & Linn, S. (2004). Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annual Review of Biochemistry, 73, 39–85.
Senavirathne, G., London, J., Gardner, A., Fishel, R., & Yoder, K. E. (2023). DNA strand breaks andgaps target retroviral intasome binding and integration. Nature Communications, 14(1), 7072. https://doi.org/10.1038/s41467-023-42641-4
Sharpless, N. E., & DePinho, R. A. (2007). How stem cells age and why this makes us grow old. Nature Reviews Molecular Cell Biology, 8(9), 703–713.
Smith, J. A., & Martin, L. (1973). Do cells cycle? Proceedings of the National Academy of Sciences, 70(4), 1263–1267.
Stambrook, P. J. (2007). An ageing question: Do embryonic stem cells protect their genomes? Mechanisms of Ageing and Development, 128(1), 31–35.
Tanke, H. J., Florijn, R. J., Wiegant, J., Raap, A. K., & Vrolijk, J. (1995). CCD microscopy and image analysis of cells and chromosomes stained by fluorescence in situ hybridization. Histochemical Journal, 27, 4–14.
Uzonur, I., Abasiyanik, M. F., Bostanci, B., Eyidemir, M., Ocba, N., Yanik, C., & Petek, M. (2004). Re-exploring planaria as a model organism for genotoxicity monitoring by an ’Improved Random Amplified Polymorphic DNA’ approach. Fresenius Environmental Bulletin, 13(12a), 1420–1426.
Wolf, R. (2007). Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 447.
Wood, R. D. (1996). DNA repair in eukaryotes. Annual Review of Biochemistry, 65, 135–167.
Wood, R. D. (1997). Nucleotide excision repair ın mammalian cells. The Journal of Biological Chemistry, 272(38), 23465–23468.
Yoshida, K., & Miki, Y. (2004). Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Science, 95(11), 866–871.
Acknowledgements
I would also like to show gratitude to my supervisor, including Ms. I. Irem Uzonur. She died of blood cancer. Her teaching style and enthusiasm for the topic made a strong impression on me and I have always carried positive memories of her classes with me.
Author information
Authors and Affiliations
Molecular Biology and Genetic, Faculty of Science, Bulent Ecevit University, Zonguldak, Turkey
Arzu Erol
Corresponding author
Correspondence to Arzu Erol.
Ethics declarations
Conflict of interest
The author has no financial or proprietary interests.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article
Cite this article
Erol, A. Genomic instability evaluation in different cell line by random amplified polymorphic DNA-PCR analysis. GENOME INSTAB. DIS. (2024). https://doi.org/10.1007/s42764-024-00122-y
Received25 December 2023
Revised08 January 2024
Accepted15 January 2024
Published04 March 2024
DOIhttps://doi.org/10.1007/s42764-024-00122-y
Share this article
Anyone you share the following link with will be able to read this content:
Get shareable link
用户登录
还没有账号?
立即注册