






The silent guardian: unraveling the roles of H3K9me3 in genome maintenance
Review Article
Published: 02 July 2024
Zhiming Li & Zhiguo Zhang
Volume 5, pages 133–153, (2024)
Abstract
Genomic integrity is a fundamental prerequisite for all living organisms, ensuring the accurate transmission of genetic information and stability of cellular functions across generations. The maintenance of genome integrity relies on a meticulously orchestrated network encompassing a variety of DNA repair factors and pathways. In eukaryotic cells, genomic DNA is assembled into chromatin, a highly organized complex of proteins and DNA. Therefore, chromatin and epigenetic factors have emerged as crucial guardians against genotoxic stress. In this review, we provide a comprehensive summary of the multifaceted roles of H3K9me3, a histone mark associated with silent chromatin, in DNA damage repair and genome maintenance, elucidating its dynamic participation in chromatin organization, silencing of repetitive DNA sequences, and modulation of DNA repair pathways. Importantly, we discuss the potential contribution of H3K9me3 to epigenetic memory following DNA damage, which introduces an additional layer of complexity to our comprehension of genomic surveillance. Finally, we explore the implications of H3K9me3 dysregulation in human cancers and the promising therapeutic avenues that may arise from a nuanced understanding of its function in maintaining genomic stability.
Introduction
The integrity of the genetic material is constantly threatened by a myriad of endogenous and exogenous factors, such as ionizing radiation, ultraviolet light, chemical mutagens, and errors during DNA replication, leading to various forms of DNA damage and even cell death if left unattended. To counteract these assaults, cells have evolved highly sophisticated and tightly controlled DNA damage repair pathways, which form a dynamic network of hundreds of factors to safeguard the fidelity of the genome (Giglia-Mari et al., 2011; Sancar et al., 2004; Scully et al., 2019). The regulation of DNA damage repair is governed by cellular surveillance mechanisms, which can detect DNA lesions and activate DNA damage checkpoints that temporarily halt the cell cycle, providing time for repair before cell division (Branzei & Foiani, 2008; Cimprich & Cortez, 2008; Marechal & Zou, 2013; Sancar et al., 2004). Depending on the types of lesions, DNA damage is repaired through different pathways, including base excision repair (BER), nucleotide excision repair (NER), and double-strand break repair (DSBR) (Sancar et al., 2004). DSBs are the most cytotoxic due to their highly deleterious nature, which are typically repaired by homologous recombination (HR) and non-homologous end joining (NHEJ) pathways (Ceccaldi et al., 2016; Chapman et al., 2012; Scully et al., 2019; Shrivastav et al., 2008). Defects in these repair processes are associated with a range of human disorders, including cancer predisposition, aging and neurodegenerative diseases, underscoring the critical role of DNA damage repair in maintaining cellular health (Curtin, 2012; Hoeijmakers, 2009; Schumacher et al., 2021; Tubbs & Nussenzweig, 2017; Wilson et al., 2023; Zhao et al., 2023b).
In eukaryotic cells, the genomic DNA is packaged into chromatin, with the nucleosome consisting of 147 bp DNA wrapped around a histone octamer, as the basic repeating unit. Chromatin is further organized into different functional domains demarcated by various types of covalent histone post-translational modifications (PTMs). For example, histone acetylation, which creates a more relaxed chromatin architecture and facilitates the recruitment of transcriptional activators via acetyl-lysine recognizing bromodomains, promotes the assembly of the transcriptional machinery, thus leading to increased gene transcription (Grunstein, 1997; Struhl, 1998). Contrary to the permissive nature of histone acetylation, H3K27me3, a marker of facultative heterochromatin by the Polycomb complex PRC2, inhibits transcription by inducing chromatin compaction and promoting the loading of repressor complexes (Barski et al., 2007; Croce & Helin, 2013; Margueron & Reinberg, 2011; Mikkelsen et al., 2007).
It has become increasingly clear that chromatin regulators are critical players in coordinating cellular responses to DNA damage and maintaining genome stability (Hauer & Gasser, 2017; Lukas et al., 2011b; Soria et al., 2012; Yasuhara & Zou, 2021). Indeed, the dynamic and reversible nature of histone modifications extends their influence beyond transcription to encompass DNA damage repair and genome stability (Altaf et al., 2007; Cao et al., 2016; Kim et al., 2019). For example, one of the most prominent histone modifications involved in DNA damage repair is phosphorylation on serine 139 of H2AX, a histone H2A variant, namely γ-H2AX (Fernandez-Capetillo et al., 2004; Mah et al., 2010). In response to DSBs, γ-H2AX is rapidly installed by PI3K-like kinases, including ATM, ATR, and DNA-PK, which serves as an anchor point for the subsequent cascade of response and repair factors that are enlisted to resolve the DNA lesions, covering megabase (Mb)-sized genomic regions (Blackford & Jackson, 2017; Li et al., 2020a). H4K20me2, an abundant histone mark, also plays key roles in dictating cellular response to DSBs (Jorgensen et al., 2013; Kohi et al., 2022). In particular, H4K20me2 forms a bivalent chromatin docking site with H2AK15 ubiquitylation that recruits 53BP1 (p53 binding protein 1), a crucial component in DSB signaling and repair choices (Botuyan et al., 2006; Fradet-Turcotte et al., 2013; Mattiroli et al., 2012; Pei et al., 2011; Stewart et al., 2009; Wilson et al., 2016). Therefore, gaining a deeper comprehension of how different histone modifications participate in the response and repair of DNA damage holds the potential to enhance our knowledge of maintaining genome integrity and identifying possible targets for therapeutic interventions in diseases.
In this review, we will focus on the various mechanisms employed by H3K9me3 to modulate DNA damage response/repair and maintain genome integrity (Fig. 1). Among the vast array of histone modifications that have been extensively delineated, H3K9me3 is a key repressive mark at constitutive heterochromatin that is composed of mainly repetitive DNA sequences. In addition, H3K9me3 is increasingly acknowledged for its versatile involvement in numerous cellular processes, such as embryonic development and stem cell differentiation, through both classic and alternative mechanisms. For example, H3K9me3 decorating a subset of cell-type specific genes, which are subjected to profound rearrangements and lose H3K9me3 signals during lineage commitment to allow for their transcriptional activation (Nicetto & Zaret, 2019; Nicetto et al., 2019), marks a pivotal role of H3K9me3 in cell fate determination. Ectopic H3K9me3 deposition by forced expression of SUV39h enzymes leads to compromised differentiation and epigenetic reprogramming (Burton et al., 2020). In addition, the revelation of bivalent domains containing H3K9me3 and other active marks, such as H3K4me3 and H3K36me3 (Barral et al., 2022; Matsumura et al., 2015), further indicates that H3K9me3 functions in a chromatin context-dependent manner. Our recent study indicate that parental H3K9me3 asymmetrically distribute at head-on LINE elements at the leading strands during DNA replication (Li et al., 2023). These findings underscore the multilayered roles entrusted to H3K9me3 as active defenders of the eukaryotic genome.
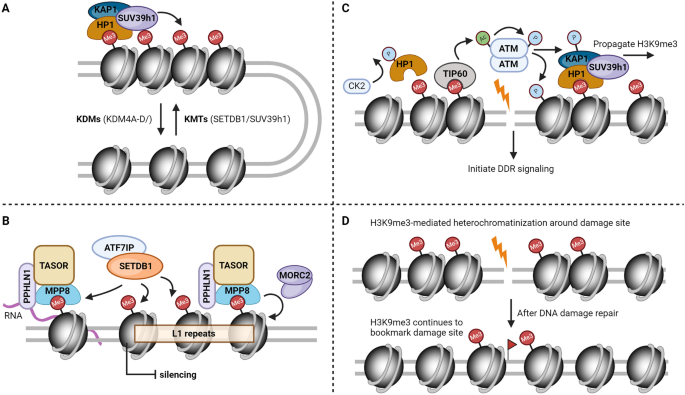
Simplified schematic models of the roles of H3K9me3 in maintaining genome stability. A H3K9me3 in the establishment and maintenance of heterochromatin. B An example of H3K9me3 in the repression of transposable elements (L1). C H3K9me3 in the regulation of cellular response to DNA damage. D Potential involvement of H3K9me3 in bookmarking DNA breaks and forming cellular memory of DNA damage
H3K9me3 writers, erasers and readers
H3K9me3 stands out as the unmistakable hallmark of heterochromatin. Like many other histone methylation marks, H3K9me3 is regulated by its writers, erasers and readers. Early studies in Drosophila identified factors involved in suppressing the position effect variegation (PEV) [Su(var)], which were the first known lysine methyltransferases (KMTs) for H3K9 in human/murine (SUV39H1/Suv39h1), Drosophila [Su(var)3–9] and yeast (clr4) (Nakayama et al., 2001; Rea et al., 2000). Several more conserved H3K9 KMTs were later identified, including ESET/SETDB1, G9a/GLP and Prdm3/Prdm16 (Pinheiro et al., 2012; Schultz et al., 2002; Tachibana et al., 2001). Although there are some redundancies among these KMTs in cells, they generally have different substrate preferences, targeting genomic regions and binding partners (Padeken et al., 2022). For example, while both SUV39h1 and SETDB1 can catalyze H3K9 from mono-methylation to tri-methylation status in vivo, G9a and GLP are the primary enzymes for H3K9me1 and H3K9me2, which play vital roles in transcription silencing and embryogenesis (Shinkai & Tachibana, 2011; Tachibana et al., 2005, 2008). Prdm3 and Prdm16, on the other hand, are only responsible for cytosolic H3K9me1 that aids in the establishment and reinforcement of heterochromatin mediated by SUV39h enzymes (Pinheiro et al., 2012). Cellular levels of H3K9me3 are balanced by the actions of lysine demethylases (KDMs), including the KDM4 subfamily (rph1 in yeast) that targets H3K9me2/3 and KDM3 that targets H3K9me1/2 (Whetstine et al., 2006; Yamane et al., 2006). KDM1A/LSD1 and KDM7 (KDM7A, PHF8 and PHF2) can also demethylate H3K9me1/2, albeit with weaker specificity (Metzger et al., 2005; Tsukada et al., 2010). By reversing the histone methylation marks installed by KMTs, these KDMs counteract the repressive effects imposed by H3K9 methylation and generally facilitate a more permissive chromatin environment (Greer & Shi, 2012; Kooistra & Helin, 2012). For example, H3K9 demethylation mediated by LSD1 activates androgen-receptor-dependent transcription of target genes (Metzger et al., 2005; Shi et al., 2004). KDM4D-dependent demethylation of H3K9me3 promotes the formation of pre-initiative complex, which is required for efficient DNA replication (Wu et al., 2017). Together, by installing and removing H3K9 methylation in a highly spatiotemporal manner, these KMTs and KDMs ensure its precise and dynamic distribution throughout the genome (Fig. 1A).
H3K9me3 recruits a number of readers, a dedicated group of effector proteins harboring methyl-lysine binding domains to exert its downstream functions. Among the growing list of readers, heterochromatin protein 1 (HP1) was the first to be identified to recognize and bind H3K9me3 through its N-terminal chromodomain, which is essential for heterochromatin formation and transcription repression (Bannister et al., 2001; Jacobs et al., 2001; Lachner et al., 2001; Nakayama et al., 2001). HP1 in turn recruits SUV39h1, which spreads H3K9me3 to flanking nucleosomes, forming a positive feedback loop that consolidates the repressive chromatin domain over a long stretch of genomic DNA (Aagaard et al., 1999; Bannister et al., 2001). HP1 chromatin loading is further dynamically regulated by the mitotic H3K9/S10 ‘methyl/phospho switch’ (Fischle et al., 2005; Hirota et al., 2005). UHRF1 (ubiquitin-like PHD and RING finger domain-containing 1) is another well-characterized H3K9me3 reader through its tandem Tudor domain, which is required for the maintenance of DNA methylation by DNMT1 (DNA methyltransferase 1) (Bostick et al., 2007; Liu et al., 2013; Rothbart et al., 2012; Sharif et al., 2007). Later studies suggest that DNMT1 can directly read H3K9me3 through its RFTS (replication foci targeting sequence) domain, which boosts DNMT1 activity stimulated by H3 ubiquitylation and faithful inheritance of DNA methylation patterns (Ren et al., 2020). A recent study revealed that TNRC18 (trinucleotide-repeat-containing 18) reads H3K9me3 through its C-terminal BAH domain, which is required for silencing of several families of endogenous retroelements (Zhao et al., 2023a). In addition, H3K9 methylation has also been reportedly recognized by plant homeodomain (PHD) fingers (Mansfield et al., 2011; Tencer et al., 2017), WD40 domain (Chan & Zhang, 2012), MBT domain (Klymenko et al., 2006) and ankyrin repeats (Collins et al., 2008). Interestingly, many H3K9me3 writers also contain these functional domains (Zhang et al., 2015a). For example, SUV39h1/2 (Clr4 in yeast) contains a chromodomain at its N terminus that binds H3K9me3, which is required for SUV39h/Clr4 enzymatic activity, H3K9me3 propagation and stable inheritance (Al-Sady et al., 2013; Melcher et al., 2000; Shirai et al., 2017; Wang et al., 2012; Zhang et al., 2008). These results suggest a direct ‘read-write’ mechanism utilized by H3K9me3 writers for its precise restoration of H3K9me3 on nucleosomes formed with newly synthesized histones using parental and existing H3K9me3 as templates following DNA replication. Importantly, the interplay between H3K9me3 readers and enzymes that regulate other chromatin marks creates an opportunity for epigenetic crosstalk between H3K9me3 and other histone modifications (Lee et al., 2010; Suganuma & Workman, 2008). For example, H3K9me3-anchored HP1 recruits SUV4-20h enzymes, which generate H4K20me3, another characteristic heterochromatic mark (Schotta et al., 2004).
H3K9me3 in the silencing of repetitive DNA sequences
A substantial portion of the eukaryotic genome is made up of repetitive DNA elements, which are generally organized into heterochromatin (Consortium, 2012; Lander et al., 2001; Nurk et al., 2022). The formation of heterochromatic domains is essential to prevent homologous recombination and abnormal expression of these DNA repeats (Allshire & Madhani, 2018; Janssen et al., 2018). As a critical regulator of the establishment and maintenance of heterochromatin states, H3K9me3 plays an important role in maintaining the stability of these repetitive elements in the genome (Allshire & Madhani, 2018; Grewal & Jia, 2007; Janssen et al., 2018; Padeken et al., 2022). Indeed, epigenomic profiling of H3K9me3 reveals its enrichment at repetitive DNA sequences (Barski et al., 2007; Gershman et al., 2022; Mikkelsen et al., 2007; Roadmap Epigenomics et al., 2015). Consistent with the importance of H3K9me3 at repetitive DNA sequences, double knockout of SUV39h1 and SUV39h2, which predominantly target satellite repeats at the pericentric heterochromatin, leads to weakened heterochromatin formation, reduced viability and developmental potential, spermatogenic failure and heightened risk of malignant transformation (Peters et al., 2001). Notably, the overall organization and clustering of heterochromatin remain largely intact in these cells (Peters et al., 2001), likely due to the compensating roles of other H3K9 KMTs and H3K9me3-independent mechanisms (Allshire et al., 2018; Janssen et al., 2018; Padeken et al., 2022). Besides its fundamental roles in silencing DNA repeats, H3K9me3 also suppresses transposable elements that scatter throughout the genome, including euchromatin. For example, together with histone variant H3.3, H3K9me3, deposited primarily by SETDB1, potentiates the silencing of endogenous retroviruses (ERVs) and evolutionarily young long interspersed elements (LINEs) that are widely dispersed in the open chromatin compartment (Elsasser et al., 2015; Karimi et al., 2011; Liu et al., 2018; Matsui et al., 2010; Robbez-Masson et al., 2018; Tchasovnikarova et al., 2015; Yang et al., 2015). SUV39h-dependent H3K9me3 also selectively targets a group of intact LINEs for their transcriptional silencing (Bulut-Karslioglu et al., 2014). G9a and GLP, on the other hand, play a more specific role in repressing class III endogenous retroviruses (MERVLs) and MERVL LTR-driven genic transcripts in two-cell stage embryos (Maksakova et al., 2013). Finally, progressive loss of all the six methyltransferases associated with H3K9me3, including G9a/GLP, SUV39h1/SUV39h2 and SETDB1/SETDB2, completely abolishes H3K9me3, resulting in activation of almost all families of repeat elements, dissolution of heterochromatin and massive genomic instability (Montavon et al., 2021).
Of note, while the activities of retrotransposons pose a threat to genome stability, they are also required for normal embryonic development (Dopkins & Nixon, 2024; Guo et al., 2024; Senft & Macfarlan, 2021). Mechanistically, transposable elements rewire the core transcription network of stem cells by providing transcription factor binding sites, cis-regulatory elements, alternative splicing sites and non-coding RNA that regulate pluripotency genes (Bourque et al., 2018; Fueyo et al., 2022). During early stages of mammalian embryo development, H3K9me3 undergoes dynamic reprogramming and large-scale re-allocation to selectively tune the stage-specific expression of lineage-specific genes, long terminal repeats (LTRs) and hominoid-specific SVA (SINE-VNTR-Alu) elements, which is critical for lineage commitment and embryogenesis (Wang et al., 2018; Xu et al., 2022a; Yu et al., 2022). As a result, disrupting H3K9me3 by deletion of SETDB1 and other KMTs, histone chaperones (e.g., CAF1), or certain transcription regulators (e.g., DUX and ZFP51) caused varying degrees of faulty development and embryonic lethality (Dodge et al., 2004; Nicetto et al., 2019; Wang et al., 2018; Xu et al., 2022a). In addition, genomic regions resistant to epigenetic reprogramming by iPS or somatic cell nuclear transfer (SCNT) are enriched with repeat elements, which are marked by H3K9me3 (Becker et al., 2016).
In general, H3K9me3 is deposited to specific repetitive DNA sequences by its methyltransferases through sequence-specific regulators as well as local transcription. Particularly, low levels of transcription within heterochromatin, which produces small non-coding RNAs, such as small interfering RNAs (siRNAs) and PIWI-interacting RNAs (piRNAs), is crucial for the recruitment of silencing factors and subsequent heterochromatin formation (Castel & Martienssen, 2013; Motamedi et al., 2004; Reinhart & Bartel, 2002; Verdel et al., 2004). Importantly, these suppressors coordinate with H3K9me3 to achieve sequence-specific targeting and robust silencing. For example, KRAB-ZFP (Kruppel-associated box zinc finger protein) transcription factors can recognize the primer binding site (PBS) of ERVs and initiate their heterochromatic silencing by assembling the repressor complex containing HP1, SETDB1 and KAP1 (Geis & Goff, 2020; Yang et al., 2017a). One prominent example is ZFP809, which bridges KAP1/TRIM28 and the PBS sequences of integrated MLVs (murine leukaemia viruses) to mediate their transcriptional silencing in stem cells (Wolf & Goff, 2007, 2009; Wolf et al., 2015). The HUSH (human silencing hub) complex, consisted of TASOR (transcription activation suppressor), MPP8 (M-phase phosphoprotein 8), and periphilin, is another active defender against repetitive DNA and plays an important role for the establishment of silencing at LINE elements (Muller & Helin, 2024; Seczynska & Lehner, 2023). MPP8 binds H3K9me3 through its chromodomain, which helps maintain HUSH at evolutionarily young and intact L1 retrotransposons, where HUSH tethers SETDB1 to deposit H3K9me3 and repress these L1s and intronless mobile elements (Douse et al., 2020; Liu et al., 2018; Prigozhin et al., 2020; Robbez-Masson et al., 2018; Seczynska et al., 2022; Tchasovnikarova et al., 2015). HUSH also recruits other cofactors, such as MORC2 (microrchidia CW-type zinc finger 2) and the NEXT (nuclear exosome targeting) complex to facilitate silencing of retroelements (Garland et al., 2022; Tchasovnikarova et al., 2017). As a result, HUSH depletion leads to widespread L1 activation and transcription deregulation of coding genes, which causes embryonic lethality and compromised stem cell self-renewal (Seczynska & Lehner, 2023). De-repression of L1s also generates double-stranded RNAs that induce interferon-stimulated genes and inflammatory reactions (Tunbak et al., 2020), which present novel therapeutic opportunities in anti-tumor immunotherapy (see more discussions below). Our recent results further suggest that HUSH cooperates with DNA polymerase Pol ε to mediate repression of L1 expression and retrotransposition during DNA replication (Li et al., 2023), revealing a role of the HUSH complex in the maintenance of L1 silencing. Importantly, the fact that only evolutionarily young and transcriptionally active L1s are targeted by HUSH suggests that HUSH represents an effective mechanism of genome surveillance against foreign nucleic acids (Liu et al., 2018; Robbez-Masson et al., 2018). Together, these studies underscore the central roles of H3K9me3 in defending against mobile DNA elements and maintaining genome integrity (Fig. 1B).
Direct involvement of H3K9me3 in DNA damage repair
One of the first pieces of evidence supporting the direct involvement of H3K9me3 in DNA damage response and repair comes from the observation that H3K9me3 is accumulated at the damage sites (Ayrapetov et al., 2014; Sun et al., 2009). Later studies further confirm the enrichment of several H3K9me3 methyltransferases, including SUV39h1, G9a, GLP and SETDB1, at damaged chromatin or DNA lesion sites (Alagoz et al., 2015; Ayrapetov et al., 2014; Ginjala et al., 2017; Li et al., 2020a; Lu et al., 2019; Yang et al., 2017b). By sensing the chromatin perturbations arising from DNA damage, H3K9me3 is either exposed by casein kinase 2 (CK2)-catalyzed phosphorylation and removal of HP1β or deposited to lesion sites within the first few seconds by SETDB1 and SUV39h1 to facilitate transient local chromatin compaction (Alagoz et al., 2015; Ayoub et al., 2008; Ayrapetov et al., 2014), which remodels the damaged chromatin to create an efficient template for the DNA repair machinery. Interestingly, such transient and dynamic H3K9me3 deposition is also recently reported as vital for the protection and restart of stalled replication forks during replication stress (Gaggioli et al., 2023), suggesting that H3K9me3-mediated heterochromatinization serves a more general purpose in safeguarding the genome from genotoxic stress.
While chromatin undergoes a swift cascade of dynamic epigenetic modifications upon DNA damage, H3K9me3 is regarded as a key signal to attract a multitude of factors essential for the initial response. For example, H3K9me3 recruits TIP60, a crucial histone acetyltransferase, through binding of its N-terminal chromodomain to DSB sites, by which TIP60 is supercharged to acetylate and activate ATM (Sun et al., 2005, 2009), an apical kinase in DNA damage response. TIP60 also acetylates histones H4 and H2AX, promoting chromatin remodeling and γ-H2AX turnover, both of which are important for effective DNA repair (Ikura et al., 2007; Murr et al., 2006). In addition, a repressive complex comprising of KAP1, HP1 and/or SUV39h1 is loaded onto H3K9me3-marked DSB lesions through the chromodomain of HP1, which initiates a cycle that quickly propagates H3K9me3 across a long stretch of DNA flanking the damage sites (Ayoub et al., 2008; Ayrapetov et al., 2014; Baldeyron et al., 2011; Luijsterburg et al., 2009) (Fig. 1C). Intriguingly, efficient SUV39h1 recruitment and H3K9me3 deposition are later found to be contingent on TIP60 (Tu et al., 2020), or H4 monoufmylation by UFM1 specific ligase 1 (UFL1), a newly discovered histone PTM, and its reader serine/threonine kinase 38 (STK38) (Qin et al., 2019, 2020). Upon the initial activation induced by chromatin compaction, ATM in turn phosphorylates KAP1, leading to the disassembly of the repressive KAP1/HP1/SUV39h1 complex, which restores DNA accessibility and facilitates the recruitment of additional repair factors, thereby establishing a negative feedback loop (Ayrapetov et al., 2014).
Besides its well-illustrated role in the TIP60-ATM axis, H3K9me3 is also instrumental in the pathway choices to resolve DSB, which are governed by 53BP1 and BRCA1, critical factors in DNA repair mediated by non-homologous end joining (NHEJ) and homologous recombination (HR), respectively (Panier & Boulton, 2014). While not a direct reader of H3K9me3, 53BP1 recruitment to the DNA lesions is indirectly regulated by H3K9me3. For example, TIP60, stimulated by H3K9me3, acetylates histone H2A and H4, which prevents 53BP1 loading to damaged chromatin through its bivalent recognition of histone modifications, thus favoring the HR pathway (Jacquet et al., 2016; Tang et al., 2013). However, later studies confirmed the interaction and co-localization between 53BP1 and H3K9me3 in cells (Zhang et al., 2022), suggesting a more intertwined relationship. In addition, chromatin condensation created by the cooperation between H3K9 methyltransferases, histone variant macroH2A1 and HP1 facilitates the recruitment and retention of BRCA1, which antagonizes the actions of 53BP1 and promotes DNA end resection (Khurana et al., 2014; Lee et al., 2013; Wu et al., 2015). These reports resonate with findings that heterochromatic DSBs, while complicated due to the limited accessibility and high repetitiveness, are largely repaired by HR (Chiolo et al., 2011). However, it should be noted that the NHEJ pathway is preferentially utilized for DSB repair in both heterochromatin and euchromatin when cells are mostly in G0/G1 and HR is inhibited (Janssen et al., 2016; Tsouroula et al., 2016). Moreover, the effects of H3K9me3-associated writes and readers in DSB repair choices can go both ways, exemplified by the different HP1 isoforms (HP1α/β promote HR while HP1γ is inhibitory) (Soria & Almouzni, 2013) and GLP/G9a (GLP promotes NHEJ while G9a favors HR) (Lu et al., 2019; Yang et al., 2017b), through varying mechanisms. These distinctions, likely stemming from the differences in genetic backgrounds, DNA damage types, local chromatin context of damage sites, availability of repair factors and/or cell cycle stages, highlight the importance to examine the role of H3K9me3 in DNA damage repair from a context-dependent perspective.
Heterochromatin constitutes a structural obstacle to efficient DNA repair by hindering the access of repair factors to damage sites (Hauer & Gasser, 2017; Soria et al., 2012), which necessitates the timely removal of H3K9me3 once it has accomplished its mission. In fact, most damage-elicited epigenetic marks are temporary and compromising their reversal generates sustained DNA damage response (Polo & Jackson, 2011), which is detrimental to cell survival. For example, deacetylation of ATM by histone deacetylase SIRT7 is required for ATM dephosphorylation and deactivation, facilitating an orderly exit as soon as DNA repair is finished (Tang et al., 2019). Therefore, persistent ATM acetylation by SIRT7 deletion or acetylation-mimic point mutation sequesters the repair apparatus at the damage sites, preventing the seamless completion of DNA damage repair (Tang et al., 2019). Analogously, demethylation of H3K9me3 by its erasers, KDM4/JMJD2/JHDM3, is also consequential for DNA repair. Both KDM4B and KDM4D have been reported to be mobilized to DNA damage sites, which depends on the activity of poly (ADP-ribose) polymerase 1 (PARP1) (Khoury-Haddad et al., 2014; Young et al., 2013), suggesting a rather early engagement of H3K9me3 demethylation in this process. KDM4D-mediated H3K9me3 demethylation is required for efficient DNA replication and repair (Khoury-Haddad et al., 2014; Wu et al., 2017), both of which are important aspects of guarding the genome. The reports regarding KDM4A in DNA damage repair, however, have been conflicting (Fnu et al., 2011; Mallette et al., 2012), likely due to its dual ability to erase H3K9/K36 methylation and read H4K20me2. Altogether, these research efforts support a model whereby the dynamics of H3K9me3 at DNA damage sites not only configurates a restrictive local chromatin microenvironment, but also creates a docking platform for a number of repair factors in a highly coordinated manner.
Inheritance of epigenetic information following DNA damage repair
Given the disruptive and short-lived nature of the epigenetic changes triggered by DNA damage, it is counterintuitive to imagine that cells would form any type of memory of DNA damage. Previous epidemiologic studies in the offsprings of people working in or living close to nuclear power plants and survivors of the atomic bombs have generated inconsistent results (Gardner et al., 1990; Kodaira et al., 1995, 2010; Little et al., 2013; Morton et al., 2021; Yeager et al., 2021). However, both short-term and long-term epigenetic memory, that is, chromatin signatures that can be preserved throughout mitosis and passed on to progeny cells independent of the initial assault, have been documented. One of the first clues came from a phenomenon termed radiation-induced bystander effect (RIBE), which refers to the chromosomal instability observed in non-exposed cells that are in close proximity or even distant from the site of initial radiation (Mothersill & Seymour, 2004). Nonirradiated hemopoietic stem cells that were transplanted into mouse bone marrow sustained genomic instability over many generations for up to 24 months, conceivably transmitted from their irradiated counterparts (Watson et al., 2000). While the underlying mechanisms remain enigmatic, altered DNA methylation, histone modifications and non-coding RNAs have been implicated in this process (Ilnytskyy et al., 2009; Koturbash et al., 2007). γ-H2AX, a widely used marker of DSB repair efficiency, is found to linger for up to several months (Siddiqui et al., 2015), well beyond when DNA lesions are normally expected to be remedied. Other repair factors, such as 53BP1, have also been reported to persist through mitotic cell divisions (Markova et al., 2007). Although it's possible that low doses of unresolved or irreparable breaks tolerated by cells lead to the persistence of γ-H2AX and its associated factors (Banath et al., 2010; Fumagalli et al., 2012), they provide evidence of relatively long-lived epigenetic perturbations caused by DNA lesions. Functionally, unprogrammed DSBs induce transcriptional silencing, instigated by DNA damage sensing kinases (ATM and DNA-PK) and PARP1 (Kruhlak et al., 2007; Shanbhag et al., 2010), which can be stably inherited for more than 30 passages in certain groups of cells. Within the complex array of factors at play, altered epigenomic profiles have been increasingly recognized to consolidate the repressive chromatin organization and contribute significantly to the heritable gene silencing patterns (Machour & Ayoub, 2020). These studies, in which the chromatin landscape needs to be more closely monitored following DNA damage, do suggest that the epigenome may not always be restored identically after the completion of DNA repair, probably reflecting the delicate balance between epigenetic fidelity and plasticity. This heritable epigenetic scar hence serves as a fingerprint that likely facilitates continuous surveillance against secondary chromosomal defects or future genotoxic events, resembling the immunological memory.
Among the DNA damage-induced epigenetic changes, histone modifications can bookmark chromatin status and persist over many generations, in a process termed epigenetic inheritance (Du et al., 2022). In both yeast and mammalian cells, pre-existing parental histones retain the positional memory after its transfer to replicating DNA for the formation of nascent chromatin, at least in closed chromatin regions (Escobar et al., 2019; Schlissel & Rine, 2019), an imperative for the re-establishment of functional epigenomes in the daughter cells. (Escobar et al., 2021; Stewart-Morgan et al., 2020). During mitotic cell divisions, parental histones are recycled in a strand-specific fashion, guided by POLE3-POLE4 and MCM2-Pol α, to the leading and lagging strands, respectively (Gan et al., 2018; Li et al., 2020b; Petryk et al., 2018; Xu et al., 2022b; Yu et al., 2018a). Disruption of these pathways, not surprisingly, alters DNA damage response and repair. For example, in yeast, mutations of these two pathways invariably impaired the error-free repair by HR (Dolce et al., 2022; Gonzalez-Garrido & Prado, 2023; Karri et al., 2024). The impact on HR is likely a result of unbalanced parental histone distribution at the nascent strands or increased free histones that cannot be properly deposited (Gonzalez-Garrido & Prado, 2023; Karri et al., 2024). In human cells, POLE3 depletion impedes both HR and NHEJ (Mao et al., 2023). Together, these studies hinted a link between histone modifications and epigenetic memory of DNA damage.
Potential role of H3K9me3 in DNA damage-induced epigenetic memory
The intertwined relationship between DNA repair and factors involved in epigenetic inheritance raises the possibility that cellular memory of DNA damage is stored by histone modifications. Indeed, a recent breakthrough study demonstrated that faithful DNA repair gradually erodes the epigenetic landscape, which advances aging at both the molecular and physiological levels (Yang et al., 2023). Using the ICE (inducible changes to the epigenome) system, non-mutagenic double-strand breaks (DSBs) introduced by the I-PpoI endonuclease were quickly repaired both in vitro and in vivo. Over time, this process initiates the loss of epigenetic information and ultimately drives mammalian aging, supporting the existence of damage-induced epigenetic memory. However, contrary to the discussions above, this information theory of aging portraits a different outcome of such memory, whereby the aberrant chromatin features exacerbate epigenetic instability and render cells more vulnerable to DNA damage, setting up a positive feedback loop that accelerates the aging process (Lu et al., 2023; Yang et al., 2023). Alternatively, one can argue that this mechanism, caught up in the dilemma of a genotoxic catastrophe, likely prevents other more deleterious consequences, such as apoptosis and carcinogenesis. Importantly, H3K9me3 plays a major regulatory role in the aging process driven by erosion of epigenetic integrity. Epigenetic alterations, including global loss of core histones and repressive chromatin marks, have been considered a hallmark of aging (Kane & Sinclair, 2019; Lopez-Otin et al., 2023). Catalytically inactive mutation of SUV39h1 recapitulates the syndromes observed in Werner syndrome (WS), an accelerated aging disorder, which can be rescued by overexpression of HP1 (Zhang et al., 2015b). Another groundbreaking study in C. elegans provides more compelling evidence to support the role of H3K9me3 as a mediator of DNA damage memory (Wang et al., 2023). In this elegant design, DNA damage induced lethality is inherited to the next generation produced from irradiated male and healthy female worms. Mechanistically, DNA damage-induced heterochromatin formation redirects the germline cells to utilize a more error-prone repair pathway, effectively overcoming the biological checkpoints that would typically pause embryonic development. Removal of linker histone (HIS-24) or heterochromatin protein-1 (HPL-1) in the male worms reduces H3K9me3 and enables the high-fidelity HR pathway, thus reversing the transgenerational embryonic lethality (Wang et al., 2023). Taken together, these results lay the groundwork for future studies to further delineate the role of H3K9me3 in bookmarking DNA damage (Fig. 1D).
It remains an open question whether and how H3K9me3- and heterochromatin-marked epigenetic memory of DNA damage exists in the long-term or in other higher eukaryotic species. As possibly the most stable histone modification, H3K9me3 has been proven to perpetuate through mitosis and meiosis in many systems and organisms (Du et al., 2022). Despite the lack of definitive evidence, there are, however, a few indications that we can speculate on. Firstly, H3K9me3 is continuously engaged in DNA damage response and repair, from the very beginning to beyond the completion of DNA repair, a prerequisite for H3K9me3 to pass on any heritable information. Secondly, besides its role in cell fate determination during development, H3K9me3 also drives the long-term effects of stress response, such as therapy- or oncogene-induced senescence that alters tumor progression and treatment outcomes (Braig et al., 2005; Di Micco et al., 2011; Dorr et al., 2013; Milanovic et al., 2018; Schleich et al., 2020; Yu et al., 2018b). Thirdly, H3K9me3 extensively marks repetitive sequences, which are more susceptible to endogenous damage associated with replication stress (Janssen et al., 2018). This susceptibility could lead to repeated cycles of H3K9me3 deposition, which protects these sequences and creates a permanent long-term memory through millions of years of evolution. Alternatively, due to the unique challenges posed by heterochromatin organization, H3K9me3 bookmarks the damaged loci, which allows them to be amended in a more confined nuclear compartment, or in a slower yet more accurate fashion, thus serving as short-term memory of DNA damage. In line with this hypothesis, increased mobility and relocation of DSBs (Hauer & Gasser, 2017; Lamm et al., 2021) and chromatin compartmentalization (Arnould et al., 2021, 2023), have been observed. H3K9me3-enriched chromatin domains also exhibit slower kinetics of DNA repair (Adar et al., 2016; Han et al., 2016). Paradoxically, H3K9me3-occupied regions are characterized with increased mutation burden and decreased DNA repair by MMR (mismatch repair) and NER (nucleotide excision repair) in cancers (Supek & Lehner, 2015; Zheng et al., 2014). Finally, H3K9me3 positions the repressive chromatin domains at the back end of replication timing, the deregulation of which disrupts the epigenome and causes replication stress (Klein et al., 2021), leading to further chromosomal aberrations and DNA damage that can be inherited to the unstressed daughter cells (Techer et al., 2017). While several pathways have been proposed to control DNA damage provoked by disordered replication timing (Lukas et al., 2011a; Spies et al., 2019; Wu et al., 2023), it would be tempting to investigate how H3K9me3 takes part in this process.
H3K9me3 as an anti-tumor therapeutic target
Epigenetic alterations have been considered a crucial driver in the onset and progression of a growing number of human diseases, such as cancer, neurodegeneration, auto-immune diseases and aging-related disorders. In fact, epigenetic reprogramming is regarded as one of the hallmarks of cancer and targeting the cancer epigenome has emerged as a promising anti-tumor approach (Cheng et al., 2019; Davalos & Esteller, 2023; Hanahan, 2022; Suva et al., 2013; Zhao et al., 2021). The first of such attempts, employing inhibitors of histone deacetylases and DNA methylation, alone or in combination, dubbed ‘epigenetic therapy’, effectively and selectively kills a variety of malignant cells (Cameron et al., 1999; Yoo & Jones, 2006; Zhu & Otterson, 2003), and has paved the way for a handful of FDA-approved drugs for tumor treatment. Later reports of histone demethylases put histone methylation on the map and invigorated the efforts to target these marks that were originally thought to be irreversible (Shi et al., 2004; Tsukada et al., 2006). Recent landmark discoveries of recurrent oncogenic histone (onco-histone) mutations in pediatric glioma, chondroblastoma, sarcoma and several other specific cancer types further highlight the driving roles of the epigenetic paradigm in tumorigenesis (Behjati et al., 2013; Chan et al., 2013a; Fang et al., 2016; Lewis et al., 2013; Lu et al., 2016; Nacev et al., 2019; Papillon-Cavanagh et al., 2017; Schwartzentruber et al., 2012; Wu et al., 2012). Importantly, the genetic vulnerabilities and dependencies on particular epigenetic factors conferred by these onco-histones, such as chromatin remodelers and modifiers (Grasso et al., 2015; Hashizume et al., 2014; Mo et al., 2022; Nagaraja et al., 2017; Panditharatna et al., 2022; Piunti et al., 2017), present a unique therapeutic opportunity to cure some of these deadly tumors. While lysine-to-methionine substitutions at K9 have not been documented in cancer cohorts, introduction of H3K9M reprograms the global epigenome in mammalian cells (Brumbaugh et al., 2019; Chan et al., 2013b; Lewis et al., 2013), fruit fly (Herz et al., 2014) and fission yeast (Shan et al., 2016), in a manner akin to the K27M and K36M mutations.
Genome instability, another fundamental cancer hallmark (Hanahan, 2022; Hanahan & Weinberg, 2000, 2011), in which H3K9me3 plays a central regulatory role, has put the spotlight on H3K9me3 and its associated factors for devising novel anti-tumor strategies. Profiling of cancer epigenomes suggests a global loss of H3K9me3 in some cancers, such as prostate, bladder, breast and colorectal cancer (Ellinger et al., 2010, 2014; Leszinski et al., 2012; Zhao et al., 2016; Zhou et al., 2022), yet elevated levels of H3K9me3 have been documented in others, including liver cancer and liposarcoma (Keung et al., 2015; Qian et al., 2020), suggesting a context-dependent relationship. H3K9me3 levels are also correlated with the TNM stage, prognosis and recurrence in various cancer types (Benard et al., 2014; Chen et al., 2013; Elsheikh et al., 2009; Lam-Ubol & Phattarataratip, 2022; Muller-Tidow et al., 2010; Park et al., 2008; Song et al., 2012; Xia et al., 2013), acting as a valuable theranostic marker in anti-tumor therapy. Interestingly, almost all enzymes regulating H3K9me3, including both methyltransferases and demethylases, are reportedly oncogenic and highly druggable (Bhat et al., 2021; Husmann & Gozani, 2019; Morera et al., 2016). For example, overexpression of G9a is observed in many malignancies (Casciello et al., 2015). Gain-of-function mutations of G9a drive melanoma formation through activation of the WNT signaling pathway (Kato et al., 2020). Similarly, ectopically overexpressed SUV39h1 drives tumor growth and metastasis (Yokoyama et al., 2013). Upon DNA damage, SET7/9-mediated methylation of SUV39h1 attenuated its activity, which led to genomic instability and specific killing of tumor cells (Wang et al., 2013), offering new ideas to target H3K9me3 and SUV39h1 in cancer. Surprisingly, in Ras-driven lymphoma models, SUV39h1 keeps lymphomagenesis in check through H3K9me3-related cellular senescence and functions as a tumor suppressor (Braig et al., 2005), underpinning the intricate roles of H3K9me3 in determining the fate of damaged cells between senescence and oncogenesis. On the other hand, all members of the KDM4 demethylases are overexpressed in multiple tumors (Berry & Janknecht, 2013; Lee et al., 2020). In ovarian cancer, KDM4A amplification promotes re-replication and copy gain of specific genomic regions, through interacting with the replication machinery (Black et al., 2013). Inhibition of JAK2 and KDM4C, both of which are frequently amplified in a subgroup of lymphoma, induces cooperative epigenetic changes, including upregulation of H3K9me3 and formation of repressive chromatin, leading to MYC silencing and elimination of lymphoma cells (Rui et al., 2010).
An emerging aspect of the implication of H3K9me3 in cancer therapeutics lies in its prospects for dictating tumor response to immunotherapy. As a promising frontier in the battle against cancer, anti-tumor immunotherapy is a breakthrough of the century, yet serious challenges remain, including limited efficacy and low response rates in certain tumors, lack of personalized targets and immune escape (Hegde & Chen, 2020; Mellman et al., 2011; Pardoll, 2012). Recently, targeting of H3K9me3 and its enzymes has shown potential to overcome some of these hurdles. Inhibition of SUV39h1 activates repetitive elements that are otherwise silenced by H3K9me3, which stimulates the accumulation of cytosolic dsRNA and dsDNA, activates the antiviral pathways and interferon signaling, and enhances cancer cell immunogenicity (Shen et al., 2021). In multiple cancer models, genetic ablation of SUV39h1 markedly improves the long-term persistence and overall anti-tumor efficacy of human chimeric antigen receptor (CAR) T cells, protecting mice from tumor relapse and rechallenges (Jain et al., 2024; Lopez-Cobo et al., 2024). In melanoma, SETDB1-mediated epigenetic silencing represents a strategy for tumors to evade immune surveillance (Griffin et al., 2021). Loss of SETDB1 derepresses latent retroelements, immunostimulatory genes, and TE-encoded viral antigens, which further triggers cytotoxic T cell responses and enhances the anti-tumor potency of immune checkpoint blockade (ICB) (Griffin et al., 2021; Zhang et al., 2021a). Inhibition of G9a, or simultaneous targeting of G9a and other epigenetic repressors have been reported to enhance tumor immune clearance by ICB in bladder cancer (Segovia et al., 2019), melanoma (Kelly et al., 2021) and ovarian serous carcinoma (Spiliopoulou et al., 2022). In squamous cell carcinoma, deletion of KDM4A potentiates the effects of PD1 neutralizing antibodies by inflicting replication stress and DNA damage, which activates tumor-cell-intrinsic immune response (Zhang et al., 2021b). In colorectal cancer, KDM4D co-activates transcription factor SP-1 to enhance the interferon pathway and PD-L1 expression, which promotes tumor immune escape (Chen et al., 2022). These reports underpin the critical roles played by H3K9me3 in reshaping the tumor immune microenvironment.
The fact that blocking either H3K9me3 methyltransferases or demethylases is beneficial for tumor patients suggests that a healthy dose of H3K9me3 is essential to keep cancers at bay. On the one hand, high levels of H3K9me3 drives chromatin compaction and epigenetic silencing of tumor suppressor genes or retrovirus-derived regulatory elements, which enables cancerous transformation and suppresses innate immune response. Such mechanisms are common strategies of the so-called ‘immune-cold” tumors to bypass immune surveillance, as discussed above, or to develop drug resistance during therapy-induced epigenetic reprogramming (Torrano et al., 2019). On the other hand, aberrant downregulation of H3K9me3 creates a permissive chromatin environment that allows for the accumulation of spontaneous oncogenic activation and chromosomal abnormalities. These mutations further grant growth advantage during successive clonal expansion and eventually result in tumorigenesis. The narrow window of H3K9me3 fluctuations underlines the tight control over its levels by a plethora of coordinating factors. Under pathological conditions, such duality agrees with the reversibility and plasticity of the epigenetic framework, which opens up avenues for developing targeted and more personalized disease-prevention tactics to treat cancers and beyond.
Concluding remarks
In summary, H3K9me3, a master regulator of heterochromatin, has also been proven multi-talented in DNA damage response and repair, which bears major implications for stress response, the etiology of human diseases and their clinical management. Within the cascade of molecular events upon DNA damage, H3K9me3 fine-tunes the response and repair process through an expanding array of reader proteins, in the context of chromatin microenvironment and potential crosstalk with other epigenetic marks. The spatiotemporal mechanisms underlying the dynamics of H3K9me3 and the ensuing chromatin rearrangements remain ambiguous, the understanding of which could yield conceptual advancement in the epigenetic regulation of genome integrity. The emerging perspectives of H3K9me3 in retaining and transmitting short-term or long-term cellular memory to stress response position it as a multifaceted molecular switch of cell fate transitions, including differentiation, programmed cell death, senescence, carcinogenesis, and others. These outstanding questions warrant continuous efforts to dissect the mechanistic details and functional significance in both physiological and pathological settings. Importantly, the translational and pre-clinical developments aimed at enzymes and effectors in connection with H3K9me3 have been promising, either alone or combining with other anti-tumor regimes. H3K9me3, therefore, plays the role of a silent guardian to preserve genetic and epigenetic stability against genotoxic stress.
References
Aagaard, L., Laible, G., Selenko, P., Schmid, M., Dorn, R., Schotta, G., Kuhfittig, S., Wolf, A., Lebersorger, A., Singh, P. B., Reuter, G., & Jenuwein, T. (1999). Functional mammalian homologues of the PEV-modifier encode centromere-associated proteins which complex with the heterochromatin component M31. EMBO Journal, 18, 1923–1938. https://doi.org/10.1093/emboj/18.7.1923
Adar, S., Hu, J., Lieb, J. D., & Sancar, A. (2016). Genome-wide kinetics of DNA excision repair in relation to chromatin state and mutagenesis. Proceedings of National Academy Science USA, 113(15), E2124-2133. https://doi.org/10.1073/pnas.1603388113
Alagoz, M., Katsuki, Y., Ogiwara, H., Ogi, T., Shibata, A., Kakarougkas, A., & Jeggo, P. (2015). SETDB1, HP1 and SUV39 promote repositioning of 53BP1 to extend resection during homologous recombination in G2 cells. Nucleic Acids Research, 43(16), 7931–7944. https://doi.org/10.1093/nar/gkv722
Allshire, R. C., & Madhani, H. D. (2018). Ten principles of heterochromatin formation and function. Nature Reviews Molecular Cell Biology, 19(4), 229–244. https://doi.org/10.1038/nrm.2017.119
Al-Sady, B., Madhani, H. D., & Narlikar, G. J. (2013). Division of labor between the chromodomains of HP1 and Suv39 methylase enables coordination of heterochromatin spread. Molecular Cell, 51(1), 80–91. https://doi.org/10.1016/j.molcel.2013.06.013
Altaf, M., Saksouk, N., & Cote, J. (2007). Histone modifications in response to DNA damage. Mutation Research, 618(1–2), 81–90. https://doi.org/10.1016/j.mrfmmm.2006.09.009
Arnould, C., Rocher, V., Finoux, A. L., Clouaire, T., Li, K., Zhou, F., Caron, P., Mangeot, P. E., Ricci, E. P., Mourad, R., Haber, J. E., Noordermeer, D., & Legube, G. (2021). Loop extrusion as a mechanism for formation of DNA damage repair foci. Nature, 590(7847), 660–665. https://doi.org/10.1038/s41586-021-03193-z
Arnould, C., Rocher, V., Saur, F., Bader, A. S., Muzzopappa, F., Collins, S., Lesage, E., Le Bozec, B., Puget, N., Clouaire, T., Mangeat, T., Mourad, R., Ahituv, N., Noordermeer, D., Erdel, F., Bushell, M., Marnef, A., & Legube, G. (2023). Chromatin compartmentalization regulates the response to DNA damage. Nature, 623(7985), 183–192. https://doi.org/10.1038/s41586-023-06635-y
Ayoub, N., Jeyasekharan, A. D., Bernal, J. A., & Venkitaraman, A. R. (2008). HP1-beta mobilization promotes chromatin changes that initiate the DNA damage response. Nature, 453(7195), 682–686. https://doi.org/10.1038/nature06875
Ayrapetov, M. K., Gursoy-Yuzugullu, O., Xu, C., Xu, Y., & Price, B. D. (2014). DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proceedings of National Academy Science USA, 111(25), 9169–9174. https://doi.org/10.1073/pnas.1403565111
Baldeyron, C., Soria, G., Roche, D., Cook, A. J., & Almouzni, G. (2011). HP1alpha recruitment to DNA damage by p150CAF-1 promotes homologous recombination repair. Journal of Cell Biology, 193(1), 81–95. https://doi.org/10.1083/jcb.201101030
Banath, J. P., Klokov, D., MacPhail, S. H., Banuelos, C. A., & Olive, P. L. (2010). Residual gammaH2AX foci as an indication of lethal DNA lesions. BMC Cancer, 10, 4. https://doi.org/10.1186/1471-2407-10-4
Bannister, A. J., Zegerman, P., Partridge, J. F., Miska, E. A., Thomas, J. O., Allshire, R. C., & Kouzarides, T. (2001). Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature, 410(6824), 120–124. https://doi.org/10.1038/35065138
Barral, A., Pozo, G., Ducrot, L., Papadopoulos, G. L., Sauzet, S., Oldfield, A. J., Cavalli, G., & Dejardin, J. (2022). SETDB1/NSD-dependent H3K9me3/H3K36me3 dual heterochromatin maintains gene expression profiles by bookmarking poised enhancers. Molecular Cell, 82(4), 816–832. https://doi.org/10.1016/j.molcel.2021.12.037
Barski, A., Cuddapah, S., Cui, K., Roh, T. Y., Schones, D. E., Wang, Z., Wei, G., Chepelev, I., & Zhao, K. (2007). High-resolution profiling of histone methylations in the human genome. Cell, 129(4), 823–837. https://doi.org/10.1016/j.cell.2007.05.009
Becker, J. S., Nicetto, D., & Zaret, K. S. (2016). H3K9me3-dependent heterochromatin: barrier to cell fate changes. Trends in Genetics, 32(1), 29–41. https://doi.org/10.1016/j.tig.2015.11.001
Behjati, S., Tarpey, P. S., Presneau, N., Scheipl, S., Pillay, N., Van Loo, P., Wedge, D. C., Cooke, S. L., Gundem, G., Davies, H., Nik-Zainal, S., Martin, S., McLaren, S., Goodie, V., Robinson, B., Butler, A., Teague, J. W., Halai, D., Khatri, B., et al. (2013). Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nature Genetics, 45(12), 1479–1482. https://doi.org/10.1038/ng.2814
Benard, A., Goossens-Beumer, I. J., van Hoesel, A. Q., de Graaf, W., Horati, H., Putter, H., Zeestraten, E. C., van de Velde, C. J., & Kuppen, P. J. (2014). Histone trimethylation at H3K4, H3K9 and H4K20 correlates with patient survival and tumor recurrence in early-stage colon cancer. BMC Cancer, 14, 531. https://doi.org/10.1186/1471-2407-14-531
Berry, W. L., & Janknecht, R. (2013). KDM4/JMJD2 histone demethylases: Epigenetic regulators in cancer cells. Cancer Research, 73(10), 2936–2942. https://doi.org/10.1158/0008-5472.CAN-12-4300
Bhat, K. P., Umit Kaniskan, H., Jin, J., & Gozani, O. (2021). Epigenetics and beyond: Targeting writers of protein lysine methylation to treat disease. Nature Reviews Drug Discovery, 20(4), 265–286. https://doi.org/10.1038/s41573-020-00108-x
Black, J. C., Manning, A. L., Van Rechem, C., Kim, J., Ladd, B., Cho, J., Pineda, C. M., Murphy, N., Daniels, D. L., Montagna, C., Lewis, P. W., Glass, K., Allis, C. D., Dyson, N. J., Getz, G., & Whetstine, J. R. (2013). KDM4A lysine demethylase induces site-specific copy gain and rereplication of regions amplified in tumors. Cell, 154(3), 541–555. https://doi.org/10.1016/j.cell.2013.06.051
Blackford, A. N., & Jackson, S. P. (2017). ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Molecular Cell, 66(6), 801–817. https://doi.org/10.1016/j.molcel.2017.05.015
Bostick, M., Kim, J. K., Esteve, P. O., Clark, A., Pradhan, S., & Jacobsen, S. E. (2007). UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science, 317(5845), 1760–1764. https://doi.org/10.1126/science.1147939
Botuyan, M. V., Lee, J., Ward, I. M., Kim, J. E., Thompson, J. R., Chen, J., & Mer, G. (2006). Structural basis for the methylation state-specific recognition of histone H4–K20 by 53BP1 and Crb2 in DNA repair. Cell, 127(7), 1361–1373. https://doi.org/10.1016/j.cell.2006.10.043
Bourque, G., Burns, K. H., Gehring, M., Gorbunova, V., Seluanov, A., Hammell, M., Imbeault, M., Izsvak, Z., Levin, H. L., Macfarlan, T. S., Mager, D. L., & Feschotte, C. (2018). Ten things you should know about transposable elements. Genome Biology, 19(1), 199. https://doi.org/10.1186/s13059-018-1577-z
Braig, M., Lee, S., Loddenkemper, C., Rudolph, C., Peters, A. H., Schlegelberger, B., Stein, H., Dorken, B., Jenuwein, T., & Schmitt, C. A. (2005). Oncogene-induced senescence as an initial barrier in lymphoma development. Nature, 436(7051), 660–665. https://doi.org/10.1038/nature03841
Branzei, D., & Foiani, M. (2008). Regulation of DNA repair throughout the cell cycle. Nature Reviews Molecular Cell Biology, 9(4), 297–308. https://doi.org/10.1038/nrm2351
Brumbaugh, J., Kim, I. S., Ji, F., Huebner, A. J., Di Stefano, B., Schwarz, B. A., Charlton, J., Coffey, A., Choi, J., Walsh, R. M., Schindler, J. W., Anselmo, A., Meissner, A., Sadreyev, R. I., Bernstein, B. E., Hock, H., & Hochedlinger, K. (2019). Inducible histone K-to-M mutations are dynamic tools to probe the physiological role of site-specific histone methylation in vitro and in vivo. Nature Cell Biology, 21(11), 1449–1461. https://doi.org/10.1038/s41556-019-0403-5
Bulut-Karslioglu, A., De La Rosa-Velazquez, I. A., Ramirez, F., Barenboim, M., Onishi-Seebacher, M., Arand, J., Galan, C., Winter, G. E., Engist, B., Gerle, B., O’Sullivan, R. J., Martens, J. H., Walter, J., Manke, T., Lachner, M., & Jenuwein, T. (2014). Suv39h-dependent H3K9me3 marks intact retrotransposons and silences LINE elements in mouse embryonic stem cells. Molecular Cell, 55(2), 277–290. https://doi.org/10.1016/j.molcel.2014.05.029
Burton, A., Brochard, V., Galan, C., Ruiz-Morales, E. R., Rovira, Q., Rodriguez-Terrones, D., Kruse, K., Le Gras, S., Udayakumar, V. S., Chin, H. G., Eid, A., Liu, X., Wang, C., Gao, S., Pradhan, S., Vaquerizas, J. M., Beaujean, N., Jenuwein, T., & Torres-Padilla, M. E. (2020). Heterochromatin establishment during early mammalian development is regulated by pericentromeric RNA and characterized by non-repressive H3K9me3. Nature Cell Biology, 22(7), 767–778. https://doi.org/10.1038/s41556-020-0536-6
Cameron, E. E., Bachman, K. E., Myohanen, S., Herman, J. G., & Baylin, S. B. (1999). Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nature Genetics, 21(1), 103–107. https://doi.org/10.1038/5047
Cao, L. L., Shen, C., & Zhu, W. G. (2016). Histone modifications in DNA damage response. Science China Life Science, 59(3), 257–270. https://doi.org/10.1007/s11427-016-5011-z
Casciello, F., Windloch, K., Gannon, F., & Lee, J. S. (2015). Functional Role of G9a Histone Methyltransferase in Cancer. Frontiers in Immunology, 6, 487. https://doi.org/10.3389/fimmu.2015.00487
Castel, S. E., & Martienssen, R. A. (2013). RNA interference in the nucleus: Roles for small RNAs in transcription, epigenetics and beyond. Nature Reviews Genetics, 14(2), 100–112. https://doi.org/10.1038/nrg3355
Ceccaldi, R., Rondinelli, B., & D’Andrea, A. D. (2016). Repair Pathway Choices and Consequences at the Double-Strand Break. Trends in Cell Biology, 26(1), 52–64. https://doi.org/10.1016/j.tcb.2015.07.009
Chan, K. M., Fang, D., Gan, H., Hashizume, R., Yu, C., Schroeder, M., Gupta, N., Mueller, S., James, C. D., Jenkins, R., Sarkaria, J., & Zhang, Z. (2013a). The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Device, 27(9), 985–990. https://doi.org/10.1101/gad.217778.113
Chan, K. M., Han, J., Fang, D., Gan, H., & Zhang, Z. (2013b). A lesson learned from the H3.3K27M mutation found in pediatric glioma: a new approach to the study of the function of histone modifications in vivo? Cell Cycle, 12(16), 2546–2552. https://doi.org/10.4161/cc.25625
Chan, K. M., & Zhang, Z. G. (2012). Leucine-rich repeat and WD repeat-containing Protein 1 is recruited to pericentric heterochromatin by Trimethylated Lysine 9 of Histone H3 and maintains heterochromatin silencing. Journal of Biological Chemistry, 287(18), 15024–15033. https://doi.org/10.1074/jbc.M111.337980
Chapman, J. R., Taylor, M. R., & Boulton, S. J. (2012). Playing the end game: DNA double-strand break repair pathway choice. Molecular Cell, 47(4), 497–510. https://doi.org/10.1016/j.molcel.2012.07.029
Chen, Q., Zhuang, S., Hong, Y., Yang, L., Guo, P., Mo, P., Peng, K., Li, W., Xiao, N., & Yu, C. (2022). Demethylase JMJD2D induces PD-L1 expression to promote colorectal cancer immune escape by enhancing IFNGR1-STAT3-IRF1 signaling. Oncogene, 41(10), 1421–1433. https://doi.org/10.1038/s41388-021-02173-x
Chen, Y. W., Kao, S. Y., Wang, H. J., & Yang, M. H. (2013). Histone modification patterns correlate with patient outcome in oral squamous cell carcinoma. Cancer, 119(24), 4259–4267. https://doi.org/10.1002/cncr.28356
Cheng, Y., He, C., Wang, M., Ma, X., Mo, F., Yang, S., Han, J., & Wei, X. (2019). Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduction and Targeted Therapy, 4, 62. https://doi.org/10.1038/s41392-019-0095-0
Chiolo, I., Minoda, A., Colmenares, S. U., Polyzos, A., Costes, S. V., & Karpen, G. H. (2011). Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell, 144(5), 732–744. https://doi.org/10.1016/j.cell.2011.02.012
Cimprich, K. A., & Cortez, D. (2008). ATR: An essential regulator of genome integrity. Nature Reviews Molecular Cell Biology, 9(8), 616–627. https://doi.org/10.1038/nrm2450
Collins, R. E., Northrop, J. P., Horton, J. R., Lee, D. Y., Zhang, X., Stallcup, M. R., & Cheng, X. (2008). The ankyrin repeats of G9a and GLP histone methyltransferases are mono- and dimethyllysine binding modules. Nature Structural & Molecular Biology, 15(3), 245–250. https://doi.org/10.1038/nsmb.1384
Consortium. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature, 489(7414), 57–74. https://doi.org/10.1038/nature11247
Curtin, N. J. (2012). DNA repair dysregulation from cancer driver to therapeutic target. Nature Reviews Cancer, 12(12), 801–817. https://doi.org/10.1038/nrc3399
Davalos, V., & Esteller, M. (2023). Cancer epigenetics in clinical practice. CA: A Cancer Journal for Clinicians, 73(4), 376–424. https://doi.org/10.3322/caac.21765
Di Croce, L., & Helin, K. (2013). Transcriptional regulation by Polycomb group proteins. Nature Structural & Molecular Biology, 20(10), 1147–1155. https://doi.org/10.1038/nsmb.2669
Di Micco, R., Sulli, G., Dobreva, M., Liontos, M., Botrugno, O. A., Gargiulo, G., dal Zuffo, R., Matti, V., d’Ario, G., Montani, E., Mercurio, C., Hahn, W. C., Gorgoulis, V., Minucci, S., & d’Adda di Fagagna, F. (2011). Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nature Cell Biology, 13(3), 292–302. https://doi.org/10.1038/ncb2170
Dodge, J. E., Kang, Y. K., Beppu, H., Lei, H., & Li, E. (2004). Histone H3–K9 methyltransferase ESET is essential for early development. Molecular and Cellular Biology, 24(6), 2478–2486. https://doi.org/10.1128/MCB.24.6.2478-2486.2004
Dolce, V., Dusi, S., Giannattasio, M., Joseph, C. R., Fumasoni, M., & Branzei, D. (2022). Parental histone deposition on the replicated strands promotes error-free DNA damage tolerance and regulates drug resistance. Genes & Development, 36(3–4), 167–179. https://doi.org/10.1101/gad.349207.121
Dopkins, N., & Nixon, D. F. (2024). Activation of human endogenous retroviruses and its physiological consequences. Nature Reviews Molecular Cell Biology, 25(3), 212–222. https://doi.org/10.1038/s41580-023-00674-z
Dorr, J. R., Yu, Y., Milanovic, M., Beuster, G., Zasada, C., Dabritz, J. H., Lisec, J., Lenze, D., Gerhardt, A., Schleicher, K., Kratzat, S., Purfurst, B., Walenta, S., Mueller-Klieser, W., Graler, M., Hummel, M., Keller, U., Buck, A. K., Dorken, B., et al. (2013). Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature, 501(7467), 421–425. https://doi.org/10.1038/nature12437
Douse, C. H., Tchasovnikarova, I. A., Timms, R. T., Protasio, A. V., Seczynska, M., Prigozhin, D. M., Albecka, A., Wagstaff, J., Williamson, J. C., Freund, S. M. V., Lehner, P. J., & Modis, Y. (2020). TASOR is a pseudo-PARP that directs HUSH complex assembly and epigenetic transposon control. Nature Communications, 11(1), 4940. https://doi.org/10.1038/s41467-020-18761-6
Du, W., Shi, G., Shan, C. M., Li, Z., Zhu, B., Jia, S., Li, Q., & Zhang, Z. (2022). Mechanisms of chromatin-based epigenetic inheritance. Science China Life Science, 65(11), 2162–2190. https://doi.org/10.1007/s11427-022-2120-1
Ellinger, J., Bachmann, A., Goke, F., Behbahani, T. E., Baumann, C., Heukamp, L. C., Rogenhofer, S., & Muller, S. C. (2014). Alterations of global histone H3K9 and H3K27 methylation levels in bladder cancer. Urologia Internationalis, 93(1), 113–118. https://doi.org/10.1159/000355467
Ellinger, J., Kahl, P., von der Gathen, J., Rogenhofer, S., Heukamp, L. C., Gutgemann, I., Walter, B., Hofstadter, F., Buttner, R., Muller, S. C., Bastian, P. J., & von Ruecker, A. (2010). Global levels of histone modifications predict prostate cancer recurrence. Prostate, 70(1), 61–69. https://doi.org/10.1002/pros.21038
Elsasser, S. J., Noh, K. M., Diaz, N., Allis, C. D., & Banaszynski, L. A. (2015). Histone H3.3 is required for endogenous retroviral element silencing in embryonic stem cells. Nature, 522(7555), 240–244. https://doi.org/10.1038/nature14345
Elsheikh, S. E., Green, A. R., Rakha, E. A., Powe, D. G., Ahmed, R. A., Collins, H. M., Soria, D., Garibaldi, J. M., Paish, C. E., Ammar, A. A., Grainge, M. J., Ball, G. R., Abdelghany, M. K., Martinez-Pomares, L., Heery, D. M., & Ellis, I. O. (2009). Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Research, 69(9), 3802–3809. https://doi.org/10.1158/0008-5472.CAN-08-3907
Escobar, T. M., Loyola, A., & Reinberg, D. (2021). Parental nucleosome segregation and the inheritance of cellular identity. Nature Reviews Genetics, 22(6), 379–392. https://doi.org/10.1038/s41576-020-00312-w
Escobar, T. M., Oksuz, O., Saldana-Meyer, R., Descostes, N., Bonasio, R., & Reinberg, D. (2019). Active and repressed chromatin domains exhibit distinct nucleosome segregation during DNA replication. Cell, 179(4), 953–963. https://doi.org/10.1016/j.cell.2019.10.009
Fang, D., Gan, H., Lee, J. H., Han, J., Wang, Z., Riester, S. M., Jin, L., Chen, J., Zhou, H., Wang, J., Zhang, H., Yang, N., Bradley, E. W., Ho, T. H., Rubin, B. P., Bridge, J. A., Thibodeau, S. N., Ordog, T., Chen, Y., et al. (2016). The histone H3.3K36M mutation reprograms the epigenome of chondroblastomas. Science, 352(6291), 1344–1348. https://doi.org/10.1126/science.aae0065
Fernandez-Capetillo, O., Lee, A., Nussenzweig, M., & Nussenzweig, A. (2004). H2AX: The histone guardian of the genome. DNA Repair (amst), 3(8–9), 959–967. https://doi.org/10.1016/j.dnarep.2004.03.024
Fischle, W., Tseng, B. S., Dormann, H. L., Ueberheide, B. M., Garcia, B. A., Shabanowitz, J., Hunt, D. F., Funabiki, H., & Allis, C. D. (2005). Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature, 438(7071), 1116–1122. https://doi.org/10.1038/nature04219
Fnu, S., Williamson, E. A., De Haro, L. P., Brenneman, M., Wray, J., Shaheen, M., Radhakrishnan, K., Lee, S. H., Nickoloff, J. A., & Hromas, R. (2011). Methylation of histone H3 lysine 36 enhances DNA repair by nonhomologous end-joining. Proceedings of the National Academy Science USA, 108(2), 540–545. https://doi.org/10.1073/pnas.1013571108
Fradet-Turcotte, A., Canny, M. D., Escribano-Díaz, C., Orthwein, A., Leung, C. C. Y., Huang, H., Landry, M. C., Kitevski-LeBlanc, J., Noordermeer, S. M., Sicheri, F., & Durocher, D. (2013). 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature, 499(7456), 50–54. https://doi.org/10.1038/nature12318
Fueyo, R., Judd, J., Feschotte, C., & Wysocka, J. (2022). Roles of transposable elements in the regulation of mammalian transcription. Nature Reviews Molecular Cell Biology, 23(7), 481–497. https://doi.org/10.1038/s41580-022-00457-y
Fumagalli, M., Rossiello, F., Clerici, M., Barozzi, S., Cittaro, D., Kaplunov, J. M., Bucci, G., Dobreva, M., Matti, V., Beausejour, C. M., Herbig, U., Longhese, M. P., & d’Adda di Fagagna, F. (2012). Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nature Cell Biology, 14(4), 355–365. https://doi.org/10.1038/ncb2466
Gaggioli, V., Lo, C. S. Y., Reveron-Gomez, N., Jasencakova, Z., Domenech, H., Nguyen, H., Sidoli, S., Tvardovskiy, A., Uruci, S., Slotman, J. A., Chai, Y., Goncalves, J., Manolika, E. M., Jensen, O. N., Wheeler, D., Sridharan, S., Chakrabarty, S., Demmers, J., Kanaar, R., et al. (2023). Dynamic de novo heterochromatin assembly and disassembly at replication forks ensures fork stability. Nature Cell Biology, 25(7), 1017–1032. https://doi.org/10.1038/s41556-023-01167-z
Gan, H., Serra-Cardona, A., Hua, X., Zhou, H., Labib, K., Yu, C., & Zhang, Z. (2018). The Mcm2-Ctf4-Polalpha axis facilitates parental histone H3–H4 transfer to lagging strands. Molecular Cell, 72(1), 140–151. https://doi.org/10.1016/j.molcel.2018.09.001
Gardner, M. J., Snee, M. P., Hall, A. J., Powell, C. A., Downes, S., & Terrell, J. D. (1990). Results of case-control study of leukaemia and lymphoma among young people near Sellafield nuclear plant in West Cumbria. BMJ, 300(6722), 423–429. https://doi.org/10.1136/bmj.300.6722.423
Garland, W., Muller, I., Wu, M., Schmid, M., Imamura, K., Rib, L., Sandelin, A., Helin, K., & Jensen, T. H. (2022). Chromatin modifier HUSH co-operates with RNA decay factor NEXT to restrict transposable element expression. Molecular Cell, 82(9), 1691–1707. https://doi.org/10.1016/j.molcel.2022.03.004
Geis, F. K., & Goff, S. P. (2020). Silencing and Transcriptional Regulation of Endogenous Retroviruses: An Overview. Viruses, 12(8), 884. https://doi.org/10.3390/v12080884
Gershman, A., Sauria, M. E. G., Guitart, X., Vollger, M. R., Hook, P. W., Hoyt, S. J., Jain, M., Shumate, A., Razaghi, R., Koren, S., Altemose, N., Caldas, G. V., Logsdon, G. A., Rhie, A., Eichler, E. E., Schatz, M. C., O’Neill, R. J., Phillippy, A. M., Miga, K. H., et al. (2022). Epigenetic patterns in a complete human genome. Science, 376(6588), 5089. https://doi.org/10.1126/science.abj5089
Giglia-Mari, G., Zotter, A., & Vermeulen, W. (2011). DNA damage response. Cold Spring Harbor Perspectives in Biology, 3(1), a000745. https://doi.org/10.1101/cshperspect.a000745
Ginjala, V., Rodriguez-Colon, L., Ganguly, B., Gangidi, P., Gallina, P., Al-Hraishawi, H., Kulkarni, A., Tang, J., Gheeya, J., Simhadri, S., Yao, M., Xia, B., & Ganesan, S. (2017). Protein-lysine methyltransferases G9a and GLP1 promote responses to DNA damage. Science and Reports, 7(1), 16613. https://doi.org/10.1038/s41598-017-16480-5
Gonzalez-Garrido, C., & Prado, F. (2023). Parental histone distribution and location of the replication obstacle at nascent strands control homologous recombination. Cell Reports, 42(3), 112174. https://doi.org/10.1016/j.celrep.2023.112174
Grasso, C. S., Tang, Y., Truffaux, N., Berlow, N. E., Liu, L., Debily, M. A., Quist, M. J., Davis, L. E., Huang, E. C., Woo, P. J., Ponnuswami, A., Chen, S., Johung, T. B., Sun, W., Kogiso, M., Du, Y., Qi, L., Huang, Y., Hutt-Cabezas, M., et al. (2015). Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nature Medicine, 21(6), 555–559. https://doi.org/10.1038/nm.3855
Greer, E. L., & Shi, Y. (2012). Histone methylation: A dynamic mark in health, disease and inheritance. Nature Reviews Genetics, 13(5), 343–357. https://doi.org/10.1038/nrg3173
Grewal, S. I., & Jia, S. (2007). Heterochromatin revisited. Nature Reviews Genetics, 8(1), 35–46. https://doi.org/10.1038/nrg2008
Griffin, G. K., Wu, J., Iracheta-Vellve, A., Patti, J. C., Hsu, J., Davis, T., Dele-Oni, D., Du, P. P., Halawi, A. G., Ishizuka, J. J., Kim, S. Y., Klaeger, S., Knudsen, N. H., Miller, B. C., Nguyen, T. H., Olander, K. E., Papanastasiou, M., Rachimi, S., Robitschek, E. J., et al. (2021). Epigenetic silencing by SETDB1 suppresses tumour intrinsic immunogenicity. Nature, 595(7866), 309–314. https://doi.org/10.1038/s41586-021-03520-4
Grunstein, M. (1997). Histone acetylation in chromatin structure and transcription. Nature, 389(6649), 349–352. https://doi.org/10.1038/38664
Guo, Y., Li, T. D., Modzelewski, A. J., & Siomi, H. (2024). Retrotransposon renaissance in early embryos. Trends in Genetics, 40(1), 39–51. https://doi.org/10.1016/j.tig.2023.10.010
Han, C., Srivastava, A. K., Cui, T., Wang, Q. E., & Wani, A. A. (2016). Differential DNA lesion formation and repair in heterochromatin and euchromatin. Carcinogenesis, 37(2), 129–138. https://doi.org/10.1093/carcin/bgv247
Hanahan, D. (2022). Hallmarks of Cancer: New Dimensions. Cancer Discovery, 12(1), 31–46. https://doi.org/10.1158/2159-8290.CD-21-1059
Hanahan, D., & Weinberg, R. A. (2000). The hallmarks of cancer. Cell, 100(1), 57–70. https://doi.org/10.1016/s0092-8674(00)81683-9
Hanahan, D., & Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell, 144(5), 646–674. https://doi.org/10.1016/j.cell.2011.02.013
Hashizume, R., Andor, N., Ihara, Y., Lerner, R., Gan, H., Chen, X., Fang, D., Huang, X., Tom, M. W., Ngo, V., Solomon, D., Mueller, S., Paris, P. L., Zhang, Z., Petritsch, C., Gupta, N., Waldman, T. A., & James, C. D. (2014). Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nature Medicine, 20(12), 1394–1396. https://doi.org/10.1038/nm.3716
Hauer, M. H., & Gasser, S. M. (2017). Chromatin and nucleosome dynamics in DNA damage and repair. Genes & Development, 31(22), 2204–2221. https://doi.org/10.1101/gad.307702.117
Hegde, P. S., & Chen, D. S. (2020). Top 10 challenges in cancer immunotherapy. Immunity, 52(1), 17–35. https://doi.org/10.1016/j.immuni.2019.12.011
Herz, H. M., Morgan, M., Gao, X., Jackson, J., Rickels, R., Swanson, S. K., Florens, L., Washburn, M. P., Eissenberg, J. C., & Shilatifard, A. (2014). Histone H3 lysine-to-methionine mutants as a paradigm to study chromatin signaling. Science, 345(6200), 1065–1070. https://doi.org/10.1126/science.1255104
Hirota, T., Lipp, J. J., Toh, B. H., & Peters, J. M. (2005). Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature, 438(7071), 1176–1180. https://doi.org/10.1038/nature04254
Hoeijmakers, J. H. (2009). DNA damage, aging, and cancer. New England Journal of Medicine, 361(15), 1475–1485. https://doi.org/10.1056/NEJMra0804615
Husmann, D., & Gozani, O. (2019). Histone lysine methyltransferases in biology and disease. Nature Structural & Molecular Biology, 26(10), 880–889. https://doi.org/10.1038/s41594-019-0298-7
Ikura, T., Tashiro, S., Kakino, A., Shima, H., Jacob, N., Amunugama, R., Yoder, K., Izumi, S., Kuraoka, I., Tanaka, K., Kimura, H., Ikura, M., Nishikubo, S., Ito, T., Muto, A., Miyagawa, K., Takeda, S., Fishel, R., Igarashi, K., et al. (2007). DNA damage-dependent acetylation and ubiquitination of H2AX enhances chromatin dynamics. Molecular and Cellular Biology, 27(20), 7028–7040. https://doi.org/10.1128/MCB.00579-07
Ilnytskyy, Y., Koturbash, I., & Kovalchuk, O. (2009). Radiation-induced bystander effects in vivo are epigenetically regulated in a tissue-specific manner. Environmental and Molecular Mutagenesis, 50(2), 105–113. https://doi.org/10.1002/em.20440
Jacobs, S. A., Taverna, S. D., Zhang, Y., Briggs, S. D., Li, J., Eissenberg, J. C., Allis, C. D., & Khorasanizadeh, S. (2001). Specificity of the HP1 chromo domain for the methylated N-terminus of histone H3. EMBO Journal, 20(18), 5232–5241. https://doi.org/10.1093/emboj/20.18.5232
Jacquet, K., Fradet-Turcotte, A., Avvakumov, N., Lambert, J. P., Roques, C., Pandita, R. K., Paquet, E., Herst, P., Gingras, A. C., Pandita, T. K., Legube, G., Doyon, Y., Durocher, D., & Cote, J. (2016). The TIP60 Complex Regulates Bivalent Chromatin Recognition by 53BP1 through Direct H4K20me Binding and H2AK15 Acetylation. Molecular Cell, 62(3), 409–421. https://doi.org/10.1016/j.molcel.2016.03.031
Jain, N., Zhao, Z., Koche, R. P., Antelope, C., Gozlan, Y., Montalbano, A., Brocks, D., Lopez, M., Dobrin, A., Shi, Y., Gunset, G., Giavridis, T., & Sadelain, M. (2024). Disruption of SUV39H1-mediated H3K9 methylation sustains CAR T cell function. Cancer Discovery, 14(1), 142–157. https://doi.org/10.1158/2159-8290.CD-22-1319
Janssen, A., Breuer, G. A., Brinkman, E. K., van der Meulen, A. I., Borden, S. V., van Steensel, B., Bindra, R. S., LaRocque, J. R., & Karpen, G. H. (2016). A single double-strand break system reveals repair dynamics and mechanisms in heterochromatin and euchromatin. Genes & Development, 30(14), 1645–1657. https://doi.org/10.1101/gad.283028.116
Janssen, A., Colmenares, S. U., & Karpen, G. H. (2018). Heterochromatin: guardian of the genome. Annual Review of Cell and Developmental Biology, 34, 265–288. https://doi.org/10.1146/annurev-cellbio-100617-062653
Jorgensen, S., Schotta, G., & Sorensen, C. S. (2013). Histone H4 lysine 20 methylation: Key player in epigenetic regulation of genomic integrity. Nucleic Acids Research, 41(5), 2797–2806. https://doi.org/10.1093/nar/gkt012
Kane, A. E., & Sinclair, D. A. (2019). Epigenetic changes during aging and their reprogramming potential. Critical Reviews in Biochemistry and Molecular Biology, 54(1), 61–83. https://doi.org/10.1080/10409238.2019.1570075
Karimi, M. M., Goyal, P., Maksakova, I. A., Bilenky, M., Leung, D., Tang, J. X., Shinkai, Y., Mager, D. L., Jones, S., Hirst, M., & Lorincz, M. C. (2011). DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell, 8(6), 676–687. https://doi.org/10.1016/j.stem.2011.04.004
Karri, S., Yang, Y., Zhou, J., Dickinson, Q., Jia, J., Huang, Y., Wang, Z., Gan, H., & Yu, C. (2024). Defective transfer of parental histone decreases frequency of homologous recombination by increasing free histone pools in budding yeast. Nucleic Acids Research. https://doi.org/10.1093/nar/gkae205
Kato, S., Weng, Q. Y., Insco, M. L., Chen, K. Y., Muralidhar, S., Pozniak, J., Diaz, J. M. S., Drier, Y., Nguyen, N., Lo, J. A., van Rooijen, E., Kemeny, L. V., Zhan, Y., Feng, Y., Silkworth, W., Powell, C. T., Liau, B. B., Xiong, Y., Jin, J., et al. (2020). Gain-of-function genetic alterations of G9a drive oncogenesis. Cancer Discovery, 10(7), 980–997. https://doi.org/10.1158/2159-8290.CD-19-0532
Kelly, G. M., Al-Ejeh, F., McCuaig, R., Casciello, F., Ahmad Kamal, N., Ferguson, B., Pritchard, A. L., Ali, S., Silva, I. P., Wilmott, J. S., Long, G. V., Scolyer, R. A., Rao, S., Hayward, N. K., Gannon, F., & Lee, J. S. (2021). G9a inhibition enhances checkpoint inhibitor blockade response in melanoma. Clinical Cancer Research, 27(9), 2624–2635. https://doi.org/10.1158/1078-0432.CCR-20-3463
Keung, E. Z., Akdemir, K. C., Al Sannaa, G. A., Garnett, J., Lev, D., Torres, K. E., Lazar, A. J., Rai, K., & Chin, L. (2015). Increased H3K9me3 drives dedifferentiated phenotype via KLF6 repression in liposarcoma. The Journal of Clinical Investigation, 125(8), 2965–2978. https://doi.org/10.1172/JCI77976
Khoury-Haddad, H., Guttmann-Raviv, N., Ipenberg, I., Huggins, D., Jeyasekharan, A. D., & Ayoub, N. (2014). PARP1-dependent recruitment of KDM4D histone demethylase to DNA damage sites promotes double-strand break repair. Proceeding of National Academy Science USA, 111(7), 728–737. https://doi.org/10.1073/pnas.1317585111
Khurana, S., Kruhlak, M. J., Kim, J., Tran, A. D., Liu, J., Nyswaner, K., Shi, L., Jailwala, P., Sung, M. H., Hakim, O., & Oberdoerffer, P. (2014). A macrohistone variant links dynamic chromatin compaction to BRCA1-dependent genome maintenance. Cell Reports, 8(4), 1049–1062. https://doi.org/10.1016/j.celrep.2014.07.024
Kim, J. J., Lee, S. Y., & Miller, K. M. (2019). Preserving genome integrity and function: The DNA damage response and histone modifications. Critical Reviews in Biochemistry and Molecular Biology, 54(3), 208–241. https://doi.org/10.1080/10409238.2019.1620676
Klein, K. N., Zhao, P. A., Lyu, X., Sasaki, T., Bartlett, D. A., Singh, A. M., Tasan, I., Zhang, M., Watts, L. P., Hiraga, S. I., Natsume, T., Zhou, X., Baslan, T., Leung, D., Kanemaki, M. T., Donaldson, A. D., Zhao, H., Dalton, S., Corces, V. G., et al. (2021). Replication timing maintains the global epigenetic state in human cells. Science, 372(6540), 371–378. https://doi.org/10.1126/science.aba5545
Klymenko, T., Papp, B., Fischle, W., Kocher, T., Schelder, M., Fritsch, C., Wild, B., Wilm, M., & Muller, J. (2006). A Polycomb group protein complex with sequence-specific DNA-binding and selective methyl-lysine-binding activities. Genes & Development, 20(9), 1110–1122. https://doi.org/10.1101/gad.377406
Kodaira, M., Ryo, H., Kamada, N., Furukawa, K., Takahashi, N., Nakajima, H., Nomura, T., & Nakamura, N. (2010). No evidence of increased mutation rates at microsatellite loci in offspring of A-bomb survivors. Radiation Research, 173(2), 205–213. https://doi.org/10.1667/RR1991.1
Kodaira, M., Satoh, C., Hiyama, K., & Toyama, K. (1995). Lack of effects of atomic bomb radiation on genetic instability of tandem-repetitive elements in human germ cells. American Journal of Human Genetics, 57(6), 1275–1283.
Kohi, S. M., Feng, T., Tian, Y., & Zhu, W.-G. (2022). Biological function and regulation of histone 4 lysine 20 methylation in DNA damage response. Genome Instability & Disease, 3, 33–46.
Kooistra, S. M., & Helin, K. (2012). Molecular mechanisms and potential functions of histone demethylases. Nature Reviews Molecular Cell Biology, 13(5), 297–311. https://doi.org/10.1038/nrm3327
Koturbash, I., Boyko, A., Rodriguez-Juarez, R., McDonald, R. J., Tryndyak, V. P., Kovalchuk, I., Pogribny, I. P., & Kovalchuk, O. (2007). Role of epigenetic effectors in maintenance of the long-term persistent bystander effect in spleen in vivo. Carcinogenesis, 28(8), 1831–1838. https://doi.org/10.1093/carcin/bgm053
Kruhlak, M., Crouch, E. E., Orlov, M., Montano, C., Gorski, S. A., Nussenzweig, A., Misteli, T., Phair, R. D., & Casellas, R. (2007). The ATM repair pathway inhibits RNA polymerase I transcription in response to chromosome breaks. Nature, 447(7145), 730–734. https://doi.org/10.1038/nature05842
Lachner, M., O’Carroll, D., Rea, S., Mechtler, K., & Jenuwein, T. (2001). Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature, 410(6824), 116–120. https://doi.org/10.1038/35065132
Lamm, N., Rogers, S., & Cesare, A. J. (2021). Chromatin mobility and relocation in DNA repair. Trends in Cell Biology, 31(10), 843–855. https://doi.org/10.1016/j.tcb.2021.06.002
Lam-Ubol, A., & Phattarataratip, E. (2022). Distinct histone H3 modification profiles correlate with aggressive characteristics of salivary gland neoplasms. Science and Reports, 12(1), 15063. https://doi.org/10.1038/s41598-022-19174-9
Lander, E. S., Linton, L. M., Birren, B., Nusbaum, C., Zody, M. C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh, W., Funke, R., Gage, D., Harris, K., Heaford, A., Howland, J., Kann, L., Lehoczky, J., LeVine, R., McEwan, P., et al. (2001). Initial sequencing and analysis of the human genome. Nature, 409(6822), 860–921. https://doi.org/10.1038/35057062
Lee, D. H., Kim, G. W., Jeon, Y. H., Yoo, J., Lee, S. W., & Kwon, S. H. (2020). Advances in histone demethylase KDM4 as cancer therapeutic targets. The FASEB Journal, 34(3), 3461–3484. https://doi.org/10.1096/fj.201902584R
Lee, J. S., Smith, E., & Shilatifard, A. (2010). The language of histone crosstalk. Cell, 142(5), 682–685. https://doi.org/10.1016/j.cell.2010.08.011
Lee, Y. H., Kuo, C. Y., Stark, J. M., Shih, H. M., & Ann, D. K. (2013). HP1 promotes tumor suppressor BRCA1 functions during the DNA damage response. Nucleic Acids Research, 41(11), 5784–5798. https://doi.org/10.1093/nar/gkt231
Leszinski, G., Gezer, U., Siegele, B., Stoetzer, O., & Holdenrieder, S. (2012). Relevance of histone marks H3K9me3 and H4K20me3 in cancer. Anticancer Research, 32(5), 2199–2205.
Lewis, P. W., Muller, M. M., Koletsky, M. S., Cordero, F., Lin, S., Banaszynski, L. A., Garcia, B. A., Muir, T. W., Becher, O. J., & Allis, C. D. (2013). Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science, 340(6134), 857–861. https://doi.org/10.1126/science.1232245
Li, Z., Chen, Y., Tang, M., Li, Y., & Zhu, W.-G. (2020a). Regulation of DNA damage-induced ATM activation by histone modifications. Genome Instability & Disease, 1, 20–33.
Li, Z., Duan, S., Hua, X., Xu, X., Li, Y., Menolfi, D., Zhou, H., Lu, C., Zha, S., Goff, S. P., & Zhang, Z. (2023). Asymmetric distribution of parental H3K9me3 in S phase silences L1 elements. Nature, 623(7987), 643–651. https://doi.org/10.1038/s41586-023-06711-3
Li, Z., Hua, X., Serra-Cardona, A., Xu, X., Gan, S., Zhou, H., Yang, W. S., Chen, C. L., Xu, R. M., & Zhang, Z. (2020b). DNA polymerase alpha interacts with H3–H4 and facilitates the transfer of parental histones to lagging strands. Science Advances, 6(35), eabb5820. https://doi.org/10.1126/sciadv.abb5820
Little, M. P., Goodhead, D. T., Bridges, B. A., & Bouffler, S. D. (2013). Evidence relevant to untargeted and transgenerational effects in the offspring of irradiated parents. Mutation Research, 753(1), 50–67. https://doi.org/10.1016/j.mrrev.2013.04.001
Liu, N., Lee, C. H., Swigut, T., Grow, E., Gu, B., Bassik, M. C., & Wysocka, J. (2018). Selective silencing of euchromatic L1s revealed by genome-wide screens for L1 regulators. Nature, 553(7687), 228–232. https://doi.org/10.1038/nature25179
Liu, X., Gao, Q., Li, P., Zhao, Q., Zhang, J., Li, J., Koseki, H., & Wong, J. (2013). UHRF1 targets DNMT1 for DNA methylation through cooperative binding of hemi-methylated DNA and methylated H3K9. Nature Communications, 4, 1563. https://doi.org/10.1038/ncomms2562
Lopez-Cobo, S., Fuentealba, J. R., Gueguen, P., Bonte, P. E., Tsalkitzi, K., Chacon, I., Glauzy, S., Bohineust, A., Biquand, A., Silva, L., Gouveia, Z., Goudot, C., Perez, F., Saitakis, M., & Amigorena, S. (2024). SUV39H1 ablation enhances Long-Term CAR-T function in solid tumors. Cancer Discovery, 14(1), 120–141. https://doi.org/10.1158/2159-8290.CD-22-1350
Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M., & Kroemer, G. (2023). Hallmarks of aging: An expanding universe. Cell, 186(2), 243–278. https://doi.org/10.1016/j.cell.2022.11.001
Lu, C., Jain, S. U., Hoelper, D., Bechet, D., Molden, R. C., Ran, L., Murphy, D., Venneti, S., Hameed, M., Pawel, B. R., Wunder, J. S., Dickson, B. C., Lundgren, S. M., Jani, K. S., De Jay, N., Papillon-Cavanagh, S., Andrulis, I. L., Sawyer, S. L., Grynspan, D., et al. (2016). Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science, 352(6287), 844–849. https://doi.org/10.1126/science.aac7272
Lu, X., Tang, M., Zhu, Q., Yang, Q., Li, Z., Bao, Y., Liu, G., Hou, T., Lv, Y., Zhao, Y., Wang, H., Yang, Y., Cheng, Z., Wen, H., Liu, B., Xu, X., Gu, L., & Zhu, W. G. (2019). GLP-catalyzed H4K16me1 promotes 53BP1 recruitment to permit DNA damage repair and cell survival. Nucleic Acids Research, 47(21), 10977–10993. https://doi.org/10.1093/nar/gkz897
Lu, Y. R., Tian, X., & Sinclair, D. A. (2023). The Information Theory of Aging. Nat Aging, 3(12), 1486–1499. https://doi.org/10.1038/s43587-023-00527-6
Luijsterburg, M. S., Dinant, C., Lans, H., Stap, J., Wiernasz, E., Lagerwerf, S., Warmerdam, D. O., Lindh, M., Brink, M. C., Dobrucki, J. W., Aten, J. A., Fousteri, M. I., Jansen, G., Dantuma, N. P., Vermeulen, W., Mullenders, L. H., Houtsmuller, A. B., Verschure, P. J., & van Driel, R. (2009). Heterochromatin protein 1 is recruited to various types of DNA damage. Journal of Cell Biology, 185(4), 577–586. https://doi.org/10.1083/jcb.200810035
Lukas, C., Savic, V., Bekker-Jensen, S., Doil, C., Neumann, B., Pedersen, R. S., Grofte, M., Chan, K. L., Hickson, I. D., Bartek, J., & Lukas, J. (2011a). 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nature Cell Biology, 13(3), 243–253. https://doi.org/10.1038/ncb2201
Lukas, J., Lukas, C., & Bartek, J. (2011b). More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nature Cell Biology, 13(10), 1161–1169. https://doi.org/10.1038/ncb2344
Machour, F. E., & Ayoub, N. (2020). Transcriptional regulation at DSBs: mechanisms and consequences. Trends in Genetics, 36(12), 981–997. https://doi.org/10.1016/j.tig.2020.01.001
Mah, L. J., El-Osta, A., & Karagiannis, T. C. (2010). gammaH2AX: A sensitive molecular marker of DNA damage and repair. Leukemia, 24(4), 679–686. https://doi.org/10.1038/leu.2010.6
Maksakova, I. A., Thompson, P. J., Goyal, P., Jones, S. J., Singh, P. B., Karimi, M. M., & Lorincz, M. C. (2013). Distinct roles of KAP1, HP1 and G9a/GLP in silencing of the two-cell-specific retrotransposon MERVL in mouse ES cells. Epigenetics & Chromatin, 6(1), 15. https://doi.org/10.1186/1756-8935-6-15
Mallette, F. A., Mattiroli, F., Cui, G., Young, L. C., Hendzel, M. J., Mer, G., Sixma, T. K., & Richard, S. (2012). RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO Journal, 31(8), 1865–1878. https://doi.org/10.1038/emboj.2012.47
Mansfield, R. E., Musselman, C. A., Kwan, A. H., Oliver, S. S., Garske, A. L., Davrazou, F., Denu, J. M., Kutateladze, T. G., & Mackay, J. P. (2011). Plant homeodomain (PHD) fingers of CHD4 are histone H3-binding modules with preference for unmodified H3K4 and methylated H3K9. Journal of Biological Chemistry, 286(13), 11779–11791. https://doi.org/10.1074/jbc.M110.208207
Mao, X., Wu, J., Zhang, Q., Zhang, S., Chen, X., Liu, X., Wei, M., Wan, X., Qiu, L., Zeng, M., Lei, X., Liu, C., & Han, J. (2023). Requirement of WDR70 for POLE3-mediated DNA double-strand breaks repair. Science Advances, 9(36), eadh2358. https://doi.org/10.1126/sciadv.adh2358
Marechal, A., & Zou, L. (2013). DNA damage sensing by the ATM and ATR kinases. Cold Spring Harbor Perspectives in Biology. https://doi.org/10.1101/cshperspect.a012716
Margueron, R., & Reinberg, D. (2011). The Polycomb complex PRC2 and its mark in life. Nature, 469(7330), 343–349. https://doi.org/10.1038/nature09784
Markova, E., Schultz, N., & Belyaev, I. Y. (2007). Kinetics and dose-response of residual 53BP1/gamma-H2AX foci: Co-localization, relationship with DSB repair and clonogenic survival. International Journal of Radiation Biology, 83(5), 319–329. https://doi.org/10.1080/09553000601170469
Matsui, T., Leung, D., Miyashita, H., Maksakova, I. A., Miyachi, H., Kimura, H., Tachibana, M., Lorincz, M. C., & Shinkai, Y. (2010). Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature, 464(7290), 927–931. https://doi.org/10.1038/nature08858
Matsumura, Y., Nakaki, R., Inagaki, T., Yoshida, A., Kano, Y., Kimura, H., Tanaka, T., Tsutsumi, S., Nakao, M., Doi, T., Fukami, K., Osborne, T. F., Kodama, T., Aburatani, H., & Sakai, J. (2015). H3K4/H3K9me3 bivalent chromatin domains targeted by lineage-specific DNA methylation pauses adipocyte differentiation. Molecular Cell, 60(4), 584–596. https://doi.org/10.1016/j.molcel.2015.10.025
Mattiroli, F., Vissers, J. H., van Dijk, W. J., Ikpa, P., Citterio, E., Vermeulen, W., Marteijn, J. A., & Sixma, T. K. (2012). RNF168 ubiquitinates K13–15 on H2A/H2AX to drive DNA damage signaling. Cell, 150(6), 1182–1195. https://doi.org/10.1016/j.cell.2012.08.005
Melcher, M., Schmid, M., Aagaard, L., Selenko, P., Laible, G., & Jenuwein, T. (2000). Structure-function analysis of SUV39H1 reveals a dominant role in heterochromatin organization, chromosome segregation, and mitotic progression. Molecular and Cellular Biology, 20(10), 3728–3741. https://doi.org/10.1128/MCB.20.10.3728-3741.2000
Mellman, I., Coukos, G., & Dranoff, G. (2011). Cancer immunotherapy comes of age. Nature, 480(7378), 480–489. https://doi.org/10.1038/nature10673
Metzger, E., Wissmann, M., Yin, N., Muller, J. M., Schneider, R., Peters, A. H., Gunther, T., Buettner, R., & Schule, R. (2005). LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature, 437(7057), 436–439. https://doi.org/10.1038/nature04020
Mikkelsen, T. S., Ku, M., Jaffe, D. B., Issac, B., Lieberman, E., Giannoukos, G., Alvarez, P., Brockman, W., Kim, T. K., Koche, R. P., Lee, W., Mendenhall, E., O’Donovan, A., Presser, A., Russ, C., Xie, X., Meissner, A., Wernig, M., Jaenisch, R., et al. (2007). Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature, 448(7153), 553–560. https://doi.org/10.1038/nature06008
Milanovic, M., Fan, D. N. Y., Belenki, D., Dabritz, J. H. M., Zhao, Z., Yu, Y., Dorr, J. R., Dimitrova, L., Lenze, D., Monteiro Barbosa, I. A., Mendoza-Parra, M. A., Kanashova, T., Metzner, M., Pardon, K., Reimann, M., Trumpp, A., Dorken, B., Zuber, J., Gronemeyer, H., et al. (2018). Senescence-associated reprogramming promotes cancer stemness. Nature, 553(7686), 96–100. https://doi.org/10.1038/nature25167
Mo, Y., Duan, S., Zhang, X., Hua, X., Zhou, H., Wei, H. J., Watanabe, J., McQuillan, N., Su, Z., Gu, W., Wu, C. C., Vakoc, C. R., Hashizume, R., Chang, K., & Zhang, Z. (2022). Epigenome programming by H3.3K27M mutation creates a dependence of pediatric glioma on SMARCA4. Cancer Discovery, 12(12), 2906–2929. https://doi.org/10.1158/2159-8290.CD-21-1492
Montavon, T., Shukeir, N., Erikson, G., Engist, B., Onishi-Seebacher, M., Ryan, D., Musa, Y., Mittler, G., Meyer, A. G., Genoud, C., & Jenuwein, T. (2021). Complete loss of H3K9 methylation dissolves mouse heterochromatin organization. Nature Communications, 12(1), 4359. https://doi.org/10.1038/s41467-021-24532-8
Morera, L., Lubbert, M., & Jung, M. (2016). Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clinical Epigenetics, 8, 57. https://doi.org/10.1186/s13148-016-0223-4
Morton, L. M., Karyadi, D. M., Stewart, C., Bogdanova, T. I., Dawson, E. T., Steinberg, M. K., Dai, J., Hartley, S. W., Schonfeld, S. J., Sampson, J. N., Maruvka, Y. E., Kapoor, V., Ramsden, D. A., Carvajal-Garcia, J., Perou, C. M., Parker, J. S., Krznaric, M., Yeager, M., Boland, J. F., et al. (2021). Radiation-related genomic profile of papillary thyroid carcinoma after the Chernobyl accident. Science, 372(6543), eabg2538. https://doi.org/10.1126/science.abg2538
Motamedi, M. R., Verdel, A., Colmenares, S. U., Gerber, S. A., Gygi, S. P., & Moazed, D. (2004). Two RNAi complexes, RITS and RDRC, physically interact and localize to noncoding centromeric RNAs. Cell, 119(6), 789–802. https://doi.org/10.1016/j.cell.2004.11.034
Mothersill, C., & Seymour, C. B. (2004). Radiation-induced bystander effects–implications for cancer. Nature Reviews Cancer, 4(2), 158–164. https://doi.org/10.1038/nrc1277
Muller, I., & Helin, K. (2024). Keep quiet: The HUSH complex in transcriptional silencing and disease. Nature Structural & Molecular Biology, 31(1), 11–22. https://doi.org/10.1038/s41594-023-01173-7
Muller-Tidow, C., Klein, H. U., Hascher, A., Isken, F., Tickenbrock, L., Thoennissen, N., Agrawal-Singh, S., Tschanter, P., Disselhoff, C., Wang, Y., Becker, A., Thiede, C., Ehninger, G., et al. (2010). Profiling of histone H3 lysine 9 trimethylation levels predicts transcription factor activity and survival in acute myeloid leukemia. Blood, 116(18), 3564–3571. https://doi.org/10.1182/blood-2009-09-240978
Murr, R., Loizou, J. I., Yang, Y. G., Cuenin, C., Li, H., Wang, Z. Q., & Herceg, Z. (2006). Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nature Cell Biology, 8(1), 91–99. https://doi.org/10.1038/ncb1343
Nacev, B. A., Feng, L., Bagert, J. D., Lemiesz, A. E., Gao, J., Soshnev, A. A., Kundra, R., Schultz, N., Muir, T. W., & Allis, C. D. (2019). The expanding landscape of “oncohistone” mutations in human cancers. Nature, 567(7749), 473–478. https://doi.org/10.1038/s41586-019-1038-1
Nagaraja, S., Vitanza, N. A., Woo, P. J., Taylor, K. R., Liu, F., Zhang, L., Li, M., Meng, W., Ponnuswami, A., Sun, W., Ma, J., Hulleman, E., Swigut, T., Wysocka, J., Tang, Y., & Monje, M. (2017). Transcriptional dependencies in diffuse intrinsic pontine glioma. Cancer Cell, 31(5), 635–652. https://doi.org/10.1016/j.ccell.2017.03.011
Nakayama, J., Rice, J. C., Strahl, B. D., Allis, C. D., & Grewal, S. I. (2001). Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science, 292(5514), 110–113. https://doi.org/10.1126/science.1060118
Nicetto, D., Donahue, G., Jain, T., Peng, T., Sidoli, S., Sheng, L., Montavon, T., Becker, J. S., Grindheim, J. M., Blahnik, K., Garcia, B. A., Tan, K., Bonasio, R., Jenuwein, T., & Zaret, K. S. (2019). H3K9me3-heterochromatin loss at protein-coding genes enables developmental lineage specification. Science, 363(6424), 294–297. https://doi.org/10.1126/science.aau0583
Nicetto, D., & Zaret, K. S. (2019). Role of H3K9me3 heterochromatin in cell identity establishment and maintenance. Current Opinion in Genetics & Development, 55, 1–10. https://doi.org/10.1016/j.gde.2019.04.013
Nurk, S., Koren, S., Rhie, A., Rautiainen, M., Bzikadze, A. V., Mikheenko, A., Vollger, M. R., Altemose, N., Uralsky, L., Gershman, A., Aganezov, S., Hoyt, S. J., Diekhans, M., Logsdon, G. A., Alonge, M., Antonarakis, S. E., Borchers, M., Bouffard, G. G., Brooks, S. Y., et al. (2022). The complete sequence of a human genome. Science, 376(6588), 44–53. https://doi.org/10.1126/science.abj6987
Padeken, J., Methot, S. P., & Gasser, S. M. (2022). Establishment of H3K9-methylated heterochromatin and its functions in tissue differentiation and maintenance. Nature Reviews Molecular Cell Biology, 23(9), 623–640. https://doi.org/10.1038/s41580-022-00483-w
Panditharatna, E., Marques, J. G., Wang, T., Trissal, M. C., Liu, I., Jiang, L., Beck, A., Groves, A., Dharia, N. V., Li, D., Hoffman, S. E., Kugener, G., Shaw, M. L., Mire, H. M., Hack, O. A., Dempster, J. M., Lareau, C., Dai, L., Sigua, L. H., et al. (2022). BAF complex maintains glioma stem cells in pediatric H3K27M glioma. Cancer Discovery, 12(12), 2880–2905. https://doi.org/10.1158/2159-8290.CD-21-1491
Panier, S., & Boulton, S. J. (2014). Double-strand break repair: 53BP1 comes into focus. Nature Reviews Molecular Cell Biology, 15(1), 7–18. https://doi.org/10.1038/nrm3719
Papillon-Cavanagh, S., Lu, C., Gayden, T., Mikael, L. G., Bechet, D., Karamboulas, C., Ailles, L., Karamchandani, J., Marchione, D. M., Garcia, B. A., Weinreb, I., Goldstein, D., Lewis, P. W., Dancu, O. M., Dhaliwal, S., Stecho, W., Howlett, C. J., Mymryk, J. S., Barrett, J. W., et al. (2017). Impaired H3K36 methylation defines a subset of head and neck squamous cell carcinomas. Nature Genetics, 49(2), 180–185. https://doi.org/10.1038/ng.3757
Pardoll, D. M. (2012). The blockade of immune checkpoints in cancer immunotherapy. Nature Reviews Cancer, 12(4), 252–264. https://doi.org/10.1038/nrc3239
Park, Y. S., Jin, M. Y., Kim, Y. J., Yook, J. H., Kim, B. S., & Jang, S. J. (2008). The global histone modification pattern correlates with cancer recurrence and overall survival in gastric adenocarcinoma. Annals of Surgical Oncology, 15(7), 1968–1976. https://doi.org/10.1245/s10434-008-9927-9
Pei, H. D., Zhang, L., Luo, K. T., Qin, Y. X., Chesi, M., Fei, F., Bergsagel, P. L., Wang, L. W., You, Z. S., & Lou, Z. K. (2011). MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature, 470(7332), 124-U144. https://doi.org/10.1038/nature09658
Peters, A. H., O’Carroll, D., Scherthan, H., Mechtler, K., Sauer, S., Schofer, C., Weipoltshammer, K., Pagani, M., Lachner, M., Kohlmaier, A., Opravil, S., Doyle, M., Sibilia, M., & Jenuwein, T. (2001). Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell, 107(3), 323–337. https://doi.org/10.1016/s0092-8674(01)00542-6
Petryk, N., Dalby, M., Wenger, A., Stromme, C. B., Strandsby, A., Andersson, R., & Groth, A. (2018). MCM2 promotes symmetric inheritance of modified histones during DNA replication. Science, 361(6409), 1389–1392. https://doi.org/10.1126/science.aau0294
Pinheiro, I., Margueron, R., Shukeir, N., Eisold, M., Fritzsch, C., Richter, F. M., Mittler, G., Genoud, C., Goyama, S., Kurokawa, M., Son, J., Reinberg, D., Lachner, M., & Jenuwein, T. (2012). Prdm3 and Prdm16 are H3K9me1 methyltransferases required for mammalian heterochromatin integrity. Cell, 150(5), 948–960. https://doi.org/10.1016/j.cell.2012.06.048
Piunti, A., Hashizume, R., Morgan, M. A., Bartom, E. T., Horbinski, C. M., Marshall, S. A., Rendleman, E. J., Ma, Q., Takahashi, Y. H., Woodfin, A. R., Misharin, A. V., Abshiru, N. A., Lulla, R. R., Saratsis, A. M., Kelleher, N. L., James, C. D., & Shilatifard, A. (2017). Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nature Medicine, 23(4), 493–500. https://doi.org/10.1038/nm.4296
Polo, S. E., & Jackson, S. P. (2011). Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes & Development, 25(5), 409–433. https://doi.org/10.1101/gad.2021311
Prigozhin, D. M., Douse, C. H., Farleigh, L. E., Albecka, A., Tchasovnikarova, I. A., Timms, R. T., Oda, S. I., Adolf, F., Freund, S. M. V., Maslen, S., Lehner, P. J., & Modis, Y. (2020). Periphilin self-association underpins epigenetic silencing by the HUSH complex. Nucleic Acids Research, 48(18), 10313–10328. https://doi.org/10.1093/nar/gkaa785
Qian, Y., Li, Y., Zheng, C., Lu, T., Sun, R., Mao, Y., Yu, S., Fan, H., & Zhang, Z. (2020). High methylation levels of histone H3 lysine 9 associated with activation of hypoxia-inducible factor 1alpha (HIF-1alpha) predict patients’ worse prognosis in human hepatocellular carcinomas. Cancer Genetics, 245, 17–26. https://doi.org/10.1016/j.cancergen.2020.04.077
Qin, B., Yu, J., Nowsheen, S., Wang, M., Tu, X., Liu, T., Li, H., Wang, L., & Lou, Z. (2019). UFL1 promotes histone H4 ufmylation and ATM activation. Nature Communications, 10(1), 1242. https://doi.org/10.1038/s41467-019-09175-0
Qin, B., Yu, J., Nowsheen, S., Zhao, F., Wang, L., & Lou, Z. (2020). STK38 promotes ATM activation by acting as a reader of histone H4 ufmylation. Science Advances, 6(23), eaxx8214. https://doi.org/10.1126/sciadv.aax8214
Rea, S., Eisenhaber, F., O’Carroll, D., Strahl, B. D., Sun, Z. W., Schmid, M., Opravil, S., Mechtler, K., Ponting, C. P., Allis, C. D., & Jenuwein, T. (2000). Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature, 406(6796), 593–599. https://doi.org/10.1038/35020506
Reinhart, B. J., & Bartel, D. P. (2002). Small RNAs correspond to centromere heterochromatic repeats. Science, 297(5588), 1831. https://doi.org/10.1126/science.1077183
Ren, W., Fan, H., Grimm, S. A., Guo, Y., Kim, J. J., Yin, J., Li, L., Petell, C. J., Tan, X. F., Zhang, Z. M., Coan, J. P., Gao, L., Cai, L., Detrick, B., Cetin, B., Cui, Q., Strahl, B. D., Gozani, O., Wang, Y., et al. (2020). Direct readout of heterochromatic H3K9me3 regulates DNMT1-mediated maintenance DNA methylation. Proc Natl Acad Sci U S A, 117(31), 18439–18447. https://doi.org/10.1073/pnas.2009316117
Roadmap Epigenomics, C., Kundaje, A., Meuleman, W., Ernst, J., Bilenky, M., Yen, A., Heravi-Moussavi, A., Kheradpour, P., Zhang, Z., Wang, J., Ziller, M. J., Amin, V., Whitaker, J. W., Schultz, M. D., Ward, L. D., Sarkar, A., Quon, G., Sandstrom, R. S., Eaton, M. L., et al. (2015). Integrative analysis of 111 reference human epigenomes. Nature, 518(7539), 317–330. https://doi.org/10.1038/nature14248
Robbez-Masson, L., Tie, C. H. C., Conde, L., Tunbak, H., Husovsky, C., Tchasovnikarova, I. A., Timms, R. T., Herrero, J., Lehner, P. J., & Rowe, H. M. (2018). The HUSH complex cooperates with TRIM28 to repress young retrotransposons and new genes. Genome Research, 28(6), 836–845. https://doi.org/10.1101/gr.228171.117
Rothbart, S. B., Krajewski, K., Nady, N., Tempel, W., Xue, S., Badeaux, A. I., Barsyte-Lovejoy, D., Martinez, J. Y., Bedford, M. T., Fuchs, S. M., Arrowsmith, C. H., & Strahl, B. D. (2012). Association of UHRF1 with methylated H3K9 directs the maintenance of DNA methylation. Nature Structural & Molecular Biology, 19(11), 1155–1160. https://doi.org/10.1038/nsmb.2391
Rui, L., Emre, N. C., Kruhlak, M. J., Chung, H. J., Steidl, C., Slack, G., Wright, G. W., Lenz, G., Ngo, V. N., Shaffer, A. L., Xu, W., Zhao, H., Yang, Y., Lamy, L., Davis, R. E., Xiao, W., Powell, J., Maloney, D., Thomas, C. J., et al. (2010). Cooperative epigenetic modulation by cancer amplicon genes. Cancer Cell, 18(6), 590–605. https://doi.org/10.1016/j.ccr.2010.11.013
Sancar, A., Lindsey-Boltz, L. A., Unsal-Kacmaz, K., & Linn, S. (2004). Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annual Review of Biochemistry, 73, 39–85. https://doi.org/10.1146/annurev.biochem.73.011303.073723
Schleich, K., Kase, J., Dorr, J. R., Trescher, S., Bhattacharya, A., Yu, Y., Wailes, E. M., Fan, D. N. Y., Lohneis, P., Milanovic, M., Lau, A., Lenze, D., Hummel, M., Chapuy, B., Leser, U., Reimann, M., Lee, S., & Schmitt, C. A. (2020). H3K9me3-mediated epigenetic regulation of senescence in mice predicts outcome of lymphoma patients. Nature Communications, 11(1), 3651. https://doi.org/10.1038/s41467-020-17467-z
Schlissel, G., & Rine, J. (2019). The nucleosome core particle remembers its position through DNA replication and RNA transcription. Proceeding of National Academy Science USA, 116(41), 20605–20611. https://doi.org/10.1073/pnas.1911943116
Schotta, G., Lachner, M., Sarma, K., Ebert, A., Sengupta, R., Reuter, G., Reinberg, D., & Jenuwein, T. (2004). A silencing pathway to induce H3–K9 and H4–K20 trimethylation at constitutive heterochromatin. Genes & Development, 18(11), 1251–1262. https://doi.org/10.1101/gad.300704
Schultz, D. C., Ayyanathan, K., Negorev, D., Maul, G. G., & Rauscher, F. J. (2002). SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes & Development, 16(8), 919–932. https://doi.org/10.1101/gad.973302
Schumacher, B., Pothof, J., Vijg, J., & Hoeijmakers, J. H. J. (2021). The central role of DNA damage in the ageing process. Nature, 592(7856), 695–703. https://doi.org/10.1038/s41586-021-03307-7
Schwartzentruber, J., Korshunov, A., Liu, X. Y., Jones, D. T., Pfaff, E., Jacob, K., Sturm, D., Fontebasso, A. M., Quang, D. A., Tonjes, M., Hovestadt, V., Albrecht, S., Kool, M., Nantel, A., Konermann, C., Lindroth, A., Jager, N., Rausch, T., Ryzhova, M., et al. (2012). Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature, 482(7384), 226–231. https://doi.org/10.1038/nature10833
Scully, R., Panday, A., Elango, R., & Willis, N. A. (2019). DNA double-strand break repair-pathway choice in somatic mammalian cells. Nature Reviews Molecular Cell Biology, 20(11), 698–714. https://doi.org/10.1038/s41580-019-0152-0
Seczynska, M., Bloor, S., Cuesta, S. M., & Lehner, P. J. (2022). Genome surveillance by HUSH-mediated silencing of intronless mobile elements. Nature, 601(7893), 440–445. https://doi.org/10.1038/s41586-021-04228-1
Seczynska, M., & Lehner, P. J. (2023). The sound of silence: Mechanisms and implications of HUSH complex function. Trends in Genetics, 39(4), 251–267. https://doi.org/10.1016/j.tig.2022.12.005
Segovia, C., San Jose-Eneriz, E., Munera-Maravilla, E., Martinez-Fernandez, M., Garate, L., Miranda, E., Vilas-Zornoza, A., Lodewijk, I., Rubio, C., Segrelles, C., Valcarcel, L. V., Rabal, O., Casares, N., Bernardini, A., Suarez-Cabrera, C., Lopez-Calderon, F. F., Fortes, P., Casado, J. A., Duenas, M., et al. (2019). Inhibition of a G9a/DNMT network triggers immune-mediated bladder cancer regression. Nature Medicine, 25(7), 1073–1081. https://doi.org/10.1038/s41591-019-0499-y
Senft, A. D., & Macfarlan, T. S. (2021). Transposable elements shape the evolution of mammalian development. Nature Reviews Genetics, 22(11), 691–711. https://doi.org/10.1038/s41576-021-00385-1
Shan, C. M., Wang, J., Xu, K., Chen, H., Yue, J. X., Andrews, S., Moresco, J. J., Yates, J. R., Nagy, P. L., Tong, L., & Jia, S. (2016). A histone H3K9M mutation traps histone methyltransferase Clr4 to prevent heterochromatin spreading. eLife. https://doi.org/10.7554/eLife.17903
Shanbhag, N. M., Rafalska-Metcalf, I. U., Balane-Bolivar, C., Janicki, S. M., & Greenberg, R. A. (2010). ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell, 141(6), 970–981. https://doi.org/10.1016/j.cell.2010.04.038
Sharif, J., Muto, M., Takebayashi, S., Suetake, I., Iwamatsu, A., Endo, T. A., Shinga, J., Mizutani-Koseki, Y., Toyoda, T., Okamura, K., Tajima, S., Mitsuya, K., Okano, M., & Koseki, H. (2007). The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature, 450(7171), 908–912. https://doi.org/10.1038/nature06397
Shen, J. Z., Qiu, Z., Wu, Q., Finlay, D., Garcia, G., Sun, D., Rantala, J., Barshop, W., Hope, J. L., Gimple, R. C., Sangfelt, O., Bradley, L. M., Wohlschlegel, J., Rich, J. N., & Spruck, C. (2021). FBXO44 promotes DNA replication-coupled repetitive element silencing in cancer cells. Cell, 184(2), 352–369. https://doi.org/10.1016/j.cell.2020.11.042
Shi, Y., Lan, F., Matson, C., Mulligan, P., Whetstine, J. R., Cole, P. A., Casero, R. A., & Shi, Y. (2004). Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell, 119(7), 941–953. https://doi.org/10.1016/j.cell.2004.12.012
Shinkai, Y., & Tachibana, M. (2011). H3K9 methyltransferase G9a and the related molecule GLP. Genes & Development, 25(8), 781–788. https://doi.org/10.1101/gad.2027411
Shirai, A., Kawaguchi, T., Shimojo, H., Muramatsu, D., Ishida-Yonetani, M., Nishimura, Y., Kimura, H., Nakayama, J. I., & Shinkai, Y. (2017). Impact of nucleic acid and methylated H3K9 binding activities of Suv39h1 on its heterochromatin assembly. eLife. https://doi.org/10.7554/eLife.25317
Shrivastav, M., De Haro, L. P., & Nickoloff, J. A. (2008). Regulation of DNA double-strand break repair pathway choice. Cell Research, 18(1), 134–147. https://doi.org/10.1038/cr.2007.111
Siddiqui, M. S., Francois, M., Fenech, M. F., & Leifert, W. R. (2015). Persistent gammaH2AX: A promising molecular marker of DNA damage and aging. Mutation Research, Reviews in Mutation Research, 766, 1–19. https://doi.org/10.1016/j.mrrev.2015.07.001
Song, J. S., Kim, Y. S., Kim, D. K., Park, S. I., & Jang, S. J. (2012). Global histone modification pattern associated with recurrence and disease-free survival in non-small cell lung cancer patients. Pathology International, 62(3), 182–190. https://doi.org/10.1111/j.1440-1827.2011.02776.x
Soria, G., & Almouzni, G. (2013). Differential contribution of HP1 proteins to DNA end resection and homology-directed repair. Cell Cycle, 12(3), 422–429. https://doi.org/10.4161/cc.23215
Soria, G., Polo, S. E., & Almouzni, G. (2012). Prime, repair, restore: The active role of chromatin in the DNA damage response. Molecular Cell, 46(6), 722–734. https://doi.org/10.1016/j.molcel.2012.06.002
Spies, J., Lukas, C., Somyajit, K., Rask, M. B., Lukas, J., & Neelsen, K. J. (2019). 53BP1 nuclear bodies enforce replication timing at under-replicated DNA to limit heritable DNA damage. Nature Cell Biology, 21(4), 487–497. https://doi.org/10.1038/s41556-019-0293-6
Spiliopoulou, P., Spear, S., Mirza, H., Garner, I., McGarry, L., Grundland-Freile, F., Cheng, Z., Ennis, D. P., Iyer, N., McNamara, S., Natoli, M., Mason, S., Blyth, K., Adams, P. D., Roxburgh, P., Fuchter, M. J., Brown, B., & McNeish, I. A. (2022). Dual G9A/EZH2 inhibition stimulates antitumor immune response in ovarian high-grade serous carcinoma. Molecular Cancer Therapeutics, 21(4), 522–534. https://doi.org/10.1158/1535-7163.MCT-21-0743
Stewart, G. S., Panier, S., Townsend, K., Al-Hakim, A. K., Kolas, N. K., Miller, E. S., Nakada, S., Ylanko, J., Olivarius, S., Mendez, M., Oldreive, C., Wildenhain, J., Tagliaferro, A., Pelletier, L., Taubenheim, N., Durandy, A., Byrd, P. J., Stankovic, T., Taylor, A. M., et al. (2009). The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell, 136(3), 420–434. https://doi.org/10.1016/j.cell.2008.12.042
Stewart-Morgan, K. R., Petryk, N., & Groth, A. (2020). Chromatin replication and epigenetic cell memory. Nature Cell Biology, 22(4), 361–371. https://doi.org/10.1038/s41556-020-0487-y
Struhl, K. (1998). Histone acetylation and transcriptional regulatory mechanisms. Genes & Development, 12(5), 599–606. https://doi.org/10.1101/gad.12.5.599
Suganuma, T., & Workman, J. L. (2008). Crosstalk among histone modifications. Cell, 135(4), 604–607. https://doi.org/10.1016/j.cell.2008.10.036
Sun, Y., Jiang, X., Chen, S., Fernandes, N., & Price, B. D. (2005). A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proceeding National Academy Science USA, 102(37), 13182–13187. https://doi.org/10.1073/pnas.0504211102
Sun, Y., Jiang, X., Xu, Y., Ayrapetov, M. K., Moreau, L. A., Whetstine, J. R., & Price, B. D. (2009). Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nature Cell Biology, 11(11), 1376–1382. https://doi.org/10.1038/ncb1982
Supek, F., & Lehner, B. (2015). Differential DNA mismatch repair underlies mutation rate variation across the human genome. Nature, 521(7550), 81–84. https://doi.org/10.1038/nature14173
Suva, M. L., Riggi, N., & Bernstein, B. E. (2013). Epigenetic reprogramming in cancer. Science, 339(6127), 1567–1570. https://doi.org/10.1126/science.1230184
Tachibana, M., Matsumura, Y., Fukuda, M., Kimura, H., & Shinkai, Y. (2008). G9a/GLP complexes independently mediate H3K9 and DNA methylation to silence transcription. EMBO Journal, 27(20), 2681–2690. https://doi.org/10.1038/emboj.2008.192
Tachibana, M., Sugimoto, K., Fukushima, T., & Shinkai, Y. (2001). SET domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. Journal of Biology Chemistry, 276(27), 25309–25317. https://doi.org/10.1074/jbc.M101914200
Tachibana, M., Ueda, J., Fukuda, M., Takeda, N., Ohta, T., Iwanari, H., Sakihama, T., Kodama, T., Hamakubo, T., & Shinkai, Y. (2005). Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3–K9. Genes & Development, 19(7), 815–826. https://doi.org/10.1101/gad.1284005
Tang, J., Cho, N. W., Cui, G., Manion, E. M., Shanbhag, N. M., Botuyan, M. V., Mer, G., & Greenberg, R. A. (2013). Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nature Structural & Molecular Biology, 20(3), 317–325. https://doi.org/10.1038/nsmb.2499
Tang, M., Li, Z., Zhang, C., Lu, X., Tu, B., Cao, Z., Li, Y., Chen, Y., Jiang, L., Wang, H., Wang, L., Wang, J., Liu, B., Xu, X., Wang, H., & Zhu, W. G. (2019). SIRT7-mediated ATM deacetylation is essential for its deactivation and DNA damage repair. Science Advances, 5(3), eeav1118. https://doi.org/10.1126/sciadv.aav1118
Tchasovnikarova, I. A., Timms, R. T., Douse, C. H., Roberts, R. C., Dougan, G., Kingston, R. E., Modis, Y., & Lehner, P. J. (2017). Hyperactivation of HUSH complex function by Charcot-Marie-Tooth disease mutation in MORC2. Nature Genetics, 49(7), 1035–1044. https://doi.org/10.1038/ng.3878
Tchasovnikarova, I. A., Timms, R. T., Matheson, N. J., Wals, K., Antrobus, R., Gottgens, B., Dougan, G., Dawson, M. A., & Lehner, P. J. (2015). GENE SILENCING. Epigenetic silencing by the HUSH complex mediates position-effect variegation in human cells. Science, 348(6242), 1481–1485. https://doi.org/10.1126/science.aaa7227
Techer, H., Koundrioukoff, S., Nicolas, A., & Debatisse, M. (2017). The impact of replication stress on replication dynamics and DNA damage in vertebrate cells. Nature Reviews Genetics, 18(9), 535–550. https://doi.org/10.1038/nrg.2017.46
Tencer, A. H., Cox, K. L., Di, L., Bridgers, J. B., Lyu, J., Wang, X., Sims, J. K., Weaver, T. M., Allen, H. F., Zhang, Y., Gatchalian, J., Darcy, M. A., Gibson, M. D., Ikebe, J., Li, W., Wade, P. A., Hayes, J. J., Strahl, B. D., Kono, H., et al. (2017). Covalent modifications of histone H3K9 promote binding of CHD3. Cell Reports, 21(2), 455–466. https://doi.org/10.1016/j.celrep.2017.09.054
Torrano, J., Al Emran, A., Hammerlindl, H., & Schaider, H. (2019). Emerging roles of H3K9me3, SETDB1 and SETDB2 in therapy-induced cellular reprogramming. Clinical Epigenetics, 11(1), 43. https://doi.org/10.1186/s13148-019-0644-y
Tsouroula, K., Furst, A., Rogier, M., Heyer, V., Maglott-Roth, A., Ferrand, A., Reina-San-Martin, B., & Soutoglou, E. (2016). Temporal and spatial uncoupling of DNA double strand break repair pathways within mammalian heterochromatin. Molecular Cell, 63(2), 293–305. https://doi.org/10.1016/j.molcel.2016.06.002
Tsukada, Y., Fang, J., Erdjument-Bromage, H., Warren, M. E., Borchers, C. H., Tempst, P., & Zhang, Y. (2006). Histone demethylation by a family of JmjC domain-containing proteins. Nature, 439(7078), 811–816. https://doi.org/10.1038/nature04433
Tsukada, Y. I., Ishitani, T., & Nakayama, K. I. (2010). KDM7 is a dual demethylase for histone H3 Lys 9 and Lys 27 and functions in brain development. Genes & Development, 24(5), 432–437. https://doi.org/10.1101/gad.1864410
Tu, B., Bao, Y., Tang, M., Zhu, Q., Lu, X., Wang, H., Hou, T., Zhao, Y., Zhang, P., & Zhu, W.-G. (2020). TIP60 recruits SUV39H1 to chromatin to maintain heterochromatin genome stability and resist hydrogen peroxide-induced cytotoxicity. Genome Instability & Disease, 1, 339–355.
Tubbs, A., & Nussenzweig, A. (2017). Endogenous DNA damage as a source of genomic instability in cancer. Cell, 168(4), 644–656. https://doi.org/10.1016/j.cell.2017.01.002
Tunbak, H., Enriquez-Gasca, R., Tie, C. H. C., Gould, P. A., Mlcochova, P., Gupta, R. K., Fernandes, L., Holt, J., van der Veen, A. G., Giampazolias, E., Burns, K. H., Maillard, P. V., & Rowe, H. M. (2020). The HUSH complex is a gatekeeper of type I interferon through epigenetic regulation of LINE-1s. Nature Communications, 11(1), 5387. https://doi.org/10.1038/s41467-020-19170-5
Verdel, A., Jia, S., Gerber, S., Sugiyama, T., Gygi, S., Grewal, S. I., & Moazed, D. (2004). RNAi-mediated targeting of heterochromatin by the RITS complex. Science, 303(5658), 672–676. https://doi.org/10.1126/science.1093686
Wang, C., Liu, X., Gao, Y., Yang, L., Li, C., Liu, W., Chen, C., Kou, X., Zhao, Y., Chen, J., Wang, Y., Le, R., Wang, H., Duan, T., Zhang, Y., & Gao, S. (2018). Reprogramming of H3K9me3-dependent heterochromatin during mammalian embryo development. Nature Cell Biology, 20(5), 620–631. https://doi.org/10.1038/s41556-018-0093-4
Wang, D., Zhou, J., Liu, X., Lu, D., Shen, C., Du, Y., Wei, F. Z., Song, B., Lu, X., Yu, Y., Wang, L., Zhao, Y., Wang, H., Yang, Y., Akiyama, Y., Zhang, H., & Zhu, W. G. (2013). Methylation of SUV39H1 by SET7/9 results in heterochromatin relaxation and genome instability. Proceedings of the National Academy of Science USA, 110(14), 5516–5521. https://doi.org/10.1073/pnas.1216596110
Wang, S., Meyer, D. H., & Schumacher, B. (2023). Inheritance of paternal DNA damage by histone-mediated repair restriction. Nature, 613(7943), 365–374. https://doi.org/10.1038/s41586-022-05544-w
Wang, T., Xu, C., Liu, Y., Fan, K., Li, Z., Sun, X., Ouyang, H., Zhang, X., Zhang, J., Li, Y., Mackenzie, F., Min, J., & Tu, X. (2012). Crystal structure of the human SUV39H1 chromodomain and its recognition of histone H3K9me2/3. PLoS ONE, 7(12), e52977. https://doi.org/10.1371/journal.pone.0052977
Watson, G. E., Lorimore, S. A., Macdonald, D. A., & Wright, E. G. (2000). Chromosomal instability in unirradiated cells induced in vivo by a bystander effect of ionizing radiation. Cancer Research, 60(20), 5608–5611.
Whetstine, J. R., Nottke, A., Lan, F., Huarte, M., Smolikov, S., Chen, Z., Spooner, E., Li, E., Zhang, G., Colaiacovo, M., & Shi, Y. (2006). Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell, 125(3), 467–481. https://doi.org/10.1016/j.cell.2006.03.028
Wilson, D. M., 3rd., Cookson, M. R., Van Den Bosch, L., Zetterberg, H., Holtzman, D. M., & Dewachter, I. (2023). Hallmarks of neurodegenerative diseases. Cell, 186(4), 693–714. https://doi.org/10.1016/j.cell.2022.12.032
Wilson, M. D., Benlekbir, S., Fradet-Turcotte, A., Sherker, A., Julien, J. P., McEwan, A., Noordermeer, S. M., Sicheri, F., Rubinstein, J. L., & Durocher, D. (2016). The structural basis of modified nucleosome recognition by 53BP1. Nature, 536(7614), 100–103. https://doi.org/10.1038/nature18951
Wolf, D., & Goff, S. P. (2007). TRIM28 mediates primer binding site-targeted silencing of murine leukemia virus in embryonic cells. Cell, 131(1), 46–57. https://doi.org/10.1016/j.cell.2007.07.026
Wolf, D., & Goff, S. P. (2009). Embryonic stem cells use ZFP809 to silence retroviral DNAs. Nature, 458(7242), 1201–1204. https://doi.org/10.1038/nature07844
Wolf, G., Yang, P., Fuchtbauer, A. C., Fuchtbauer, E. M., Silva, A. M., Park, C., Wu, W., Nielsen, A. L., Pedersen, F. S., & Macfarlan, T. S. (2015). The KRAB zinc finger protein ZFP809 is required to initiate epigenetic silencing of endogenous retroviruses. Genes & Development, 29(5), 538–554. https://doi.org/10.1101/gad.252767.114
Wu, G., Broniscer, A., McEachron, T. A., Lu, C., Paugh, B. S., Becksfort, J., Qu, C., Ding, L., Huether, R., Parker, M., Zhang, J., Gajjar, A., Dyer, M. A., Mullighan, C. G., Gilbertson, R. J., Mardis, E. R., Wilson, R. K., Downing, J. R., Ellison, D. W., et al. (2012). Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nature Genetics, 44(3), 251–253. https://doi.org/10.1038/ng.1102
Wu, J., Liu, Y., Zhangding, Z., Liu, X., Ai, C., Gan, T., Liang, H., Guo, Y., Chen, M., Liu, Y., Yin, J., Zhang, W., & Hu, J. (2023). Cohesin maintains replication timing to suppress DNA damage on cancer genes. Nature Genetics, 55(8), 1347–1358. https://doi.org/10.1038/s41588-023-01458-z
Wu, R., Wang, Z., Zhang, H., Gan, H., & Zhang, Z. (2017). H3K9me3 demethylase Kdm4d facilitates the formation of pre-initiative complex and regulates DNA replication. Nucleic Acids Research, 45(1), 169–180. https://doi.org/10.1093/nar/gkw848
Wu, W., Nishikawa, H., Fukuda, T., Vittal, V., Asano, M., Miyoshi, Y., Klevit, R. E., & Ohta, T. (2015). Interaction of BARD1 and HP1 is required for BRCA1 retention at sites of DNA damage. Cancer Research, 75(7), 1311–1321. https://doi.org/10.1158/0008-5472.CAN-14-2796
Xia, R., Zhou, R., Tian, Z., Zhang, C., Wang, L., Hu, Y., Han, J., & Li, J. (2013). High expression of H3K9me3 is a strong predictor of poor survival in patients with salivary adenoid cystic carcinoma. Archives of Pathology and Laboratory Medicine, 137(12), 1761–1769. https://doi.org/10.5858/arpa.2012-0704-OA
Xu, R., Li, S., Wu, Q., Li, C., Jiang, M., Guo, L., Chen, M., Yang, L., Dong, X., Wang, H., Wang, C., Liu, X., Ou, X., & Gao, S. (2022a). Stage-specific H3K9me3 occupancy ensures retrotransposon silencing in human pre-implantation embryos. Cell Stem Cell, 29(7), 1051–1066. https://doi.org/10.1016/j.stem.2022.06.001
Xu, X., Duan, S., Hua, X., Li, Z., He, R., & Zhang, Z. (2022b). Stable inheritance of H3.3-containing nucleosomes during mitotic cell divisions. Nature Communication, 13(1), 2514. https://doi.org/10.1038/s41467-022-30298-4
Yamane, K., Toumazou, C., Tsukada, Y., Erdjument-Bromage, H., Tempst, P., Wong, J., & Zhang, Y. (2006). JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell, 125(3), 483–495. https://doi.org/10.1016/j.cell.2006.03.027
Yang, B. X., El Farran, C. A., Guo, H. C., Yu, T., Fang, H. T., Wang, H. F., Schlesinger, S., Seah, Y. F., Goh, G. Y., Neo, S. P., Li, Y., Lorincz, M. C., Tergaonkar, V., Lim, T. M., Chen, L., Gunaratne, J., Collins, J. J., Goff, S. P., Daley, G. Q., et al. (2015). Systematic identification of factors for provirus silencing in embryonic stem cells. Cell, 163(1), 230–245. https://doi.org/10.1016/j.cell.2015.08.037
Yang, J. H., Hayano, M., Griffin, P. T., Amorim, J. A., Bonkowski, M. S., Apostolides, J. K., Salfati, E. L., Blanchette, M., Munding, E. M., Bhakta, M., Chew, Y. C., Guo, W., Yang, X., Maybury-Lewis, S., Tian, X., Ross, J. M., Coppotelli, G., Meer, M. V., Rogers-Hammond, R., et al. (2023). Loss of epigenetic information as a cause of mammalian aging. Cell, 186(2), 305–326. https://doi.org/10.1016/j.cell.2022.12.027
Yang, P., Wang, Y., & Macfarlan, T. S. (2017a). The role of KRAB-ZFPs in transposable element repression and mammalian evolution. Trends in Genetics, 33(11), 871–881. https://doi.org/10.1016/j.tig.2017.08.006
Yang, Q., Zhu, Q., Lu, X., Du, Y., Cao, L., Shen, C., Hou, T., Li, M., Li, Z., Liu, C., Wu, D., Xu, X., Wang, L., Wang, H., Zhao, Y., Yang, Y., & Zhu, W. G. (2017b). G9a coordinates with the RPA complex to promote DNA damage repair and cell survival. Proceedings of the National Academy Science USA, 114(30), 6054–6063. https://doi.org/10.1073/pnas.1700694114
Yasuhara, T., & Zou, L. (2021). Impacts of chromatin dynamics and compartmentalization on DNA repair. DNA Repair (amst), 105, 103162. https://doi.org/10.1016/j.dnarep.2021.103162
Yeager, M., Machiela, M. J., Kothiyal, P., Dean, M., Bodelon, C., Suman, S., Wang, M., Mirabello, L., Nelson, C. W., Zhou, W., Palmer, C., Ballew, B., Colli, L. M., Freedman, N. D., Dagnall, C., Hutchinson, A., Vij, V., Maruvka, Y., Hatch, M., et al. (2021). Lack of transgenerational effects of ionizing radiation exposure from the Chernobyl accident. Science, 372(6543), 725–729. https://doi.org/10.1126/science.abg2365
Yokoyama, Y., Hieda, M., Nishioka, Y., Matsumoto, A., Higashi, S., Kimura, H., Yamamoto, H., Mori, M., Matsuura, S., & Matsuura, N. (2013). Cancer-associated upregulation of histone H3 lysine 9 trimethylation promotes cell motility in vitro and drives tumor formation in vivo. Cancer Science, 104(7), 889–895. https://doi.org/10.1111/cas.12166
Yoo, C. B., & Jones, P. A. (2006). Epigenetic therapy of cancer: Past, present and future. Nature Reviews Drug Discovery, 5(1), 37–50. https://doi.org/10.1038/nrd1930
Young, L. C., McDonald, D. W., & Hendzel, M. J. (2013). Kdm4b histone demethylase is a DNA damage response protein and confers a survival advantage following gamma-irradiation. Journal of Biological Chemistry, 288(29), 21376–21388. https://doi.org/10.1074/jbc.M113.491514
Yu, C., Gan, H., Serra-Cardona, A., Zhang, L., Gan, S., Sharma, S., Johansson, E., Chabes, A., Xu, R. M., & Zhang, Z. (2018a). A mechanism for preventing asymmetric histone segregation onto replicating DNA strands. Science, 361(6409), 1386–1389. https://doi.org/10.1126/science.aat8849
Yu, H., Chen, M., Hu, Y., Ou, S., Yu, X., Liang, S., Li, N., Yang, M., Kong, X., Sun, C., Jia, S., Zhang, Q., Liu, L., Hurst, L. D., Li, R., Wang, W., & Wang, J. (2022). Dynamic reprogramming of H3K9me3 at hominoid-specific retrotransposons during human preimplantation development. Cell Stem Cell, 29(7), 1031–1050. https://doi.org/10.1016/j.stem.2022.06.006
Yu, Y., Schleich, K., Yue, B., Ji, S., Lohneis, P., Kemper, K., Silvis, M. R., Qutob, N., van Rooijen, E., Werner-Klein, M., Li, L., Dhawan, D., Meierjohann, S., Reimann, M., Elkahloun, A., Treitschke, S., Dorken, B., Speck, C., Mallette, F. A., et al. (2018b). Targeting the senescence-overriding cooperative activity of structurally unrelated H3K9 demethylases in melanoma. Cancer Cell, 33(2), 322–336. https://doi.org/10.1016/j.ccell.2018.01.002
Zhang, K., Mosch, K., Fischle, W., & Grewal, S. I. (2008). Roles of the Clr4 methyltransferase complex in nucleation, spreading and maintenance of heterochromatin. Nature Structural & Molecular Biology, 15(4), 381–388. https://doi.org/10.1038/nsmb.1406
Zhang, L., Geng, X., Wang, F., Tang, J., Ichida, Y., Sharma, A., Jin, S., Chen, M., Tang, M., Pozo, F. M., Wang, W., Wang, J., Wozniak, M., Guo, X., Miyagi, M., Jin, F., Xu, Y., Yao, X., & Zhang, Y. (2022). 53BP1 regulates heterochromatin through liquid phase separation. Nature Communications, 13(1), 360. https://doi.org/10.1038/s41467-022-28019-y
Zhang, S. M., Cai, W. L., Liu, X., Thakral, D., Luo, J., Chan, L. H., McGeary, M. K., Song, E., Blenman, K. R. M., Micevic, G., Jessel, S., Zhang, Y., Yin, M., Booth, C. J., Jilaveanu, L. B., Damsky, W., Sznol, M., Kluger, H. M., Iwasaki, A., et al. (2021a). KDM5B promotes immune evasion by recruiting SETDB1 to silence retroelements. Nature, 598(7882), 682–687. https://doi.org/10.1038/s41586-021-03994-2
Zhang, T., Cooper, S., & Brockdorff, N. (2015a). The interplay of histone modifications - writers that read. EMBO Reports, 16(11), 1467–1481. https://doi.org/10.15252/embr.201540945
Zhang, W., Li, J., Suzuki, K., Qu, J., Wang, P., Zhou, J., Liu, X., Ren, R., Xu, X., Ocampo, A., Yuan, T., Yang, J., Li, Y., Shi, L., Guan, D., Pan, H., Duan, S., Ding, Z., Li, M., et al. (2015b). Aging stem cells. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science, 348(6239), 1160–1163. https://doi.org/10.1126/science.aaa1356
Zhang, W., Liu, W., Jia, L., Chen, D., Chang, I., Lake, M., Bentolila, L. A., & Wang, C. Y. (2021b). Targeting KDM4A epigenetically activates tumor-cell-intrinsic immunity by inducing DNA replication stress. Molecular Cell, 81(10), 2148–2165. https://doi.org/10.1016/j.molcel.2021.02.038
Zhao, Q. Y., Lei, P. J., Zhang, X., Zheng, J. Y., Wang, H. Y., Zhao, J., Li, Y. M., Ye, M., Li, L., Wei, G., & Wu, M. (2016). Global histone modification profiling reveals the epigenomic dynamics during malignant transformation in a four-stage breast cancer model. Clinical Epigenetics, 8, 34. https://doi.org/10.1186/s13148-016-0201-x
Zhao, S., Allis, C. D., & Wang, G. G. (2021). The language of chromatin modification in human cancers. Nature Reviews Cancer, 21(7), 413–430. https://doi.org/10.1038/s41568-021-00357-x
Zhao, S., Lu, J., Pan, B., Fan, H., Byrum, S. D., Xu, C., Kim, A., Guo, Y., Kanchi, K. L., Gong, W., Sun, T., Storey, A. J., Burkholder, N. T., Mackintosh, S. G., Kuhlers, P. C., Edmondson, R. D., Strahl, B. D., Diao, Y., Tackett, A. J., et al. (2023a). TNRC18 engages H3K9me3 to mediate silencing of endogenous retrotransposons. Nature, 623(7987), 633–642. https://doi.org/10.1038/s41586-023-06688-z
Zhao, Y., Simon, M., Seluanov, A., & Gorbunova, V. (2023b). DNA damage and repair in age-related inflammation. Nature Reviews Immunology, 23(2), 75–89. https://doi.org/10.1038/s41577-022-00751-y
Zheng, C. L., Wang, N. J., Chung, J., Moslehi, H., Sanborn, J. Z., Hur, J. S., Collisson, E. A., Vemula, S. S., Naujokas, A., Chiotti, K. E., Cheng, J. B., Fassihi, H., Blumberg, A. J., Bailey, C. V., Fudem, G. M., Mihm, F. G., Cunningham, B. B., Neuhaus, I. M., Liao, W., et al. (2014). Transcription restores DNA repair to heterochromatin, determining regional mutation rates in cancer genomes. Cell Reports, 9(4), 1228–1234. https://doi.org/10.1016/j.celrep.2014.10.031
Zhou, M., Yan, J. Q., Chen, Q. X., Yang, Y. Z., Li, Y. L., Ren, Y. X., Weng, Z. J., Zhang, X. F., Guan, J. X., Tang, L. Y., & Ren, Z. F. (2022). Association of H3K9me3 with breast cancer prognosis by estrogen receptor status. Clinical Epigenetics, 14(1), 135. https://doi.org/10.1186/s13148-022-01363-y
Zhu, W. G., & Otterson, G. A. (2003). The interaction of histone deacetylase inhibitors and DNA methyltransferase inhibitors in the treatment of human cancer cells. Current Medicinal Chemistry: Anti-Cancer Agents, 3(3), 187–199. https://doi.org/10.2174/1568011033482440
Acknowledgements
The authors apologize for not being able to include all relevant literature due to space limitations. Z. L. and Z. Z. wrote the manuscript. This work is supported by NIH grants R35GM118015 (to Z. Z.).
Author information
Authors and Affiliations
Institute for Cancer Genetics, Columbia University Irving Medical Center, New York, NY, 10032, USA
Zhiming Li & Zhiguo Zhang
Herbert Irving Comprehensive Cancer Center, Columbia University Irving Medical Center, New York, NY, 10032, USA
Zhiming Li & Zhiguo Zhang
Department of Pediatrics, Columbia University Irving Medical Center, New York, NY, 10032, USA
Zhiming Li & Zhiguo Zhang
Department of Genetics and Development, Columbia University Irving Medical Center, New York, NY, 10032, USA
Zhiming Li & Zhiguo Zhang
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article
Cite this article
Li, Z., Zhang, Z. The silent guardian: unraveling the roles of H3K9me3 in genome maintenance. GENOME INSTAB. DIS. 5, 133–153 (2024). https://doi.org/10.1007/s42764-024-00131-x
Received
Revised
Accepted
Published
Issue Date
DOIhttps://doi.org/10.1007/s42764-024-00131-x
Share this article
Anyone you share the following link with will be able to read this content:
用户登录
还没有账号?
立即注册